

Abstract
Fluorescent proteins offer exceptional labeling specificity in living cells and organisms. Unfortunately, their photophysical properties remain far from ideal for long-term imaging of low-abundance cellular constituents, in large part because of their poor photostability. Despite widespread engineering efforts, improving the photostability of fluorescent proteins remains challenging due to lack of appropriate high-throughput selection methods. Here, we use molecular dynamics guided mutagenesis in conjunction with a recently developed microfluidic-based platform, which sorts cells based on their fluorescence photostability, to identify red fluorescent proteins with decreased photobleaching from a HeLa cell-based library. The identified mutant, named Kriek, has 2.5- and 4-fold higher photostability than its progenitor, mCherry, under widefield and confocal illumination, respectively. Furthermore, the results provide insight into mechanisms for enhancing photostability and their connections with other photophysical processes, thereby providing direction for ongoing development of fluorescent proteins with improved single-molecule and low-copy imaging capabilities.
Insight, innovation, integration
Fluorescent proteins enable imaging in situ, throughout the visible spectrum, with superb molecular specificity and single-molecule sensitivity. Unfortunately, when compared to leading small-molecule fluorophores (e.g., Cy3), fluorescent proteins, suffer from accelerated photobleaching and poor integrated photon output. This results from a lack of appropriate high-throughput methods for improving the photostability of fluorescent proteins, as well as a poor molecular understanding of fluorescent protein photobleaching. Here, we report the first application of a recently developed microfluidic cell-sorter to identify fluorescent proteins from a mCherry-derived library with improved photostability. The results provide insight into fluorescent protein photophysics, greatly accelerate identification of improved mutants, and can be applied to both genetically encoded and small-molecule fluorophores.
Introduction
Fluorescent proteins (FPs) are powerful tools for cellular imaging and have been instrumental in providing many critical insights in cell biology. However, FPs still have limitations and many experimental systems would benefit from brighter and more photostable probes.6 For example, red fluorescent proteins (RFPs) are in particularly high demand due to decreased autofluorescence when using longer excitation wavelengths. However, currently RFPs emit 10-100x less photons than small-molecule fluorophores, and therefore remain difficult to use in experiments that focus on low-abundance or single-molecule imaging.7, 8 Moreover, as the scientific community continues to push the boundaries of fluorescence imaging by expressing FP-tagged proteins at low copy numbers via genome-editing techniques, tracking single molecules in cells, and imaging over extended periods of time, limitations of the existing palette of FPs become more apparent. Thus, it is perhaps not surprising that FPs are constantly being subjected to mutagenesis and selection in an effort to create FPs with more favorable properties.
The current suite of FPs has resulted from a combination of discovery of new FPs from marine organisms and protein engineering to optimize the biochemical, spectroscopic, and photophysical properties. To date, most FP engineering efforts have employed error-prone PCR, site-directed mutagenesis, or gene-shuffling to generate a library of mutants, followed by bacterial colony screening and/or fluorescence-activated cell sorting (FACS).9-11 While this approach has been highly successful and is responsible for the vast majority of FPs used today, there are limitations, particularly for efforts aimed at targeting more sophisticated photophysical properties such as photoswitching, photostability, and dark state conversion. The vast majority of screens use brightness as the sole selection criterion. Unfortunately, the brightness of an FP-expressing cell is a parameter that depends upon a combination of factors, including cell size, genome integration site, copy number, the rate of protein-folding and degradation, chromophore maturation, extinction coefficient, and various photophysical processes that compete with fluorescence, such as transitions to non-fluorescent states and fluorescence quenching. To overcome these challenges, researchers are increasingly turning towards more complex, quantitative, multiparameter screening strategies.12
Photophysical parameters, such as extinction coefficient, quantum yield, photostability, and dark state conversion, are key determinants that define the signal-to-noise ratio and maximum imaging duration. Improved quantification and isolation of such parameters allows greater precision in spectroscopic tuning of FPs for state-of-the-art imaging applications. For example, Goedhartet al. used fluorescence lifetime to screen cyan-emitting FPs (CFPs) in a low-throughput colony based assay.13 To the extent that the fluorescence lifetime depends on the non-radiative relaxation rate from the excited state, the lifetime provides a concentration-independent measure of the fluorescence quantum yield. After screening a semi-saturated mutagenesis library at position T65 in SCFP3A (originally evolved from enhanced CFP), the T65S mutation was found to increase the quantum yield from 0.56 to 0.84. Remarkably, subsequent structure-guided mutagenesis coupled with excited-state calculations resulted in mTurquoise2, a CFP with a quantum yield of 0.93.14
In addition to quantum yield, irreversible photobleaching is another important property for which few screens have been carried out. Photobleaching is light-induced inactivation of a fluorescent moiety and, in live-cell imaging, is commonly responsible for diminished fluorescence signal and limited observation times.15 Improving the photostability of FPs remains challenging due to the lack of high-throughput selection strategies that can operate over a wide range of intensity regimes (~ 0.1-100 kW/cm2). Tsien and colleagues identified improved FPs by exposing bacterial colonies to a solar illuminator,16 but this technique is limited to illumination intensities below those necessary for low-copy or single-molecule imaging. Given that photobleaching mechanisms are illumination-dependent,4 it would be valuable to develop a screening approach in which the excitation intensity could be tuned over a wide range. Additionally, selection within the mammalian context is preferred given that some FPs identified in E. coli fail to fold productively in eukaryotes.13
In this work, we demonstrate the application of a recently derived microfluidic flow cytometer to sort cells expressing a library of RFPs on the basis of irreversible photobleaching.3, 5 This approach offers several advantages. Compared to colony screening, single-cell microfluidic analysis is more sensitive, quantitative, and versatile, enabling screens for photostability over a wider range of excitation intensities (e.g., 0.1-100 kW cm-2). In addition, we demonstrate that a screen for photostability can be implemented simultaneously with screening based on the net cellular brightness.3, 5 Furthermore, by performing the selection on RFP-expressing mammalian cells, the RFPs must remain non-cytotoxic, and be compatible with the eukaryotic protein expression machinery. As an application of this approach, we use molecular dynamics (MD) guided mutagenesis of mCherry and microfluidic cell sorting to identify variants with decreased rates of photobleaching.
Results
Library Design
In our efforts to screen a library based on photostability, mCherry was selected as a template for mutagenesis given its wide use in cell biology, the availability of a crystal structure to inform library design, and reasonably high photostability under laser excitation.4 Similar to other FPs, the crystal structure of mCherry reveals excellent interstrand hydrogen-bonding (e.g., amide nitrogen-carbonyl oxygen distance < 3 Å), indicative of a rigid beta-barrel structure. However, the well-ordered hydrogen-bonding is disrupted between β-strands 7 and 10 (Figure 1A).1 MD simulations suggest that relative to Citrine, a yellow FP derived from Aequoriavictoria, β-strands 7 and 10 in mCherry are more dynamic and subject to increased oxygen permeability.2, 17, 18 Although the mechanisms of photobleaching remain poorly understood in FPs,19, 20 one proposed pathway for photodegradation is the excited state-mediated formation of, and subsequent reaction with, reactive oxygen species. Therefore, we hypothesized that decreasing interstrand dynamics could improve mCherry photostability by minimizing access of 3O2 to the chromophore. In support of this hypothesis, imaging under anaerobic conditions was found to decrease the rate of photobleaching for mCherry (Figure S1). Consequently, we decided to pursue a MD-guided library design, with mutations targeted to β-7/10 strand interface. MD simulations recommended three mutations (W143K, Q163R, and R164E) that decreased interstrand dynamics in silico, but incorporation of these mutations into mCherry resulted in a non-fluorescent variant.
Figure 1.
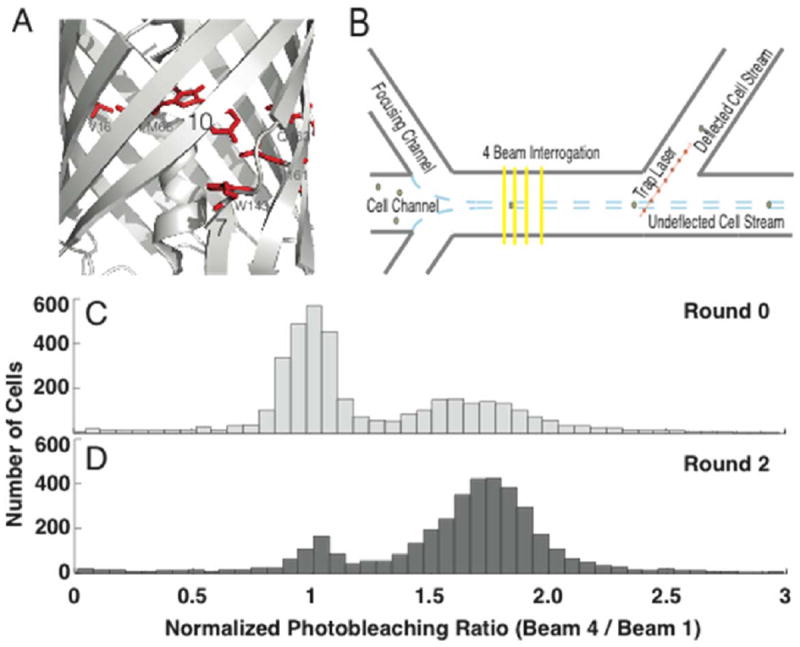
Beta-strand 7/10 dynamics, microfluidic selection strategy, and photostability population shift. (A) Crystal structure (PDB 2H5Q) of mCherry, highlighting the targeted region between β-strands 7 and 10 1, 2, including residues targeted for mutagenesis, indicated in red (V16, M66, W143, I161, and Q163). (B) Cells expressing a single genomically integrated copy of a mutant FP are hydrodynamically focused to a velocity of ~ 6-8 mm/s and subjected to four illumination events 3. Each illumination event is ~ 2 ms in duration, and it takes ~ 8 ms for the cell to travel to the next beam, thereby providing sufficient time to allow recovery from reversible dark-states 3-5. Photobleaching is quantitated by the quotient of the fluorescence intensity from the fourth and first excitation events. Cells exhibiting improved photostability (e.g., higher photobleaching ratio) are deflected to a separate output channel by applying an optical force with a 1064 nm focused laser. (C) Histogram of the photobleaching ratio (Beam 4 / Beam 1) normalized to the photostability of mCherry for the library before, and (D) after being subjected to two rounds of sorting. Ratios at higher values represent cells expressing mutant FPs with improved photostability.
To identify adjacent positions that could recover fluorescence, a history of reported mutations was compiled for the mFruit family (e.g., monomeric descendants of DsRed). Approximately 1 in every 3 positions within the mFruit family has been mutated. Candidate positions were identified based upon their tolerance to mutagenesis. The most commonly mutated positions in the mFruits are V16, M66, W143, I161, Q163, I197, and A217, two of which were positions that were also identified as significant in the MD simulations. Given the agreement between the MD simulations and the literature analysis, we decided to pursue a library strategy that targeted mutations at a subset of these positions (16, 66, 143, 161, 163). Mutation type was guided by reported mutations, and included both saturated (positions 143 and 163) and semi-saturated (positions 16, 66, and 161) mutagenesis.21 The third position identified in the MD study, R164, was excluded because of its role in disrupting the A/C dimerization interface,22 and I197 and A217 were excluded to limit the size of the library. The final library size was 144,000 mutants, and fluorescent colony screening of the library in E. coli revealed that ~ 10 % of the library was fluorescent.
Microfluidic Sorting Based Upon Irreversible Photobleaching
To screen and sort the library based on irreversible photobleaching, we used a recently developed microfluidic photobleaching cytometer. The cytometer enables quantitative and high-throughput measurements for irreversible photobleaching on single mammalian cells, and can be configured in 3 beam, 4 beam, and 8 beam modes.3, 5 A greater number of beams increases the extent of photobleaching, but decreases throughput due to the greater duration of time necessary for the cell to traverse the interrogation region. Accordingly, because the 4 beam geometry provides a compromise between photobleaching and throughput, it was employed to assess the spectroscopic diversity present in the library. The extent of photobleaching was quantified from the ratio of the fluorescence signals from the first and fourth beams, and was normalized to the photostability of mCherry (Figure 1B).3, 5 Time between excitation events is determined by the spacing of the beams and the microfluidic flow rate, and was 8 ms in our platform to permit relaxation from reversible dark-states and isolate the irreversible photobleaching component.3, 5 Details regarding microfluidic operation can be found in the Experimental section. With laser powers of ~ 2 kW/cm2 for each excitation beam, a coefficient of variation (the standard deviation divided by the mean) of ~ 18 % was common for a single-FP cell population, with greater uncertainty arising with low-expression cells. However, for HeLa cells virally transduced with the library, a coefficient of variation of 186 % was observed, indicating greater photophysical diversity. Importantly, given that mCherry has a normalized photobleaching ratio of ~ 1, a fraction of cells exhibited improved photostability (Figure 1C).
Given the promising diversity in photostability, the library was subjected to two rounds of selection. In each round, enough cells were screened to provide 3-fold coverage of the library, and the top-third most photostable population was gated for sorting. Consequently, 42,000 and 14,000 cells were screened in the first and second rounds, and are referred to as Sort1 and Sort2.1, respectively. After two rounds, the sorted cells were passed through the microfluidic cytometer a third time to measure their photostability, and it was evident that a significant fraction of the library had shifted to higher photostability (Figure 1D). For comparison, FACS-based sorts often require > 7 rounds to converge on a population, likely because of cell-to-cell and round-to-round variability in fluorescence brightness.23 In contrast, our system measured an intensive property (i.e., the ratio of fluorescence intensities, which does not depend on chromophore concentration) and appears to be less sensitive to cell-to-cell or round-to-round variability in fluorescence brightness.3, 5
Interestingly, we observed that the mean fluorescence intensity of Sort2.1 was ~ 28 % dimmer than the initial unsorted library population. We hypothesized that this could result from diminished cellular health following the two rounds of optical sorting, or from gradual enrichment of mutants with decreased fluorescence quantum yields and/or extinction coefficients. Therefore, an alternative selection was performed on Sort1, involving both photostability and net fluorescence brightness as measured at beam 1, and was referred to as Sort2.2. Nevertheless, despite restricting cell selection to those exhibiting greater net brightness and improved photostability, Sort2.2 was also found to be dimmer than the original library.
Mutations and Spectroscopic Properties of Enriched Variants
The microfluidic cytometer identified clones that appeared to be dimmer but more photostable than the parent mCherry. To characterize these clones, we first isolated and sequenced the clones to identify the mutations that gave rise to altered photophysical properties. For these studies, cells from both Sort2.1 and Sort2.2 were harvested, the FPs were cloned into a bacterial expression system using standard molecular biology protocols, and thirty two fluorescent clones were selected at random and sequenced. mCherry was identified in 12 clones, and is likely the cause of the residual mCherry-like population in Figure 1D. Of the remaining 20 mutants, amino-acid mutations found in different proteins tended to share similar chemical attributes (Table S1). For example, only 3 FPs had mutated V16, and of those, all were mutated to an alanine. Additionally, H17R was always observed in mutants with V16A, likely arising from an error in the synthesis of the DNA oligonucleotide used to generate the library. It was also observed that M66, the first amino acid within the chromophore forming tripeptide, was mutated to a glutamine in 4 of 20 mutants. A glutamine in this position is also found in DsRed,11, 24, 25 mRFP1,22 mHoneydew,9 select green-fluorescent proteins and non-fluorescent chromoproteins,26-28 as well as the photosensitizing KillerRed. 29, 30 The majority of mutations at W143 were hydrophobic in nature, and included alanine (11/20), methionine (2/20), isoleucine (1/20), although polar residues were also observed, including serine (5/20) and cysteine (1/20). At position I161, only non-polar residues were discovered, including leucine (10/20), valine (8/20), and methionine (1/20). Lastly, Q163 was found to be mutated to isoleucine (10/20), leucine (6/20), threonine (1/20), valine (1/20), and tryptophan (1/20).
To characterize the resulting FPs more quantitatively, we carried out spectroscopic measurements on purified protein. Proteins from the 6 brightest mutants (S2a-f) were grown under identical conditions in E. coli and purified. In order to obtain sufficient quantities of protein for analysis, twice the volume of bacteria were harvested for the purification of S2d, S2e, and S2f relative to S2a, S2b, and S2c. Absorption spectra were measured and normalized to the 230 nm absorption peak to quantify protein expression and chromophore maturation. mCherry showed the largest A590nm/A230nm ratio, indicating the largest extent of protein folding and/or chromophore maturation, followed by S2a, S2c, S2b, S2d, S2e, then S2f (Figure 2A). Although A230 nm is sensitive to all of the aromatic amino-acids (W/Y/F) present, including impurities, and some FPs harboured aromatic mutations (e.g., S2c, W143I), this result is consistent with the decreased brightness of the bacterial colonies and pellets for mutants S2d, S2e, and S2f. Additionally, a population of the “GFP-like” chromophore, λabs ~ 510 nm, was present in all three of these mutants (Figure 2B).31-33 These results are consistent with poor protein folding and chromophore formation in S2d, S2e, and S2f. To evaluate the photostability of the mutants, octanolmicrodroplets containing purified protein were photobleached with a laser-scanning confocal microscope.34 S2a, S2b, S2c, S2d, and S2f showed improvement in photostability over mCherry, with S2c showing the greatest improvement (Figure S2). However, given the poor extent of maturation of S2d, S2e, and S2f, as well as the modest improvements in photostability for S2a and S2b, only S2c was investigated further. S2c was named Kriek, after the Belgian beer that involves fermentation of cherries.
Figure 2.
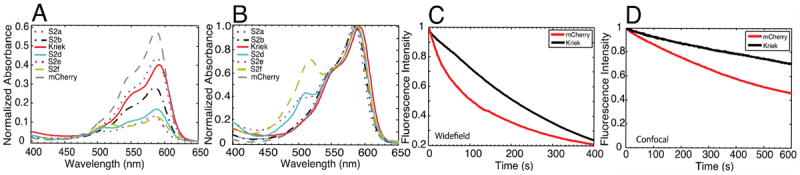
Spectral and Photobleaching Properties of Isolated Mutants. S2c and Kriek are the same FP variant (mCherry W143I I161M Q163V). (A) Absorption spectra of Sort2 mutants normalized at λabs ~ 230 nm and (B) λabs ~ 590 nm. (C) Widefield (λex = 520-560 nm) and (D) laser-scanning confocal (λex = 561 nm) photobleaching decays for Kriek (Black) and mCherry (Red).
To measure the photostability of Kriek under more physiologically-relevant conditions, widefield photobleaching was carried out on immobilized E. coli. For each measurement, the decrease in fluorescence was fit to a monoexponential decay curve, and the time constant (e.g., the inverse rate constant) of the decay was used to quantitatively compare photostability. Under continuously exposed widefield illumination, mCherry and Kriek had average photostability time-constants of 110 and 280 seconds, respectively, representing a 2.5-fold higher photostability (e.g., Kriek / mCherry). For laser scanning confocal imaging, which was performed on nuclear-localized FPs within adherent HeLa cells and required a biexponential fit, mCherry and Kriek had average photostability time-constants 430 and 1700 seconds, respectively, thus Kriek had 4-fold the photostability of mCherry (Figure 2D). Under these conditions even after 10 minutes of continual confocal illumination, Kriek only decreased ~ 20 % in fluorescence intensity (Figure 2D).
Interestingly, the improved performance of Kriek under confocal illumination, as opposed to widefield illumination, could be a direct consequence of the selection criteria used in our microfluidic cytometer. In widefield microscopy, the sample is illuminated with low-intensity (~ 100 W/cm2) incoherent light for 10-100’s of milliseconds with a high-duty cycle (little time between exposures). In contrast, confocal microscopy illuminates the sample with intense (~ 1 kW/cm2) diffraction-limited and coherent light for ~ 1 μs at a low duty-cycle (often ~ 1 second between exposures). As such, our cytometer operates under conditions more similar to a confocal microscope, and would likely identify FP variants adept at sustaining high-intensity low-duty cycleimaging conditions.
In addition to the photostability, the extinction coefficient (λabs), fluorescence quantum yield (Φfl), and peak absorption (λabs) and emission wavelengths (λem), are important determinants in the overall fluorophore functionality. The fluorescence quantum yield and extinction coefficient were determined as described previously,4 and the photophysical properties for Kriek and mCherry are summarized in Table 1. Consistent with previous measurements performed at pH=7.4, mCherry was found to have an extinction coefficient and quantum yield of 97,000 M-1 cm-1 and 0.23, respectively9, while Kriek had an extinction coefficient and quantum yield of 100,000 M-1 cm-1 and 0.08, respectively. It follows from these values that the brightness of Kriek (i.e., the product of the quantum yield and extinction coefficient) is therefore ~ 3-fold smaller than that of mCherry. As for the excitation and emission spectra, the absorption peak of Kriek (λabs =592 nm) is slightly red-shifted relative to mCherry (λabs =587 nm) and the fluorescence shows an enhanced Stokes shift (711 cm-1vs. 588 cm-1). Analysis of individual mutants harboring permutations of mutations found in Kriek suggests that I161M is responsible for the red-shift in the absorption and emission spectra (Table S2). The I161M mutation was also observed in the screening of libraries in which the applied selection pressure was a red-shift in the fluorescence emission wavelength.35, 36
Table 1.
Spectral Properties of mCherry and Kriek.
| RFP | λabs(n m) | λex(n m) | λem(n m) | Φfl | εmax M-1 cm-1 | Widefield Photobleaching Time-Constant(s) | Confocal Photobleaching Time-Constant(s) |
|---|---|---|---|---|---|---|---|
| mCherry | 587 | 591 | 608 | 0.23 | 97,000 | 110 | 470 (75 %) 300 (25 %) |
| Kriek | 592 | 593 | 618 | 0.08 | 100,000 | 280 | 1890 (88 %) 45 (12 %) |
Φfl is the fluorescence quantum yield, and εmax is the extinction coefficient at the absorption maximum. The extinction coefficients for mCherry and Kriek have a standard error of the mean of 9,000 and 13,000, respectively, and are statistically insignificant (paired sample t-test, p = 0.22). Widefield photobleaching was performed on immobilized E. coli, and confocal photobleaching was performed on nuclear-localized FPs in adherent HeLa cells. For widefield illumination, the gradual decrease in fluorescence intensity was fit to a monoexponential decay. For confocal illumination a biexponential fit was necessary, and the time-constants and their pre-exponential factor in parentheses are reported. The weighted time-constants of confocal photobleaching are 1700 and 600 seconds, for Kriek and mCherry, respectively.
MD Simulations of Kriek and mCherry
Our original mutation strategy was designed to rigidify the β-7/10 strand interface, thus restricting access to 3O2. To test whether the strategy altered β-7/10 strand dynamics, 100 ns MD simulations were performed for both Kriek and mCherry. The time-dependent distance between the α-carbons on residues A145 (β-7) and K198 (β-10) enabled quantification of β-7/10 interstrand dynamics (Figure 3A). Both mCherry and Kriek fluctuated between ‘closed’ (< 6 Å) and ‘open’ (> 6 Å) conformations (Figure 3B and C). Kriek was 3.5-fold more likely to exist in the ‘closed’ state than mCherry, corresponding to an increased β-7/10 stability of ΔG° ~ 3.53 J mol-1 K-1, thus largely obstructing 3O2 access. Consequently, the mutations in Kriek appear to obstruct one major pathway for 3O2 to the chromophore. However, recent MD simulations identified an additional 3O2 pathway through the bottom of the β-barrel that were largely dictated by the conformational changes of residues Q64 and F99.37 A similar analysis on Kriek suggested that Q64 and F99 were more frequently in an ‘open’ configuration than mCherry. Consequently, mitigation of β-7/10 interstrand dynamics resulted in increased 3O2 access via a separate and independent diffusion pathway.
Figure 3.
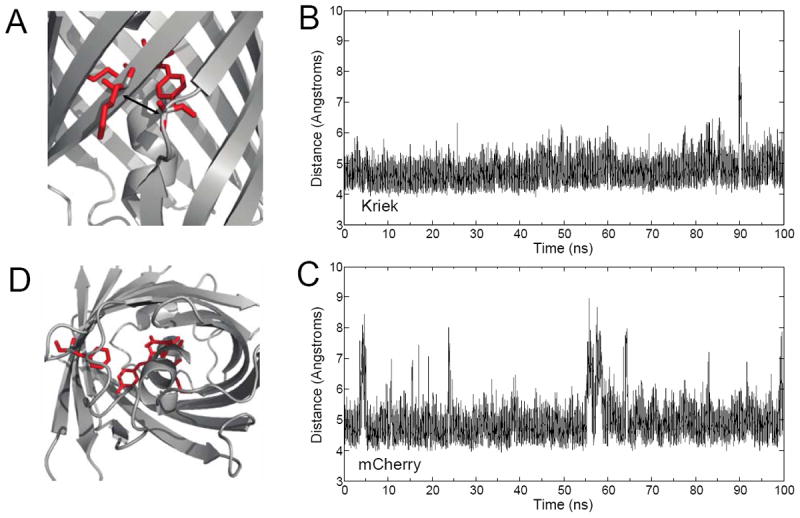
Comparative Molecular Dynamics of Kriek and mCherry. (A) Interstrand dynamics were quantitated by measuring the time-dependent distance between the α-carbons on A145 (β-7) and K198 (β-10), highlighted in red. (B) 100 ns time-trajectory of interstrand dynamics for Kriek and (C) mCherry. (D) Alternative 3O2 pathway, and ’gatekeeper’ residues, Q64, and F99, highlighted in red.
Oxygen Dependent Photobleaching and Fluorescence Lifetime
Given the results from the MD simulations, as well as the proximity of the three mutations to the 7/10 β-strand interface, we sought to evaluate our original hypothesis that decreased oxygen accessibility to the chromophore would account for the improvement in photostability. To evaluate this possibility, E. coli were transformed and allowed to adhere to poly-l-lysine coated coverslips. Photobleaching was measured in a widefield illumination mode before and after continuous purging of the environment for ~ 30 minutes with 100 % N2 for anaerobic measurements. Under anaerobic conditions, Kriek showed decreased rates of photobleaching, highlighting the importance of molecular oxygen in photodegradation processes (Figure 4A). These results suggest that photobleaching still depends on 3O2, perhaps due to access through the channel at the base of the FP as identified in the MD simulations. Time-correlated single-photon counting measurements were performed in vitro to obtain the excited-state lifetimes. Whereas mCherry has a fluorescence lifetime of 1.64 ns, Kriek has a fluorescence lifetime of 0.87 ns, almost a factor of two smaller (Figure 4B). Interestingly, because fluorescence lifetime is linearly proportional to quantum yield for fluorophores with the same radiative rate (as determined from the extinction coefficient), by this metric, Kriek is ~1.9-fold less bright than mCherry.
Figure 4.
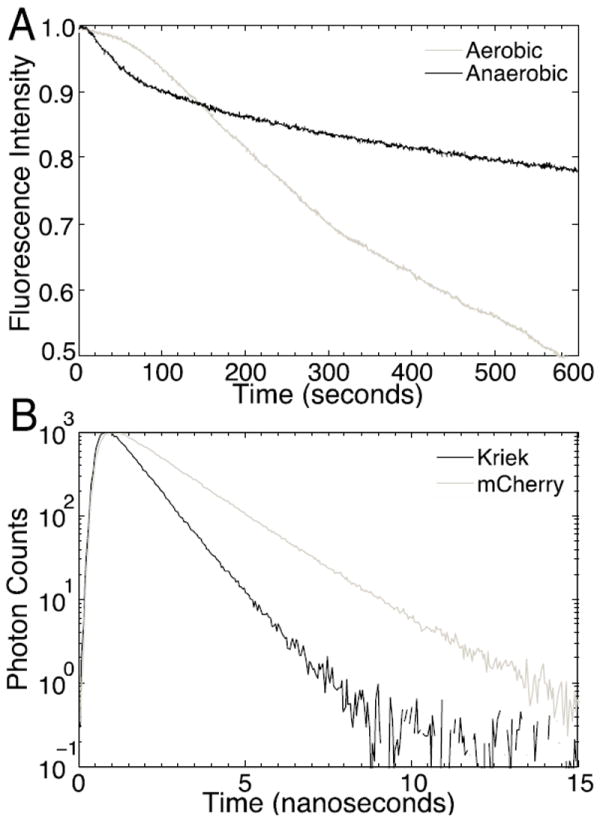
Kriek Oxygen Dependent Photobleaching and Fluorescence Lifetime. (A) Photobleaching of Kriek expressing E. coli before (aerobic) and after (anaerobic) environmental purging with N2. Purging of O2 significantly decreased the rate of photobleaching. The initial non-exponential phase of photobleaching for Kriek is attributed to fluorescent protein kindling. (B) Fluorescence lifetime measurement by time-correlated single-photon counting. Kriek has a lifetime of 0.87 ns, whereas mCherry has a lifetime of 1.64 ns.
Discussion
Here, we applied a newly developed microfluidic cytometer to screen a FP library derived from mCherry on the basis of increased photostability on exposure to 2 kW/cm2 illumination. The selection successfully revealed clones with increased photostability compared to the parent FP, demonstrating the value of expanding the repertoire of selection methods in library screening. However, Kriek also exhibited a 3- decreased quantum yield and 1.9-fold decreased fluorescence lifetime. We believe that fluorescence lifetime measurement is less sensitive to experimental error and provides a more precise assessment of the fluorescence efficiency (Note S1). Given these values, Kriek emits 30% more photons than mCherry under widefield illumination and twice as many photons than mCherry under confocal illumination. Nevertheless, the decreased quantum yield is undesirable, highlighting the challenge of simultaneously optimizing photostability and brightness, and revealing the importance of implementing multi-parameter screening tools to ensure that overall photophysical functionality is improved.
Given the success of the selection in producing an FP with higher photostability and reduced dynamics of the β-7/10 region of the barrel, our findings suggest that further selections aiming to rigidify the barrel dynamics would be productive. It is interesting to note that although the MD simulations show that β-7/10 interstrand dynamics are reduced in Kriek relative to mCherry, the observation of a larger Stokes shift in the mutant suggests that structural fluctuations coupled to the electronic transition are actually larger in Kriek than in mCherry, as the Stokes shift is a measure of the capability of the environment to reorganize in response to the electronic perturbation associated with the change in structure and electron distribution in a chromophore. The increased Stokes shift and concomitant larger structural flexibility of the environment around the chromophore in Kriek is the likely cause of the reduced excited-state lifetime. Attempts to generate red FPs with large Stokes shift, e.g. mPlum and tagRFP675, have generally resulted in chromophores with reduced fluorescence quantum yield relative to the parent FP.38-40 Thus, reducing the structural fluctuations in an FP to obtain high fluorescence quantum yield and high phostability, will likely lead to a trade-off in terms of reduced Stokes shift.
There are several potential mechanisms that could give rise to increased photostability. For example, a decrease in the lifetime of either the first excited state or the triplet state would be expected to decrease photobleaching, which typically proceeds when the chromophore is in an excited state. Other potential mechanisms include reduced excited-state absorption to higher-order singlet and triplet states, attenuated excited-state reaction pathways, including photoionization and oxidation by molecular oxygen41-48 or conversion to a photoprotective dark-state.4 Some of these mechanisms for increasing photostability would be expected to lead to a concomitant decrease in fluorescence. For example, a decrease in the lifetime of the first excited state usually results from an increase in non-radiative decay mechanisms (e.g., internal conversion), that decreases the quantum yield and hence overall brightness. On the other hand, a decrease in the triplet lifetime not only improves photostability, but also improves the fluorescence brightness, as it decreases the time spent within a dark state. Similarly, if a decrease in fluorescence lifetime is due only to an increase in the radiative decay rate, both the fluorescence brightness and the photostability will improve. Indeed, metal-enhanced fluorescence in GFP was recently shown to increase the radiative rate, improving both the brightness and photostability 6- and 10-fold, respectively.49
To examine whether the decreased fluorescence lifetime of Kriek can entirely account for improvement in photostability, a numerical simulation was performed on a simple two-state model comprised of ground and excited singlet-states, and a photodegradation path out of the first excited singlet-state (Fig. S3). In addition, the rates of excitation and photodegradation were fixed, and the rate of photodegradation was examined as a function of fluorescence lifetime. The simulation predicts that for a 2-state system with rates of photobleaching < 104 s-1 from the excited state, photobleaching scales approximately linearly with the excited-state fluorescence lifetime. These rates of photobleaching are consistent with previous work that analysed a panel of closely related red fluorescent proteins using pulsed and continuous laser illumination.4 Consequently, if all other photophysical parameters are kept constant, we would expect Kriek to photobleach ~ 2x slower than mCherry, since it has an excited-state lifetime 2x shorter than that of mCherry. Therefore, the decreased fluorescence lifetime only accounts for 50% of the gain in photostability under confocal illumination (and 80% under widefield), suggesting that under these illumination conditions, the Kriek mutations do supress photodegradation pathways compared to mCherry.
The work presented here illustrates that the microfluidic selection method offers several advantages over previous methodology. Photobleaching has intensity dependent pathways, and the ability to rapidly and quantitatively measure photobleaching in a high-throughput manner for laser intensities appropriate for confocal or single-molecule imaging is unique. Manual screening of 56,000 cells (e.g., single mammalian cells or bacterial colonies) at such high excitation intensities would be time intensive, and might be expected to result in a similar outcome. The results from this study emphasize that single parameter selections may optimize one parameter (e.g., photostability) at the expense of another (e.g., quantum yield) and that sorting based on an intrinsic parameter is more powerful than using a parameter that depends on the level of expression, such as overall brightness. It is worth emphasizing however, that our microfluidic platform is adaptable and allows for the selection parameters to increase in number and complexity as our knowledge of FP photophysics advances.
Library design is critical to the success of a selection and/or directed-evolution process. The library discussed here was guided by MD simulations and mutations reported in the literature, and predominantly involved amino-acids proximal to β-strands 7 and 10.2 However, in retrospect it seems that this region may influence fluorescence brightness and photobleaching. Future libraries should seek to identify positions that alter the photostability in a manner that is independent of the fluorescence quantum yield, or that synergistically improve photostability and fluorescence quantum yield. Efficient identification of such positions would benefit from an improved understanding of the molecular mechanisms governing irreversible photobleaching. Although further work is necessary to conclusively determine pathways by which oxygen may access the chromophore, evidence suggests that oxygen access to the chromophore pocket is a key contributor to RFP photodegradation. Unfortunately, the oxygen sensitivity of photobleaching depends not only on the rate of molecular collisions with oxygen, but also on the lifetime and occupancy of oxygen-sensitive states (e.g., the triplet state). Once improved mutants are discovered, detailed spectroscopic analysis (e.g., transient-absorption, fluorescence correlation spectroscopy, etc.) in the presence and absence of molecular oxygen could provide valuable insight into the molecular and kinetic details governing these principles.50, 51
To avoid decreased quantum yield in future selections, more emphasis should be placed on the rate of photons emitted prior to photobleaching. Total photon output depends upon the extinction coefficient, quantum yield of fluorescence, and quantum yield of photobleaching. Furthermore, time spent in dark-states also contributes to a decreased fluorescence photon flux, and thus limits signal.41, 52Consequently, to generate FPs with overall increased photon output, selections should ideally measure the quantum yield of fluorescence, the percent dark-state conversion, and the rate of irreversible photobleaching. Incorporation of additional selection pressures is anticipated to permit unequivocal advances in RFP photophysical properties.
Experimental
Microfluidic Chip, Optical Design, and Operation
Microfluidics were custom designed and commercially fabricated in an all borosilicate glass chip (Micronit). Detailed descriptions of the microfluidic chip, laser illumination, sample loading, and optical-force sorting of cells is discussed by Daviset al.3, 5 Briefly, microfluidic chips were sonicated for 1 hour with deionized water and placed in a custom polytetrafluoroethylene microfluidic manifold designed with 5 fluid reservoirs (one sample, two sheath, and two output), 1/8 inch outer diameter pneumatic quick disconnect interfaces, and mounted on an inverted microscope (Olympus IX-71). Flow was driven at the input reservoirs with high-resolution pressure controllers (Pneutronics, OEM EPS10-5-0-2), with the outlet reservoirs maintained at atmospheric pressure. To prevent non-specific cellular adhesion to the glass surfaces, microfluidic channels were passivated for 20 minutes with 0.2 μM filtered Ca2+, Mg2+, PO43- free HHBSS with 1 % bovine serum albumin (BSA, Sigma-Aldrich). To prevent obstruction of the microfluidic channels, the instrument (including microscope, microfluidic, optics, etc.) was maintained in an actively filtered HEPA environment.
For sorting of mutant FP libraries, HeLa suspension cells expressing FPs were trypsinized and placed in a solution of phenol-free media (MEM-Alpha, Gibco) at a final density of 2.5 million cells/mL with the addition of 10 % fetal bovine serum, 1 % penicillin-streptomycin, and 14 % Opti-Prep. To eliminate multicellular aggregates, cells were filtered through a 40 μm nylon mesh cell-strainer (BD Biosciences) prior to being deposited directly in the microfluidic manifold. The microfluidic sheath channels were filled with 0.2 μm filtered Ca2+, Mg2+, PO43- free HHBSS with 1 % bovine serum albumin (BSA, Sigma-Aldrich). Cells were sorted directly into conditioned DMEM with 30 % fetal bovine serum and 1 % penicillin-streptomycin, and placed into a 96-well plate for maintenance at 37° Celsius and 5 % CO2.
Library Construction
The original DNA template for mCherry9 was PCR amplified with gene-specific primers (Integrated DNA Technologies), recombined into pDonr221 (Gateway, Life Technologies), and commercially sequenced (Quintara Biosciences). Mutations were introduced using site-overlap extension with primers specific to the pDonr221 backbone (M13Fwd & Rev) and mCherry, and the mutated mCherry gene was recombined into a custom retroviral vector (pCLNCX, Imgenex; Gateway Conversion Kit, Life Technologies).21 Following electroporation of E. coli (Life Technologies, MAX Efficiency DH10B) with the DNA library, serial dilutions (10-3 and 10-4) were plated on agarose supplemented with 100 μg/mL ampicillin to estimate the electroporation efficiency. The remainder of the library was grown overnight and the plasmid DNA isolated (Midi-Prep Kit, Qiagen).
Cell Maintenance, Infection, and Enrichment of Fluorescent FPs
HeLa suspension cells were cultured in Dulbecco’s modified essential medium (DMEM) supplemented with 10 % fetal bovine serum and 1% penicillin-streptomycin, as previously described.3-5 HEK-293 FT cells were cultured similarly, using Dulbecco’s modified essential medium supplemented with 10 % fetal bovine serum (FBS), 1 % penicillin-streptomycin, sodium pyruvate, and L-glutamate. For generation of retrovirus, HEK-293 FT cells were cotransfected with pCL-Ampho, pVSV-G, and the pCLNCX-library using standard transfection reagents (Opti-MEM, Life Technologies; TransIT-LT1, Mirus Bio LLC). After 24 hours, the media was gently replaced, and after 48 hours, the virus containing supernatant was carefully filtered through a 0.45 μm cellulose acetate syringe-filter and titrated onto HeLa suspension cells in the presence of 12 μg/mL hexadimethrine bromide (Sigma-Aldrich). The multiplicity of infection was kept below 10 % to decrease the likelihood of multiple viral integrations per cell. Two days post-infection, 80,000 fluorescent HeLa suspension cells were enriched using a DakoCytomation Mo-Flo cytometer.
Identification of FP Mutants
To identify the mutant RFPs, mRNA was isolated (RNeasy Micro Kit, Qiagen), cDNA prepared using a random hexamer primer (SuperScript II, Life Technologies), and PCR amplification of the cDNA library was performed to clone the sorted RFP variants into a bacterial expression vector (pBad, Life Technologies). In each case, individual clones were isolated and submitted for commercial sequencing (Quintara Biosciences).
In Vitro Analysis of FPs
All in vitro measurements were performed as previously described with the exception that a 15 mM MOPS, 100 mMKCl, pH=7.4 (as opposed to pH=7.0) buffer was used.4
Photobleaching Measurements
Confocal photobleaching measurements were performed on a laser-scanning system (Nikon, A1R) with feedback for axial sample drift. A diffraction-limited 561 nm laser was triggered with an acousto-optic tunable filter to excite the sample, the fluorescence was collected in the epi-direction, focused through a 1.2 Airy unit pinhole, filtered with a 600/50 bandpass filter, and detected with a photomultiplier tube. The laser power at the sample plane was ~ 600 μW. The image size was 512 ×512, and images were collected in a bidirectional scanning mode with the non-resonant galvanometer-based scan-head. For widefield photobleaching measurements, illumination was provided with a metal halide lamp equipped with a shutter for rapid exposure and a liquid light guide for uniform illumination. Excitation was spectrally filtered through a 562/40 bandpass filter (Semrock) and reflected off of a 593 nm dichroic (Semrock) into the back-aperture of the objective; fluorescence was collected in the epi-direction, and emission filtered with a 624/40 bandpass filter (Semrock) prior to imaging with a EMCCD detector (iXon X3 DU897, Andor). In each case, the signal was corrected for background and normalized. For anaerobic measurements, E. coli were immobilized within a glass-bottomed 96-well plate using poly-l-lysine, and the environment surrounding the plate was purged with N2 for 30 minutes using a stage-top environmental chamber (Pathology Devices). During the course of experiments, replacement of the microscope lamp for widefield photobleaching measurements (from an arc to a high-efficiency lamp) rendered small differences in the observed photobleaching decay (Figures 2C and 4A).
MD Simulations
Time series trajectories were obtained from explicit solvent, all-atom simulations using the NAMD molecular dynamics package17 with the CHARMM27 force field.18 The initial X-ray crystallographic structure of mCherry was obtained from the Protein Data Bank (PDB code 2H5Q) and the missing residues were inserted using MODELLER.53 The Kriek structure was obtained by replacing three residues W143I, I161M, and Q163V in mCherry. Force field parameters for the mature chromophore were adopted from the anionic GFP chromophore developed by Reuter et al.54 and from CHARMM27 parameters for acylimine nitrogen. The deprotonated, anionic form of the chromophore in the ground state was used. In addition, E215 was protonated using a patch. The VMD package55 was used to setup the system for simulations. The initial structures of mCherry and Kriek with crystallographic water molecules were solvated using VMD with box cutoff set to 10 Å, which resulted in a simulation box of dimensions 83.3×75.6×63.4 Å for each variant. Each solvated system was electrically neutralized by randomly adding six Na+ ions in the bulk water. The particle mesh Ewald method56 was used to treat long-range interactions with a 12 Å nonbonded cutoff. A 100-ps energy minimization was performed using the conjugate gradient and line search algorithm. The system was then heated for 90 ps with a linear gradient of 20K/6 ps from 20 to 300 K. At 300 K, each system was equilibrated for 910 ps with a 2 fs integration time step in the NVT (constant number, volume, and temperature) ensemble, followed by a 100-ns production run using NVT dynamics with 2 fs time steps. Langevin dynamics was used to maintain the temperature at 300 K.
Supplementary Material
Acknowledgments
The work presented here received generous funding from the National Institutes of Health (GM083849, A.E.P. and R.J., and GM096903 to P.C.) and the National Science Foundation (NSF) Physics Frontier Center at JILA. K.M.D. and J.L.L. were individually supported by the University of Colorado Molecular Biophysics Training Grant (T32 G-065103) and K.M.D. received additional support through the NSF Computational Optical Sensing and Imaging Integrative Graduate Education and Research Traineeship (0801680). R.J. is a staff member in the Quantum Physics Division of the National Institute of Standards and Technology (NIST). We would like to thank Dr Roger Tsien for kindly providing us with the DNA for mCherry. Furthermore, we would like to thank Patrick Konold and Dr Erik Holmstrom for their assistance with the fluorescence emission and lifetime measurements, respectively. Experimental equipment and reagents identified within this paper are provided so that this work may be replicated elsewhere and are not recommended or endorsed by NIST. Renditions of the protein structures were created using PyMOL.
References
- 1.Shu X, Shaner NC, Yarbrough CA, Tsien RY, Remington SJ. Biochemistry. 2006;45:9639–9647. doi: 10.1021/bi060773l. [DOI] [PubMed] [Google Scholar]
- 2.Chapagain PP, Regmi CK, Castillo W. The Journal of chemical physics. 2011;135:235101. doi: 10.1063/1.3660197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis LM, Lubbeck JL, Dean KM, Palmer AE, Jimenez R. Lab on a chip. 2013;13:2320–2327. doi: 10.1039/c3lc50191d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dean KM, Lubbeck JL, Binder JK, Schwall LR, Jimenez R, Palmer AE. Biophysical journal. 2011;101:961–969. doi: 10.1016/j.bpj.2011.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lubbeck JL, Dean KM, Ma H, Palmer AE, Jimenez R. Analytical chemistry. 2012;84:3929–3937. doi: 10.1021/ac202825z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sengupta P, van Engelenburg SB, Lippincott-Schwartz J. Chemical reviews. 2014;114:3189–3202. doi: 10.1021/cr400614m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harms GS, Cognet L, Lommerse PH, Blab GA, Schmidt T. Biophysical journal. 2001;80:2396–2408. doi: 10.1016/S0006-3495(01)76209-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eggeling C, Widengren J, Rigler R, Seidel CA. Analytical chemistry. 1998;70:2651–2659. doi: 10.1021/ac980027p. [DOI] [PubMed] [Google Scholar]
- 9.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Nature biotechnology. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 10.Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. The Journal of biological chemistry. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- 11.Bevis BJ, Glick BS. Nature biotechnology. 2002;20:83–87. doi: 10.1038/nbt0102-83. [DOI] [PubMed] [Google Scholar]
- 12.Subach FV, Piatkevich KD, Verkhusha VV. Nature methods. 2011;8:1019–1026. doi: 10.1038/nmeth.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goedhart J, van Weeren L, Hink MA, Vischer NO, Jalink K, Gadella TW., Jr Nature methods. 2010;7:137–139. doi: 10.1038/nmeth.1415. [DOI] [PubMed] [Google Scholar]
- 14.Goedhart J, von Stetten D, Noirclerc-Savoye M, Lelimousin M, Joosen L, Hink MA, van Weeren L, Gadella TW, Jr, Royant A. Nature communications. 2012;3:751. doi: 10.1038/ncomms1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiolka R, Shao L, Rego EH, Davidson MW, Gustafsson MG. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5311–5315. doi: 10.1073/pnas.1119262109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, Tsien RY. Nature methods. 2008;5:545–551. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Journal of computational chemistry. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. Journal of computational chemistry. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Habuchi S, Cotlet M, Gensch T, Bednarz T, Haber-Pohlmeier S, Rozenski J, Dirix G, Michiels J, Vanderleyden J, Heberle J, De Schryver FC, Hofkens J. Journal of the American Chemical Society. 2005;127:8977–8984. doi: 10.1021/ja047023o. [DOI] [PubMed] [Google Scholar]
- 20.Adam V, Carpentier P, Violot S, Lelimousin M, Darnault C, Nienhaus GU, Bourgeois D. Journal of the American Chemical Society. 2009;131:18063–18065. doi: 10.1021/ja907296v. [DOI] [PubMed] [Google Scholar]
- 21.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 22.Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Engelenburg SB, Nahreini T, Palmer AE. Chembiochem : a European journal of chemical biology. 2010;11:489–493. doi: 10.1002/cbic.200900689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov SA. Nature biotechnology. 1999;17:969–973. doi: 10.1038/13657. [DOI] [PubMed] [Google Scholar]
- 25.Baird GS, Zacharias DA, Tsien RY. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11984–11989. doi: 10.1073/pnas.97.22.11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alieva NO, Konzen KA, Field SF, Meleshkevitch EA, Hunt ME, Beltran-Ramirez V, Miller DJ, Wiedenmann J, Salih A, Matz MV. PloS one. 2008;3:e2680. doi: 10.1371/journal.pone.0002680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai M, Fisher HE, Temirov J, Kiss C, Phipps ME, Pavlik P, Werner JH, Bradbury AR. Protein engineering, design & selection : PEDS. 2007;20:69–79. doi: 10.1093/protein/gzl056. [DOI] [PubMed] [Google Scholar]
- 28.Ward WW, Cormier MJ. The Journal of biological chemistry. 1979;254:781–788. [PubMed] [Google Scholar]
- 29.Carpentier P, Violot S, Blanchoin L, Bourgeois D. FEBS letters. 2009;583:2839–2842. doi: 10.1016/j.febslet.2009.07.041. [DOI] [PubMed] [Google Scholar]
- 30.Bulina ME, Chudakov DM, Britanova OV, Yanushevich YG, Staroverov DB, Chepurnykh TV, Merzlyak EM, Shkrob MA, Lukyanov S, Lukyanov KA. Nature biotechnology. 2006;24:95–99. doi: 10.1038/nbt1175. [DOI] [PubMed] [Google Scholar]
- 31.Tubbs JL, Tainer JA, Getzoff ED. Biochemistry. 2005;44:9833–9840. doi: 10.1021/bi0472907. [DOI] [PubMed] [Google Scholar]
- 32.Strack RL, Strongin DE, Mets L, Glick BS, Keenan RJ. Journal of the American Chemical Society. 2010;132:8496–8505. doi: 10.1021/ja1030084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pletnev S, Subach FV, Dauter Z, Wlodawer A, Verkhusha VV. Journal of the American Chemical Society. 2010;132:2243–2253. doi: 10.1021/ja908418r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kremers GJ, Hazelwood KL, Murphy CS, Davidson MW, Piston DW. Nature methods. 2009;6:355–358. doi: 10.1038/nmeth.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chica RA, Moore MM, Allen BD, Mayo SL. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20257–20262. doi: 10.1073/pnas.1013910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Jackson WC, Steinbach PA, Tsien RY. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:16745–16749. doi: 10.1073/pnas.0407752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Regmi CK, Bhandari YR, Gerstman BS, Chapagain PP. The journal of physical chemistry B. 2013;117:2247–2253. doi: 10.1021/jp308366y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konold P, Regmi CK, Chapagain PP, Gerstman BS, Jimenez R. The Journal of Physical Chemistry B. 2014;118:2940–2948. doi: 10.1021/jp412371y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L, Jackson WC, Steinbach PA, Tsien RY. Proceedings of the National Academy of Sciences. 2004;101:16745–16749. doi: 10.1073/pnas.0407752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piatkevich KD, Malashkevich VN, Morozova KS, Nemkovich NA, Almo SC, Verkhusha VV. Scientific reports. 2013;3:1847. doi: 10.1038/srep01847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donnert G, Eggeling C, Hell SW. Nature methods. 2007;4:81–86. doi: 10.1038/nmeth986. [DOI] [PubMed] [Google Scholar]
- 42.Widengren J, Chmyrov A, Eggeling C, Lofdahl PA, Seidel CA. The journal of physical chemistry A. 2007;111:429–440. doi: 10.1021/jp0646325. [DOI] [PubMed] [Google Scholar]
- 43.Greenbaum L, Rothmann C, Lavie R, Malik Z. Biological chemistry. 2000;381:1251–1258. doi: 10.1515/BC.2000.153. [DOI] [PubMed] [Google Scholar]
- 44.Jimenez-Banzo A, Nonell S, Hofkens J, Flors C. Biophysical journal. 2008;94:168–172. doi: 10.1529/biophysj.107.107128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vogelsang J, Kasper R, Steinhauer C, Person B, Heilemann M, Sauer M, Tinnefeld P. Angewandte Chemie. 2008;47:5465–5469. doi: 10.1002/anie.200801518. [DOI] [PubMed] [Google Scholar]
- 46.Eggeling C, Volkmer A, Seidel CAM. ChemPhysChem. 2005;6:791–804. doi: 10.1002/cphc.200400509. [DOI] [PubMed] [Google Scholar]
- 47.Schuster Jr, Cichos F, von Borczyskowski C. Applied Physics Letters. 2005;87:051915. [Google Scholar]
- 48.Zondervan R, Kulzer F, Orlinskii SB, Orrit M. The Journal of Physical Chemistry A. 2003;107:6770–6776. [Google Scholar]
- 49.Fu Y, Zhang J, Lakowicz JR. Biochemical and biophysical research communications. 2008;376:712–717. doi: 10.1016/j.bbrc.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heikal AA, Hess ST, Baird GS, Tsien RY, Webb WW. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11996–12001. doi: 10.1073/pnas.97.22.11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hendrix J, Flors C, Dedecker P, Hofkens J, Engelborghs Y. Biophysical journal. 2008;94:4103–4113. doi: 10.1529/biophysj.107.123596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dickson RM, Cubitt AB, Tsien RY, Moerner WE. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 53.Eswar N, Webb B, MartiRenom MA, Madhusudhan MS, Eramian D, Shen M-y, Pieper U, Sali A. 2006 doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reuter N, Lin H, Thiel W. The Journal of Physical Chemistry B. 2002;106:6310–6321. [Google Scholar]
- 55.Humphrey W, Dalke A, Schulten K. Journal of molecular graphics. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 56.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. The Journal of chemical physics. 1995;103:8577. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.