

ABSTRACT
Apicomplexa are obligate intracellular parasites that cause important diseases in humans and animals. Manipulating the pathogen genome is the most direct way to understand the functions of specific genes in parasite development and pathogenesis. In Toxoplasma gondii, nonhomologous recombination is typically highly favored over homologous recombination, a process required for precise gene targeting. Several approaches, including the use of targeting vectors that feature large flanks to drive site-specific recombination, have been developed to overcome this problem. We have generated a new large-insert repository of T. gondii genomic DNA that is arrayed and sequenced and covers 95% of all of the parasite’s genes. Clones from this fosmid library are maintained at single copy, which provides a high level of stability and enhances our ability to modify the organism dramatically. We establish a robust recombineering pipeline and show that our fosmid clones can be easily converted into gene knockout constructs in a 4-day protocol that does not require plate-based cloning but can be performed in multiwell plates. We validated this approach to understand gene function in T. gondii and produced a conditional null mutant for a nucleolar protein belonging to the NOL1/NOP2/SUN family, and we show that this gene is essential for parasite growth. We also demonstrate a powerful complementation strategy in the context of chemical mutagenesis and whole-genome sequencing. This repository is an important new resource that will accelerate both forward and reverse genetic analysis of this important pathogen.
IMPORTANCE
Toxoplasma gondii is an important genetic model to understand intracellular parasitism. We show here that large-insert genomic clones are effective tools that enhance homologous recombination and allow us to engineer conditional mutants to understand gene function. We have generated, arrayed, and sequenced a fosmid library of T. gondii genomic DNA in a copy control vector that provides excellent coverage of the genome. The fosmids are maintained in a single-copy state that dramatically improves their stability and allows modification by means of a simple and highly scalable protocol. We show here that modified and unmodified fosmid clones are powerful tools for forward and reverse genetics.
INTRODUCTION
Toxoplasma gondii is an obligate intracellular parasite that belongs to the phylum Apicomplexa, which includes numerous important pathogens, such as Plasmodium, Cryptosporidium, Eimeria, Neospora, and Theileria, that cause diseases in humans and animals. Among apicomplexans, T. gondii has emerged as the experimentally most tractable organism and is now used by many investigators as a genetic model to understand parasite biology (1). The ability to introduce transgenic reporters and to ablate or modify parasite genes has driven experimental work on apicomplexans over the last 2 decades. A variety of approaches have been developed to generate and introduce the DNA molecules that bring about these changes. Initially, this was based largely on mini-gene plasmids that place the coding sequence of a gene, typically obtained from cDNA, into the context of a promoter and suitable 5′ and 3′ untranslated regions (2, 3). These tools are easily constructed and allow researchers to study the expression and localization of proteins by appending an epitope tag, a fluorescent protein, or an enzyme reporter (4). These vectors can also be used for conditional gene expression in combination with regulatable promoters, such as those recognized by the tetracycline-regulated transactivator system or protein destabilization domains, which can be modulated with small-molecule ligands (5, 6). A limitation of this approach is that it removes the gene from its natural expression context in the genome. This can result in protein expression at an inappropriate level or time, which may obscure the true location or function of the protein or produce dominant negative effects that make it more difficult to interpret the results. Targeting the modification directly to the genomic locus of the gene can mitigate some of these problems. Typically, this is achieved by single- or double-crossover homologous recombination using sequences derived from genomic DNA to target the recombination event to the desired locus. T. gondii uses homologous as well as a nonhomologous end-joining DNA repair systems, and typically, nonhomologous insertion is highly favored, which can make gene targeting challenging for some genes. The development of ΔKu80 mutant strains overcomes this by drastically reducing nonhomologous recombination and thus increasing the proportion of transgenics derived by homologous recombination in a population of transfected cells. This allows gene localization and gene replacement to occur under the control of endogenous regulatory elements (7, 8). Another advancement has been the development of tetracycline-regulated transactivator TATi/ΔKu80 strains for creating conditional gene knockouts in the parasite (9). These combine superior efficiency of homologous recombination (due to deletion of Ku80) with the tetracycline-regulatable promoter system. Most recently, clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9-induced double-strand breaks have also been shown to yield higher crossover frequencies (10, 11).
A third strategy uses the massive flanking sequences afforded by large-insert genomic constructs to enhance homologous-recombination events; this is independent of mutations in repair mechanisms or the induction of genome injury and can be used in wild-type (wt) parasite strains. These large genomic inserts are not amenable to restriction cloning but can readily be modified in Escherichia coli via recombination-based genetic engineering (recombineering) to convert them into gene-tagging or gene knockout constructs (12, 13). The two large-insert cosmid libraries (TOX and PSB) available for T. gondii have been used to study the functions of genes in various biological processes, such as cell division, egress from host cells, isoprenoid biosynthesis, fatty acid synthesis, and apicoplast and mitochondrial function (1, 9, 14–20). Recombineering is a widely used platform to quickly and cost-effectively modify large DNA to produce large numbers of vectors for genome-wide functional analysis in mice (21, 22). However, cosmids are maintained at 50 copies per bacterial cell, and the presence of multiple copies can result in the modification of only a subset of cosmids. Also, activation of the phage recombination system in the context of multicopy constructs can produce illicit recombination and rearrangements, creating deletions or chimeric molecules.
Here we report the construction of a new fosmid resource of genomic DNA from the highly virulent RH strain of T. gondii. The library uses a copy control vector that overcomes many of the above-mentioned complications and offers a robust and fast route to genetic modification. The fosmid clones are maintained as a single copy per bacterial cell, which dramatically improves their stability during storage and through the recombineering process. Upon completion of the modification, a second high-copy-number origin of replication can be triggered using an inducer molecule to produce bulk DNA. We describe a powerful recombineering approach using these fosmids to modify the parasite’s genome by homologous recombination. Given our interest in understanding the role of nuclear compartmentalization in the regulation of the cell cycle in T. gondii (23), we decided to test the utility of the fosmid approach to understand the role of a previously uncharacterized parasite nucleolar protein belonging to the NOL1/NOP2/SUN family. We report here the creation of a conditional knockout mutant for this essential nucleolar protein via promoter replacement by the fosmid approach. The T. gondii model not only permits reverse genetic modification of parasites but also offers exciting forward genetic possibilities. Such approaches have been used to map the genetic loci underlying the differences in strain virulence and have led to the discovery of how secreted rhoptry kinases allow the parasite to evade attachment by innate and acquired host immunity (24–26). Similarly, chemical mutagenesis in combination with screens for temperature sensitivity has produced important insights into host cell invasion and parasite replication (27, 28). Here we show the potential of the new fosmid resource for genetic complementation analysis using the example of a cell cycle mutant. We tested fosmids covering six nonsynonymous single nucleotide polymorphisms (SNPs) predicted by whole-genome sequence analysis and identified the key mutation in the gene encoding regulator of chromatin condensation 1 (RCC1), responsible for the phenotype.
RESULTS
The fosmid library provides high coverage of the Toxoplasma gondii genome.
Our goal was to establish a resource for genome engineering in T. gondii that is stable, is amenable to efficient and scalable manipulation, and provides access to the entire genome. We constructed a fosmid library by hydroshearing T. gondii RH genomic DNA into ~40-kb fragments (Fig. 1A). Hydroshearing to produce a random unbiased library based on physical breakage and size fragmentation was used rather than the traditional approach of partial Sau3AI digestion of DNA. To construct the library, we chose a copy control vector (pCC2FOS) that contains an E. coli F-factor single origin of replication as well as an inducible high-copy-number oriV gene. This system offers the advantage of stably maintaining fosmid clones in EPI300-T1R phage-resistant E. coli at a single copy per cell, thus avoiding the undesired recombination that we experienced with previously constructed cosmid libraries for a subset of clones. The fosmid clones could be amplified to high copy numbers (10 to 200 copies/cell) when desired since the EPI300-T1R E. coli cells provided the product of the trfA replication initiation gene under the tight control of an arabinose-inducible promoter for initiation of replication from oriV.
FIG 1 .

Characterization of the Toxoplasma gondii RH strain fosmid genomic DNA library. (A) CHEF gel showing the size comparison of native high-molecular-weight T. gondii genomic DNA with hydrosheared DNA used for library preparation. The positions and sizes (kb) for the midrange II PFG marker are shown. (B) Size distribution (kb) of the 5,509 T. gondii fosmid library clones. The mean size of ~35 kb is indicated as a red dashed line. (C) Coverage of T. gondii genes as fosmid clone number increases. The line plateaus at 2,212 fosmids, covering 7,816 T. gondii genes.
The T. gondii fosmid library contained 200,000 independent clones that were maintained as a glycerol stock at −80°C. A random subset of 10,000 clones was picked and end sequenced using the vector-specific primers PC1F and PC1R (primer sequences are provided in Table S1 in the supplemental material). Sequences from clones (n = 8,408) that produced high-quality reads from both ends were used as queries in a BLAST search against the T. gondii ME49 genome to obtain the start and end coordinates of each of the clones in the genome. Although all 8,408 clones mapped to the T. gondii genome, only 7,046 fosmids mapped to annotated chromosomes; the remaining (n = 1,362) mapped to unassembled regions, such as genomic scaffolds or assemblies. Of the 7,046 clones that mapped to Toxoplasma chromosomes, 1,537 clones showed multiple BLAST hits on different chromosomes and were excluded from our analysis to avoid ambiguity. The genome-wide set of the mapped fosmid clones (n = 5,509) is publicly available under the Genome Browser track of ToxoDB (http://toxodb.org/cgi-bin/gbrowse/toxodb/).
The insert size distribution of the sequenced and mapped fosmid library is shown in Fig. 1B, with 98.5% (5,426 out of 5,509) of the clone inserts in the 20- to 50-kb range; the average insert size is 35 kb. We next wanted to determine the number of genes that were covered by these fosmid clones (Fig. 1C). As expected, the number of genes covered increased with the number of clones, but the curve plateaus at 2,212 fosmids. These 2,212 fosmids covered ~95% (7,861 out of 8,317) of the genes on the T. gondii chromosomes (Fig. 1C), suggesting that we reached saturation with respect to the number of T. gondii genes covered.
We also wanted to compare the coverage of the fosmids to that of the existing clones from two cosmid libraries (Fig. 2). For this purpose, we used the start and end coordinates of the clones and generated a map of their coverage across all 14 Toxoplasma ME49 chromosomes, based on the current genome assembly and annotation. The 5,509 fosmid clones were found to provide deeper coverage of the 14 T. gondii chromosomes than did the 7,773 cosmid clones (Fig. 2A). We also performed an analysis to compare the numbers of genes covered per chromosome by cosmid (Fig. 2B) and fosmid clones (Fig. 2C). The fosmid clones, even though fewer, covered 79 more genes in total than the cosmids (also see Table S2 in the supplemental material).
FIG 2 .

Comparison of coverages of T. gondii chromosomes by cosmids and fosmids. (A) Ideogram showing the regions of the T. gondii chromosomes covered by cosmids (red) and fosmids (blue). White spaces indicate no coverage. (B and C) Cosmids (B) and fosmids (C) mapping to genes on the 14 T. gondii chromosomes. The genes covered on each chromosome are shown in coral, whereas those that are not covered are shown in turquoise.
Modification of fosmid DNA via recombineering.
We wanted to optimize a recombineering strategy to modify our set of arrayed and sequenced fosmids for transfection into the parasite to create gene knockouts. Given our interest in understanding the role of nucleolar proteins, we picked fosmid RHfos05J01 (TGME49_chrIX, positions 2582982 to 2625679; size, 42.69 kb) as an example. This clone covers the T. gondii gene (TGME49_288530) that encodes a protein belonging to the NOL1/NOP2/SUN family. To create a conditional knockout for the SUN gene, we grew bacteria carrying fosmid RHfos05J01 recovered from our frozen clone collection and used the gentamicin-dihydrofolate reductase (DHFR)-T7S4 modification cassette (9) to replace the endogenous promoter in the fosmid with a tetracycline-regulatable promoter (Fig. 3A). This cassette was amplified with long primers (SUN_PR_F and SUN_PR_R) that amplify the cassette and also contain homology flanks that target 50 bp 5′ of the promoter and 3′ of the initiation codon of the SUN gene (primer sequences are provided in Table S1 in the supplemental material). The RHfos05J01 fosmid was grown in chloramphenicol, and recombination was induced after electroporation with plasmid pSC101gbaArec. The amplified SUN promoter replacement cassette was then introduced, and recombination led to the replacement of the SUN gene promoter with the T7S4 promoter. The fosmids were selected on chloramphenicol and gentamicin plates, and the colonies that grew were screened by PCR using primers P3 and P4. No growth was observed on chloramphenicol and gentamicin plates for clones in which the recombination machinery was not switched on and for no-DNA controls. The copy number of the modified SUN fosmid clone was induced using l-arabinose to produce bulk DNA for parasite transfection.
FIG 3 .

Fosmid modification by recombineering. (A) Steps of fosmid modification. In step I, the fosmid containing the gene of interest (GOI) is grown overnight in the presence of chloramphenicol (chl) at 37°C. Plasmid pSC101gbaArec, a plasmid carrying recombination Red γ, β, and α proteins is transformed into the cells. This plasmid has a tetracycline (tet) resistance marker and a temperature-sensitive origin of replication that allows growth only at 30°C, and the recombinase genes under the tight control of an arabinose-regulatable promoter. GENT, gentamicin. In step II, addition of l-arabinose (ara) switches on the recombination machinery. Cells are transformed with the gentamicin-DHFR-T7S4 promoter PCR cassette containing 50-bp regions of homology flanking the 5′ and 3′ ends of the cassette. In step III recombination occurs, and the cassette is integrated, leading to replacement of the endogenous promoter with the T7S4 promoter, at which time cells are grown with chloramphenicol and gentamicin. Overnight growth at 37°C leads to removal of the pSC101gbaArec recombination protein expression plasmid, preventing further rearrangement of the construct. The modified fosmid is induced to high copy numbers by addition of l-arabinose. Modified fosmid DNA can now be used directly for transfection into T. gondii, and the parasites are selected with pyrimethamine (Pyr). After homologous recombination in the parasite, the T7S4 promoter replaces the endogenous promoter and gene expression can be regulated upon addition of anhydrotetracycline (ATc). (B) PCR detection of modification of multiple fosmids performed using liquid recombineering. The results of PCR screening of five different fosmid clones to detect promoter replacement (PR) with the tetracycline-regulatable promoter or insertion (PI) are shown. No band is seen for the unmodified fosmids (UM). The PCR results shown here are an inverted image of an ethidium-bromide-stained agarose gel. The sequences of the primers used for PCR amplification are shown in Table S1, and further detail on the fosmid clones and the genes that they cover are provided in Table S3 in the supplemental material.
We also wanted to optimize a liquid recombineering protocol in deep 96-well plates. This protocol may be faster, as it avoids plates and colony picking, and the recombineering procedure can be performed in a high-throughput manner to allow modification of multiple fosmids in parallel. We repeated the modification of the SUN gene with two different targeting cassettes (resulting in promoter replacement or insertion) and picked four additional fosmids (see Table S3 in the supplemental material). Ten independent fosmid modifications were conducted in parallel. Eight of the 10 targeted constructs were modified successfully using liquid recombineering in a 96-well plate in the first attempt (see Table S3 in the supplemental material) as determined by PCR screening (Fig. 3B). No bacterial growth was observed for experiments in which the recombination machinery was not induced or in which a modification cassette was not supplied, indicating a negligible background. Our results suggest that the liquid recombineering procedure is very robust and fast. A targeting construct with large flanks on both sides could be obtained in 4 days; 7 days is required for modification when bacteria are plated and cloned at each step.
Modified fosmid DNA efficiently replaces the endogenous promoter of the T. gondii SUN gene.
We tested the utility of modified fosmids to create conditional knockout mutants of T. gondii using the locus of the T. gondii SUN (TgSUN) protein. We first introduced an HA3 epitope tag into this locus in a TATi ΔKu80 background using single homologous recombination (7) to be able to later follow the fate of the gene and protein. Analyzing the resulting drug-resistant clones by immunofluorescence assay, we found that TgSUN is a nucleolar protein that is robustly expressed in the tachyzoite stage and formed a ring-shape structure in the nucleolus of the parasite (Fig. 4A). To further describe the nucleolar compartment to which this protein localizes, we used an antibody to fibrillarin (to stain the dense fibrillar compartment) and found that TgSUN surrounds this protein (Fig. 4B). Thus, the SUN protein is localized to the outermost granular compartment of the nucleolus. Western blot analysis revealed that the tagged protein was of the predicted molecular mass of ~91 kDa (87.6-kDa SUN protein plus the 3.17-kDa epitope tag), and no reactive band was observed in the TATiΔKu80 parental line (Fig. 4C).
FIG 4 .

Localization and expression of the T. gondii SUN protein. (A) Fluorescence microscopy of the C-terminally HA-tagged SUN protein using anti-HA (green) and IMC1 (red [to outline parasite cells]) antibodies shows its nucleolar localization. Nuclei are stained with DAPI (4′,6-diamidino-2-phenylindole) (blue). (B) SUN protein is associated with the granular compartment (outer) of the nucleolus. Fibrillarin antibody staining of the dense fibrillar compartment is shown in red, and the HA-tagged SUN protein (green) forms a ring-shaped structure around the fibrillarin. (C) Western blot of parasite pellets showing expression of HA3-tagged SUN protein using anti-HA antibody. The TATi ΔKu80 parental line showing no expression is included as a control. The lower panel shows the loading control using anti-α-tubulin antibody.
We then transfected this tagged parasite line with the modified fosmid DNA and selected for pyrimethamine resistance conferred by the DHFR marker that we introduced into the fosmid. Clones (n = 48) were screened by PCR for double homologous recombination and replacement of the endogenous promoter with the regulatable T7S4 promoter (Fig. 5A). Since the fosmid insert is large (42 kb), it was impractical to amplify a diagnostic PCR product that was anchored by a primer in the modification cassette on one side and by a primer in a region outside the fosmid on the other. Therefore, we performed a PCR screen to detect the loss of the endogenous promoter due to replacement by the modification cassette (Fig. 5B, amplification with primers P4 and P5) and found no amplification in the promoter replacement clone, while amplification was seen in the TATi ΔKu80 parental line. A success rate of 35% for homologous recombination was obtained, as 17 of the 48 clones screened showed replacement of the endogenous promoter with the T7S4 promoter. We also used primers flanking the bacterial chloramphenicol acetyltransferase (CAT) resistance marker found on the backbone of the fosmid. Double homologous recombination of the modification cassette into the chromosomal locus would eliminate this sequence; in contrast, episomal maintenance of the fosmid would preserve it. No amplification for the CAT gene using primers P6 and P7 was observed, and we therefore conclude that the promoter was successfully replaced in the parasite (Fig. 5B).
FIG 5 .

Fosmid-based generation of the conditional SUN2 mutant by promoter replacement. (A) Scheme of replacement of the endogenous promoter with an inducible tetracycline-regulatable promoter (T7S4) via double homologous recombination in the TATi ΔKu80 HA-tagged line using fosmid DNA. The fosmid DNA is modified by recombineering in E. coli to introduce the gentamicin-DHFR-T7S4 cassette in place of the endogenous promoter for the SUN gene as described in the legend of Fig. 2. The modified fosmid DNA is then transfected into T. gondii for homologous recombination and promoter replacement to occur. The genes upstream (TGME49_288500, _288510, and _288520) and downstream (TGME49_288540, _288550, _288560, and _288570) of SUN are shown as unfilled boxes. The HA3 tag and the chloramphenicol resistance marker in the parasite are displayed in pink and light blue, respectively. (B) PCR mapping of the SUN promoter replacement clone using primers indicated in panel A. The integration at the 5′ (P1 and P2 primers) and 3′ (P3 and P4 primers) ends of the promoter replacement is documented. PCR amplification with a primer set (P4, P5) that amplified within the endogenous promoter region in TATi ΔKu80, but not in the iΔSUN-KO line, is also shown. Primers (P6, P7) flanking the CAT resistance marker on the fosmid backbone resulted in a 930-bp band in the modified fosmid (shown as a control) but no amplification in the iΔSUN-KO and TATi ΔKu80 parasite lines. The primer sequences are provided in Table S1 in the supplemental material. The PCR results shown here are an inverted color image of an ethidium-bromide-stained agarose gel.
Modified fosmid provides tight regulation of protein expression.
The tetracycline-inducible SUN knockout clone (iΔSUN-KO) was grown in the presence of anhydrotetracycline (ATc) to assess the regulation provided by the introduced modified fosmid cassette. Western blotting using anti-HA antibody revealed that the expression of the SUN protein was markedly downregulated over the course of 48 h of ATc treatment (Fig. 6A). This reduction was also apparent in immunofluorescence assays performed after 24 h of ATc treatment; these parasites also showed a more condensed labeling that had lost its typical ring shape. The remaining protein was now localized to the dense fibrillar component and colocalized with fibrillarin (Fig. 6B, +ATc, 24-h panels). After 48 h of ATc treatment, nucleolar TgSUN was no longer detectable (Fig. 6B, +ATc, 48-h panels). No change in nucleolar morphology was observed in the iΔSUN-KO and parental line in the absence of ATc (Fig. 6B, −ATc panels).
FIG 6 .
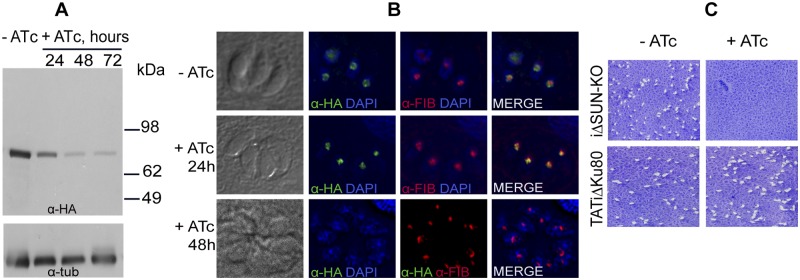
Regulation of SUN protein expression. (A) Western blot using anti-HA antibody protein lysates of the iΔSUN-KO line obtained from cells grown in the absence (−ATc) or presence (+ATc) of ATc for different lengths of time. Note the rapid decrease in expression of the SUN protein after the addition of ATc. The lower panel shows labeling with an anti-α-tubulin antibody as a loading control. (B) Fluorescence microscopy of T. gondii iΔSUN-KO parasites grown in the absence and presence of ATc for 24 and 48 h. The nucleus is stained with DAPI (blue); anti-HA (green) and anti-fibrillarin (red) antibodies show the nucleolar localization of the SUN protein. In the absence of ATc, the ring-like nucleolar localization pattern is indistinguishable from the native pattern shown in Fig. 3B, where the SUN protein surrounds the fibrillarin-labeled zone. In the presence of ATc, initially, the remaining SUN protein seems condensed, losing its ring-like pattern, and instead colocalizes with fibrillarin (24 h), whereas at 48 h of ATc treatment, staining for the SUN protein is no longer observed. (C) Plaque assays in the absence (−) or presence (+) of ATc. No plaques are seen in the iΔSUN-KO parasite line upon addition of ATc and after growth for 7 days, indicating that this gene is essential for parasite growth. The TATi ΔKu80 HA-tagged parental line grown in the presence or absence of ATc is shown as a control. Quantification of plaque size is shown in Fig. S1 in the supplemental material.
Next we measured the growth of iΔSUN-KO and its TATi ΔKu80 HA-tagged parental line in the presence and absence of ATc by plaque assays. We did not observe plaques when the promoter replacement parasites were grown in the presence of ATc for 7 days, indicating that the protein is essential for parasite growth (Fig. 6C). The parental line continued to grow efficiently in the presence of ATc and is shown as a control (Fig. 6C). These results suggest that fosmid recombineering is suitable for modifying essential genes in the T. gondii parasite. In the absence of ATc, the plaques observed for the iΔSUN-KO promoter replacement line were smaller in size than those for the TATi ΔKu80-HA parental line (see Fig. S1 in the supplemental material; the difference was moderate yet statistically significant). This may be due to differences between the level of expression of the SUN protein driven by the T7S4 promoter and the level of expression of the protein driven by its endogenous promoter.
Fosmid complementation of a ts mutant identifies a new variant of the Toxoplasma RCC1 ortholog as the key protein responsible for conditional growth arrest.
Large-insert clones of genomic DNA are ideal for complementation analysis. We explored the potential of this new library for genetic complementation of temperature-sensitive (ts) mutants. Specifically we tested the ts mutant 13-136A8 strain isolated in a previously described large chemical mutagenesis screen (28). This mutant was found to grow normally at 34°C but showed a severe growth defect when cultured at 40°C. Phenotypic characterization revealed that the majority of the cells were unable to complete a second round of the division (Fig. 7A, 40°C panel) and were arrested in the premitotic stage, as evidenced by costaining with the nuclear centrocone marker MORN1 and apicoplast protein Atrx1. Neither expansion of the centrocone that normally occurs in the S/M phase (Fig. 7A, 34°C, anti-MORN1 staining) nor duplication of the apicoplast was detected in the 13-136A8 mutant parasites grown at 40°C. These phenotypic features together with the absence of budding pointed toward a likely S-phase arrest of the ts mutant 13-136A8. We observed irregular DNA staining, with evident relaxation of the chromatin in the proximal part of the nucleus (Fig. 7A, graphs, blue DAPI line) and overcondensation in the apical region, although the overcondensation region was close to but clearly segregated from plastid DNA (Fig. 7A). This mutant represents the first S-phase mutant of Toxoplasma characterized.
FIG 7 .

Growth defect and complementation of the ts mutant 13-136A8 using fosmids. (A) Mutant parasites were grown for 24 h at 34°C or 40°C and stained with anti-Atrx1 (left panel) and anti-MORN1 (right panel) to detect apicoplasts and nuclear centrocone, respectively. Cells were costained with DAPI (blue) and anti-IMC1 (red). The white arrows indicate diffuse distribution of the nuclear chromatin in the parasites arrested at 40°C, whereas DNA condensation was not affected in the cells grown at 34°C. Quantification of the DAPI fluorescent signal across the nucleus (white line) is presented on the graphs. Incubation at 40°C resulted in the overcondensation of the chromatin at the apical end (overlapping green and blue peaks [40°C]) and DNA relaxation at the proximal end (blue line, right shoulder). Note that at 40°C, the mutant was unable to replicate the plastid or the nuclear centrocone, which is consistent with a premitotic arrest in S phase. (B) Genetic complementation of the ts mutant 13-136A8 was performed using fosmids in three sequential steps. A pool of fosmids covering predicted SNPs were used in the series 1 complementation. In series 2, the fosmid pool was divided into two sets, and in the third and final round, three fosmids were tested individually. Successful growth of the ts mutant at 40°C is shown in green. (С) The genomic locus spanned by the insert of fosmid clone RHfos19E05 resulted in rescue of the high temperature sensitivity of the ts mutant 13-136A8. Fosmid RHfos19E05 contains six genes: TGGT1_213880, TGGT1_213885, TGGT1_213890, TGGT1_213900, TGGT1_213910, and TGGT1_213920. The V728G mutation identified by whole-genome sequencing in the RCC1/BLIP-II domain of TGGT1_213900 is indicated with an arrow. (D) HA-tagged TgRCC1 localizes to the parasite nucleus, consistent with the S-phase phenotype of the 13-136A8 ts mutant. Costaining of the TgRCC1 protein with anti-HA antibody (green), DAPI (blue) (nuclear staining), and anti-IMC1 antibody (red) is shown.
Previously, we developed a genetic protocol to identify the defective gene in ts mutants that employs primary complementation with a cosmid genomic library followed by marker rescue, secondary complementation with single cosmids to resolve the locus, and, finally, sequencing to identify the point mutation (28). While successful, this protocol is low throughput, labor-intensive, and time-consuming. To identify the mutation responsible for this growth defect, we developed an updated strategy taking advantage of the reduced cost of whole-genome sequencing and the fosmid library. We first performed whole-genome comparison of this mutant and its parent RH strain and identified SNPs resulting in nonsynonymous mutations in eight predicted proteins (see Table S4 in the supplemental material). We prioritized candidates further by requiring genes to show expression in tachyzoites (>15th-percentile expression in tachyzoites) and a mutation leading to a nonconservative amino acid change. This narrowed the field to six genes that were selected for genetic complementation of the 13-136A8 mutant. Six corresponding fosmid clones spanning each gene were identified in the library and used for DNA preparation. We used a stepwise protocol to avoid the pitfall of reversion (Fig. 7B). The initial transformation was carried out using a pool of all six fosmids; this rescued the growth of the 13-136A8 mutant at 40°C. Next, the mutant was transformed with two sets of three fosmids each (set 1, RHfos20F20, RHfos01J09, and RHfos21J15, and set 2, RHfos07K16, RHfos19E05, and RHfos10E09). Only set 2 was able to restore growth at the restrictive temperature. When fosmids from set 2 were individually tested, only fosmid RHfos19E05 resulted in phenotypic complementation (Fig. 7B). This 39.45-kb fosmid (chromosome V, positions 1401130 to 1440586) spans six genes, including TGGT1_213900, bearing the mutation V728G identified by whole-genome sequencing in a conserved RCC1/BLIP-II region (Fig. 7C). The complementing gene TgRCC1 encodes a predicted regulator of chromosome condensation, and the role of this factor in nuclear trafficking in T. gondii has been previously reported (29). Consistently with the S-phase phenotype of this mutant, endogenous HA3 epitope tagging of RCC1 revealed a granular nuclear localization (Fig. 7D), which validates the results reported previously for this protein (29). Nuclear localization and conserved function in chromatin organization explain the DNA relaxation observed in the TgRCC1-deficient cells, which likely contributes to the temperature-sensitive arrest of parasites in the premitotic stage, which in other eukaryotes requires proper DNA condensation. Taken together, whole-genome sequencing followed by serial pool complementation with sets of fosmid clones rapidly reduces the effort and time required to pinpoint the mutations responsible for conditional growth in ts mutants.
DISCUSSION
We describe an improved system for genetic analysis and modification to study the biology of the parasite T. gondii. The main resource generated in this effort is an arrayed and end-sequenced fosmid library that was produced by random shearing, with an average insert size of 35 kb. This library yielded excellent coverage of the parasite’s genome, exceeding and extending the available resources (14, 30). The sequenced clones cover ~95% of the genes that often provide researchers multiple fosmids, covering a particular gene of interest to choose from. We demonstrate several important applications of this resource.
The deep coverage of the library and the high stability of its clones make it an excellent resource for complementation assays. Forward genetic screens are very powerful tools of discovery, as they do not require preconceived notions of mechanism and function. While mutants are easily generated by chemical mutagenesis (31), linking mutation to phenotype has required significant effort (28). Improvements in sequencing technology now permit mutant identification by whole-genome sequencing (27, 32), and this technology will likely further improve and become more affordable. However, mutagenesis typically produces multiple changes per genome that have to be validated one by one. We demonstrate here that the arrayed genome resource can be used to quickly and rigorously reduce that complexity.
The arrayed genome also provides avenues for genome engineering and reverse genetic analysis. The optimized liquid recombineering pipeline in E. coli that we describe here for T. gondii fosmid modification is highly efficient and scalable and thus can be used to modify numerous clones in parallel (using the deep, 96-well plate format). This robust recombineering procedure also substantially reduces the time required to create constructs for gene knockouts. This platform can also be used for gene tagging, the introduction of point mutations at desired sites, and parasite complementation experiments. The power of liquid recombineering was first described to generate green fluorescent protein (GFP)-tagged transgenes from Caenorhabditis briggsae bacterial artificial chromosome (BAC) genomic clones (33). This technology has successfully been applied to engineer fosmid clones in Drosophila (34) and to the bacteriophage N15-based library for Plasmodium berghei, albeit with a smaller average insert size of 9 kb due to the AT-rich nature of the Plasmodium genome (13). Importantly, this resource is easily combined with other approaches. The use of CRISPR/Cas9 has recently been described for T. gondii for introducing double-strand breaks and allows efficient gene disruption (10, 11). The collection of the fosmid clones and the robust recombineering pipeline make the library an attractive system to provide homologous recombination donor constructs for modification experiments. Furthermore, while yielding exciting efficiency, CRISPR/Cas9 has some limitation stemming from significant off-target effects (35–37). Having available a technology that relies on simple homologous recombination without additional mutation may be a strength in certain settings.
In this study, we document the utility of the fosmid modification approach to create a conditional knockout for the essential nucleolar SUN gene in T. gondii via a promoter replacement strategy. The high efficiency of recombination that occurs due to the long homology arms of the fosmid made it easy to isolate stable transgenic clones in the parasite. This is the first SUN protein described for any apicomplexan parasite. The first SUN domain protein (Fmu) was described in E. coli and methylates C-967 in the 16S rRNA, while other members of the family containing this domain are involved in the methylation of tRNAs (38). The best studied among them is the Myc-induced SUN domain-containing protein (Misu or NSun2), a nucleolar tRNA methyltransferase important for c-Myc-induced proliferation in skin that is required for proper mitotic spindle assembly and cell cycle progression (39–41). Our results indicate the essential role of the T. gondii SUN protein in nucleolar stability and parasite growth.
In conclusion, the arrayed and sequenced T. gondii fosmid clone library is a valuable resource to the parasitology community, enabling and enhancing both forward and reverse genetic analysis.
MATERIALS AND METHODS
Toxoplasma gondii fosmid library preparation.
High-molecular-weight genomic DNA was extracted from Toxoplasma gondii RH strain parasites. Briefly, parasites from four T-175 parasite flasks were harvested and filtered through 3-µm polycarbonate membrane filters, followed by DNase I treatment to remove host cell DNA contamination. DNase I was heat inactivated at 75°C for 20 min, and the parasite pellet was suspended in 10 ml of 10 mM Tris, 1 mM EDTA (TE), pH 8.0. Parasites were lysed in 0.2% SDS and RNase A (0.04 µg/ml) for 4 h at 37°C. Proteinase K was added to the lysate and incubated at 37°C for 1 h, followed by overnight digestion at 56°C. DNA was extracted with phenol-chloroform-isoamyl alcohol (25:24:1) followed by chloroform extraction and precipitation using 0.2 volumes of 10 M ammonium acetate and 2 volumes of 100% ethanol. Precipitated DNA was spooled out of the tube and resuspended in TE overnight at 4°C.
The extracted DNA was aliquoted into tubes (20 µg per tube) and subjected to fragmentation using a HydroShear device employing a large shearing assembly (fragment size, 4 kb to >50 kb; Digilab Inc., MA). DNA shearing was monitored by pulse-field gel electrophoresis (PFGE) on a 1% agarose gel in 0.5× Tris-borate-EDTA (TBE) on a contour-clamped homogeneous electric field (CHEF) and crossed-field gel electrophoresis mapper system (Bio-Rad, CA) using the following parameters: 4 V/cm, an included angle of 120°, a switch time linearly ramped from 5 to 25 s, and 16 h at 16°C. The gel was stained with ethidium bromide and visualized on a UV transilluminator and compared with the Midrange II PFG marker (New England Biolabs, MA) to visualize the zone of DNA fragments in the ~40-kb range. Shearing was optimized in a series of pilot experiments and carried out at a speed code of 16 for 25 cycles, with a retraction speed of 20 for library construction.
DNA of the desired fragment length (~40 kb) was end repaired to generate blunt phosphorylated 5′ ends, ligated with the linearized pCC2Fos copy control vector, packaged using MaxPlax lambda packaging extracts, and transformed into phage EPI300-T1R E. coli cells according to the manufacturer’s instructions (Epicentre Biotechnologies, WI). Different dilutions of the package phage were plated to count colonies and determine the size of the library. The fosmid library contained 200,000 independent clones, and glycerol stocks of the library were prepared and frozen at −80°C.
Fosmid library clone picking, sequencing, and mapping to the T. gondii genome.
To characterize the library, 20 random fosmid clones were picked in 12.5 µg/ml chloramphenicol and induced to high copy numbers using 0.2% arabinose for miniprep DNA isolation. The clones were digested with NotI, recognition sites for which flank the insert on the fosmid, and insert size was determined by gel electrophoresis. The clones were also sequenced using vector-specific primers, PC1F and PC1R (primer sequences can be found in Table S1 in the supplemental material). A dilution of library glycerol stock was plated onto large 22- by 22-cm rectangular QTrays containing 12.5 µg/ml chloramphenicol and incubated at 37°C overnight. A QBot robotic device (Genetix Inc., USA) was used to pick clones and array them into 384-well plates; the device was also used to replicate the plates. The clones were deposited into freezing medium [36 mM K2HPO4 (anhydrous), 13.2 mM KH2PO4, 1.7 mM sodium citrate, 0.4 mM MgSO4·7H2O, 6.8 mM (NH4)2SO4, 4.4% (vol/vol) glycerol in LB containing 12.5 µg/ml chloramphenicol], and plates were stored at −80°C. A set of plates was sent for end sequencing to Lucigen Corporation (WI) using primers PC1F and PC1R. The sequences obtained for each clone were subjected to BLAST search in the Toxoplasma database (http://www.toxodb.org), version 8.2, and mapped to the annotated ME49 genome.
High-throughput modification of fosmids in Escherichia coli via recombineering.
The fosmid clones containing the gene of interest were picked from the 384-well plates and grown overnight at 37°C in 12.5 µg/ml chloramphenicol either in tubes (single fosmids) or in 2-ml-deep 96-well plates (for high-throughput modification of multiple fosmids). The overnight cultures were diluted, and a secondary inoculation was allowed to grow at 37°C with shaking at 250 rpm in the incubator (for tubes) or 900 rpm in a BioShake iQ ThermoMixer (Quantifoil Instruments GmBH, Germany) until the optical density at 600 nm (OD600) reached 0.6 to 0.8. The tube or plate was chilled on ice for 15 min, and electrocompetent cells were prepared by pelleting and washing them three times in chilled sterile water. After the final washing step, 10 ng of recombination plasmid pSC101gbaArec (a kind gift from Oliver Billker, Wellcome Trust Sanger Institute, United Kingdom) was added. This is a low-copy-number plasmid and has a temperature-sensitive origin of replication and a tetracycline resistance marker. The resuspended pellet was transferred to a chilled 1-mm cuvette. Cells were electroporated with a BTX ECM 630 system (1,800 V, 25 µF, 200 Ω) and allowed to recover at 30°C for 70 min in antibiotic-free media. Chloramphenicol and tetracycline were added to the media to final concentrations of 12.5 µg/ml and 5 µg/ml, respectively, and cells were grown overnight at 30°C.
The overnight cultures were used for secondary inoculation and grown at 30°C to an OD of 0.3 to 0.4. The expression of recombination genes was induced by adding l-arabinose to a final concentration of 0.2%, and temperature was raised to 37°C for 40 min. The tubes/plates were chilled for 15 min, and electrocompetent cells were prepared. A recombineering PCR cassette was amplified with proofreading PrimeSTAR HS DNA polymerase (TaKaRa Bio Inc., Japan) using long primers (50-bp gene-specific overhangs and 25 bp matching the cassette). The PCR product was treated with DpnI to destroy the template plasmid prior to gel purification. Five hundred nanograms of this PCR recombineering cassette was delivered by electroporation as described above. After recovery, cultures were grown overnight at 37°C in media or plated on LB plates with 12.5 µg/ml chloramphenicol (for the fosmid) and 10 µg/ml gentamicin (for the cassette). The modification of fosmid clones was confirmed by PCR. Also, the confirmed modified fosmids were grown in chloramphenicol and gentamicin and induced with 0.2% arabinose (to increase fosmid copy number), and isolated miniprep DNA was subjected to Sanger sequencing.
Construction of a tagged reporter parasite line.
T. gondii NOL1/NOP2/SUN (ToxoDB gene identifier, TGGT1_288530) and wild-type (wt) T. gondii RCC1 (TgRCC1) (ToxoDB gene identifier, TGGT1_213900) were tagged with a C-terminal 3-hemagglutinin (HA3) epitope through modification of their respective genomic locus. For the SUN gene, a 2,726-bp region of the genomic sequence preceding the stop codon was amplified from T. gondii genomic DNA using primers SUN-LICF and SUN-LICR (see Table S1 in the supplemental material). The amplified product was introduced by ligation-independent cloning (42) into the vector pLIC-HA3-CAT (7, 8). T. gondii TATi ΔKu80 parasites (9) grown in human foreskin fibroblasts (HFF) were transfected with the linearized pLIC-SUN-HA3-CAT plasmid and selected on chloramphenicol, and clonal parasite lines were isolated, as previously described (4). For the TgRCC1 gene, a 1,329-bp PCR DNA fragment was amplified using the RCC_LICF and RCC_LICR primers (see Table S1 in the supplemental material), encompassing the 3′ end of TgRCC1, to construct the plasmid pLIC-TgRCC1-HA3x/HxGPRC. This plasmid was electroporated into ΔKu80 parasites and subjected to positive selection on mycophenolic acid and xanthine.
Transfection of modified fosmids into tagged reporter lines and isolation of conditional mutants.
The SUN fosmid was modified to replace the endogenous promoter with a tetracycline-regulatable promoter, tetO7sag4 (T7S4), using the recombineering procedure described above. The gentamicin-DHFR-T7S4 cassette described by Sheiner et al. (9) was used as a template to amplify the cassette with 50-bp homology flanks using primers SUN_PR_F and SUN_PR_R. The modified fosmid was transfected into TATi ΔKu80 parasites and selected on pyrimethamine. Clones were screened for promoter replacement, 5′ and 3′ integration, and the presence of the circulating fosmid. Growth of the conditional SUN mutant was measured by plaque assay in the presence and absence of 0.5 µM anhydrotetracycline (ATc). The area of plaques was quantified using ImageJ v1.84 software.
Whole-genome sequencing and genetic complementation.
The ts mutant 13-136A8 and parental RH Δhxgprt strain were grown at 34°C, and genomic DNA was extracted using the DNeasy blood and tissue kit (Qiagen GmbH, Hilden, Germany). Paired-end sequencing of the purified 13-136A8 and RH Δhxg prt DNAs was performed at the Oklahoma Medical Research Foundation sequencing facility on the Illumina HiSeq 2500 platform using the chemistry and protocol recommended by the manufacturer (Illumina Inc., San Diego, CA). Coverages of 139 times (97 million reads) and 113 times (85 million reads), respectively, were achieved for the 13-136A8 and RH strains. The sequences were first filtered in PRINSEQ version 0.20.3 (43) to remove low-quality reads and were mapped against the T. gondii GT1 reference genome (ToxoDB version 9.0) using BWA short-read aligner version 0.7.2 (44). The single nucleotide polymorphisms (SNPs) were identified using the Genome Analysis tool kit (GATK) UnifiedGenotyper (45, 46). The SNPs detected in the 13-136A8 mutant were compared with SNPs in wild-type RH to identify the SNPs specific to the mutant. These SNPs were also manually verified by visualizing the alignment files in the Integrated Genome Viewer (IGV) tool (47).
Genetic complementation of the ts mutant 13-136A8 was performed using the pooled fosmid sets covering the SNPs identified by the whole-genome sequence comparison. In preliminary experiments, we measured the spontaneous frequency of temperature resistance in the 13-136A8 mutant to be well below 10−7. In our complementation tests, we thus scored sustained growth at the restricted temperature of 40°C as a positive rescue. Differences between the abilities of fosmids to complement were readily observable as growth after transfection or a lack thereof. First, a complete set of six fosmids followed by two sets of three fosmids each were tested for complementation of the ts mutant 13-136A8. Finally, individual fosmids from the complemented set were used to confirm complementation and growth of the mutant at 40°C.
Fluorescence microscopy.
For immunofluorescence assays, HFF were grown on coverslips and infected with parasites. After 24 h of infection, coverslips were fixed with 4% paraformaldehyde, permeabilized with 0.25% Triton X-100 in phosphate-buffered saline (PBS), and blocked overnight in 4% bovine serum albumin (BSA) in PBS. Primary antibodies used were rat anti-HA (clone 3F10; Roche Applied Science, IN; 1:200 dilution), mouse monoclonal antifibrillarin (17C12, a gift from Michael Terns, University of Georgia; 1:1,000 dilution), mouse monoclonal anti-Atrx1 (a gift from Peter Bradley, University of California, Los Angeles, CA; 1:2,000 dilution), rabbit anti-MORN1 (kindly provided by Marc-Jan Gubbels, Boston, MA; 1:2,000 dilution), and mouse and rabbit anti-IMC1 (kindly provided by Gary Ward, University of Vermont, VT; 1:2,000 dilution). Secondary antibodies used were Alexa Fluor 488-, 546-, and 594-conjugated antibodies (Molecular Probes, Life Technologies, NY) at a dilution of 1:200. Images were collected on an Applied Precision Delta Vision inverted epifluorescence microscope using an Olympus UPlans APO 100×/1.40 oil lens and on a Carl Zeiss Axio Observer.Z1 inverted microscope with a Plan-Apochromat 100×/1.40 oil differential interference contrast (DIC) lens, deconvolved, and adjusted for contrast using SoftWoRx and AxioVs40 v4.8.1.0, respectively.
Western blotting.
Parasites were harvested, filtered, counted, washed with PBS, and pelleted by centrifugation. Parasites were suspended in lysis and loading buffer, heated to 100°C for 3 min, and run on a 4 to 12% Tris-glycine-SDS Mini-Protean precast gel (Bio-Rad, CA). Western blotting was performed as previously described (48). Monoclonal rat anti-HA tag (clone 3F10; Roche Applied Science, IN) and mouse anti-α-tubulin (12G10, a gift of Jacek Gaertig, University of Georgia) antibodies were used at 1:500 and 1:2,000 dilutions, respectively, to probe the blots. Horseradish peroxidase (HRP)-conjugated anti-rat or anti-mouse secondary antibody (Pierce, Thermo Scientific Inc., IL) was used at a 1:10,000 dilution and detected by chemiluminescence using the ECL Western blotting substrate (Pierce, Thermo Scientific Inc., IL).
SUPPLEMENTAL MATERIAL
Quantification of the area of plaques formed by the iΔSUN-KO and TATi ΔKu80-HA parasite lines after 7 days of growth on HFF monolayers. The average area of 25 plaques was assessed for each line. The mean areas ± standard deviations (SD) are plotted for 3 independent experiments. In the absence of ATc, the plaques formed by the iΔSUN-KO line were significantly smaller in size than those of the parental TATi ΔKu80-HA line (*, P < 0.0001, Student’s t test). ATc, anhydrotetracycline. Download
Sequences of the primers used in the study.
Numbers of genes covered by the mapped cosmids (n = 7,773) and fosmids (n = 5,509) per Toxoplasma gondii chromosome.
Fosmids modified using liquid recombineering.
Single nucleotide polymorphisms (SNPs) in the Toxoplasma gondii ts mutant 13-136A8 identified by whole-genome sequencing.
ACKNOWLEDGMENTS
This work was supported in part by U.S. National Institutes of Health RO1 grants AI064671, AI084415 (to B.S.), AI077662, and A109843 (to M.W.W.). We thank Markus Meissner (Wellcome Trust Center for Molecular Parasitology, University of Glasgow, United Kingdom) for providing additional funding to sequence the fosmid library.
We thank Cornelia Lemke (Plant Genome Mapping Laboratory, University of Georgia) for her help with automated colony picking, replication, and arraying of the fosmid library and the EuPathDB team for adding the fosmid data set to the ToxoDB.
Footnotes
Citation Vinayak S, Brooks CF, Naumov A, Suvorova ES, White MW, Striepen B. 2014. Genetic manipulation of the Toxoplasma gondii genome by fosmid recombineering. mBio 5(6):e02021-14. doi:10.1128/mBio.02021-14.
REFERENCES
- 1. Weiss LM, Kim K. 2013. Toxoplasma gondii. The model apicomplexan: perspectives and methods, 2nd Éditions Elsevier Academic Press, Burlington, VT. [Google Scholar]
- 2. Donald RG, Roos DS. 1993. Stable molecular transformation of Toxoplasma gondii: a selectable dihydrofolate reductase-thymidylate synthase marker based on drug-resistance mutations in malaria. Proc. Natl. Acad. Sci. U. S. A. 90:11703–11707. 10.1073/pnas.90.24.11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Soldati D, Boothroyd JC. 1993. Transient transfection and expression in the obligate intracellular parasite Toxoplasma gondii. Science 260:349–352. 10.1126/science.8469986. [DOI] [PubMed] [Google Scholar]
- 4. Striepen B, Soldati D. 2007. Genetic manipulation of Toxoplasma gondii, p 391–418 In Weiss LM, Kim K. (ed), Toxoplasma gondii. The model apicomplexan: perspectives and methods. Academic Press, Elsevier, London, United Kingdom. [Google Scholar]
- 5. Herm-Götz A, Agop-Nersesian C, Münter S, Grimley JS, Wandless TJ, Frischknecht F, Meissner M. 2007. Rapid control of protein level in the apicomplexan Toxoplasma gondii. Nat. Methods 4:1003–1005. 10.1038/nmeth1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meissner M, Schlüter D, Soldati D. 2002. Role of Toxoplasma gondii myosin A in powering parasite gliding and host cell invasion. Science 298:837–840. 10.1126/science.1074553. [DOI] [PubMed] [Google Scholar]
- 7. Huynh MH, Carruthers VB. 2009. Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot. Cell 8:530–539. 10.1128/EC.00358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fox BA, Ristuccia JG, Gigley JP, Bzik DJ. 2009. Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end joining. Eukaryot. Cell 8:520–529. 10.1128/EC.00357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sheiner L, Demerly JL, Poulsen N, Beatty WL, Lucas O, Behnke MS, White MW, Striepen B. 2011. A systematic screen to discover and analyze apicoplast proteins identifies a conserved and essential protein import factor. PLoS Pathog. 7:e1002392. 10.1371/journal.ppat.1002392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shen B, Brown KM, Lee TD, Sibley LD. 2014. Efficient gene disruption in diverse strains of Toxoplasma gondii using CRISPR/CAS9. mBio 5(3):e01114-14. 10.1128/mBio.01114-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sidik SM, Hackett CG, Tran F, Westwood NJ, Lourido S. 2014. Efficient genome engineering of Toxoplasma gondii using CRISPR/Cas9. PLoS One 9:e100450. 10.1371/journal.pone.0100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Court DL, Sawitzke JA, Thomason LC. 2002. Genetic engineering using homologous recombination. Annu. Rev. Genet. 36:361–388. 10.1146/annurev.genet.36.061102.093104. [DOI] [PubMed] [Google Scholar]
- 13. Pfander C, Anar B, Schwach F, Otto TD, Brochet M, Volkmann K, Quail MA, Pain A, Rosen B, Skarnes W, Rayner JC, Billker O. 2011. A scalable pipeline for highly effective genetic modification of a malaria parasite. Nat. Methods 8:1078–1082. 10.1038/nmeth.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brooks CF, Johnsen H, van Dooren GG, Muthalagi M, Lin SS, Bohne W, Fischer K, Striepen B. 2010. The Toxoplasma apicoplast phosphate translocator links cytosolic and apicoplast metabolism and is essential for parasite survival. Cell Host Microbe 7:62–73. 10.1016/j.chom.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ramakrishnan S, Docampo MD, Macrae JI, Pujol FM, Brooks CF, van Dooren GG, Hiltunen JK, Kastaniotis AJ, McConville MJ, Striepen B. 2012. Apicoplast and endoplasmic reticulum cooperate in fatty acid biosynthesis in apicomplexan parasite Toxoplasma gondii. J. Biol. Chem. 287:4957–4971. 10.1074/jbc.M111.310144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nair SC, Brooks CF, Goodman CD, Sturm A, Strurm A, McFadden GI, Sundriyal S, Anglin JL, Song Y, Moreno SN, Striepen B. 2011. Apicoplast isoprenoid precursor synthesis and the molecular basis of fosmidomycin resistance in Toxoplasma gondii. J. Exp. Med. 208:1547–1559. 10.1084/jem.20110039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Francia ME, Jordan CN, Patel JD, Sheiner L, Demerly JL, Fellows JD, de Leon JC, Morrissette NS, Dubremetz JF, Striepen B. 2012. Cell division in apicomplexan parasites is organized by a homolog of the striated rootlet fiber of algal flagella. PLoS Biol. 10:e1001444. 10.1371/journal.pbio.1001444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brooks CF, Francia ME, Gissot M, Croken MM, Kim K, Striepen B. 2011. Toxoplasma gondii sequesters centromeres to a specific nuclear region throughout the cell cycle. Proc. Natl. Acad. Sci. U. S. A. 108:3767–3772. 10.1073/pnas.1006741108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McCoy JM, Whitehead L, van Dooren GG, Tonkin CJ. 2012. TgCDPK3 regulates calcium-dependent egress of Toxoplasma gondii from host cells. PLoS Pathog. 8:e1003066. 10.1371/journal.ppat.1003066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin SS, Gross U, Bohne W. 2011. Two internal type II NADH dehydrogenases of Toxoplasma gondii are both required for optimal tachyzoite growth. Mol. Microbiol. 82:209–221. 10.1111/j.1365-2958.2011.07807.x. [DOI] [PubMed] [Google Scholar]
- 21. Copeland NG, Jenkins NA, Court DL. 2001. Recombineering: a powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2:769–779. 10.1038/35093556. [DOI] [PubMed] [Google Scholar]
- 22. Adams DJ, Quail MA, Cox T, van der Weyden L, Gorick BD, Su Q, Chan WI, Davies R, Bonfield JK, Law F, Humphray S, Plumb B, Liu P, Rogers J, Bradley A. 2005. A genome-wide, end-sequenced 129Sv BAC library resource for targeting vector construction. Genomics 86:753–758. 10.1016/j.ygeno.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 23. Suvorova ES, Radke JB, Ting LM, Vinayak S, Alvarez CA, Kratzer S, Kim K, Striepen B, White MW. 2013. A nucleolar AAA-NTPase is required for parasite division. Mol. Microbiol. 90:338–355. 10.1111/mmi.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taylor S, Barragan A, Su C, Fux B, Fentress SJ, Tang K, Beatty WL, Hajj HE, Jerome M, Behnke MS, White M, Wootton JC, Sibley LD. 2006. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314:1776–1780. 10.1126/science.1133643. [DOI] [PubMed] [Google Scholar]
- 25. Saeij JP, Boyle JP, Coller S, Taylor S, Sibley LD, Brooke-Powell ET, Ajioka JW, Boothroyd JC. 2006. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314:1780–1783. 10.1126/science.1133690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hunter CA, Sibley LD. 2012. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 10:766–778. 10.1038/nrmicro2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Farrell A, Thirugnanam S, Lorestani A, Dvorin JD, Eidell KP, Ferguson DJ, Anderson-White BR, Duraisingh MT, Marth GT, Gubbels MJ. 2012. A DOC2 protein identified by mutational profiling is essential for apicomplexan parasite exocytosis. Science 335:218–221. 10.1126/science.1210829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gubbels MJ, Lehmann M, Muthalagi M, Jerome ME, Brooks CF, Szatanek T, Flynn J, Parrot B, Radke J, Striepen B, White MW. 2008. Forward genetic analysis of the apicomplexan cell division cycle in Toxoplasma gondii. PLoS Pathog. 4:e36. 10.1371/journal.ppat.0040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frankel MB, Mordue DG, Knoll LJ. 2007. Discovery of parasite virulence genes reveals a unique regulator of chromosome condensation 1 ortholog critical for efficient nuclear trafficking. Proc. Natl. Acad. Sci. U. S. A. 104:10181–10186. 10.1073/pnas.0701893104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Behnke MS, Khan A, Wootton JC, Dubey JP, Tang K, Sibley LD. 2011. Virulence differences in Toxoplasma mediated by amplification of a family of polymorphic pseudokinases. Proc. Natl. Acad. Sci. U. S. A. 108:9631–9636. 10.1073/pnas.1015338108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pfefferkorn ER, Pfefferkorn LC. 1976. Toxoplasma gondii: isolation and preliminary characterization of temperature-sensitive mutants. Exp. Parasitol. 39:365–376. 10.1016/0014-4894(76)90040-0. [DOI] [PubMed] [Google Scholar]
- 32. Brown KM, Suvorova E, Farrell A, McLain A, Dittmar A, Wiley GB, Marth G, Gaffney PM, Gubbels MJ, White M, Blader IJ. 2014. Forward genetic screening identifies a small molecule that blocks Toxoplasma gondii growth by inhibiting both host- and parasite-encoded kinases. PLoS Pathog. 10:e1004180. 10.1371/journal.ppat.1004180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sarov M, Schneider S, Pozniakovski A, Roguev A, Ernst S, Zhang Y, Hyman AA, Stewart AF. 2006. A recombineering pipeline for functional genomics applied to Caenorhabditis elegans. Nat. Methods 3:839–844. 10.1038/nmeth933. [DOI] [PubMed] [Google Scholar]
- 34. Ejsmont RK, Bogdanzaliewa M, Lipinski KA, Tomancak P. 2011. Production of fosmid genomic libraries optimized for liquid culture recombineering and cross-species transgenesis. Methods Mol. Biol. 772:423–443. 10.1007/978-1-61779-228-1_25. [DOI] [PubMed] [Google Scholar]
- 35. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. 2013. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31:822–826. 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. 2013. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31:827–832. 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. 2013. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 31:839–843. 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tscherne JS, Nurse K, Popienick P, Michel H, Sochacki M, Ofengand J. 1999. Purification, cloning, and characterization of the 16S RNA m5C967 methyltransferase from Escherichia coli. Biochemistry (Mosc.) 38:1884–1892. 10.1021/bi981880l. [DOI] [PubMed] [Google Scholar]
- 39. Hussain S, Benavente SB, Nascimento E, Dragoni I, Kurowski A, Gillich A, Humphreys P, Frye M. 2009. The nucleolar RNA methyltransferase Misu (NSun2) is required for mitotic spindle stability. J. Cell Biol. 186:27–40. 10.1083/jcb.200810180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frye M, Watt FM. 2006. The RNA methyltransferase Misu (NSun2) mediates Myc-induced proliferation and is upregulated in tumors. Curr. Biol. 16:971–981. 10.1016/j.cub.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 41. Sakita-Suto S, Kanda A, Suzuki F, Sato S, Takata T, Tatsuka M. 2007. Aurora-B regulates RNA methyltransferase NSUN2. Mol. Biol. Cell 18:1107–1117. 10.1091/mbc.E06-11-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aslanidis C, de Jong PJ. 1990. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18:6069–6074. 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43:491–498. 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The genome Analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14:178–192. 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Van Dooren GG, Tomova C, Agrawal S, Humbel BM, Striepen B. 2008. Toxoplasma gondii Tic20 is essential for apicoplast protein import. Proc. Natl. Acad. Sci. U. S. A. 105:13574–13579. 10.1073/pnas.0803862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Quantification of the area of plaques formed by the iΔSUN-KO and TATi ΔKu80-HA parasite lines after 7 days of growth on HFF monolayers. The average area of 25 plaques was assessed for each line. The mean areas ± standard deviations (SD) are plotted for 3 independent experiments. In the absence of ATc, the plaques formed by the iΔSUN-KO line were significantly smaller in size than those of the parental TATi ΔKu80-HA line (*, P < 0.0001, Student’s t test). ATc, anhydrotetracycline. Download
Sequences of the primers used in the study.
Numbers of genes covered by the mapped cosmids (n = 7,773) and fosmids (n = 5,509) per Toxoplasma gondii chromosome.
Fosmids modified using liquid recombineering.
Single nucleotide polymorphisms (SNPs) in the Toxoplasma gondii ts mutant 13-136A8 identified by whole-genome sequencing.