

Abstract
Neuronal pentraxin receptor (NPR) is a synaptic protein implicated in AMPA receptor trafficking at excitatory synapses. Since glutamate neurotransmission is disrupted in Alzheimer’s disease (AD), NPR levels measured from plasma represent a potential biomarker for synaptic dysfunction associated with AD. We sought to determine the relationship between AD pathology and brain and plasma NPR levels by examining age-associated NPR levels in these compartments in a transgenic APP/PS1 rat model of AD. NPR levels in cortical homogenate were similar in wild-type (Wt) and APP/PS1 rats at 3 months of age (prior to Aβ plaque deposition), but significantly increased in APP/PS1 rats by 9 and 18-20 months of age (after the onset of plaque deposition). These age-dependent differences were driven by proportional increases in NPR in membrane-associated cortical fractions. Genotype-related differences in NPR expression were also seen in the hippocampus, which exhibits significant Aβ pathology, but not in the cerebellum, which does not. Plasma analyses revealed increased levels of a 26 kDa NPR fragment in APP/PS1 rats relative to Wt rats by 18-20 months of age, which correlated with the levels of full-length NPR in cortex. Our findings indicate that cerebral accumulation of NPR and Aβ occurs with similar temporal and regional patterns in the APP/PS1 model, and suggest that a 26 kDa plasma NPR fragment may represent a peripheral biomarker of this process.
Keywords: plasma, cortex, hippocampus, biomarker, neuronal pentraxin, transgenic, Alzheimer’s disease, synaptic function
INTRODUCTION
Neuronal pentraxin receptor (NPR) is a membrane-associated protein that is almost exclusively produced in the brain (Dodds et al., 1997). NPR is primarily found in excitatory synapses (Cho et al., 2008), and exhibits particularly high levels of expression in the cerebral cortex, hippocampus, and cerebellum (Dodds et al., 1997). It has been implicated in the regulation of glutamate neurotransmission in excitatory neurons. For example, in synapses, NPR binds to other members of the neuronal pentraxin protein family, including neuronal pentraxin 1 (NP1) and neuronal pentraxin 2 (NP2) (Kirkpatrick et al., 2000), in order to recruit and cluster AMPA-type glutamate receptors (Xu et al., 2003; Sia et al., 2007;Koch and Ullian, 2010). Furthermore, a key mechanistic step in the down-regulation of AMPA receptor neurotransmission required for long-term depression (LTD) is the cleavage of NPR by tumor necrosis factor-α converting enzyme (TACE) (Cho et al., 2008). NPR may also mediate the uptake and clearance of synaptic debris in the process of synaptic remodeling (Bjartmar et al., 2006; Dodds et al., 1997; Kirkpatrick et al., 2000).
The prominent role of NPR in synaptic plasticity raises the possibility that its expression may be altered in neurodegenerative conditions, such as Alzheimer’s disease (AD), that are characterized by synaptic dysfunction and loss (Selkoe, 2002). In particular, given the widespread disruption of glutamate neurotransmission seen with AD neuropathology (Revett et al., 2013), NPR levels in cerebrospinal fluid (CSF) or peripheral blood may represent accessible markers of underlying dysfunction at excitatory synapses.
However, the relationship between AD pathology and CSF NPR levels remains uncertain. Proteomic studies of CSF indicate that NPR levels are altered in sporadic late-onset AD patients relative to controls with normal cognition or other neurological conditions, with one group reporting increased levels in AD (Yin et al., 2009) and others reporting decreased levels in AD (Abdi et al., 2006; Finehout et al., 2007; Hu et al., 2007). These apparently conflicting data reflect the possibility that CSF NPR levels may be modulated by underlying disease severity. One recent study that compared CSF samples from small groups of individuals with normal cognition or mild cognitive impairment (MCI), which often represents incipient AD, found higher NPR levels in MCI, though this result did not reach statistical significance (Wildsmith et al., 2014). However, in the same study, the investigators also examined longitudinal CSF NPR levels in a larger set of individuals diagnosed with AD. Not only were baseline CSF NPR levels in the AD group numerically lower than in the MCI group, they progressively declined over time, suggesting a potential negative correlation with disease progression at more advanced stages (Wildsmith et al., 2014). Similarly, amongst carriers of mutations in the presenilin 1 (PSEN1) or amyloid precursor protein (APP) genes, which are associated with autosomal dominant familial AD, elevated CSF NPR levels are seen in pre-symptomatic, but not in cognitively impaired or demented individuals (Ringman et al., 2012). Taken together, these findings raise the possibility that CSF NPR may have utility as an AD biomarker, with higher levels seen at earlier disease stages and lower levels seen at later disease stages.
Serum NPR levels, which are more accessible than CSF NPR levels, have also been shown to be elevated in MCI and AD patients relative to cognitively normal elderly controls (Yin et al., 2009). Unlike the plethora of other putative plasma and serum biomarkers of AD have been identified through plasma proteomics (Doecke et al., 2012; Hu et al., 2012; O’Bryant et al., 2011; Ray et al., 2007), NPR expression is predominantly restricted to the brain, potentially increasing its specificity for examining underlying neurodegenerative disease. There are relatively few reports regarding proteins of predominantly neuronal origin that are consistently detected in plasma or serum. One notable exception is microtubule-associated protein tau, a component of the axonal cytoskeleton. Although plasma/serum tau levels are elevated in AD (Fiandaca et al., 2014; Zetterberg et al., 2013), they are also elevated after traumatic (Neselius et al., 2013) or hypoxic (Randall et al., 2013) brain injury. Although synaptic proteins, such as SNAP-25, have also been identified in serum (Marshall et al., 2014), their levels have yet to be rigorously investigated in specific disease states. Other synaptic proteins that are specifically expressed in the central nervous system, such as NPR, may have utility as potential biomarkers indicative of synaptic dysfunction that are measurable in plasma and/or serum.
Despite the intriguing results suggesting that NPR levels in CSF or blood could be a useful marker of underlying AD progression, little is known about the effects of AD neuropathology on NPR expression and compartmentalization in the brain. Earlier work demonstrated increased NP1 levels in brain tissue from both human AD patients and a transgenic APP/PS1 mouse model (Abad et al., 2006), suggesting that β-amyloid (Aβ) pathology can modulate the expression of other neuronal pentraxins. Therefore, we examined NPR expression in both brain and plasma in a transgenic APP/PS1 rat model of AD. This model exhibits regionally specific age-associated accumulation of Aβ (Flood et al., 2009; Liu et al., 2008; Teng et al., 2011), which allowed us to determine the relationship between Aβ and NPR. Furthermore, since the vast majority of NPR synthesis occurs in the brain, we sought to determine whether NPR levels measured from plasma correlate with those measured in brain.
METHODS
Experimental Animals
Plasma and brain samples were obtained from transgenic and wild-type Sprague-Dawley rats. The transgenic (Tg) rat model is homozygous for three gene constructs: 1) human APP 695 with the K670N/M671L mutation (rat synapsin-1 promoter); 2) human APP minigene with the K670N/M671L and V717F mutations (platelet derived growth factor β promoter); and 3) human PS-1 with the M146V mutation (rat synapsin-1 promoter) that are expressed on a Sprague-Dawley background (Flood et al., 2009). The time-course of Aβ accumulation in this model has previously been described in detail (Flood et al., 2009; Liu et al., 2008; Teng et al., 2011). Sparse parenchymal Aβ plaques begin to appear between 7 to 9 months of age, with progressive age-associated increases in plaque density seen in cortical and hippocampal regions, but not the cerebellum. Breeding pairs obtained from Cephalon Inc. (West Chester, PA) were bred and aged at the UCLA School of Medicine vivarium facility. Wild-type (Wt) Sprague-Dawley rats were obtained from outside vendors (Harlan Laboratories, Indianapolis, IN; Charles River, Wilmington, MA). All animals were housed under a 12-hour light/dark cycle and had access to standard rat chow ad libitum. The UCLA Chancellor’s Animal Research Committee approved this study and all animal experiments were conducted in compliance with its guidelines.
Behavioral testing
A subset of Tg and Wt rats were tested on the Morris water maze (MWM) (Morris et al., 1982) at 18-19 months of age. Animals were placed in a circular water tank 6 feet in diameter, and were trained for 4 trials per day over 10 days to learn to locate and swim to a hidden escape platform submerged below the surface of the water using distal cues in the periphery of the room. The outcome measure was latency to finding the hidden platform, with a maximum of 90 seconds permitted for each trial. Trials where the animal failed to find the platform were assigned a latency of 90 seconds.
Rat brain and plasma processing
One set of Tg and Wt animals was euthanized via carbon dioxide overdose at 3, 9, or 20 months of age. These rats were immediately decapitated, and their brains were removed and dissected into several anatomical regions, including the hippocampus, association cortex (parietal, somatosensory, motor, and visual cortex, denoted as “mixed cortex” below), and cerebellum. The dissected tissue was minced, slowly frozen in 0.32 M sucrose and stored at −80°C. This cryopreserved tissue subsequently underwent gentle mechanical homogenization in ice-cold buffer [0.32 M sucrose, 10 mM TRIS pH 7.5, plus protease inhibitors: pepstatin (4 μg/ml), aprotinin (5 μg/ml), trypsin inhibitor (20 μg/ml), EDTA (2 mM), EGTA (2 mM), PMSF (0.2 mM), leupeptin (4 μg/ml)] as previously described (Arold et al., 2012).
Separate sets of Tg and Wt animals were terminally sedated at 3, 9, and 18-20 months of age with pentobarbital. The chest cavity was opened and 3 mL of blood was collected from the right ventricle into K2EDTA coated tubes. Blood samples were centrifuged at room temperature at 2000 g for 15 minutes. The plasma component was aliquoted into polypropylene tubes, snap frozen in liquid nitrogen, and stored at −80° C. Aliquots of a subset of these samples were depleted of highly abundant plasma proteins prior to subsequent analyses using Seppro IgY-R7 spin columns (Sigma-Aldrich; St. Louis, MO), which incorporate avian polyclonal IgY antibodies to remove rat albumin, α1-antitrypsin, transferrin, fibrinogen, IgG, IgM, and haptoglobin.
Animals were then perfused with a HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer containing protease [leupeptin (1 μg/ml), aprotinin (1 μg/ml), pepstatin (1 μg/ml), and PMSF (50 μg/ml)] and phosphatase [sodium vanadate (1 mM), sodium pyrophosphate (1 mM), and sodium fluoride (50 mM)] inhibitors as previously described (Calon et al., 2004). Brains were removed immediately, dissected as described above, flash frozen in liquid nitrogen, and stored at −80° C. This tissue was subsequently serially extracted into tris-buffered saline (TBS) and lysis fractions (pH for both buffers=8.0) as previously described (Calon et al., 2004; Yang et al., 2005).
Western Blotting
Western blots were performed on brain homogenate, brain extract, and plasma samples. Protein assays were performed using BCA protein assay kits (Thermo Scientific Pierce; Rockford IL) per the manufacturer’s protocol. Equal amounts of protein (20 μg) from each sample were mixed with 1x Novex Tris-glycine SDS sample buffer (Invitrogen; Carlsbad CA) and 100mM dithiothreitol (DTT; Expedeon, Harston UK), then boiled, resolved by SDS-PAGE on a 10-20% Tris-glycine gradient gel (Invitrogen; Carlsbad CA) and transferred to a PVDF membrane (Millipore; Billerica MA). To insure equal protein loading and transfer quality, membranes were stained with Ponceau S; only membranes with equal loading were quantified. After blocking with 3% BSA/PBS (1 hr at RT), membranes were probed with polyclonal sheep anti-NPR antibody raised against recombinant human NPR [amino acids (aa’s) 24-499, which have 87% homology between human and rodent sequences; AF4414, R&D Systems; Minneapolis, MN], polyclonal rabbit anti-TACE antibody (QED Bioscience; San Diego, CA) or polyclonal rabbit anti-phospho-TACE antibody (Thr735, Assay Biotechnology; Sunnyvale, CA) in 0.1% Tween 20/PBS with 0.05% sodium azide (ON, 4°C). A subset of rat plasma samples was also probed with a polyclonal rabbit anti-NPR antibody targeting the C-terminus (AB15522, Millipore; Billerica MA) or the mid-portion (ab55903*, Abcam; Cambridge UK) of the protein in the same fashion. Membranes were then washed 3 times in 0.1% Tween 20/PBS and incubated with HRP-conjugated anti-sheep IgG (R&D Systems; Minneapolis, MN) or anti-rabbit IgG (Jackson ImmunoResearch Laboratories; West Grove, PA) in 0.1% Tween 20/PBS (1 hr at RT) and washed again. Chemiluminescent signals were detected with Super Signal West Femto substrate (Thermo Scientific Pierce; Rockford, IL) using a BioSpectrum 600 imaging system (UVP; Upland, CA) or on x-ray films (Switzer Medical; Fresno, CA) using a Kodak X-OMAT 2000A processor and quantified using VisionWorks software (UVP; Upland, CA).
Aβ ELISAs
Total Aβ levels were measured by a sandwich ELISA as previously described (Lim et al., 2000). The anti-Aβ antibodies used in this assay are 4G8 (recognizes aa’s 17-24 and reacts to both human and rat Aβ; Covance; Princeton, NJ), for capture and biotinylated 10G4 (recognizes aa’s 5-13 and is presumed to be human specific; Yang et al., 1994) for detection. ELISAs using mixed cortex extracts were loaded with 20 μg of total protein per well. ELISAs using cerebellar extracts were loaded with up to 80 μg of total protein per well.
Data Analyses
Statistical analyses were performed using SPSS 22 for Macintosh (IBM, Armonk NY). Comparisons of NPR, Aβ, and TACE expression between Wt and Tg animals were performed using unpaired t-tests. Comparisons of NPR expression between TBS and lysis fractions were performed using paired t-tests. Correlations between plasma and brain NPR expression were calculated using Pearson’s correlation coefficient.
RESULTS
NPR in rat mixed cortex homogenate
Western blotting was performed using AF4414 antibody on mixed cortex homogenate from Wt and Tg rats at 3, 9, and 20 months of age. In Tg rats, Aβ plaques are absent at 3 months of age, are sparsely present by 9 months of age, and exhibit extensive deposition by 20 months of age (Flood et al., 2009; Liu et al., 2008; Teng et al., 2011). The two full-length NPR species that have previously been identified in brain homogenate are 55 kDa (non-glycosylated) and 65 kDa (glycosylated) in size (Dodds et al., 1997). We quantified the relative abundance of these species in Wt and Tg rats in each age group (Figure 1). At 3 months of age, similar levels of both the 55 kDa [t(12)=0.22, p=0.83] and 65 kDa [t(12)=−1.61, p=0.13] NPR species were seen across genotypes. However, higher levels of both NPR species were seen in Tg rats relative to Wt rats at both 9 months [55 kDa: t(10)=−3.02, p=0.013; 65 kDa: t(10)=−3.40, p=0.007] and 20 months [55 kDa: t(10)=−2.97, p=0.013; 65 kDa: t(10)=−2.75, p=0.002] of age. These data indicate that increases in cortical NPR levels in Tg relative to Wt animals emerge in an age-dependent fashion that parallels the emergence of Aβ plaque pathology.
Figure 1.

Representative Western blots and quantification of NPR levels in mixed cortex homogenate from Wt and Tg rats at 3 months, 9 months, and 20 months of age. Dotted line represents normalized optical density of bands from Wt animals at each age (3 months: n=5; 9 and 20 months: n=6). The numbers of Tg animals at each age are shown in parentheses. Error bars represent standard error of the mean (SEM). *p<0.05 vs. Wt.
NPR in soluble and membrane-associated brain fractions
Prior work has suggested that cleavage of NPR by tumor necrosis factor-α converting enzyme (TACE) shifts NPR from a membrane-associated compartment to a soluble compartment to modulate synaptic plasticity (Cho et al., 2008). Since Tg rats demonstrate deficits in synaptic plasticity relative to Wt rats (Liu et al., 2008), we subsequently sought to determine whether NPR compartmentalization also shifted with age. At 3 and 9 months of age, examination of the mixed cortex TBS fraction revealed greater expression of both the 55 kDa [3 months: t(12)=−3.17, p=0.008; 9 months: t(12)=−2.55, p=0.025] and 65 kDa [3 months: t(12)=−2.53, p=0.026; 9 months: t(12)=−3.20, p=0.008] NPR species in Tg relative to Wt rats (Figure 2a). However, in the mixed cortex lysis fraction, similar levels of the 55 kDa [3 months: t(12)=−0.89, p=0.39; 9 months: t(12)=−1.45, p=0.17] and 65 kDa [3 months: t(12)=−1.71, p=0.11; 9 months: t(12)=−0.91, p=0.38] NPR species were seen at both time-points (Figure 2b). In contrast, at 18-20 months of age, the expression of the 55 kDa species in Tg relative to Wt rats was significantly lower in the TBS fraction [Figure 2a; t(12)=2.28, p=0.042], but significantly higher in the lysis fraction [Figure 2b; t(12)=−3.77, p=0.003]. Likewise, while expression of the 65 kDa species at this age was similar across genotypes in the TBS fraction [Figure 2a; t(12)=1.07, p=0.31], significantly higher expression was seen in Tg relative to Wt animals in the lysis fraction [Figure 2b; t(12)=−3.02, p=0.011]. In Figure 2, optimal exposure times for the TBS blots were longer than those for the lysis blots due to lower NPR concentrations in the TBS fraction. We subsequently quantified the relative expression of the 55 and 65 kDa NPR species in cortical lysis and TBS fractions measured from the same blots and calculated lysis/TBS ratios for each band. In Wt and Tg animals, higher NPR concentrations were seen in lysis relative to TBS fractions for both the 55 kDa [Wt: ratio=3.6, SD=2.0, t(5)=−4.71, p=0.005; Tg: ratio=9.1, SD=3.5, t(7)=−18.85, p<0.001] and 65 kDa [Wt: ratio=5.5, SD=1.7, t(5)=−10.03, p<0.001; Tg: ratio=9.4, SD=4.5, t(7)=−7.61, p<0.001] species. Relative to Wt animals, the lysis/TBS ratios for Tg animals were significantly higher for the 55 kDa band [t(11)=−3.45, p=0.005] and marginally higher for the 65 kDa band [t(11)=−1.98, p=0.074].
Figure 2.

Representative Western blots and quantification of NPR levels in A) soluble and B) membrane-associated mixed cortex fractions from Wt and Tg rats at 3 months, 9 months, and 18-20 months of age. Dotted line represents normalized optical density of bands from Wt animals at each age (n=7 in all age groups). The numbers of Tg animals at each age are shown in parentheses. Error bars represent standard error of the mean (SEM). *p<0.05 vs. Wt.
Given the age-related accumulation of NPR in the membrane-associated compartment seen in cortical fractions from Tg rats, we also used the AF4414 antibody to examine genotype effects on NPR expression at 18-20 months of age in two other neuroanatomical regions that also demonstrate high levels of NPR expression (Dodds et al., 1997) but have differential expression of Aβ pathology (Teng et al., 2011): hippocampus (which exhibits marked age-associated Aβ plaque deposition) and cerebellum (which does not demonstrate any age-associated Aβ plaque deposition). In 18-20 month old hippocampal lysis fractions, the expression of both the 55 kDa [t(12)=−3.37, p=0.006] and 65 kDa [t(12)=−3.88, p=0.002] NPR species were significantly increased in Tg relative to Wt rats (Figure 3a). In contrast, Western blots for NPR using 18-20 month old cerebellar lysis fractions showed no differences between Tg and Wt rats in the expression of either the 55 kDa [t(9)=−0.92, p=0.38] or 65 kDa [t(9)=−0.77, p=0.46] NPR species (Figure 3b).
Figure 3.

Representative Western blots and quantification of NPR levels in membrane-associated A) hippocampal and B) cerebellar fractions from Wt and Tg rats at 18-20 months of age. Error bars represent standard error of the mean (SEM). *p<0.05 vs. Wt.
Soluble Aβ levels in Tg and Wt brain
We further sought to clarify whether the regional differences in NPR expression in Tg animals were related to soluble Aβ levels in the TBS fraction as measured by ELISA. In mixed cortex TBS fractions from 18-20 month old animals, there was the expected marked elevation of total Aβ levels in Tg relative to Wt animals [Figure 4; t(12)=−3.14, p=0.008]. However, in cerebellar TBS fractions from the same animals, total Aβ levels remained undetectable in both genotypes, even after increasing the sample loading from 20 to 80 μg of total protein per well (data not shown). These results suggest that increased expression of NPR species in membrane-associated fractions occurs only in regions where increased Aβ accumulation is seen. However, soluble Aβ levels in mixed cortex in these animals did not directly correlate with expression of the full-length 55 kDa [r(14)=0.27, p=0.35] or 65 kDa [r(14)=0.38, p=0.19] NPR species in the membrane-associated fraction.
Figure 4.

Total Aβ levels in soluble mixed cortex fractions from Wt and Tg rats at 18-20 months of age. The numbers of animals in each group are shown in parentheses. Error bars represent standard error of the mean (SEM). *p<0.05 vs. Wt.
Brain NPR levels and behavioral performance
Previous work with this Tg rat model has demonstrated age-associated deficits on the MWM (Liu et al., 2008), which is particularly sensitive to hippocampal dysfunction (Morris et al., 1982). A subset of Wt and Tg rats were tested on the MWM at 18-20 months of age. Tg animals exhibited significantly longer latencies to finding the hidden platform across training days 6 through 10 than Wt animals [Figure 5a; t(12)=−3.55, p=0.004]. These animals were euthanized shortly after completing MWM testing. Subsequent analyses indicated that significantly poorer MWM performance was seen in animals with higher levels of the 65 kDa full-length NPR species in membrane-associated hippocampal fractions [Figure 5b; r(14)=0.836, p<0.001], suggesting that regional NPR accumulation may contribute to the behavioral deficits seen in Tg rats.
Figure 5.
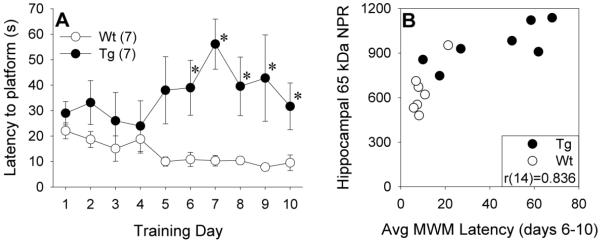
A) Morris water maze (MWM) platform latency in 18-20 month old Wt and Tg rats across training days. Error bars represent standard error of the mean (SEM). *p<0.05 vs. Wt. B) Scatterplot of average MWM platform latency across training days 6-10 versus membrane-associated NPR levels in hippocampal fractions from 18-20 month old Wt and Tg rats. Hippocampal NPR levels are expressed in arbitrary units of optical density.
Phospho-TACE levels in Tg and Wt brain
The age-associated increase in NPR localization in the membrane-associated compartment in Tg animals may be due to reductions in TACE activity that are mediated by Aβ accumulation. Previous work in hippocampal neurons cultured from Tg2576 mice suggests that Aβ accumulation increases phosphoinositide-dependent kinase-1 (PDK-1) activity and decreases TACE activity via Thr735 phosphorylation (Pietri et al., 2013). Other reports indicate that inactive TACE is maintained in a dimeric state (Xu et al., 2012). Therefore, we performed Western blots to assess total and phosphorylated TACE levels in membrane-associated mixed cortex fractions from rats at 3 months and 20 months of age (Figure 6). At 3 months of age, when Tg rats express similar levels of membrane-associated NPR and increased levels of soluble NPR relative to Wt rats, total TACE levels were similar between Wt and Tg rats in the monomeric (~75 kDa) band [t(12)=0.26, p=0.80]. Monomeric phospho-TACE expression was also similar between Wt and Tg animals at this age [t(12)=1.55, p=0.15], but Tg animals had marginally lower expression of dimeric (~160 kDa) phospho-TACE [t(12)=1.91, p=0.08] relative to Wt animals. At 20 months of age, similar total TACE levels were again seen in the Wt and Tg groups in the monomeric band [t(11)=−0.42, p=0.68]. While monomeric phospho-TACE expression was again similar between Wt and Tg animals [t(12)=−1.47, p=0.17], significantly higher expression of dimeric phospho-TACE was seen in Tg animals [t(12)=−7.32, p<0.001]. These results are consistent with greater TACE inactivation in older Tg animals and represent a potential Aβ-mediated mechanism for the age-associated accumulation and compartmental shifts seen with NPR in this model.
Figure 6.

A) Quantification of Western blots for total and phospho-TACE levels in membrane-associated mixed cortex fractions in 3 and 18-20 month old Wt and Tg rats. Dotted line represents normalized optical density of bands from Wt animals at each age (n=7 for both age groups). The numbers of Tg animals at each age are shown in parentheses. Error bars represent standard error of the mean (SEM). *p<0.05 vs. Wt. B) Representative Western blots for monomeric (~75 kDa) and dimeric (~160 kDa) phospho-TACE levels in 18-20 month old Wt and Tg rats.
NPR levels in Tg and Wt rat plasma samples
A prior report suggested that elevated NPR levels were seen in serum samples from human subjects meeting criteria for AD or mild cognitive impairment relative to cognitively normal aged controls (Yin et al., 2009). Therefore, we performed Western blots using AF4414 antibody to determine whether age-associated increases in relative NPR expression in Tg rat cortex were paralleled by similar changes in NPR expression in the plasma. The dominant NPR species seen in plasma was the 55 kDa species, but its expression did not differ by genotype at 3, 9, or 18-20 months of age (data not shown). The 65 kDa NPR species was not seen in plasma. However, an immunoreactive band at 26 kDa demonstrated age- and genotype-associated changes (Figure 7). This 26 kDa plasma NPR species showed similar expression in Wt and Tg animals at 3 months [t(12)=0.15, p=0.89] and 9 months [t(12)=0.15, p=0.89] of age. However, by 18-20 months of age, plasma expression of this 26 kDa fragment was significantly higher in Tg animals [t(12)=−2.70, p=0.019]. We repeated Western blots with plasma samples from 18-20 month animals after spin column depletion of high abundance proteins, which include immunoglobulins. Expression of the 26 kDa NPR species in the depleted samples remained significantly higher in Tg versus Wt animals [t(12)=−4.70, p=0.001]. The 26 kDa NPR species was not detected in Western blots using antibodies targeting the C-terminus or mid-portions of the protein (data not shown), suggesting that it represents a region of the protein that lies closer to the N-terminus and neighboring transmembrane domain. This NPR fragment was only inconsistently seen by Western blot in mixed cortex homogenate, TBS, or lysis fractions. In brain samples from 18-20 month animals, much longer exposure times were needed to visualize this 26 kDa band relative to the aforementioned 55 kDa and 65 kDa full length bands, and the expression of this band (when visualized) did not consistently exhibit genotype effects.
Figure 7.

Representative Western blots and quantification of the 26 kDa NPR species in plasma from Wt and Tg rats at 18-20 months of age. Depleted samples underwent removal of high abundance plasma proteins prior to Western blotting. Error bars represent standard error of the mean (SEM). *p<0.05 vs. Wt.
Correlations between the brain and plasma NPR expression
Correlational analyses were performed in order to determine whether plasma levels of the 26 kDa NPR species reflected cortical levels of full-length NPR. Significant correlations were seen between plasma expression of the 26 kDa NPR species and mixed cortex lysis fraction expression of the 55 kDa [r(11)=0.633, p=0.037] and 65 kDa [r(11)=0.657, p=0.028] NPR species (Figure 8) which supports a possible precursor-product relationship. However, plasma expression of the 26 kDa NPR species did not correlate with mixed cortex TBS fraction expression of either the 55 kDa [r(11)=−0.027, p=0.94] or 65 kDa [r(11)=0.259, p=0.44] NPR species.
Figure 8.
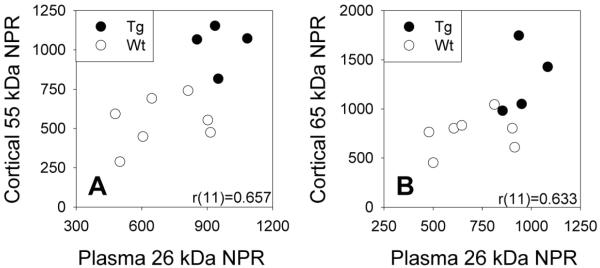
Scatterplots of plasma expression of 26 kDa NPR species versus membrane-associated mixed cortex expression of full-length A) 55 kDa and B) 65 kDa NPR species in 18-20 month old Wt and Tg rats. Plasma and cortical NPR levels are expressed in arbitrary units of optical density.
DISCUSSION
Our data demonstrate that increasing cortical and hippocampal NPR levels parallel the age-associated deposition of cerebral Aβ pathology that has previously been characterized in this transgenic APP/PS1 rat model of AD (Flood et al., 2009; Liu et al., 2008; Teng et al., 2011). The accumulation of NPR in older Tg animals is seen in the cortex and hippocampus, which both express high levels of both soluble Aβ and Aβ plaques, suggesting that NPR accumulation may be mediated by Aβ pathology. This hypothesis is supported by our results demonstrating elevated NPR levels in cortical homogenate (which includes both soluble and membrane-associated fractions) from Tg animals by 9 months of age, when sparse Aβ plaques are just starting to deposit in this model (Flood et al., 2009; Liu et al., 2008). Conversely, genotype effects on NPR expression were not observed in the cerebellum, where soluble Aβ levels were undetectable and Aβ plaques are largely absent (Teng et al., 2011). Nevertheless, since we did not find significant linear correlations between cortical Aβ and NPR levels, additional downstream mechanisms, such as oxidative damage or neuroinflammation, may be mediating Aβ regulation of NPR levels in the brain.
In its full-length forms, NPR has a short transmembrane domain near its N-terminus (Cho et al., 2008), and it is thus unsurprising that a greater proportion of cortical NPR is seen in the membrane-associated fraction. However, this finding was most pronounced in 18-20 month old Tg animals (i.e. those with the most advanced Aβ pathology), which demonstrated higher expression of membrane-associated NPR than age-matched Wt animals. This result may be mechanistically related to Aβ-related TACE inactivation (Pietri et al., 2013), since TACE-mediated cleavage of NPR is required for its release from the plasma membrane (Cho et al., 2008). TACE dimers in the cell membrane represent inactive TACE (Xu et al., 2012), and at older ages, Tg rats exhibit higher levels of phospho-TACE dimers than Wt rats. Furthermore, since NPR cleavage and release from the membrane is also needed for LTD (Cho et al., 2008), the accumulation of NPR in membrane-associated cortical fractions may contribute to the deficits in synaptic plasticity that have been previously reported in this model (Liu et al., 2008).
Age-associated TACE inactivation in Tg rats also provides a potential explanation for why these animals have lower NPR expression in soluble cortical fractions at 18-20 months of age relative to Wt controls. However, Tg rats also exhibit increased NPR expression in soluble cortical fractions relative to Wt rats at younger ages, when soluble Aβ levels are already elevated, but plaque deposition has not yet (3 months) or just begun (9 months) to occur (Flood et al., 2009; Liu et al., 2008). These results are consistent with the hypothesis that at early stages of AD progression, there is a compensatory increase in synaptic plasticity, including at glutamatergic synapses, which may represent an ultimately ineffective attempt to counter Aβ-related synaptotoxicity (Bell et al., 2007; Williams et al., 2009). Alternatively, greater levels of soluble NPR in younger Tg animals could be a consequence of relatively increased LTD, since LTD-associated cleavage of NPR by TACE (which does not yet appear to be inactivated at younger ages) would yield such soluble species and increasing levels of soluble Aβ appear to facilitate LTD in hippocampal slice preparations (Cheng et al., 2009; Shankar et al., 2008). However, other reports suggest that LTD may actually be more difficult to induce in transgenic mouse models of AD than in wild-type controls (Song et al., 2014; Tamagnini et al., 2012), which would oppose this hypothesis.
Beyond demonstrating a potential brain basis for AD-related changes in CSF NPR levels, our data also demonstrate that a 26 kDa plasma NPR immunoreactive species exhibits age- and genotype-associated changes in expression that parallel changes in full-length brain NPR expression and thus may represent a viable peripheral marker of brain NPR levels. This NPR fragment appears to be derived from the N-terminus of the full-length protein, but is present in relatively low abundance in the cortical samples. It may correspond with a proteolytically processed N-terminal fragment of similar size that has previously been detected in mouse forebrain lysate (Cho et al., 2008). NPR fragments of this approximate size have also been detected via two-dimensional gel electrophoresis from human CSF samples (Finehout et al., 2007; Yin et al., 2009). The relatively higher abundance of this fragment in plasma samples could mean that a greater degree of proteolytic breakdown may occur peripherally rather than in the brain. However, the protease(s) responsible for generating this 26 kDa fragment have not been conclusively identified (Cho et al., 2008). The significant correlation between the expression of the 26 kDa plasma NPR species and expression of the membrane-associated, but not soluble full-length cortical NPR species would appear to argue against the simple diffusion of soluble NPR across the blood-brain barrier. Instead, NPR may reach the periphery in a membrane-bound form. Although exosomes, which can cross the blood-brain barrier and represent a potential clearance mechanism for degraded proteins (Kalani et al., 2014) might be considered for this role, recent proteomic characterization of CSF suggests relative depletion rather than enrichment of NPR in extracellular vesicular fractions (Chiasserini et al., 2014).
Prior human CSF proteomic studies in AD suggest that NPR levels increase at early stages of the disease but decrease with subsequent disease progression (Ringman et al., 2012; Wildsmith et al., 2014). Therefore, our results demonstrating the greatest relative elevations in brain and plasma NPR levels in the oldest cohorts of APP/PS1 rats, which have extensive and widespread Aβ plaque pathology (Flood et al., 2009; Liu et al., 2008; Teng et al., 2011), might appear to contrast with these human CSF results. However, others have suggested that transgenic rodent models of cerebral amyloidosis, even at advanced ages, most closely model the earliest stages of AD, when cognition is intact or only mildly impaired and synaptic and neuronal losses remain relative modest (Ashe, 2005). Therefore, it seems likely that the elevated NPR levels seen in this transgenic rat model are analogous to those seen in patients at the presymptomatic (Ringman et al., 2012) or MCI (Wildsmith et al., 2014; Yin et al., 2009) stages of AD, and subsequent reductions in NPR expression, which may be associated with more advanced AD (Abdi et al., 2006; Finehout et al., 2007; Hu et al., 2007; Wildsmith et al., 2014) may only be apparent in more aggressive animal models.
When taken together, our data indicate that corresponding increases in NPR expression in brain and plasma compartments can be detected with increasing disease progression in a transgenic APP/PS1 rat model, suggesting the potential utility of plasma NPR levels as a peripheral biomarker of underlying synaptic dysfunction. Previous work has indicated that CSF NPR levels are increased in AD but not in Parkinson’s disease (PD) (Yin et al., 2009). Although our results are consistent with those findings, since they are derived from a model that overexpresses Aβ, the prominent role of the neuronal pentraxins in synaptic function raises the possibility that NPR expression may also be altered by other conditions characterized by synaptic disruption. Indeed, relative decreases have been reported in NPR RNA levels in rat brain after cerebral ischemia (Kim et al., 2002) and in a mesencephalic dopaminergic neuronal culture model of PD (Anantharam et al., 2007). Furthermore, decreased CSF NPR levels have been reported in multiple sclerosis (Kroksveen et al., 2012). Although a relative upregulation of NPR RNA in cardiac muscle has been reported after myocardial infarction (Andersson et al., 2006), the expression of NPR RNA in brain is far higher than that seen in other organs (Dodds et al., 1997). These data indicate that plasma NPR levels are likely to be primarily driven by brain NPR levels, but we cannot entirely exclude the possibility that a small proportion of plasma NPR levels may be derived from other sources.
There are two other factors that may limit the interpretation of our results. Although both the Tg and Wt animals are Sprague-Dawley rats, the Tg animals were obtained from a homozygous in-bred colony while the Wt animals were obtained from commercial outbred colonies. Therefore, we cannot completely rule out the possibility that other genetic variables beyond the APP/PS1 transgenes may have contributed to our findings. Aβ is the predominant pathology expressed by the Tg rats, though they also exhibit some hyperphosphorylated tau accumulation in dystrophic neurites (Liu et al., 2008). As such, a portion of the alterations in NPR expression seen in clinical AD populations may be driven by more prominent tau-related pathology. This question warrants further investigation in transgenic rodent models that overexpress tau.
Further translational studies of plasma NPR expression in clinical populations, particularly those that compare participants with either normal cognition or MCI that have positive or negative Aβ PET imaging, may provide additional insight into the relationship between peripheral NPR levels and cerebral Aβ pathology.
Research Highlights.
Neuronal pentraxin receptor (NPR) was measured in an Alzheimer’s disease model.
Brain and plasma NPR increased with age, paralleling β-amyloid (Aβ) accumulation.
NPR accumulates in membrane-associated brain fractions.
NPR levels measured from brain and plasma were significantly correlated.
ACKNOWLEDGMENTS
This work was supported by the National Institute on Aging (K08 AG34628 [to ET; jointly sponsored by NIA, AFAR, the John A. Hartford Foundation, the Atlantic Philanthropies, the Starr Foundation and an anonymous donor] and RC1 AG035878 [to GMC]), the National Center for Research Resources and the National Center for Advancing Translational Sciences (UL1 TR000124), and the UCLA Clinical and Translational Science Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Although this antibody is advertised as N-terminal specific, the synthetic immunogen sequence used to produce it (MADGAWDSPALLVELENA) lies in the mid-region of the rat NPR sequence, starting at amino acid 174 (Abcam, personal communication).
REFERENCES
- Abad MA, et al. Neuronal pentraxin 1 contributes to the neuronal damage evoked by amyloid-beta and is overexpressed in dystrophic neurites in Alzheimer’s brain. J Neurosci. 2006;26:12735–47. doi: 10.1523/JNEUROSCI.0575-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdi F, et al. Detection of biomarkers with a multiplex quantitative proteomic platform in cerebrospinal fluid of patients with neurodegenerative disorders. J Alzheimers Dis. 2006;9:293–348. doi: 10.3233/jad-2006-9309. [DOI] [PubMed] [Google Scholar]
- Anantharam V, et al. Microarray analysis of oxidative stress regulated genes in mesencephalic dopaminergic neuronal cells: relevance to oxidative damage in Parkinson’s disease. Neurochem Int. 2007;50:834–47. doi: 10.1016/j.neuint.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson KB, et al. Regulation of neuronal type genes in congestive heart failure rats. Acta Physiol (Oxf) 2006;186:17–27. doi: 10.1111/j.1748-1716.2005.01503.x. [DOI] [PubMed] [Google Scholar]
- Arold S, et al. Apolipoprotein E level and cholesterol are associated with reduced synaptic amyloid beta in Alzheimer’s disease and apoE TR mouse cortex. Acta Neuropathol. 2012;123:39–52. doi: 10.1007/s00401-011-0892-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe KH. Mechanisms of memory loss in Abeta and tau mouse models. Biochem Soc Trans. 2005;33:591–4. doi: 10.1042/BST0330591. [DOI] [PubMed] [Google Scholar]
- Bell KF, et al. Paradoxical upregulation of glutamatergic presynaptic boutons during mild cognitive impairment. J Neurosci. 2007;27:10810–7. doi: 10.1523/JNEUROSCI.3269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjartmar L, et al. Neuronal pentraxins mediate synaptic refinement in the developing visual system. J Neurosci. 2006;26:6269–81. doi: 10.1523/JNEUROSCI.4212-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon F, et al. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron. 2004;43:633–45. doi: 10.1016/j.neuron.2004.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, et al. Amyloid beta-protein fragments 25-35 and 31-35 potentiate long-term depression in hippocampal CA1 region of rats in vivo. Synapse. 2009;63:206–14. doi: 10.1002/syn.20599. [DOI] [PubMed] [Google Scholar]
- Chiasserini D, et al. Proteomic analysis of cerebrospinal fluid extracellular vesicles: a comprehensive dataset. J Proteomics. 2014;106:191–204. doi: 10.1016/j.jprot.2014.04.028. [DOI] [PubMed] [Google Scholar]
- Cho RW, et al. mGluR1/5-dependent long-term depression requires the regulated ectodomain cleavage of neuronal pentraxin NPR by TACE. Neuron. 2008;57:858–71. doi: 10.1016/j.neuron.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds DC, et al. Neuronal pentraxin receptor, a novel putative integral membrane pentraxin that interacts with neuronal pentraxin 1 and 2 and taipoxin-associated calcium-binding protein 49. J Biol Chem. 1997;272:21488–94. doi: 10.1074/jbc.272.34.21488. [DOI] [PubMed] [Google Scholar]
- Doecke JD, et al. Blood-based protein biomarkers for diagnosis of Alzheimer disease. Arch Neurol. 2012;69:1318–25. doi: 10.1001/archneurol.2012.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiandaca MS, et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2014 doi: 10.1016/j.jalz.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finehout EJ, et al. Cerebrospinal fluid proteomic biomarkers for Alzheimer’s disease. Ann Neurol. 2007;61:120–9. doi: 10.1002/ana.21038. [DOI] [PubMed] [Google Scholar]
- Flood DG, et al. A transgenic rat model of Alzheimer’s disease with extracellular Abeta deposition. Neurobiol Aging. 2009;30:1078–90. doi: 10.1016/j.neurobiolaging.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Hu WT, et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology. 2012;79:897–905. doi: 10.1212/WNL.0b013e318266fa70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, et al. Identification and validation of novel CSF biomarkers for early stages of Alzheimer’s disease. Proteomics Clin Appl. 2007;1:1373–84. doi: 10.1002/prca.200600999. [DOI] [PubMed] [Google Scholar]
- Kalani A, et al. Exosomes: mediators of neurodegeneration, neuroprotection and therapeutics. Mol Neurobiol. 2014;49:590–600. doi: 10.1007/s12035-013-8544-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YD, et al. DNA array reveals altered gene expression in response to focal cerebral ischemia. Brain Res Bull. 2002;58:491–8. doi: 10.1016/s0361-9230(02)00823-7. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick LL, et al. Biochemical interactions of the neuronal pentraxins. Neuronal pentraxin (NP) receptor binds to taipoxin and taipoxin-associated calcium-binding protein 49 via NP1 and NP2. J Biol Chem. 2000;275:17786–92. doi: 10.1074/jbc.M002254200. [DOI] [PubMed] [Google Scholar]
- Koch SM, Ullian EM. Neuronal pentraxins mediate silent synapse conversion in the developing visual system. J Neurosci. 2010;30:5404–14. doi: 10.1523/JNEUROSCI.4893-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroksveen AC, et al. Cerebrospinal fluid proteome comparison between multiple sclerosis patients and controls. Acta Neurol Scand Suppl. 2012:90–6. doi: 10.1111/ane.12029. [DOI] [PubMed] [Google Scholar]
- Lim GP, et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J Neurosci. 2000;20:5709–14. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, et al. A transgenic rat that develops Alzheimer’s disease-like amyloid pathology, deficits in synaptic plasticity and cognitive impairment. Neurobiol Dis. 2008;31:46–57. doi: 10.1016/j.nbd.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall J, et al. Creation of a federated database of blood proteins: a powerful new tool for finding and characterizing biomarkers in serum. Clin Proteomics. 2014;11:3. doi: 10.1186/1559-0275-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, et al. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–3. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Neselius S, et al. Olympic boxing is associated with elevated levels of the neuronal protein tau in plasma. Brain Inj. 2013;27:425–33. doi: 10.3109/02699052.2012.750752. [DOI] [PubMed] [Google Scholar]
- O’Bryant SE, et al. A blood-based screening tool for Alzheimer’s disease that spans serum and plasma: findings from TARC and ADNI. PLoS One. 2011;6:e28092. doi: 10.1371/journal.pone.0028092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietri M, et al. PDK1 decreases TACE-mediated alpha-secretase activity and promotes disease progression in prion and Alzheimer’s diseases. Nat Med. 2013;19:1124–31. doi: 10.1038/nm.3302. [DOI] [PubMed] [Google Scholar]
- Randall J, et al. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: results of a pilot study. Resuscitation. 2013;84:351–6. doi: 10.1016/j.resuscitation.2012.07.027. [DOI] [PubMed] [Google Scholar]
- Ray S, et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med. 2007;13:1359–62. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- Revett TJ, et al. Glutamate system, amyloid β peptides and tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatry Neurosci. 2013;38:6–23. doi: 10.1503/jpn.110190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringman JM, et al. Proteomic changes in cerebrospinal fluid of presymptomatic and affected persons carrying familial Alzheimer disease mutations. Arch Neurol. 2012;69:96–104. doi: 10.1001/archneurol.2011.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–91. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia GM, et al. Interaction of the N-terminal domain of the AMPA receptor GluR4 subunit with the neuronal pentraxin NP1 mediates GluR4 synaptic recruitment. Neuron. 2007;55:87–102. doi: 10.1016/j.neuron.2007.06.020. [DOI] [PubMed] [Google Scholar]
- Song S, et al. In vivo administration of granulocyte colony-stimulating factor restores long-term depression in hippocampal slices prepared from transgenic APP/PS1 mice. J Neurosci Res. 2014;92:975–80. doi: 10.1002/jnr.23378. [DOI] [PubMed] [Google Scholar]
- Tamagnini F, et al. Early impairment of long-term depression in the perirhinal cortex of a mouse model of Alzheimer’s disease. Rejuvenation Res. 2012;15:231–4. doi: 10.1089/rej.2011.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng E, et al. [F-18]FDDNP microPET imaging correlates with brain Abeta burden in a transgenic rat model of Alzheimer disease: effects of aging, in vivo blockade, and anti-Abeta antibody treatment. Neurobiol Dis. 2011;43:565–75. doi: 10.1016/j.nbd.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildsmith KR, et al. Identification of longitudinally dynamic biomarkers in Alzheimer’s disease cerebrospinal fluid by targeted proteomics. Mol Neurodegener. 2014;9:22. doi: 10.1186/1750-1326-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C, et al. Transcriptome analysis of synaptoneurosomes identifies neuroplasticity genes overexpressed in incipient Alzheimer’s disease. PLoS One. 2009;4:e4936. doi: 10.1371/journal.pone.0004936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, et al. Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron. 2003;39:513–28. doi: 10.1016/s0896-6273(03)00463-x. [DOI] [PubMed] [Google Scholar]
- Xu P, et al. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci Signal. 2012;5:ra34. doi: 10.1126/scisignal.2002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- Yang F, et al. Monoclonal antibody to the C-terminus of beta-amyloid. Neuroreport. 1994;5:2117–20. doi: 10.1097/00001756-199410270-00032. [DOI] [PubMed] [Google Scholar]
- Yin GN, et al. Neuronal pentraxin receptor in cerebrospinal fluid as a potential biomarker for neurodegenerative diseases. Brain Res. 2009;1265:158–70. doi: 10.1016/j.brainres.2009.01.058. [DOI] [PubMed] [Google Scholar]
- Zetterberg H, et al. Plasma tau levels in Alzheimer’s disease. Alzheimers Res Ther. 2013;5:9. doi: 10.1186/alzrt163. [DOI] [PMC free article] [PubMed] [Google Scholar]