

Abstract
Systemic sclerosis (SSc) is a polygenic, autoimmune disorder of unknown etiology, characterized by the excessive accumulation of extracellular matrix (ECM) proteins, vascular alterations, and autoantibodies. The tight skin (Tsk)2/+ mouse model of SSc demonstrates signs similar to SSc including tight skin and excessive deposition of dermal ECM proteins. By linkage analysis, we mapped the Tsk2 gene mutation to less than 3 megabases on chromosome 1. We performed both RNA sequencing of skin transcripts and genome capture DNA sequencing of the region spanning this interval in Tsk2/+ and wild-type littermates. A missense point mutation in the procollagen III amino terminal propeptide segment (PIIINP) of Col3a1 was found to be the best candidate for Tsk2, so both in vivo and in vitro genetic complementation tests were used to prove that this Col3a1 mutation is the Tsk2 gene. All previously documented mutations in the human Col3a1 gene are associated with Ehlers-Danlos syndrome, a connective tissue disorder that leads to a defect in type III collagen synthesis. To our knowledge, the Tsk2 point mutation is the first documented gain-of-function mutation associated with Col3a1, which leads instead to fibrosis. This discovery provides insight into the mechanism of skin fibrosis manifested by Tsk2/+ mice.
Introduction
There are multiple animal models of SSc (Artlett, 2010), yet none mimics all facets of SSc disease. Of the genetic models, the cause of disease in tight-skin 1 (Tsk1/+) mice is known to be a tandem duplication in the fibrillin-1 (Fbn1) gene (Siracusa et al., 1996). Other models of SSc have employed mice with individual gene deficiencies or overexpression including Fos-related antigen-2 (Fra2) (Maurer et al., 2009), endothelin-1 (Edn1) (Hocher et al., 2000; Richard et al., 2008) and Friend leukemia integration 1 transcription factor (Fli1) (Asano et al., 2010), which have proven useful for understanding the contribution of these proteins to the vasculopathy and/or lung fibrosis seen in SSc. Non-genetic models of SSc include the bleomycin-induced scleroderma model (Yamamoto et al., 1999), which has been used to study many of the initiating events involved in fibrosis.
The Tsk2/+ mouse was first described in 1986, when an offspring of a 101/H mouse exposed to the mutagenic agent ethylnitrosourea was noted to have tight-skin in the interscapular region (Peters and Ball, 1986). The mutagenized gene causing SSc-like signs in Tsk2/+ mice was reported to be located on chromosome 1 between 42.5 and 52.5 megabases (Mb) (Christner et al., 1996); however, the genetic defect was never identified. Like Tsk1, Tsk2 SSc-like traits are highly penetrant in Tsk2/+ heterozygotes and it is homozygous embryonic lethal. Tsk2/+ mice have many features of human disease including tight-skin, dysregulated dermal extracellular matrix (ECM) deposition, and evidence of an autoimmune response (Christner et al., 1995; Gentiletti et al., 2005).
Herein, we report the positional cloning and identity of the Tsk2 gene. We have discovered that Tsk2/+ mice carry a deleterious gain-of-function missense mutation in Col3a1, that exchanges a cysteine for serine in the N-terminal propeptide, PIIINP. The Tsk2/+ mouse affords a unique opportunity to examine the pathways leading to the multiple clinical parameters of fibrotic disease from birth onward.
Results
Linkage and sequencing studies reveal a SNP mutation in Col3a1
Identification of the Tsk2 gene was initiated with further mapping of the Tsk2 interval by genotyping backcross progeny of Tsk2/+ mice bred to C57Bl/6 (B6) mice. Littermate mice were genotyped for informative microsatellites (D1Mit233, D1Mit235, a microsatellite in Gls, and D1Mit18) and single nucleotide polymorphism (SNP) genotyping assays used for additional markers. Multiple recombinants were recovered that mapped the interval to between 42.53 and 52.22 Mb on chromosome 1. Recombinants were bred and then backcrossed to a consomic B6.chr 1-A/J mouse to fine-map the region by SNP typing, as A/J mice bear many known SNPs compared to B6 mice. Additional recombinants were recovered and new SNPs from the sequencing projects (see below) were used to narrow the Tsk2 interval to between 44.67 – 46.27 Mb (Fig. 1A), representing a greater than 3-fold reduction of the size of the interval bearing 101/H genomic DNA and Tsk2. There are six known genes in this interval (Fig. 1B).
Figure 1.
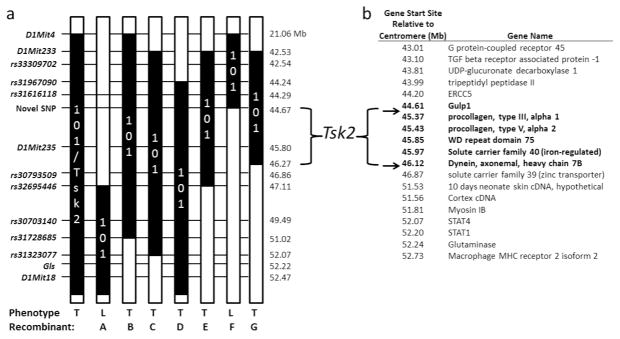
Tsk2 lies between and not including 44.67 – 46.27Mb Mb on chromosome 1
(A) The Tsk2 interval was narrowed by genotyping back-crossed mice on the B6 and B6.chr 1-A/J backgrounds. Grey bars (101/H) depict the original parental strain, bearing Tsk2. White bars depict the B6 genome. Indefinite areas between typed markers are grey. Recombinants A – G bear additional recombination sites. The phenotypes are tight (T – Tsk2/+) or loose (L – WT).
(B) With the use of additional markers (arrows, see text), the current interval comprises Col3a1, Col5a2, Wdr75, Slc40a1, part of Gulp1, and part of Dnahc7b; the five latter genes do not have coding region mutations. The elements of the Gulp1 gene above 44.67 Mb are excluded by the recombination in mouse F, and Dnahc7b below 46.27 is excluded by Mouse G.
To identify the mutation underlying Tsk2, we employed both RNA sequencing (RNA-Seq) and genome capture sequencing of the reduced genomic interval. Sequence reads were aligned to the MM9 reference genome (B6) and analyzed for polymorphisms in the Tsk2 interval. There were 265 SNPs found in both WT and Tsk2/+ littermates that represent differences between the reference B6 genome and the 101/H background; these were excluded from further study. Thirteen SNPs were found in all four Tsk2/+ mice analyzed; ten of these SNPs were also found to be in liver RNA from 101/H strain or in other non-fibrotic mouse strains (http://phenome.jax.org/), and were also ruled out as candidates for Tsk2 (Table 1). The remaining three SNPs were heterozygous and confirmed to be only in Tsk2/+ mice. One of these, in a Gulp1 intron, proved useful as an additional marker that resides outside the supported linkage interval for Tsk2/+ on the proximal end in an informative recombinant mouse (Fig. 1A). A second SNP was also found in an intron of Gulp1. The RNA-Seq data did not identify any splicing defects in Gulp1 mRNA in the Tsk2/+ mice (Supplementary Fig. 1), indicating that this SNP does not change Gulp1 mRNA splicing, and its gene expression in skin is unchanged (Fig. 2). Thus the intronic SNP in Gulp1 is unlikely to play a role in the tight skin phenotype. The remaining mutation was in Col3a1 that results in a T to A transversion at Chr1:45,378,353, causing a Cys → Ser amino acid change in the procollagen III amino terminal propeptide (PIIINP) segment, a natural cleavage product of COL3A1. The mutant protein is designated COL3A1Tsk2 (C33S).
Table 1.
Nucleotide changes between Tsk2/+ mice and 101/H or B6 mice
| Nucleotide position on chr 1 (mm9) | Genotype of Tsk2/+ | Genotype of B6 | Genotype of 101/H | Present in Other Strains? | Potential candidate for Tsk2? | Protein or mRNA containing substitution |
|---|---|---|---|---|---|---|
| SNP found by RNA-Seq | ||||||
| 44,675,490 | A | T | T | No | No, outside interval | Gulp1 Intron |
| 44,833,682* | C | T | T | No | YES | Gulp1Intron |
| 45,378,353* | A | T | T | No | YES | COL3A1 Exon (C33S) |
| 45,432,389 | C | G | Nd | Yes | No | Col5a2 3′UTR |
| 45,441,243 | C | A | C | No | No, in 101/H | Col5a2 Intron |
| 45,860,529 | G | A | G | Yes | No | Wdr75 Intron |
| 45,874,790 | T | C | T | Yes | No | Wdr75 Intron |
| 45,875,728 | C | T | C | Yes | No | Wdr75 Exon |
| 45,880,257 | CG | AC | CG | No | No, in 101/H | WDR75 Exon |
| 46,872,610 | T | G | Nd | Yes | No | Slc39a10 Intron |
| 46,874,711 | C | T | C | Yes | No | Slc39a10 Intron |
| 46,939,340 | T | C | T | Yes | No | BC040767 Intron |
| 46,939,624 | A | G | Nd | Yes | No | BC040767 Intron |
| SNP found by 454 Sequencing | ||||||
| 44,833,682* | C | T | T | No | YES | Gulp1Intron |
| 45,378,353* | A | T | T | No | YES | COL3A1 Exon (C33S) |
| 45,465,923 | A | T | T | No | YES | Col5a2 Intron |
| 46,124,856 | A | G | A | Yes | No | Dnahc76 Intron |
| 46,124,857 | A | C | T | Yes | No | Dnahc76 Intron |
| 46,268,651 | C | T | T | No | No, outside interval | Dnahc76 Intron |
All single-copy nucleotide changes found by RNA-Seq or 454 sequencing were checked for their presence in other non-fibrotic strains (http://phenome.jax.org/) or individually verified by a phototyping assay (Bunce et al., 1995) and/or resequencing to confirm the single nucleotide change. SNP that were ruled out by one of these assays are considered not to be potential candidates for Tsk2. When known, genotypes shown for 101/H are from RNA-Seq, 454 sequencing or phototyping. Nd = not determined.
seen in both assays.
Figure 2.

Col3a1 is the only interval gene expressed at high levels in the skin of Tsk2/+ mice.
(A) This graph shows gene expression for the seven Tsk2 interval genes, as determined from the RNA-Seq abundance results.
(B) Heat map for seven Tsk2 interval genes detected as transcripts in RNA-Seq.
(C + D) Distribution of nucleotide calls in heterozygous Tsk2/+ and homozygous WT mice for Col3a1 and Gulp1.
We calculated the Reads per Kilobase per Million mapped reads for each gene and found that of the genes in the reduced genomic interval, Col3a1 shows the highest absolute expression level with all other genes showing negligible expression levels. RNA-Seq results indicate that there is a trend toward higher Col3a1 mRNA abundance in 4-week old Tsk2/+ skin samples compared to WT littermates (Fig. 2A, B). The Col3a1Tsk2 (C33S) mutation is unlikely to change the expression levels of the Col3a1 mRNA directly but will result in a mutated protein that is deposited in the ECM along with the WT protein in mixed heterotrimers, and could result in activation of pathways that impinge on Col3a1 such as TGFβ (Sargent, et al., submitted). Because Tsk2/+ (affected) mice are heterozygous, the Col3a1Tsk2 (C33S) mutation should account for 50% of the reads assuming equal expression from each allele. We calculated the read count from the RNA-seq data for the reference and alternate alleles for Col3a1 at Chr1:45,378,353. In WT mice we find all reads (492 total) contain the reference T allele, whereas in Tsk2/+, we find 48% of reads (273/564 total reads) contain the WT (T) allele and 52% (291/564 total reads) contain the Col3a1Tsk2 (C33S) allele (T -> A; Fig. 2C). As a comparison, we show the intronic Gulp1 SNP at Chr1:44,833,682 has significantly lower read coverage consistent with its intronic location (11-fold coverage in Tsk2/+ and 2-fold coverage in WT). The intronic Gulp1 SNP also shows a distribution of reads consistent with heterozygosity in Tsk2/+ and with homozygosity in WT (Fig. 2D). These findings show that the Col3a1Tsk2 (C33S) locus is heterozygous as expected for the Tsk2 mutation in these animals, and expression occurs equally from each of the alleles.
Because RNA-Seq only captures variation in the transcribed regions of the genome, and thus might miss an important genomic feature that is unique to Tsk2, we sequenced captured genomic DNA samples corresponding to the minimal linkage region from B6.Tsk2/+ heterozygotes and 101/H homozygous parental strain mice. Multiple DNA differences between the Tsk2/+ mouse and its parental 101/H strain were detected. A majority of the differences observed were accounted for by non-chromosome 1 repetitive DNA sequences such as LINE, SINE and retroviral elements contained within the Tsk2 interval on chromosome 1. After filtering repetitive elements from the comparison, there were six single copy DNA sequence differences, of which three were confirmed to be Tsk2/+ specific (Table 1). Among these, there is a SNP that proved useful in demarcating the distal end of the Tsk2 linkage interval (Chr1:46,268,651; Table 1 and Fig. 1) as it was outside the linkage interval. This allowed us to eliminate the only other gene expressed at an appreciable level in the broader interval, Slc39a10. In addition, the GULP1 intronic SNP was confirmed and another SNP in an intron of Col5a2 was observed. Both these latter SNPs are deemed unrelated to the phenotype, again because of their low overall expression, and the lack of any influence on splicing or expression in the RNASeq results (Fig. 2A, B; Supplementary Fig. 1). Most important, however, the heterozygous T-to-A transversion in Col3A1 at Chr1:45,378,353 was observed in the genomic sequence comparison, and was identical to the mutation identified by RNA-Seq. There were no additional variants that could be validated on the Tsk2 chromosome within ~535,000 nucleotides proximal to the transcription start site of Col3a1 gene or closer than 59,732 nucleotides distal of the end of the Col3a1 3′ untranslated region (UTR). Selective resequencing of the 3′UTR likewise revealed no differences between Tsk2 and 101/H (not shown). Thus, this non-synonymous coding mutation is the most likely to be Tsk2 by genomic assessment as well as by RNA sequencing.
Mice bearing Col3a1Tsk2 and Col3a1KO are not viable
To prove that Tsk2 is a single nucleotide change in the Col3A1 coding region required a separate genetic test. Both Tsk2/Tsk2 (Peters and Ball, 1986) and Col3a1-knockout (KO) (Liu et al., 1997) homozygotes exhibit embryonic lethality, which is also seen in our mouse colony (Table 2). We therefore designed a genetic complementation test to determine if Col3a1Tsk2 (from Tsk2 mice) could complement and rescue the null allele for Col3a1. Conversely, this same cross would determine if any other gene in the Col3a1-homozygous knockout could serve to complement the Tsk2 mutation.
Table 2.
Progeny born from Col3a1-deficient, Col3a1-sufficient, and Tsk2/+ mice.
All progeny were assessed for chromosome 1 markers (SNPS and microsatellites) that characterize the origin of the tested allele (Tsk2 or Col3a1).
| Table 2A shows the number of mice born of each genotype and phenotype from Tsk2/+ x Tsk2/+ or Col3a1−/+ x Col3a1−/+ parents. | |||
|---|---|---|---|
| A. | Genotype and phenotype of progeny | ||
| Parents | Tsk2/+ (Tight skin) | WT/WT (Normal skin) | Tsk2/Tsk2 (lethal) |
| Tsk2/+ x Tsk2/+ | 22 | 21 | 0 |
| Col3a1+/Col3a1− | Col3a1+/Col3a1+ | Col3a1−/Col3a1− | |
| Col3a1−/+ x Col3a1−/+ | 16 | 13 | 3 |
| Table 2B shows the number of mice born of each genotype and phenotype from Tsk2/+ x Col3a1−/+ parents; note there are no compound heterozygotes (Tsk2/Col3a1-) born from this mating. | ||||
|---|---|---|---|---|
| B. | Genotype and phenotype of progeny | |||
| Parents | WT/Col3a1+ (Normal skin) | Tsk2/Col3a1+ (Tight skin) | WT/Col3a1− (Normal skin) | Tsk2/Col3a1− |
| Tsk2/+ x Col3a1−/+ | 12 | 10 | 15 | 0 |
Tsk2/+ x Col3a1−/+ mice were bred together, and 37 progeny mice (Table 2) were genotyped. If Col3a1Tsk2 (C33S) can complement the Col3a1-KO, then we would expect to find nine or ten Col3a1Tsk2/Col3a1-KO compound heterozygotes. In fact, no viable compound heterozygotes were born (Table 2, Supplementary Fig. 2). The hybrid bearing Tsk2/Col3a1-null chromosomes was not viable because the Tsk2 gene on the Tsk2-bearing chromosome cannot ‘complement’ (rescue) the loss of the Col3a1 gene on the Col3a1-KO chromosome. It bears only the allele of Col3a1Tsk2 at the Col3a1 locus, which is insufficient to provide a functional COL3A1 protein that is missing in the Col3a1-KO. The Col3a1-null chromosome likewise cannot complement the Tsk2 mutation: the remaining genes on the Col3a1-KO chromosome cannot prevent the death of (cannot ‘complement’) mice bearing the Tsk2 chromosome, whereas hybrids carrying Tsk2/Col3a1-wild type alleles are alive, but fibrotic. In fact, having the Tsk2 mutation is more damaging than not expressing COL3A1 at all, because while a few percent of Col3a1-KO homozygotes make it to birth, Tsk2/Tsk2 homozygotes (and Tsk2/Col3a1-KO) never do, and whereas Col3a1/Tsk2 mice are viable but small in stature and fibrotic, Col3a1−/+ heterozygotes are normal. Therefore, the mutation in Tsk2/+ mice lies within Col3a1 and, when homozygous, is substantially more deleterious than a complete genetic deficiency of COL3A1.
Col3a1Tsk2 induces increased COL1A1 and ECM production in vitro
Because the compound heterozygous animals do not survive to accumulate fibrotic levels of ECM, a direct in vivo test for fibrosis is impossible, so we performed an ‘in vitro complementation’ test, wherein we transfected mutant or wild-type Col3a1 cDNA into Col3a1-KO fibroblasts, harvested from a Col3a1-KO/KO homozygote at birth. Using the production of COL1A1 as a measure of fibrosis (shown to be expressed at high levels in Tsk2/+ skin and used as a marker of fibrosis (Barisic-Dujmovic et al., 2008; Christner et al., 1998)), we assessed both protein and mRNA levels in fibroblasts that received DNA from a plasmid containing a single allele of a single Col3a1 gene. In three independent experiments, COL1A1 protein was significantly elevated after 48 hours of transfection with Col3a1Tsk2 relative to transfection with Col3a1WT (Fig. 3A); mRNA for Col1a1 was likewise increased in cells transfected with mutant Col3a1Tsk2 cDNA (Fig. 3B). Transfection efficiencies were equal in each of the experiments (Fig. 3C).
Figure 3.

Mouse Col3a1-KO fibroblasts transfected with mutant Col3a1Tsk2 express a more fibrotic protein profile than Col3a1WT transfectants.
(A) Culture supernatants assayed by Western blot for COL1A1. Col3a1Tsk2 transfectants produced 34% more COL1A1 than Col3a1WT (p<0.001) or mock transfectants (p<0.0001).
(B) Col1a1 mRNA is more highly expressed in Col3a1-KO fibroblasts transfected with Col3a1Tsk2 than with Col3a1WT (p<0.0001).
(C) There was no significant difference in efficiency of plasmid transfection between Col3a1Tsk2 and Col3a1WT.
(D) Col3a1−/− fibroblasts transfected with Col3a1Tsk2 show a significant increase in Gene Ontology (GO) terms associated with fibrosis.
(E) Expression of the genes that contributed most to the ECM enrichment results in in Col3a1Tsk2 vs. Col3a1WT transfected mice fibroblasts or in 4-week old female Tsk2/+ vs. WT mice.
(F) Expression of genes that contributed to integrin binding term.
(G) Expression of genes that contributed to transmembrane receptor protein kinase activity term.
Given the observation that the production of a major indicator of fibrosis, COL1A1, is increased by the transfection of the Col3a1Tsk2 gene, we assessed the impact of the mutant gene genome-wide. RNA from the Col3a1Tsk2 and Col3a1WT transfected Col3a1-KO fibroblasts and from four week-old Tsk2/+ and WT littermate skin was analyzed by cDNA microarray. Differentially expressed pathways between the two transfections were determined by Gene Set Enrichment Analysis (GSEA). Transfection of Col3a1Tsk2 results in significant enrichment of genes associated with fibrotic Gene Ontology (GO) terms including basement membrane, extracellular matrix, integrin binding, and transmembrane receptor protein kinase activity (Fig 3D; GSEA FDR < 5%). The biological processes observed in the skin of four 4-week old female Tsk2/+ mice relative to WT littermates also shows increases in genes associated with GO terms extracellular matrix, integrin binding and basal lamina (ZL, CB, KBL, CA, EPB, MLW, manuscript in preparation). The genes that significantly contributed to the GSEA pathway enrichment in the transfected fibroblasts were extracted from microarray data of the transfections, as well as from female Tsk2/+ and WT skin at 4 weeks of age (Fig. 3E–F), and were elevated both in the fibroblasts transfected with Col3a1Tsk2 and in Tsk2/+ mouse skin. These include those genes typically associated with fibrosis including CTGF, THY1, FBN1, the collagens, laminins, TGFB1, TGFBR1, ADAMTS family genes and MMP11. In addition, there was up-regulation in Col3a1Tsk2-transfected fibroblasts and Tsk2/+ skin RNA of the VEGF-Receptors FLT1 and FLT4, as well as genes associated with PDGF signaling (PDGFRB and PDGFRL; Fig. 3F). These data indicate that expression of the Col3a1Tsk2 gene alone can induce a substantial fibrotic gene expression program.
Taken together, this means that Col3a1 and Tsk2 are almost certainly one and the same gene. Col3a1Tsk2 (C33S) is therefore deemed a deleterious gain-of-function allele of Col3a1, and the Col3a1-KO is a classical loss-of-function allele. Mice thus need at least one copy of a functional, normal Col3a1 gene.
Tsk2/+ mice have increased dermal COL3A1 protein accumulation
The behavior of Col3a1 in Tsk2/+ mice could reveal the mechanism by which this mutation causes very substantial ECM fibrosis and very tight skin. We measured the level of COL3A1 protein by histological examinations of Tsk2/+ and WT littermate skin. Reticular fibers are composed primarily of COL3A1 and are a structural element in the skin, found in the panniculus carnosus and in the dermis. COL3A1 expression in skin from two-week old mice is high and declines after birth in WT littermates, but does not decline in the Tsk2/+ mice (Fig. 4A). As Tsk2/+ mice age, the reticular fibers thicken and become more pronounced compared to their WT littermates reflecting the accumulation of COL3A1. This finding was confirmed in skin from four-week old mice by western blots, which revealed that there is significantly more COL3A1 in the skin of Tsk2/+ mice compared to age- and sex-matched WT littermates (Fig. 4B–C). We propose that the excess COL3A1 protein we observe by several measures in Tsk2/+ mice is due to a trend for excess production of Col3a1 mRNA (Fig. 2A) rather than reduced degradation of the Col3 protein. Because the PIIINP fragment is removed from the majority of Col3 molecules before natural Col3 turnover degradation takes place in the tissue, mature COL3A1 from Tsk2 is identical to mature COL3A1 from WT mice, and its natural degradation is unlikely to be affected by any changes in PIIINP. These data show there is an overall increased accumulation of mature COL3A1 protein in the Tsk2/+ mice; in addition, at least half of the type III procollagen and PIIINP trimers produced likely contain one or more strands bearing the Tsk2 (C33S) mutation.
Figure 4.
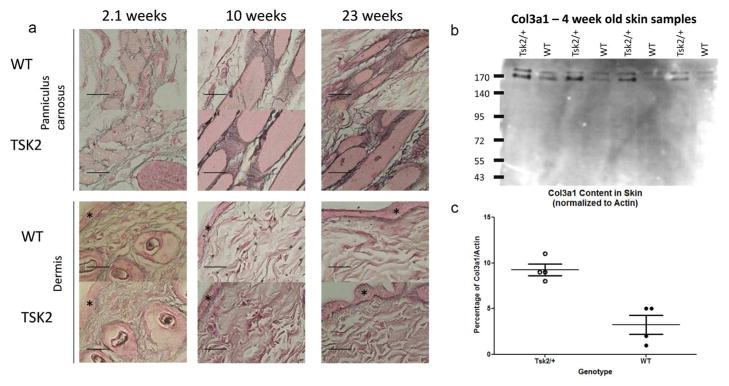
Tsk2/+ mice have increased reticular fiber accumulation and COL3A1 in skin compared WT littermates.
(A) Reticular fiber staining was performed on mice of the indicated ages (2–23 weeks). Stars mark the location of the epidermis. COL3A1 fibers (black staining) are much thicker and more abundant at each life stage in Tsk2/+ than in WT. Fibers were found to be especially pronounced in the panniculus carnosus region of the tissue; increased staining of COL3A1 in the dermis was also noted. The dermal reticular fibers are composed entirely of COL3A1 protein as this protein is receptive to silver impregnation, and they are increased in Tsk2/+ mice. All images were taken at 200X magnification. Bar size = 100 μM.
(B + C) Skin lysates were analyzed for COL3A1 content (both bands) relative to beta-actin (not shown) by western blot analysis. Tsk2/+ mouse skin has significantly more COL3A1 protein than WT mouse skin (p=0.0025, ANOVA).
Discussion
Sequencing of both expressed RNAs and the genomic region in the Tsk2/+ interval, coupled with the genetic complementation study, prove that Tsk2/+ mice harbor a deleterious coding mutation in Col3a1, leading to an amino acid change (C33S) in the N-terminal region of the protein (PIIINP). This point mutation is consistent with those expected from ethylnitrosourea-induced mutagenesis, which generates random single-base-pair point mutations by direct alkylation of nucleic acids. The most common mutations are AT-to-TA and AT-to-GC changes (Cordes, 2005; Noveroske et al., 2000); all three Tsk2-specific mutations identified here were T-to-A or T-to-C mutations. The Tsk2/+ allele is expressed in a 1:1 ratio with the WT by RNASeq indicating equal transcription and making a duplication event unlikely.
Effects of the Tsk2 mutation include: 1) accumulation of COL3A1 protein in vivo over time; 2) induction and accumulation of COL1A1 protein in vivo and in in vitro expression models; 3) a more lethal phenotype than the homozygous genetic loss of Col3a1; and 4) a more lethal compound heterozygous phenotype than that of the homozygous gene knockout. The latter two characteristics indicate that COL3A1Tsk2 (C33S) has a dominant prenatal lethal effect, although our in vitro complementation results suggest that the presence of COL3A1-C33S (or its mRNA) is not lethal to skin fibroblasts per se. A major function of the Col3a1 gene is promoting blood vessel development (Liu et al., 1997), which likely led to the lethality observed in the complementation experiment. In the Col3a1-KO, a few mice are born with the homozygous deficiency, and these mice die of rupture of the major blood vessels (Liu et al., 1997). The possibility that Col3a1Tsk2 mutation could directly induce a deleterious vascular phenotype in Tsk2/+ mice is intriguing; it is notable that genes encoding vascular features (Flt1 and Flt4, genes for VEGF receptors) are significantly up-regulated in both Col3a1Tsk2-transfected skin fibroblasts and in Tsk2/+ skin relative to WT (Fig 3G). It is possible that a complete Col3a1 deficiency could be compensated by other collagens, but the Col3a1Tsk2 mutation is a deleterious gain-of-function, and the deposition of COL3A1-C33S may actively prevent other more benign collagen alternatives from functioning in the vasculature. Thus, our theory is that two doses of a damaging protein are worse than no expression of a normal one.
To our knowledge, this is the first mutation in Col3a1 that results in a gain-of-function phenotype instead of Ehlers-Danlos-like syndromes that are due to loss-of-function or antimorphic collagen-poor phenotypes. Ehlers-Danlos is a group of connective tissue disorders characterized by highly elastic, fragile but not fibrotic skin due to a defect in collagen synthesis (Nishiyama et al., 2001). In addition, these patients have a significant risk for aneurism. Ehlers-Danlos syndrome has been associated with 337 mutations in COL3A1 (http://www.le.ac.uk/ge/collagen/), as well as mutations on COL1A1 and COL5A2. These mutations result in amino acid substitutions in the C terminus of the protein, RNA splicing alterations, deletions, or null alleles. Interestingly, in Ehlers-Danlos syndrome type IV (a very different disease than that observed in Tsk2/+ mice), studies have shown that patients bearing a mutated COL3A1 (compared to a null COL3A1) develop more severe disease and succumb to disease prematurely, whereas those with null COL3A1 were able to live a relatively normal life with limited disease (Leistritz et al., 2011). Currently, all reported COL3A1 mutations result in decreased collagen protein secretion leading to variably thinner skin and defects in the vasculature that are observed in these patients. In contrast to the mutations observed in Ehlers-Danlos, the Tsk2/+ mouse mutation results in thickened skin with no apparent evidence of aneurism. The mutation reported here occurs in the N-terminal PIIINP fragment of the protein, rather than the C-terminal region associated with Ehlers-Danlos.
The PIIINP molecule is a homotrimer with a molecular weight of approximately 42,000 daltons and comprises three domains: a cysteine-rich globular domain (Col 1) containing 79 amino acids with five intrachain disulphide bonds, a triple-helical domain (Col 3) with 12 amino acids and three interchain disulphide bonds, and a non-collagenous domain (Col 2) comprising of 39 amino acids ending with the N-telopeptide that forms a triple helical structure (Bruckner et al., 1978). The mutation in Col3a1Tsk2 substitutes a serine for the cysteine in one of the five Col 1-domain cysteines involved in disulphide bonds (Bruckner et al., 1978).
Features shared by Tsk2/+ mice and people with fibrotic diseases (scleroderma, liver fibrosis, kidney fibrosis) include the dysregulation of PIIINP (Abignano and Del Galdo, 2014; Del Galdo and Matucci-Cerinic, 2014; Majewski et al., 1999; Quillinan et al., 2014; Sondergaard et al., 1997). The PIIINP fragment is a clinically validated biomarker of liver fibrosis (Leroy et al., 2004; Rosenberg et al., 2004) and scleroderma (Majewski et al., 1999; Sondergaard et al., 1997) and it has been used as a surrogate marker of fibrosis in clinical trials of potential SSc therapies (Denton et al., 2009; Majewski et al.). Our finding of a point mutation in the protein that likely has a deleterious effect on PIIINP function is consistent with these clinical results and the fibrotic phenotype in the Tsk2/+ mouse.
Its high level in the sera of such patients may not merely be a benign biomarker. Support for this hypothesis derives from our in vitro complementation results showing that the presence of COL3A1-C33S is sufficient to up-regulate the synthesis and secretion of COL1A1, consistent with the increased activity of the Col1a1 promoter and excess production of COL1A1 in Tsk2/+ mice (Barisic-Dujmovic et al., 2008; Christner et al., 1998). It is likely that higher levels of or altered COL3A1 protein or PIIINP fragment also directly influence the composition and size of COL1A1/A2- and COL3A1-containing fibers, and that these features indirectly up-regulate TGF-β1 signaling, an important mediator of collagen production. A previous report from our laboratory has demonstrated increased dermal elastic fibers and TGF-β1 accumulation in the skin of Tsk2/+ mice beginning at two weeks of age, lending further support to our hypothesis (Long et al., 2014). In addition, our gene expression analyses show that similar global impact of the Col3a1Tsk2 gene occurs both in vitro and in vivo, and in both settings, there are fundamental changes in the extracellular matrix and in fibroblasts due to the presence of this mutation. The hypothesis that Col3a1Tsk2 (or PIIINPTsk2) directly causes dermal fibrosis and scleroderma-like characteristics is attractive: it would likely be dominant within the heterozygote, as collagen III is a homotrimeric triple helix (Ramachandran and Kartha, 1955), and the gene product of the mutant chromosome could be expected to contribute to alteration of a majority of collagen helices even in the presence of 50% normal collagen (Strachan and Read, 1999).
Materials and Methods
All studies and procedures were approved by the Institutional Animal Care and Use Committee at Drexel University College of Medicine, and conducted in accord with recommendations in the “Guide for the Care and Use of Laboratory Animals” (Institute of Laboratory Animal Resources, National Research Council, National Academy of Sciences). Detailed methods are provided in the supplemental materials.
Animals
Tsk2/+ mice were serially backcrossed the C57Bl/6J (B6) background. Recombinant B6.Tsk2/+ mice were also bred to B6.chr 1-A/J mice (Jackson Laboratory, Bar Harbor, ME) and the resulting B6.Tsk2/+ F1 mice were backcrossed to B6.chr 1-A/J mice. Wild-type littermates were used as controls.
DNA isolation from tail snips, microsatellite and SNP typing
These were performed as in our previous publications (Bunce et al., 1995; Butterfield et al., 1998) Specific locations of SNP polymorphisms between B6 (which is very similar to 101/H) and A/J were determined using Mouse Genome Informatics (www.informatics.jax.org), and Mouse Phenome Database (http://phenome.jax.org/)
Complementation analysis
Tsk2/+ mice were crossed to Col3a1−/+ mice and their progeny mated to verify that the SNP in Col3a1 is Tsk2. The resulting generations of the cross were genotyped by PCR for Tsk2/+ using microsatellites and primers specific to Col3a1 or the inserted neomycin cassette (see supplemental material).
In vitro assessment of fibrogenesis by Col3a1Tsk2
We constructed a plasmid harboring the Col3a1Tsk2 allele by introducing the Tsk2 T-to-A mutation into a wild-type Col3a1 clone (pCMV6-Kan/Neo; Origene). A Col3a1-KO line was transfected with either plasmid as described (Artlett et al., 1998). Supernatants were retained and cell lysates were harvested directly from the dish at 48 hours.
RNA isolation and real-time PCR
RNA was isolated from skin or fibroblasts using a RNA isolation kit from Clontech (Mountain View, CA), and cDNAs synthesized from 2.0 μg of total RNA using an High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA). Relative quantification of all products was measured using SYBR Green chemistry (Applied Biosystems, Foster City, CA).
RNA-Seq
Total RNA was prepared from three WT and four Tsk2/+ mice skin biopsies using Qiagen RNeasy Fibrous Tissue Mini Kit. RNA-seq sequencing libraries were prepared for the seven samples using NuGEN Ovation RNA-Seq System (NuGen, San Carlos, CA). Libraries were multiplexed and sequenced on an Illumina HiSeq 2000 platform to obtain 16.7–50.9 million 50 bp paired-end reads per sample. The raw reads were aligned to the reference mouse genome (MM9 assembly) using Tophat software with default parameters (Trapnell et al., 2012a; Trapnell et al., 2012b). Supplemental Figure 1 shows RNA-Seq read coverage for three interval genes.
454 Sequencing
Samples were captured and amplified as described in the Roche Nimblegen sequence capture manual (Version 1.0). Titanium general libraries were prepared from the captured DNAs from two 101/H mice and two Tsk2/+ mice using 5000 ng of DNA. Enriched captured fragments were sequenced as described in GS FLX Titanium emPCR and Sequencing Protocols, October, 2008. Sequence capture array probes were designed by Roche Nimblegen using the mouse genome sequence between 44,241,286 and 47,116,890 on chromosome 1 of mouse genome (mm9). Multiplexed 454 sequenced reads were assembled using Newbler v2.6 with scaffolding against the same chromosome region that the probes were derived from.
DNA microarray hybridization and data analysis
This was performed as in our previous publications (Pendergrass et al., 2012). cDNA samples were amplified and labeled using the Agilent Low Input Linear Amplification kit (Agilent Technologies, Santa Clara, CA) and were hybridized against Universal Mouse Reference (UMR) (Strategene, La Jolla, CA) to Agilent Whole Mouse Genome arrays (G4122F) (Agilent Technologies, Santa Clara, CA) in a common reference based design. Microarrays were hybridized and washed in accordance with manufacturer’s protocols and scanned using a dual laser GenePix 4000B scanner (Axon Instruments, Foster City, CA). The pixel intensities of the acquired images were then quantified using GenePix Pro 5.1 software (Axon Instruments, Foster City, CA).
Western blot analyses
Culture supernatant were collected or skin was homogenized in RIPA buffer (Sigma-Aldrich, St Louis MO) using a glass homogenizer. Total protein was measured with a Bradford assay (Sigma-Aldrich, St Louis MO), and western blots were performed as in our publications ((Sassi-Gaha et al., 2010)) Antibodies used included goat anti-COL3A1 (#sc-8781); goat anti-COL1A1 (#sc-28657) from Santa Cruz Biotechnology, Inc, Santa Cruz, CA; rabbit anti-β-Actin (#4967, Cell Signaling Technologies, Boston, MA); donkey anti-goat (#705-035-003, Jackson ImmunoResearch Laboratories, West Grove, PA); or goat anti-rabbit (#111-035-003, Jackson ImmunoResearch), and signals was developed using SuperSignal West Dura ECL reagent (Thermo Scientific Inc, Rockford, IL). Band intensities were measured using ImageQuant TL Software (GE Healthcare Life Sciences).
Reticular fiber staining
Reticular fibers were stained using the Chandler’s Precision Reticular Fiber Stain kit (American Master*Tech, Lodi CA) according to the manufacturer’s protocol.
Statistics
A two-tailed student’s t-test or a one-way ANOVA was used to determine statistical significance of collagen protein expression, as noted.
Supplementary Material
Acknowledgments
We thank Dr. Paul Christner for providing the breeding pairs of the original Tsk2/+ mice, and Dr. Xianhua Piao at Harvard University for the Col3a1-KO mice.
Funding: This work was supported by a Scleroderma Foundation Grant and awards from the NIH (AR061384) and the Department of Defense (PR100338).
Abbreviations
- Col3a1
collagen, type III, alpha 1
- B6
C57Bl/6
- ECM
extracellular matrix
- KO
knockout
- Mb
megabases
- PIIINP
procollagen III amino terminal propeptide segment
- SSc
systemic sclerosis
- SNP
single nucleotide polymorphism
- Tsk
tight-skin
- UTR
untranslated region
- WT
wild-type
Footnotes
Competing interests: The authors state no conflict of interest.
Author contributions: KBL and CMB bred and genotyped the B6.Tsk2 mice and all the derivative animals in this report; KBL, CMA, CMB and SSG conducted the histology on skin and transfections on fibroblasts; EPB was responsible for the design and interpretation of the research including the genetic analyses; ZL and MLW conducted the expression analyses and interpreted the results, VM conducted GSEA analysis, SGC constructed the plasmids containing the mutant Col3a1 cDNA; GDE, JE, RE, AA performed the genomic DNA capture and sequencing and interpreted these results; KBL, EPB, CMA and MLW wrote the paper.
References
- Abignano G, Del Galdo F. Quantitating skin fibrosis: innovative strategies and their clinical implications. Curr Rheumatol Rep. 2014;16:404. doi: 10.1007/s11926-013-0404-5. [DOI] [PubMed] [Google Scholar]
- Artlett CM. Animal models of scleroderma: fresh insights. Current opinion in rheumatology. 2010;22:677–82. doi: 10.1097/BOR.0b013e32833e307b. [DOI] [PubMed] [Google Scholar]
- Asano Y, Stawski L, Hant F, et al. Endothelial Fli1 deficiency impairs vascular homeostasis: a role in scleroderma vasculopathy. Am J Pathol. 2010;176:1983–98. doi: 10.2353/ajpath.2010.090593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barisic-Dujmovic T, Boban I, Clark SH. Regulation of collagen gene expression in the Tsk2 mouse. J Cell Physiol. 2008;215:464–71. doi: 10.1002/jcp.21319. [DOI] [PubMed] [Google Scholar]
- Bruckner P, Bachinger HP, Timpl R, et al. Three conformationally distinct domains in the amino-terminal segment of type III procollagen and its rapid triple helix leads to and comes from coil transition. European journal of biochemistry/FEBS. 1978;90:595–603. doi: 10.1111/j.1432-1033.1978.tb12640.x. [DOI] [PubMed] [Google Scholar]
- Bunce M, O’Neill CM, Barnardo MC, et al. Phototyping: comprehensive DNA typing for HLA-A, B, C, DRB1, DRB3, DRB4, DRB5 & DQB1 by PCR with 144 primer mixes utilizing sequence-specific primers (PCR-SSP) Tissue antigens. 1995;46:355–67. doi: 10.1111/j.1399-0039.1995.tb03127.x. [DOI] [PubMed] [Google Scholar]
- Butterfield RJ, Sudweeks JD, Blankenhorn EP, et al. New genetic loci that control susceptibility and symptoms of experimental allergic encephalomyelitis in inbred mice. J Immunol. 1998;161:1860–7. [PubMed] [Google Scholar]
- Christner PJ, Hitraya EG, Peters J, et al. Transcriptional activation of the alpha1(I) procollagen gene and up-regulation of alpha1(I) and alpha1(III) procollagen messenger RNA in dermal fibroblasts from tight skin 2 mice. Arthritis Rheum. 1998;41:2132–42. doi: 10.1002/1529-0131(199812)41:12<2132::AID-ART8>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Christner PJ, Peters J, Hawkins D, et al. The tight skin 2 mouse. An animal model of scleroderma displaying cutaneous fibrosis and mononuclear cell infiltration. Arthritis Rheum. 1995;38:1791–8. doi: 10.1002/art.1780381212. [DOI] [PubMed] [Google Scholar]
- Christner PJ, Siracusa LD, Hawkins DF, et al. A high-resolution linkage map of the tight skin 2 (Tsk2) locus: a mouse model for scleroderma (SSc) and other cutaneous fibrotic diseases. Mamm Genome. 1996;7:610–2. doi: 10.1007/s003359900181. [DOI] [PubMed] [Google Scholar]
- Cordes SP. N-ethyl-N-nitrosourea mutagenesis: boarding the mouse mutant express. Microbiol Mol Biol Rev. 2005;69:426–39. doi: 10.1128/MMBR.69.3.426-439.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Galdo F, Matucci-Cerinic M. The search for the perfect animal model discloses the importance of biological targets for the treatment of systemic sclerosis. Ann Rheum Dis. 2014;73:635–6. doi: 10.1136/annrheumdis-2013-203910. [DOI] [PubMed] [Google Scholar]
- Denton CP, Engelhart M, Tvede N, et al. An open-label pilot study of infliximab therapy in diffuse cutaneous systemic sclerosis. Ann Rheum Dis. 2009;68:1433–9. doi: 10.1136/ard.2008.096123. [DOI] [PubMed] [Google Scholar]
- Gentiletti J, McCloskey LJ, Artlett CM, et al. Demonstration of autoimmunity in the tight skin-2 mouse: a model for scleroderma. Journal of immunology. 2005;175:2418–26. doi: 10.4049/jimmunol.175.4.2418. [DOI] [PubMed] [Google Scholar]
- Hocher B, Schwarz A, Fagan KA, et al. Pulmonary fibrosis and chronic lung inflammation in ET-1 transgenic mice. American journal of respiratory cell and molecular biology. 2000;23:19–26. doi: 10.1165/ajrcmb.23.1.4030. [DOI] [PubMed] [Google Scholar]
- Leistritz DF, Pepin MG, Schwarze U, et al. COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet Med. 2011;13:717–22. doi: 10.1097/GIM.0b013e3182180c89. [DOI] [PubMed] [Google Scholar]
- Leroy V, Monier F, Bottari S, et al. Circulating matrix metalloproteinases 1, 2, 9 and their inhibitors TIMP-1 and TIMP-2 as serum markers of liver fibrosis in patients with chronic hepatitis C: comparison with PIIINP and hyaluronic acid. Am J Gastroenterol. 2004;99:271–9. doi: 10.1111/j.1572-0241.2004.04055.x. [DOI] [PubMed] [Google Scholar]
- Liu X, Wu H, Byrne M, et al. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci U S A. 1997;94:1852–6. doi: 10.1073/pnas.94.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long KB, Artlett CM, Blankenhorn EP. Tight skin 2 mice exhibit a novel time line of events leading to increased extracellular matrix deposition and dermal fibrosis. Matrix Biol. 2014 doi: 10.1016/j.matbio.2014.05.002. [DOI] [PubMed] [Google Scholar]
- Majewski S, Wojas-Pelc A, Malejczyk M, et al. Serum levels of soluble TNF alpha receptor type I and the severity of systemic sclerosis. Acta Derm Venereol. 1999;79:207–10. doi: 10.1080/000155599750010986. [DOI] [PubMed] [Google Scholar]
- Maurer B, Busch N, Jungel A, et al. Transcription factor fos-related antigen-2 induces progressive peripheral vasculopathy in mice closely resembling human systemic sclerosis. Circulation. 2009;120:2367–76. doi: 10.1161/CIRCULATIONAHA.109.855114. [DOI] [PubMed] [Google Scholar]
- Nishiyama Y, Nejima J, Watanabe A, et al. Ehlers-Danlos syndrome type IV with a unique point mutation in COL3A1 and familial phenotype of myocardial infarction without organic coronary stenosis. J Intern Med. 2001;249:103–8. doi: 10.1046/j.1365-2796.2001.00761.x. [DOI] [PubMed] [Google Scholar]
- Noveroske JK, Weber JS, Justice MJ. The mutagenic action of N-ethyl-N-nitrosourea in the mouse. Mamm Genome. 2000;11:478–83. doi: 10.1007/s003350010093. [DOI] [PubMed] [Google Scholar]
- Pendergrass SA, Lemaire R, Francis IP, et al. Intrinsic Gene Expression Subsets of Diffuse Cutaneous Systemic Sclerosis Are Stable in Serial Skin Biopsies. Journal of Investigative Dermatology. 2012;132:1363–73. doi: 10.1038/jid.2011.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Ball ST. Tight Skin 2 (Tsk2) Mouse News Letters. 1986:91–2. [Google Scholar]
- Quillinan NP, McIntosh D, Vernes J, et al. Treatment of diffuse systemic sclerosis with hyperimmune caprine serum (AIMSPRO): a phase II double-blind placebo-controlled trial. Ann Rheum Dis. 2014;73:56–61. doi: 10.1136/annrheumdis-2013-203674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran GN, Kartha G. Structure of collagen. Nature. 1955;176:593–5. doi: 10.1038/176593a0. [DOI] [PubMed] [Google Scholar]
- Richard V, Solans V, Favre J, et al. Role of endogenous endothelin in endothelial dysfunction in murine model of systemic sclerosis: tight skin mice 1. Fundamental & clinical pharmacology. 2008;22:649–55. doi: 10.1111/j.1472-8206.2008.00634.x. [DOI] [PubMed] [Google Scholar]
- Rosenberg WM, Voelker M, Thiel R, et al. Serum markers detect the presence of liver fibrosis: a cohort study. Gastroenterology. 2004;127:1704–13. doi: 10.1053/j.gastro.2004.08.052. [DOI] [PubMed] [Google Scholar]
- Sassi-Gaha S, Loughlin DT, Kappler F, et al. Two dicarbonyl compounds, 3-deoxyglucosone and methylglyoxal, differentially modulate dermal fibroblasts. Matrix Biol. 2010;29:127–34. doi: 10.1016/j.matbio.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Siracusa LD, McGrath R, Ma Q, et al. A tandem duplication within the fibrillin 1 gene is associated with the mouse tight skin mutation. Genome Res. 1996;6:300–13. doi: 10.1101/gr.6.4.300. [DOI] [PubMed] [Google Scholar]
- Sondergaard K, Heickendorff L, Risteli L, et al. Increased levels of type I and III collagen and hyaluronan in scleroderma skin. Br J Dermatol. 1997;136:47–53. [PubMed] [Google Scholar]
- Strachan T, Read AP. Human Molecular Genetics. 2. Wiley-Liss; New York: 1999. [Google Scholar]
- Yamamoto T, Takagawa S, Katayama I, et al. Animal model of sclerotic skin. I: Local injections of bleomycin induce sclerotic skin mimicking scleroderma. J Invest Dermatol. 1999;112:456–62. doi: 10.1046/j.1523-1747.1999.00528.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.