

Abstract
Facet joint injury induces persistent pain that may be maintained by structural plasticity in the spinal cord. Astrocyte-derived thrombospondins, especially thrombospondin-4 (TSP4), have been implicated in synaptogenesis and spinal sensitization in neuropathic pain, but the TSP4 response and its relationship to synaptic changes in the spinal cord have not been investigated for painful joint injury. This study investigates the role of TSP4 in the development and maintenance of persistent pain following injurious facet joint distraction in rats and tests the hypothesis that excitatory synaptogenesis contributes to such pain. Painful facet joint loading induces dorsal horn excitatory synaptogenesis along with decreased TSP4 in the DRG and increased astrocytic release of TSP4 in the spinal cord, all of which parallel the time course of sustained tactile allodynia. Blocking injury-induced spinal TSP4 expression with antisense oligonucleotides or reducing TSP4 activity at its neuronal receptor in the spinal cord with gabapentin treatment both attenuate the allodynia and dorsal horn synaptogenesis that develop after painful facet joint loading. Increased spinal TSP4 also facilitates the development of allodynia and spinal hyperexcitability, even after non-painful physiologic loading of the facet joint. These results suggest that spinal TSP4 plays an important role in the development and maintenance of persistent joint-mediated pain by inducing excitatory synaptogenesis and facilitating the transduction of mechanical loading of the facet joint that leads to spinal hyperexcitability.
Keywords: facet joint, allodynia, gabapentin, synaptogenesis, thrombospondin-4
Introduction
Chronic spine pain is a prevalent and costly medical problem. The cervical facet joints are common sources of chronic pain, and are susceptible to injury during neck trauma and degeneration (Hogg-Johnson et al., 2008). Abnormal motions of the cervical facets can load the joint tissues that are innervated by mechanoreceptive and nociceptive fibers, including the facet capsular ligament (Pearson et al., 2004; Siegmund et al., 2009). Facet joint loading that produces pain initiates a host of changes in the spinal cord that are characteristic of central sensitization, including hyperexcitability and increased spontaneous activity in dorsal horn neurons (Crosby et al., 2013; Lee et al., 2008; Quinn et al., 2010). Effective clinical treatment requires a better understanding of the spinal mechanisms contributing to central sensitization from joint pain.
Structural plasticity is one mechanism by which pain is maintained through the potentiation of both nociceptive and non-nociceptive pathways (Jaken et al., 2010; Latremoliere and Woolf, 2009; Woolf et al., 1992). Although increases in synapse number are observed in the dorsal horn in neuropathic pain states (Jaken et al., 2010; Lin et al., 2011; Peng et al., 2010), the spinal signals initiating synaptogenesis remain unclear. One family of extracellular matrix proteins, the thrombospondins (TSPs), promotes synaptogenesis (Christopherson et al., 2005; Eroglu et al., 2009; Lo et al., 2011). The thrombospondins are glycoproteins that are produced by numerous cell types and have widely varying roles in cell signaling (Adams, 2001; Adams and Lawler, 2004). For example, platelets, fibroblasts, skeletal muscle, endothelial cells, and astrocytes and neurons in the CNS each express one or more TSP isoforms (Adams, 2001; Arber and Caroni, 1995; Christopherson et al., 2005; Stenina et al., 2008). Expression of TSPs can be radically altered in pathophysiological conditions (Adams and Lawler, 2004; Chen et al., 2000). In particular, thrombospondin-4 (TSP4) is upregulated in spinal astrocytes following nerve ligation and enhances excitatory synaptic transmission in the dorsal horn, but spinal TSP1 and TSP2 are unchanged after that injury (Kim et al., 2012). Despite its potential involvement in the development of pain from neural trauma, the role of TSP4 in joint-mediated pain and synaptogenesis is unknown.
Given the involvement of thrombospondin-4 in spinal sensitization and neuropathic pain (Kim et al., 2012), and its potential to induce synaptogenesis (Christopherson et al., 2005), we hypothesized that TSP4 plays a role in joint-mediated pain by promoting dorsal horn excitatory synaptogenesis following painful facet joint loading. The temporal regulation of TSP4 is unknown after injurious facet joint distraction, so TSP4 expression was evaluated at day 1 and day 7. However, synaptogenesis has been reported to occur by 3–5 days after a peripheral nerve injury (Lo et al., 2011). As such, excitatory synaptogenesis was evaluated at day 7 following injurious joint loading, which corresponds to a time after the reported 3–5 day development period for new synapses and when behavioral sensitivity and spinal neuronal hyperexcitability are observed after injurious facet joint loading (Crosby et al., 2014; Quinn et al., 2010). Spinal TSP4 levels were then modulated to directly assess the role of TSP4 in synaptogenesis and joint-mediated pain after different severities of facet joint distraction that simulate injurious or physiologic loading of tissues in the joint. Findings indicate that TSP4 promotes excitatory synaptogenesis and neuronal hyperexcitability in the spinal cord, and is required for the development of persistent pain after facet joint loading that is injurious.
Materials and Methods
Facet joint distraction procedures
All surgical procedures were performed using adult male Holtzman rats (362–464g) under inhalation isoflurane anesthesia (4% induction, 2–3% maintenance). Facet joint loading was performed using a distraction of the bilateral C6/C7 facet joints, which has been described previously to produce physiologic or injurious tissue loading under different magnitudes of distraction (Dong and Winkelstein, 2010; Lee et al., 2004; Lee and Winkelstein, 2009). Briefly, the overlying paraspinal musculature was separated to expose the vertebrae from C4-T1. The C6 and C7 vertebrae were attached to a custom loading device using microforceps and the C6 vertebra was distracted rostrally to stretch the facet capsule across the C6/C7 joints (Figure 1). Distraction of 0.2mm was applied for physiologic loading and 0.7mm imposed for injurious loading, in separate rats. Bead markers were placed on the C6 and C7 vertebrae, as well as in a grid on the right C6/C7 facet capsule, and were tracked with a Phantom v4.3 CCD camera (Figure 1) (Vision Research, Wayne, IN) during distraction of the joint. Using the marker locations before joint loading and at the peak of loading, both the vertebral and facet capsule displacements were quantified, and the maximum principal capsular strain was calculated during each joint distraction (Figure 1). Sham surgery included all of the same procedures with no joint distraction applied and controlled for the effect of the surgical procedures. Following surgery, the incision was closed with 3-0 polyester suture and surgical staples, and rats recovered under supervision in room air.
Figure 1.

Characterization of the mechanical loading of the C6/C7 facet joint. (A) Injurious loading (Inj) imposes greater C6/C7 vertebral distraction and generates higher maximum principal strains (MPS) in the facet capsule than does physiologic loading (Phys). (B,C) Vertebral distraction, capsule distraction, and load across the facet joint are significantly higher during injurious loading than physiologic loading. Data are mean±SD (n=18–19 rats per group). *p<0.0001 compared by Student’s t-test. (D) Representative images of the facet joint are shown before distraction (unloaded), and at the peak of injurious loading of the joint. The maximum principal strain field is shown for the corresponding loaded facet capsule of a representative rat, with injurious joint loading inducing 0.92mm of vertebral distraction and a mean MPS of 34%.
Assessment of behavioral sensitivity
Tactile allodynia was measured in the forepaws of each rat by quantifying the paw withdrawal threshold (PWT) during application of a series of von Frey filaments, with increasing strength (1.4, 2, 4, 6, 8, 10, 15, and 26g), to the plantar surface of the forepaw (Lee and Winkelstein, 2009). Each filament weight was applied five times, and a positive response was recorded if a rat responded to von Frey stimulation by licking, shaking, or withdrawing the forepaw. If a rat displayed a positive response to two consecutive filaments, the lower weight filament was taken as the paw withdrawal threshold. Rats not responding to any of the filaments were assigned the maximum threshold of 26g. Testing was repeated in three rounds on each day, separated by at least 10 minutes, and the average threshold from the three rounds was calculated for each rat.
Western blot analysis of DRG & spinal cord tissue
Rats were anesthetized with sodium pentobarbital (65mg/kg, i.p.) and transcardially perfused with 300mL of PBS before removing the spinal cord the C6 and C7 level and the DRG at C6. Tissue samples were homogenized in lysis buffer containing 50mM Tris HCl (pH 8.0), 1% Triton X-100, 150mM NaCl, 1mM EDTA, and protease and phosphatase inhibitors (Sigma-Aldrich Corp.; St. Louis, MO). Protein samples (25μg for DRG, 50μg for spinal cord) were loaded on a polyacrylamide gel for SDS-PAGE (Invitrogen; Carlsbad, CA) and run for 75 minutes at 150V. Protein was transferred to a polyvinylidene difluoride (PVDF) membrane using an iBlot (Invitrogen; Carlsbad, CA) and was blocked for 1 hour in 5% dry-milk blocking reagent in 0.1% Tween-20 Tris-buffered saline (TBS-Tween). The membrane was incubated overnight at 4°C with rabbit anti-thrombospondin-4 (TSP4) antibody (1:1000; Santa Cruz; Dallas, TX) and mouse anti-β-tubulin (1:2000; Covance; Princeton, NJ). The PVDF membrane was washed in TBS-Tween three times for 10 minutes each time, followed by 2 hour incubation at room temperature with goat anti-rabbit 800 and goat anti-mouse 680 IRDye fluorescent secondary antibodies (1:10,000; Li-Cor Biosciences; Lincoln, NE). The membrane was imaged using an Odyssey Imaging System (Li-Cor Biosciences; Lincoln, NE). The fluorescence of the TSP4 band was analyzed using the Odyssey 2.1 software to quantify the intensity of the pixels within the bands and normalized to the corresponding β-tubulin fluorescence as a protein loading control for each sample.
Immunofluorescent labeling of spinal cord tissue
Rats were anesthetized with sodium pentobarbital (65mg/kg, i.p.) and transcardially perfused with 250mL of PBS followed by 250mL of 4% paraformaldehyde (PFA). The C6 and C7 spinal cord segments were removed and post-fixed in 4% PFA overnight, then cryopreserved in 30% sucrose in PBS for 7 days at 4°C. Samples were freeze-mounted in OCT medium (Fisher Scientific, Waltham, MA) and axial cryosections (14μm each, 5–6 per rat) were mounted on Superfrost Plus slides (Fisher Scientific) for immunolabeling. For labeling of TSP4 and glial fibrillary acidic protein (GFAP), slides were blocked in 10% normal goat serum in PBS for 2 hours at room temperature. Slides were then incubated overnight at 4°C with primary antibodies in 10% goat serum with 0.3% Triton-X PBS. Primary antibodies included chicken anti-TSP4 (1:1000; previously described in Dunkle et al. (2007)), and rabbit anti-GFAP (1:500; Dako, Denmark). Sections were incubated for 2 hours at room temperature with goat anti-chicken Alexa 488 and goat anti-rabbit Alexa 568 fluorescent secondary antibodies (1:1000, Invitrogen; Carlsbad, CA), and incubated with DAPI (1:10,000, Invitrogen) for 10 minutes. After rinsing in PBS and deionized water, slides were air-dried and cover slips were applied with Fluorogel (EMS; Hatfield, PA). Each tissue section was imaged at 20× using a Zeiss LSM510 confocal microscope. Separate images of sub-regions of the spinal cord (superficial dorsal horn, deep dorsal horn, and dorsal column) were analyzed by densitometry using MATLAB code to quantify the percent of TSP4- or GFAP-positive or colocalized pixels in each dorsal horn region (Dong et al., 2013b ; Hubbard et al., 2008).
Synapse quantification in the dorsal horn
Sections were immunolabeled as described above with mouse anti-synapsin (1:100; Synaptic Systems; Goettingen, Germany) and rabbit anti-homer antibodies (1:200; Synaptic Systems). For each spinal cord section, one image stack was acquired from each of the superficial dorsal horn (laminae I–II) and deep dorsal horn (laminae IV–VI) at 0.33μm increments up to 3μm of depth using a Zeiss LSM510 confocal microscope. The image stacks were acquired from the same locations in each section based on the cytoarchitecture of the dorsal horn, so the areas that were analyzed were consistent in anatomical location across sections and rats. The maximum intensity projection of each set of 3 sequential images was used to generate a single image (0.02mm2 tissue area) with 1μm of tissue depth. Because irregular surface topology of the spinal cord sections and lack of antibody penetration led to large variations in labeling efficiency in the outermost and innermost maximum intensity projections, only the middle projection from each spinal cord section was analyzed, corresponding to tissue at 1–2μm from the surface of the section. The Puncta Analyzer plugin for ImageJ (National Institutes of Health; Bethesda, MD) was used to identify puncta exhibiting colocalization of the pre- and post-synaptic markers in each maximum intensity projection, using previously published methods (Ippolito and Eroglu, 2010). The area of the tissue parenchyma, excluding any holes and/or gaps in the tissue sections, was quantified using a customized MATLAB code. The number of colocalized puncta in each of the superficial or deep dorsal horn was normalized to the corresponding tissue area for that region. The synapse number per area was then averaged across all rats in each treatment group.
Intrathecal injections
Rats were anesthetized with isoflurane (4% for induction, 2–3% for maintenance) and placed in a prone position. The skin over the lumbar spine was shaved and cleaned with Betadyne solution, and a 30-gauge needle attached to a microinjector (Hamilton; Reno, NV) was inserted vertically along the midline at the gap between the L5 and L6 spinous processes. The needle was angled approximately 30° to the right or left and inserted into the spinal canal between the laminae of the L5 and L6 vertebrae, with placement in the cauda equina confirmed by the presence of a reflexive tail flick or hindpaw flinch. Antisense or mismatch oligonucleotides, gabapentin, saline, recombinant TSP4, or StrepII peptide were administered in single bolus injections. Each of these injectates were delivered intrathecally in the lumbar region for safety reasons of avoiding the spinal cord, and because that method has been shown to effectively deliver agents to the cervical spinal cord in the rat (Dong et al., 2013a; Kras et al., 2013). The needle was held in place for 30–60 seconds before removing to prevent leakage from the lumbar spine. Rats were then monitored during recovery in room air.
Oligonucleotide treatments
This study used antisense oligonucleotides that were previously reported to block TSP4 expression (Kim et al., 2012). Antisense oligonucleotides were designed against a segment of the TSP4 mRNA using the sequence, CCATCATTGTTGCTATCTTCC, with stabilizing modifications as described previously (Kim et al., 2012). An equal length random sequence of nucleotides, ACCATCGTTGTTACTTTCTCC, was used as a mismatch control for the oligonucleotide treatment. Antisense (75μg/rat/day) and mismatch (75μg/rat/day) oligonucleotides were delivered by bolus 25μL intrathecal injections. Injections were administered daily beginning 3 days before injurious facet joint distraction and continuing through day 7 after the joint distraction, when spinal cord tissue was harvested.
Gabapentin treatment
Rats received either gabapentin or a saline vehicle treatment in bolus intrathecal injections. Gabapentin (4.2μg in 30μL saline) was administered 90 minutes prior to injurious facet joint distraction. A second dose was administered 1 day after injurious joint distraction, immediately following behavioral assessment on that day. Saline vehicle injections (30μL) were administered under the same paradigm to control for the effects of the injections. The gabapentin concentration and treatment times were optimized previously in dose-response studies (Dong et al., 2013a).
Recombinant TSP4 purification
Full length TSP4 protein was recombinantly expressed in 293-Ebstein-Barr nuclear antigen (EBNA) cells using previously described methods by Hansen et al. (2011) for COMP, another TSP family member. Briefly, 293-EBNA cells were stably transfected with cDNA for rat TSP4 (GenBank accession no. X89963) with a StrepII tag. Cell culture media were collected every other day and stored at −20°C with 1.2mM N-ethylmaleimide (NEM) and 1.2mM phenylmethylsulfonyl fluoride (PMSF) to inhibit protease activity. Secreted TSP4-StrepII proteins in the cell media were purified with a Streptactin affinity chromatography column (1.5mL; IBA; Goettingen, Germany), then concentrated from the elution fractions using Amicon Ultra centrifugal filters with a 50kDa threshold (Millipore; Billerica, MA) and stored at −80°C. To assess behavioral sensitivity after increases in spinal TSP4, purified recombinant TSP4 (20, 30, 45, or 60μg/rat) was administered intrathecally in bolus injections in 25μL, and allodynia was assessed for 10 days after TSP4 injection. In separate rats, the effect of increased spinal TSP4 on the development of allodynia after joint loading was tested by intrathecally injecting 20μg or 60μg TSP4 3 days before injurious or physiologic facet joint loading. In both studies, intrathecal injection of 60μg StrepII peptide was used to control for the presence of the StrepII tag on recombinant TSP4 protein.
Electrophysiological recording of spinal dorsal horn neurons
Rats were anesthetized with sodium pentobarbital (45mg/kg, i.p.) and given supplementary doses (5–10mg/kg, i.p.) as needed based on toe pinch reflexes. A bilateral laminectomy and dural resection were performed at C6 and C7 to expose the spinal cord. Rats were then immobilized on a stereotaxic frame using ear bars and a vertebral clamp at T2 to stabilize the cervical spine (David Kopf Instruments; Tujunga, CA). Core temperature was maintained at 35–37°C using a temperature controller with a rectal probe (Physitemp; Clifton, NJ). The exposed spinal cord was bathed in 37°C mineral oil for the entire period when spinal neuronal recordings were being performed to prevent drying.
Extracellular potentials were recorded from dorsal horn neurons by lowering a carbon fiber electrode (Kation; Minneapolis, MN) through the pial surface of the C6 or C7 spinal cord with a micropositioner (Narishige; Tokyo, Japan). Neurons were identified by brushing the plantar surface of the forepaw, and were then mechanically stimulated at the forepaw with 10 seconds of light brushing, five consecutive 1-second stimulations at 1-second intervals with a series of von Frey filaments (1.4g, 4g, 10g, 26g), and, finally, 10 seconds of noxious pinch by a 60g vascular clip (Crosby et al., 2013; Crosby et al., 2014; Hains et al., 2003; Quinn et al., 2010). Voltage recordings from each neuron were spike-sorted using Spike2 software (CED; Cambridge, UK) to ensure that spikes from only one neuron were considered at a time. For the brush and the pinch stimuli, spikes were summed over the 10 seconds that the stimulus was administered. For the von Frey filament stimuli, spikes were considered to be evoked by a single von Frey stimulus if they occurred during the 1-second application or the 1-second rest period immediately following its application (Quinn et al., 2010); each filament had a total of five spike counts since each filament was applied to the forepaw five times. Baseline spike counts recorded prior to stimulation were subtracted from the spike counts for each stimulus to isolate the evoked response, and spike counts for each stimulus were log-transformed due to a positive skew in the distributions of the spike totals.
Results
Facet joint distraction inducing tactile allodynia also increases excitatory synapses in the spinal dorsal horn
Rats developed tactile allodynia at day 1 that persisted through day 7 after injurious loading to the bilateral C6/C7 facet joints (Figure 2A). To determine whether the number of dorsal horn synapses was altered in rats with sustained allodynia, we quantified synapses in the C6/C7 spinal dorsal horn 7 days after painful joint distraction by immunolabeling the pre- and post-synaptic markers synapsin and homer, respectively (Ippolito and Eroglu, 2010; Ullian et al., 2001). Because synapsin is a general synaptic marker and homer is a glutamatergic synaptic marker (Brakeman et al., 1997; De Camilli et al., 1983), excitatory synapses were quantified by counting the colocalization of synaptic puncta. The number of excitatory synapses increased in the superficial dorsal horn after painful joint loading (Figures 2B–2D), supporting that injury-induced synaptogenesis in the dorsal horn is evident with sustained allodynia after facet joint injury. However, given that allodynia is observed at day 1, the temporal mechanism by which this synaptogenesis develops is not known.
Figure 2.

Tactile allodynia and synaptogenesis after painful C6/C7 facet joint loading. (A) Allodynia is evident beginning at day 1 after injurious C6/C7 facet joint loading (Injury), measured as a decrease in the paw withdrawal threshold (PWT). PWT is presented as mean±SEM (n=6–9 rats per group). *p<0.0001 compared with baseline, #p<0.024 compared with sham by repeated-measures ANOVA with post-hoc Tukey’s HSD test. (B) Quantification of excitatory synapses in the superficial or deep laminae of the C6 dorsal horn (DH). Excitatory synapse density increases in the superficial DH after painful joint injury. Groups are displayed as fold-change over sham±SEM (n=3–5 sections per rat, 4–6 rats per group). *p=0.032 compared with sham by Student’s t-test. (C,D) Immunofluorescent labeling of synapsin (red) and homer (green) in the C6/C7 superficial dorsal horn after sham (C) or painful facet joint loading (D). Colocalization (merged, yellow) represents synaptic puncta (arrows). Scale bar=20μm, inset scale bar=5μm.
Altered expression of synaptogenic TSP4 parallels development of sustained tactile allodynia
We quantified the expression profile of the synaptogenic protein, TSP4, after painful facet joint loading to determine whether TSP4 dysregulation may contribute to tactile allodynia in joint pain. Using Western blot assays, TSP4 expression was decreased in the C6 DRG and increased in the C6/C7 spinal cord at day 1, but not significantly (p=0.054 for DRG, p=0.09 for spinal cord). TSP4 was significantly decreased in the DRG and increased in the spinal cord at day 7 after painful joint distraction (Figures 3A & 3B).
Figure 3.

TSP4 protein expression after painful C6/C7 facet joint loading. (A) Expression of TSP4 decreases in the C6 dorsal root ganglion (DRG) 7 days after painful joint loading (Injury). *p=0.012 compared with sham by two-way ANOVA with post-hoc Tukey’s HSD test. (B) Spinal TSP4 expression increases 7 days after painful joint loading. *p=0.002 compared with sham by two-way ANOVA with post-hoc Bonferroni test. For Western blot quantification, TSP4 bands are normalized to β-tubulin as a loading control; data are presented as mean±SEM (n=6–9 rats per group), normalized to sham. (C–H) Immunolabeling of TSP4 (green) and GFAP (red) in the superficial dorsal horn (C,F), deep dorsal horn (D,G), and dorsal column (E,H), after sham or injurious joint distraction. Scale bar=50μm, inset scale bar=10μm. Densitometry quantification of TSP4-positive pixels (I), GFAP-positive pixels (J), and their co-localization (yellow) (K). Groups are mean±SEM (n=4–6 sections per rat, 4–6 rats per group). *p<0.024 compared with sham by Student’s t-test.
Region-specific expression of TSP4 was quantified on day 7 after painful facet joint distraction by immunofluorescent labeling in the superficial dorsal horn, deep dorsal horn, and dorsal columns of the C6/C7 spinal cord (Figures 3C–3H). The specificity of the TSP4 antibody was previously characterized (Dunkle et al., 2007). Because astrocyte-derived TSPs induce CNS synaptogenesis (Christopherson et al., 2005), and nerve injury-induced spinal TSP4 expression is mainly in astrocytes (Kim et al., 2012), spinal cord sections were co-labeled with GFAP to quantify astrocytic TSP4 expression after painful facet joint loading. The total TSP4 labeling increased after painful joint loading only in the dorsal columns (Figure 3I). The amount of co-localized GFAP and TSP4 (indicating astrocytic TSP4) increased in both the superficial dorsal horn and the dorsal columns (Figure 3K). These results support that spinal TSP4 expression is altered following injurious facet joint distraction; in particular, astrocytic expression of TSP4 increases in the dorsal regions of the spinal cord where it may potentiate signaling of the nociceptive primary afferent fibers terminating in the superficial dorsal horn.
Blocking spinal TSP4 expression prevents behavioral sensitivity & synaptogenesis after facet joint injury
To determine whether TSP4 in the spinal cord that is observed at day 7 (Figure 3) contributes to the allodynia that is also evident (Figure 2A), we blocked spinal TSP4 expression prior to painful joint loading using intrathecal delivery of antisense oligonucleotides. Rats received daily intrathecal injections of TSP4 antisense or mismatch oligonucleotides (75μg/rat/day) beginning 3 days before painful joint loading because the effectiveness of previous TSP4 antisense treatments peaked on the third day of repeated injections (Kim et al., 2012). Antisense oligonucleotides blocked the development of allodynia after injurious joint distraction, but rats receiving mismatch oligonucleotides developed tactile allodynia (Figure 4A), similar to rats receiving painful joint distraction with no treatment (Figure 2A). Western blot analysis of TSP4 on day 7 confirmed that antisense treatment reduced TSP4 expression in the spinal cord relative to TSP4 levels after mismatch treatment (Figure 4B).
Figure 4.

Blocking TSP4 expression with antisense oligonucleotides prevents tactile allodynia and spinal excitatory synaptogenesis. Daily intrathecal injection of antisense (AS) or mismatch (MM) oligonucleotides (75μg/rat/day) began 3 days before painful facet distraction. (A) AS oligonucleotides prevent the onset of tactile allodynia. Paw withdrawal threshold (PWT) is presented as mean±SEM (n=11–12 rats per group). *p<0.0001 compared with day 0 (pre-joint distraction), #p<0.0001 compared with MM on each day by repeated-measures ANOVA with post-hoc Tukey’s HSD test. (B) Daily AS oligonucleotide treatment reduces spinal TSP4 expression on day 7 after joint loading. TSP4 bands are normalized to β-tubulin as a loading control (n=5–6 rats per group). *p=0.049 compared with MM by Student’s t-test. (C) Immunolabeling of synapsin (red) and homer (green); colocalization (yellow) represents synaptic puncta (arrows). Scale bar=10μm. (D) AS oligonucleotide treatment reduces synapse density in the C6/C7 superficial dorsal horn below that of MM treatment. Groups are mean±SEM (n=6 sections per rat, 6 rats per group). *p=0.001 compared with MM by Student’s t-test. The dotted line represents synapse densities in the sham group relative to the painful joint loading group.
Given the synaptogenic properties of TSP4 and its upregulation in the same dorsal horn regions where excitatory synapses also increase in this model (Figures 2 & 3), we hypothesized that TSP4 may contribute to persistent allodynia by inducing excitatory synaptogenesis. To test this hypothesis we quantified synapses in the superficial dorsal horn after injurious joint distraction with antisense or mismatch oligonucleotide treatment. Synaptic puncta were identified in the superficial laminae of the dorsal horn after co-labeling for synapsin and homer (Figure 4C). Fewer synaptic puncta were counted after antisense treatment than the mismatch oligonucleotide treatment (Figure 4D). These data suggest that spinal TSP4 plays a role in the maintenance of allodynia after injurious joint loading, likely through the development of additional excitatory dorsal horn synapses.
Gabapentin reduces injury-induced allodynia & dorsal horn excitatory synaptogenesis
Gabapentin blocks the synaptogenic activity of TSP4 by inhibiting the binding of TSP4 to its neuronal receptor, the α2δ-1 subunit of voltage-gated calcium channels (Eroglu et al., 2009; Luo et al., 2002; Rose and Kam, 2002). We previously reported intrathecal gabapentin to attenuate the behavioral sensitivity and dorsal horn neuronal hyperexcitability that are evident after painful facet joint loading (Dong et al., 2013a); so, we tested whether gabapentin does this via reducing excitatory synaptogenesis. Rats received two administrations of intrathecal gabapentin (4.2μg in 30μL saline) or vehicle (30μL saline), as previously reported (Dong et al., 2013a) – immediately before injury and again at 1 day after injury following assessment of allodynia. Vehicle-treated rats exhibited tactile allodynia on day 7 after injurious joint distraction, but allodynia was not present at that day after gabapentin treatment (Figure 5A). Gabapentin also prevented the injury-induced increase in excitatory synapses in the superficial dorsal horn (Figures 5B & 5C). Since gabapentin blocks the synaptogenic activity of TSP4 (Eroglu et al., 2009), these results further support the role of TSP4-mediated synaptogenesis in facet joint pain.
Figure 5.
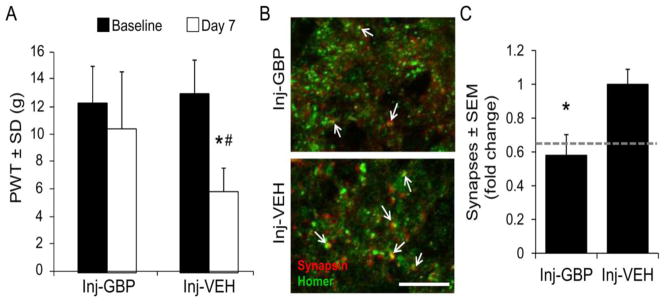
Gabapentin attenuates tactile allodynia and excitatory synaptogenesis after painful facet joint loading. (A) Allodynia on day 7 after painful joint distraction is reduced by intrathecal gabapentin (Inj-GBP, 4.2μg in 30μL saline, 90 minutes before and 1 day after distraction) compared to saline vehicle (Inj-VEH, 30μL saline, 90 minutes before and 1 day after distraction). Paw withdrawal threshold (PWT) is presented as mean±SEM (n=9 rats per group). *p=0.0001 compared with baseline, #p=0.01 compared with Inj-GBP by repeated-measures ANOVA with post-hoc Tukey’s HSD test. (B) Immunolabeling of synapsin (red) and homer (green) in the superficial dorsal horn 7 days after painful joint loading with gabapentin or vehicle treatment. Colocalization (yellow) indicates synaptic puncta (arrows). Scale bar=10μm. (C) Gabapentin prevents the injury-induced increase in excitatory synapses. Groups are mean±SEM (n=4 sections per rat, 6 rats per group). *p=0.0002 compared with Inj-GBP by Student’s t-test. The dotted line represents synapse densities in the sham group relative to the painful joint loading group.
Spinal TSP4 potentiates allodynia & neuronal hyperexcitability induced by facet loading
In addition to synaptogenesis, TSP4 may also contribute to spinal hyperexcitability by presynaptically enhancing excitatory input from primary afferent fibers at synapses in the dorsal horn (Kim et al., 2012). Since facet joint loading increases firing of the afferents that innervate the facet capsule (Lu et al., 2005), we tested whether increased spinal TSP4 levels potentiate allodynia and neuronal excitability after non-painful or painful loading of the facet joint. To identify the dose-response of spinal TSP4 and behavioral sensitivity, forepaw tactile allodynia was assessed after intrathecal administration of full-length recombinant TSP4 protein with a StrepII tag for affinity purification (Figure 6A). Intrathecal bolus injection of 30, 45, or 60μg/rat of TSP4 induced differential patterns of forepaw allodynia that peaked at 3–4 days after injection; hypersensitivity was most prolonged, lasting for 2–6 days after injection, after the 60μg/rat dose (Figure 6B). Injection of the 20μg/rat dose of TSP4 or 60μg/rat of the StrepII peptide, as a control, did not result in sensitivity. These results suggest that intrathecal TSP4 is sufficient to induce tactile allodynia in the forepaw, but sub-threshold doses can be administered that do not independently produce sensitivity. As such, two doses of TSP4 were used for additional experiments: a non-sensitizing dose (20μg/rat) and a sensitizing dose (60μg/rat).
Figure 6.

Increased spinal TSP4 potentiates allodynia and spinal hyperexcitability induced by facet capsule loading. (A) Coomassie-stained SDS-PAGE gel and Western blot membrane showing the purification StrepII-tagged TSP4 from transfected 293 EBNA cell media. (B) Naïve rats received single intrathecal injections of TSP4 (20μg, 30μg, 45μg, or 60μg TSP4 per rat) or a StrepII peptide control (60μg) (n=6 rats per group). TSP4 injections of 30μg or higher decrease the forepaw withdrawal threshold (PWT). *p<0.011 for 60μg, #p<0.014 for 45μg, ^p<0.043 for 30μg, compared with StrepII peptide control. (C,D) Rats received non-sensitizing (20μg) or sensitizing (60μg) intrathecal TSP4, or StrepII peptide (60μg) 3 days before sham (Sham), physiologic (Phys), or injurious facet joint loading (Inj) (n=6–7 rats per group). (C) TSP4 potentiates the development of tactile allodynia after joint loading. *p<0.01 60μg+Phys vs. pre-injury, #p<0.0002 20μg+Phys vs. pre-injury, ^p<0.013 vs. Strep+Sham. (D) Sensitivity induced by TSP4 is not exacerbated by the presence of any facet joint loading. *p<0.01 60μg+Phys and 60μg+Inj vs. pre-injury, #p<0.028 60μg+Sham vs. pre-injury, ^p<0.028 vs. Strep+Sham. (E) Representative neuronal firing in response to five 26g von Frey filament stimulations for a subset of groups (Strep+Sham, 20μg+Phys, 60μg+Phys, 60μg+Inj). (F) Neuronal firing in response to light brush and pinch is elevated when facet loading is preceded by non-sensitizing or sensitizing TSP4 treatment. *p<0.006 compared to Strep+Sham. (G) Neuronal firing is also elevated in response to 1.4g, 4g, 10g, and 26g von Frey filaments when facet loading is preceded by non-sensitizing or sensitizing TSP4. *p<0.008 for 20μg+Phys, #p<0.013 for 60μg+Phys, ^p<0.024 for 60μg+Inj compared with Strep+Sham.
To determine whether spinal TSP4 potentiates the development of allodynia and neuronal hyperexcitability after facet joint loading, we administered non-sensitizing or sensitizing doses of recombinant TSP4 3 days before loading the facet joint. Facet joint loading was administered using three different magnitudes of joint distraction: sham (no loading), 0.2mm joint distraction which replicates the physiologic joint loading during normal activity but does not cause allodynia (Lee and Winkelstein, 2009), or a 0.7mm injurious joint loading that induces allodynia (Figure 1). Physiologic loading of the C6/C7 facet joint induced significantly lower joint distraction and facet capsule strain than injurious loading (Figure 1). Neither injections of StrepII peptide with either sham or physiologic loading, nor injection of the non-sensitizing 20μg dose of TSP4 with sham loading, induced sensitivity (Figures 6C & 6D), confirming that non-sensitizing TSP4 and physiologic loading alone are not sufficient to induce behavioral sensitivity. The combination of either 20μg or 60μg TSP4 with physiologic facet joint loading produced sustained allodynia that persisted through day 7 (Figure 6C). However, the effects of TSP4 and facet joint loading on behavioral sensitivity were not cumulative – sensitivity after injection of 60μg TSP4 was not increased further by physiologic or injurious joint loading (Figure 6D).
Increased spinal TSP4 also potentiated the development of dorsal horn neuronal hyperexcitability 7 days after facet loading. Neuronal firing evoked by both non-noxious (light brush, 1.4g and 4g von Frey filaments) and noxious (pinch, 10g and 26g von Frey filaments) mechanical stimuli at the forepaw increased over sham when 20μg or 60μg TSP4 was followed by physiologic loading (Figures 6E–5G). Neuronal activity after either 20μg or 60μg TSP4 with physiologic loading was increased to the same level as when 60μg TSP4 was followed by injurious loading (Figures 6F & 6G). Those results indicate that the facet joint loading that occurs during normal physiologic events can induce the same sustained neuronal hyperexcitability as a painful facet joint injury, when in the presence of increased spinal TSP4. The effects of increased spinal TSP4 and facet joint loading on neuronal firing also were not additive; evoked neuronal firing after 60μg TSP4 with physiologic loading was not different from the responses after 60μg TSP4 with injurious loading (Figures 6E–6G). Together, these behavioral and electrophysiological results suggest that even non-sensitizing increases in spinal TSP4 facilitate the development of allodynia and spinal neuronal hyperexcitability after joint loading, but that the degree of such changes is not exacerbated by additional increases in spinal TSP4.
Discussion
Painful facet joint loading induces dysregulation of thrombospondin-4 (TSP4) along with the initiation of excitatory synaptogenesis in the dorsal horn paralleling the time course of sustained tactile allodynia through the duration of this study (Figures 2 & 3). Of note, although this study only evaluated behavior for 7 days, allodynia persists for nearly 6 weeks after the same injurious facet joint loading (Rothman et al., 2008); additional studies characterizing the long-term effects of painful facet joint loading on TSP4 and spinal sensitization would provide insight for chronic outcomes. Nonetheless, blocking injury-induced TSP4 expression or reducing TSP4 activity at its neuronal receptor both attenuate allodynia and dorsal horn synaptogenesis (Figures 4 & 5). Increased spinal TSP4 also facilitates the development of sensitization, even after loading of the facet joint that does not independently lead to persistent pain (Figure 6). Together, these complementary results provide the first demonstration that TSP4 and excitatory synaptogenesis in the spinal cord contribute to joint-mediated pain, and support previous reports of a more universal role for TSP4 in neuropathic pain states (Kim et al., 2012; Li et al., 2014a; Zeng et al., 2013).
The peripheral and central neurochemical changes associated with inflammatory and neuropathic pain can differ substantially (Honore et al., 2000), emphasizing the need to define those mechanisms of each pain state, especially for joint-mediated pain that includes either, or both, inflammatory and neuropathic characteristics (Bove et al., 2006; Dong and Winkelstein, 2010; Kras et al., 2013; Kras et al., 2014; Lee et al., 2008; Quinn et al., 2010; Rothman et al., 2008). Although not a direct neural tissue insult, injurious facet distraction can produce pain through the induction of secondary axonal damage and increased discharge and afterdischarge of fibers innervating the facet joint (Crosby et al., 2014; Kallakuri et al., 2008; Lu et al., 2005). In this study, although astrocytic TSP4 is upregulated in the spinal cord and TSP4 is downregulated in the DRG at day 7 after painful facet loading, non-significant trends are observed at the corresponding day 1 (Figure 3). The lack of significance in the changes observed at day 1 may be due in part to small sample sizes, but these findings are consistent with reports of astrocytic TSP4 dysregulation in neuropathic pain beginning at 2 days after lumbar spinal nerve ligation, and at later time points after spinal cord contusion and trigeminal nerve injury (Kim et al., 2012; Li et al., 2014a; Zeng et al., 2013). Although we cannot rule out that neuronal TSP4 may contribute to the increase in spinal TSP4 in this study (Figure 3) (Arber and Caroni, 1995), this is not likely since injury-induced TSP4 expression is not localized to neurons after painful nerve injury (Kim et al., 2012). The decrease in TSP4 in the DRG shows that TSP4 is differentially regulated in the periphery and the spinal cord after painful facet joint loading, and may indicate a peripheral feedback mechanism contributing to pain. However, this study focused primarily on the spinal role of TSP4, so further investigation is required to evaluate the effects of TSP4 dysregulation in the DRG, as has been recently examined after painful nerve injury (Pan et al., 2014). Overall, the similarities in the spatiotemporal regulation of TSP4 in the DRG and spinal cord after painful joint loading and neural trauma strongly suggest that there are common mechanism(s) by which TSP4 contributes to spinal sensitization and chronic pain, regardless of the inciting event.
Given the synaptogenic properties of thrombospondins (Christopherson et al., 2005), upregulation of TSP4 in the superficial dorsal horn could directly increase the number of excitatory synapses that is observed in that region (Figures 2 & 3). TSP4 is also increased in the dorsal columns, where it may potentiate firing in the low-threshold Aβ fibers that project to the brain through the columns (Kim et al., 2012) and contribute to the decreased mechanical pain threshold in the forepaw after painful joint loading. Blocking TSP4 expression or activity abolishes injury-induced allodynia and the increase in excitatory synapses (Figures 4 & 5), suggesting that increased spinal TSP4 is requisite for synaptogenesis and spinal sensitization through interaction with its neuronal receptor, α2δ-1. Of note, the relative increase in excitatory synapses (1.56-fold) in the superficial dorsal horn after painful facet joint injury (Figure 2B) is similar to the range of increases in synapses reported in that spinal region after peripheral nerve injury (Jaken et al., 2010; Li et al., 2014b; Lin et al., 2011; Peng et al., 2010). However, excitatory synaptogenesis could amplify nociception in the dorsal horn by multiple pathways, such as increasing excitatory synapses between primary afferents and nociceptive-specific neurons in the superficial laminae or increasing connections between excitatory interneurons and wide dynamic range neurons that contribute to pain signaling (Basbaum et al., 2009). Additional studies of spinal structural plasticity after joint injury are needed to determine which pre- and post-synaptic neuronal populations, if any, form aberrant connections during excitatory synaptogenesis.
Gabapentin blocks the development of behavioral sensitivity and the initiation of excitatory synaptogenesis (Figure 5), possibly by inhibiting the activity of TSP4 (Eroglu et al., 2009). Gabapentin has been shown also to reduce spinal astrocytic activation and dorsal horn neuronal hyperexcitability in this same painful joint distraction model (Dong et al., 2013a), suggesting that gabapentin may prevent TSP4 activity that is critical for the development of facet-mediated pain. Because gabapentin is short-acting and likely not active for the study duration (Field et al., 1997), it is also possible that gabapentin’s effects at the time of injury prevent the initial increase in spinal TSP4, rather than continuously blocking TSP4 activity after it is upregulated.
Increased spinal TSP4 is sufficient to induce forepaw tactile allodynia, but it also potentiates the development of dorsal horn neuronal hyperexcitability after mechanical loading of the facet joint (Figure 6). Nociceptive and low-threshold mechanosensitive afferents in the facet capsule are differentially activated by capsule strains produced during injurious and physiologic joint loading, respectively (Dong et al., 2012; Lu et al., 2005). However, in the presence of increased spinal TSP4, even physiologic joint loading is sufficient to produce allodynia and dorsal horn hyperexcitability (Figure 6). These findings implicate TSP4 sensitization of the non-nociceptive spinal pathways that are activated by physiologic joint loading, potentially by facilitating presynaptic excitatory neurotransmitter release from Aβ afferents terminating in the dorsal horn (Kim et al., 2012). The specific mechanisms by which TSP4 facilitates excitatory neurotransmission remain unclear, but TSP-family proteins regulate calcium signaling at the plasma and ER membranes (Duquette et al., 2014), so altering presynaptic calcium levels could enhance excitatory neurotransmitter release (Neher and Sakaba, 2008). Collectively, these results implicate increased spinal TSP4 in decreasing the mechanical threshold for pain from joint loading. Repeated exposure to joint loading that upregulates TSP4, or in those with higher endogenous levels of spinal TSP4, may present a higher risk for the development of persistent joint-mediated pain.
In summary, this study shows that thrombospondin-4 plays an important role in the development and maintenance of persistent pain after joint injury, and facilitates the transduction of mechanical joint loading to initiate allodynia and spinal hyperexcitability. The dysregulation of TSP4 after injury, in conjunction with behavioral sensitivity and increased excitatory synapses, indicates a relationship between TSP4 expression, spinal structural plasticity, and persistent joint pain. TSP4-mediated synaptogenesis may be important in maintaining persistent pain after injurious facet joint loading. However, evaluating excitatory synapses at later time points than those in the current study in rats with persistent behavioral sensitivity will be of particular interest to better understand such possible mechanisms of persistent pain. Nevertheless, this study supports that TSP4 is an important contributor to persistent pain after injury, and TSP4 presents a potential therapeutic target for the treatment of chronic pain.
Highlights.
Painful facet joint loading induces excitatory synaptogenesis in the spinal cord.
Painful joint loading also increases spinal TSP4 and decreases TSP4 in the DRG.
Blocking TSP4 reduces synaptogenesis and persistent pain.
Increased spinal TSP4 is sufficient to induce mechanical hyperalgesia.
Spinal TSP4 facilitates the development of spinal sensitization after joint loading.
Acknowledgments
This work was supported by grants from the National Institutes of Health (#AR056288; BAW), (#DE021847; ZDL), the Catherine Sharpe Foundation, and a fellowship from the Ashton Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JC. Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol. 2001;17:25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell B. 2004;36:961–968. doi: 10.1016/j.biocel.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber S, Caroni P. Thrombospondin-4, an extracellular matrix protein expressed in the developing and adult nervous system promotes neurite outgrowth. J Cell Biol. 1995;131:1083–1094. doi: 10.1083/jcb.131.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove SE, Laemont KD, Brooker RM, Osborn MN, Sanchez BM, Guzman RE, Hook KE, Juneau PL, Connor JR, Kilgore KS. Surgically induced osteoarthritis in the rat results in the development of both osteoarthritis-like joint pain and secondary hyperalgesia. Osteoarthr Cartilage. 2006;14:1041–1048. doi: 10.1016/j.joca.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Brakeman PR, Lanahan AA, O’Brien R, Roche K, Barnes CA, Huganir RL, Worley PF. Homer: a protein that selectively binds metabotropic glutamate receptors. Nature. 1997;386:284–288. doi: 10.1038/386284a0. [DOI] [PubMed] [Google Scholar]
- Chen YW, Zhao P, Borup R, Hoffman EP. Expression profiling in the muscular dystrophies: identification of novel aspects of molecular pathophysiology. J Cell Biol. 2000;151:1321–1336. doi: 10.1083/jcb.151.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Crosby ND, Weisshaar CL, Winkelstein BA. Spinal neuronal plasticity is evident within 1 day after a cervical facet joint injury. Neurosci Lett. 2013;542:102–106. doi: 10.1016/j.neulet.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby ND, Gilliland TM, Winkelstein BA. Early afferent activity from the facet joint after painful trauma to its capsule potentiates neuronal excitability and glutamate signaling in the spinal cord. Pain. 2014 doi: 10.1016/j.pain.2014.06.019. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Camilli P, Cameron R, Greengard P. Synapsin 1, a nerve terminal specific phosphoprotein: its general distribution in synapses of the central and peripheral nervous system demonstrated by immunofluorescence in frozen and plastic sections. J Cell Biol. 1983;96:1337–1354. doi: 10.1083/jcb.96.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Winkelstein BA. Simulated whiplash modulates expression of the glutamatergic system in the spinal cord suggesting spinal plasticity is associated with painful dynamic cervical facet loading. J Neurotraum. 2010;27:163–174. doi: 10.1089/neu.2009.0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Quindlen JC, Lipschutz DE, Winkelstein BA. Whiplash-like facet joint loading initiates glutamatergic responses in the DRG and spinal cord associated with behavioral hypersensitivity. Brain Res. 2012;1461:51–63. doi: 10.1016/j.brainres.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Crosby ND, Winkelstein BA. Gabapentin alleviates facet-mediated pain in the rat through reduced neuronal hyperexcitability and astrocytic activation in the spinal cord. J Pain. 2013a;14:1564–1572. doi: 10.1016/j.jpain.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Smith JR, Winkelstein BA. Ketorolac reduces spinal astrocytic activation and PAR1 expression associated with attenuation of pain following facet joint injury. J Neurotraum. 2013b;30:818–825. doi: 10.1089/neu.2012.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkle ET, Zaucke F, Clegg DO. Thrombospondin-4 and matrix three-dimensionality in axon growth and adhesion in the developing retina. Exp Eye Res. 2007;84:707–717. doi: 10.1016/j.exer.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Duquette M, Nadler M, Okuhara D, Thompson J, Shuttleworth T, Lawler J. Members of the thrombospondin gene family bind stromal interaction molecule 1 and regulate calcium channel activity. Matrix Biol. 2014 doi: 10.1016/j.matbio.2014.05.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor a2d-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MJ, Holloman EF, McCleary S, Hughes J, Singh L. Evaluation of gabapentin and S-(+)-3-isobutylgaba in a rat model of postoperative pain. JPET. 1997;282:1242–1246. [PubMed] [Google Scholar]
- Hains BC, Johnson KM, Eaton MJ, Willis WD, Hulsebosch CE. Serotonergic neural precursor cell grafts attenuate bilateral hyperexcitability of dorsal horn neurons after spinal hemisection in rat. Neuroscience. 2003;116:1097–1110. doi: 10.1016/s0306-4522(02)00729-7. [DOI] [PubMed] [Google Scholar]
- Hansen U, Platz N, Becker A, Bruckner P, Paulsson M, Zaucke F. A secreted variant of cartilage oligomeric matrix protein carrying a chondrodysplasia-causing mutation (p.H587R) disrupts collagen fibrillogenesis. Arthritis Rheum. 2011;63:159–167. doi: 10.1002/art.30073. [DOI] [PubMed] [Google Scholar]
- Hogg-Johnson S, van der Velde G, Carroll LJ, Holm LW, Cassidy JD, Guzman J, Cote P, Halderman S, Ammendolia C, Carragee E, Hurwitz E, Nordin M, Peloso P. The burden and determinants of neck pain in the general population. Eur Spine J. 2008;17:S39–S51. doi: 10.1097/BRS.0b013e31816454c8. [DOI] [PubMed] [Google Scholar]
- Honore P, Rogers SD, Schwei MJ, Salak-Johnson JL, Luger NM, Sabino MC, Clohisy DR, Mantyh PW. Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience. 2000;98:585–598. doi: 10.1016/s0306-4522(00)00110-x. [DOI] [PubMed] [Google Scholar]
- Hubbard RD, Chen Z, Winkelstein BA. Transient cervical nerve root compression modulates pain: load thresholds for allodynia and sustained changes in spinal neuropeptide expression. J Biomech. 2008;41(3):677–85. doi: 10.1016/j.jbiomech.2007.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito DM, Eroglu C. Quantifying synapses: an immunohistochemistry-based assay to quantify synapse number. J Vis Exp. 2010;45:2270. doi: 10.3791/2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaken RJP, Joosten EAJ, Knuwer M, Miller R, ven der Meulen I, Marcus MAE, Deumens R. Synaptic plasticity in the substantia gelatinosa in a model of chronic neuropathic pain. Neurosci Lett. 2010;469:30–33. doi: 10.1016/j.neulet.2009.11.038. [DOI] [PubMed] [Google Scholar]
- Kallakuri S, Singh A, Lu Y, Chen C, Patwardhan A, Cavanaugh JM. Tensile stretching of cervical facet joint capsule and related axonal changes. Eur Spine J. 2008;17:556–563. doi: 10.1007/s00586-007-0562-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Li KW, Boroujerdi A, Yu YP, Zhou CY, Deng P, Park J, Zhang X, Lee J, Corpe M, Sharp K, Steward O, Eroglu C, Barres B, Zaucke F, Xu ZC, Luo ZD. Thrombospondin-4 contributes to spinal sensitization and neuropathic pain states. J Neurosci. 2012;32:8977–8987. doi: 10.1523/JNEUROSCI.6494-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kras JV, Weisshaar CL, Quindlen JA, Winkelstein BA. Brain-derived neurotrophic factor is upregulated in the cervical DRG & spinal cord and contributes to the maintenance of pain from facet joint injury. J Neurosci Res. 2013;91:1312–1321. doi: 10.1002/jnr.23254. [DOI] [PubMed] [Google Scholar]
- Kras JV, Dong L, Winkelstein BA. Increased interleukin-1a and prostaglandin E2 expression in the spinal cord at day 1 after painful facet joint injury: evidence of early spinal inflammation. Spine. 2014;39:207–212. doi: 10.1097/BRS.0000000000000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KE, Davis MB, Mejilla RM, Winkelstein BA. In vivo cervical facet capsule distraction: mechanical implications for whiplash and neck pain. Stapp Car C. 2004;48:373–395. doi: 10.4271/2004-22-0016. [DOI] [PubMed] [Google Scholar]
- Lee KE, Davis MB, Winkelstein BA. Capsular ligament involvement in the development of mechanical hyperalgesia after facet joint loading: behavioral and inflammatory outcomes in a rodent model of pain. J Neurotraum. 2008;25:1383–1393. doi: 10.1089/neu.2008.0700. [DOI] [PubMed] [Google Scholar]
- Lee KE, Winkelstein BA. Joint distraction magnitude is associated with different behavioral outcomes and substance P levels for cervical facet joint loading in the rat. J Pain. 2009;10:436–445. doi: 10.1016/j.jpain.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Li KW, Kim DS, Zaucke F, Luo ZD. Trigeminal nerve injury-induced thrombospondin-4 upregulation contributes to orofacial neuropathic pain states in a rat model. Eur J Pain. 2014a;18:489–495. doi: 10.1002/j.1532-2149.2013.00396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li KW, Yu YP, Zhou C, Kim DS, Lin B, Sharp K, Steward O, Luo ZD. Calcium channel α2δ1 proteins mediate trigeminal neuropathic pain states associated with aberrant excitatory synaptogenesis. J Biol Chem. 2014b;289:7025–7037. doi: 10.1074/jbc.M114.548990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, Peng B, Yang ZW, Min S. Number of synapses increased in the rat spinal dorsal horn after sciatic nerve transection: a stereological study. Brain Res Bull. 2011;84:430–433. doi: 10.1016/j.brainresbull.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Lo FS, Zhao S, Erzurumlu RS. Astrocytes promote peripheral nerve injury-induced reactive synaptogenesis in the neonatal CNS. J Neurophysiol. 2011;106:2876–2887. doi: 10.1152/jn.00312.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Chen C, Kallakuri S, Patwardhan A, Cavanaugh JM. Neural response of cervical facet joint capsule to stretch: a study of whiplash pain mechanisms. Stapp Car C. 2005;49:49–65. doi: 10.4271/2005-22-0003. [DOI] [PubMed] [Google Scholar]
- Luo ZD, Calcutt NA, Higuera ES, Valder CR, Song YH, Svensson CI, Myers RR. Injury type-specific calcium channel a2d-1 subunit upregulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. JPET. 2002;303:1199–1205. doi: 10.1124/jpet.102.041574. [DOI] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Pan B, Yu H, Park J, Yu YP, Luo ZD, Hogan QH. Painful nerve injury upregulates thrombospondin-4 expression in dorsal root ganglia. J Neurosci Res. 2014 doi: 10.1002/jnr.23498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson AM, Ivancic PC, Ito S, Panjabi MM. Facet joint kinematics and injury mechanisms during simulated whiplash. Spine. 2004;29:390–397. doi: 10.1097/01.brs.0000090836.50508.f7. [DOI] [PubMed] [Google Scholar]
- Peng B, Lin JY, Shang Y, Yang ZW, Wang YP. Plasticity in the synaptic number associated with neuropathic pain in the rat spinal dorsal horn: a stereological study. Neurosci Lett. 2010;486:24–28. doi: 10.1016/j.neulet.2010.09.037. [DOI] [PubMed] [Google Scholar]
- Quinn KP, Dong L, Golder FJ, Winkelstein BA. Neuronal hyperexcitability in the dorsal horn after painful facet joint injury. Pain. 2010;151:414–421. doi: 10.1016/j.pain.2010.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MA, Kam PCA. Gabapentin: pharmacology and its use in pain management. Anaesthesia. 2002;57:451–462. doi: 10.1046/j.0003-2409.2001.02399.x. [DOI] [PubMed] [Google Scholar]
- Rothman SM, Hubbard RD, Lee KE, Winkelstein BA. Detection, transmission, and perception of pain. In: Slipman CW, Derby R, Simeone FA, Mayer TG, editors. Interventional spine: an algorithmic approach. Philadelphia: Saunders; 2008. pp. 29–38. [Google Scholar]
- Siegmund GP, Winkelstein BA, Ivancic PC. The anatomy and biomechanics of acute and chronic whiplash injury. Traffic Inj Prev. 2009;10:101–112. doi: 10.1080/15389580802593269. [DOI] [PubMed] [Google Scholar]
- Stenina OI, Desai SY, Krukovets I, Kight K, Janigro D, Topol EJ, Plow EF. Thrombospondin-4 and its variants: expression and differential effects on endothelial cells. Circulation. 2003;108:1514–1519. doi: 10.1161/01.CIR.0000089085.76320.4E. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Shortland P, Coggeshall RE. Peripheral nerve injury triggers central sprouting of myelinated afferents. Nature. 1992;355:75–78. doi: 10.1038/355075a0. [DOI] [PubMed] [Google Scholar]
- Zeng J, Kim D, Li KW, Sharp K, Steward O, Zaucke F, Luo ZD. Thrombospondin-4 contributes to spinal cord injury-induced changes in nociception. Eur J Pain. 2013;17:1458–1464. doi: 10.1002/j.1532-2149.2013.00326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]