

ABSTRACT
Clostridium difficile is the most common hospital-acquired pathogen, causing antibiotic-associated diarrhea in over 250,000 patients annually in the United States. Disease is primarily mediated by toxins A and B, which induce potent proinflammatory signaling in host cells and can activate an ASC-containing inflammasome. Recent findings suggest that the intensity of the host response to infection correlates with disease severity. Our lab has identified the proinflammatory cytokine interleukin-23 (IL-23) as a pathogenic mediator during C. difficile infection (CDI). The mechanisms by which C. difficile induces IL-23, however, are not well understood, and the role of toxins A and B in this process is unclear. Here, we show that toxins A and B alone are not sufficient for IL-23 production but synergistically increase the amount of IL-23 produced in response to MyD88-dependent danger signals, including pathogen-associated molecular patterns (PAMPs) and host-derived damage associated molecular patterns (DAMPs). Danger signals also enhanced the secretion of IL-1β in response to toxins A and B, and subsequent IL-1 receptor signaling accounted for the majority of the increase in IL-23 that occurred in the presence of the toxins. Inhibition of inflammasome activation in the presence of extracellular K+ likewise decreased IL-23 production. Finally, we found that IL-1β was increased in the serum of patients with CDI, suggesting that this systemic response could influence downstream production of pathogenic IL-23. Identification of the synergy of danger signals with toxins A and B via inflammasome signaling represents a novel finding in the mechanistic understanding of C. difficile-induced inflammation.
IMPORTANCE
Clostridium difficile is among the leading causes of death due to health care-associated infection, and factors determining disease severity are not well understood. C. difficile secretes toxins A and B, which cause inflammation and tissue damage, and recent findings suggest that some of this tissue damage may be due to an inappropriate host immune response. We have found that toxins A and B, in combination with both bacterium- and host-derived danger signals, can induce expression of the proinflammatory cytokines IL-1β and IL-23. Our results demonstrate that IL-1β signaling enhances IL-23 production and could lead to increased pathogenic inflammation during CDI.
INTRODUCTION
Clostridium difficile is a Gram-positive, spore-forming anaerobe and the cause of a major hospital-acquired infection, recently surpassing methicillin-resistant Staphylococcus aureus (MRSA) as the most common health care-associated infection (1). C. difficile infection (CDI) results in over $1 billion in excess medical costs each year in the United States alone (2). Ribotype 027 strains, which show enhanced transmissibility, are increasingly common, worsening the burden of CDI (3). Infection with C. difficile results in a spectrum of disease ranging from mild diarrhea to severe pseudomembranous colitis, toxic megacolon, and death (4). Antibiotic treatment predisposes individuals to CDI by disrupting commensal microbes in the gut and providing a competitive advantage for C. difficile growth as well as impairing the host mucosal immune response (5).
Toxins A and B (TcdA and TcdB) are the major virulence factors of C. difficile and are responsible for glucosylating host Rho family GTPases (6). Glucosylation blocks the exchange of GDP for GTP and prevents RhoA, Rac1, and Cdc42 from functioning, resulting in cell rounding and death (7). Toxins A and B also elicit a robust proinflammatory response via activation of multiple mitogen-activated protein kinases, which in turn activate the transcription factor nuclear factor κB (NF-κB) (8, 9). Both toxins activate an ASC-containing inflammasome and can induce secretion of interleukin-8 (IL-8), tumor necrosis factor alpha (TNF-α), and IL-6, in addition to IL-1β (10–12).
C. difficile pathogen-associated molecular patterns (PAMPs) also contribute to the host inflammatory response, including the surface layer proteins (SLPs), which activate Toll-like receptor 4 (TLR4) and flagellin, which is thought to be a TLR5 ligand (13, 14). C. difficile also possesses uncharacterized Nod1 stimulatory molecules (15). Although toxins A and B have been shown to enhance cytokine production resulting from recognition of C. difficile PAMPs, it is not well known how this occurs (14). Innate inflammatory signaling involving Nod1, TLR4, MyD88, and the inflammasome adaptor ASC are protective in mouse models of CDI (13, 15–17). Conversely, human clinical studies have shown that the host response may also be pathogenic, as IL-8 and CXCL5 levels positively predict disease severity, whereas C. difficile bacterial burden does not (18). Our lab has previously identified the proinflammatory cytokine IL-23 as a pathogenic mediator during CDI. The importance of IL-23 in these studies suggests that IL-23 may be a key regulator of the balance between bacterial eradication and host tissue damage (19). IL-23 expression is initiated by the transcription factors NF-κB and AP-1 downstream of TLR and IL-1 receptor signaling (20), leading us to hypothesize that C. difficile may be able to directly induce IL-23 expression through the action of toxins A and B. Purified SLPs as well as live C. difficile can induce IL-23 production, but the contribution of toxins A and B to this process has not been extensively studied. Additionally, it is unknown how the toxins contribute to IL-23 production in response to ribotype 027 strains, which are thought to be more proinflammatory (11).
We used murine and human dendritic cells to elucidate the regulation of IL-23 in response to C. difficile. We found that toxins A and B increased levels of IL-23 but alone were not sufficient to induce its production. Furthermore, we identified a crucial role for the host inflammasome and IL-1β signaling in toxin-enhanced IL-23 production. Finally, we demonstrate here that IL-1β is increased in the serum of patients with CDI, demonstrating that this cytokine is systemic during human infection and could potentiate IL-23 production in such patients.
RESULTS
Clostridium difficile toxins A and B are not sufficient to induce detectable IL-23.
In order to determine how C. difficile induces IL-23 expression, we treated murine bone marrow-derived dendritic cells (BMDCs) with purified toxins A and B (Fig. 1). We found that the toxins, either alone or in combination, were not sufficient to induce IL-23 production (at concentrations ranging from 2 pg/ml to 25 µg/ml) (Fig. 1 and data not shown). Toxins A and B were functionally active, as toxin A induced CXCL1 production and both toxins activated NF-κB and caused significant cell death (see Fig. S1A to C in the supplemental material). Next, we attempted to induce IL-23 production by using fresh toxin-containing culture filtrates from C. difficile strain R20291, a ribotype 027 strain responsible for a 2006 outbreak in Stoke Mandeville, United Kingdom (21). Wild-type R20291 filtrate induced significant levels of IL-23, while the isogenic toxin mutant R20291 AB- showed greatly decreased IL-23 induction as measured in an anti-IL-23p19 enzyme-linked immunosorbent assay (ELISA). Adding 2 ng/ml each of purified toxins A and B to R20291 AB- culture filtrate restored IL-23 production to levels induced by wild-type R20291 (Fig. 1A). This concentration of toxins was used to mimic physiological levels found in serum during infection (22). Interestingly, toxins A and B also increased the amount of IL-23 produced by BMDCs upon exposure to lipopolysaccharide (LPS) from Escherichia coli, a PAMP known to induce IL-23 (20). We concluded that while toxins A and B alone were not sufficient to induce IL-23, the toxins did enhance IL-23 production in the presence of bacterial products.
FIG 1 .

C. difficile toxins induce detectable IL-23 from bone marrow-derived dendritic cells only in the presence of danger signals. (A) BMDCs were treated for 24 h with purified toxins A and B (2 ng/ml), LPS (100 ng/ml), or culture filtrates from C. difficile strain R20291 or R20291 AB−. BMDC medium was assayed for IL-23 via an ELISA. (B) BMDCs were incubated with serum amyloid A (500 ng/ml) with or without toxins A and B (2 ng/ml). Student’s t test was used to determine statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data shown were combined from at least 3 experiments.
Damage-associated molecular patterns also prime cells for IL-23 production.
Although bacterial PAMPs are common priming signals for inflammasome activation, we hypothesized that host-derived danger signals capable of activating NF-κB would also potentiate IL-23 production. Serum amyloid A (SAA) is a host-derived inflammatory mediator which is expressed in colonic epithelium and can play a role in modulating dextran sodium sulfate (DSS) colitis in murine models (23). Exposing BMDCs to serum amyloid A alone did not induce detectable IL-23, but IL-23 production was greatly enhanced when toxins A and B were added (Fig. 1B). This suggests that although a danger signal is required for toxin-induced IL-23 production, this signal need not be derived from bacteria.
C. difficile toxins A and B and PAMPs induce IL-23 mRNA expression in human dendritic cells.
Next, we sought to determine whether human dendritic cells respond similarly in the presence of toxins A and B (Fig. 2). We generated monocyte-derived dendritic cells (MoDCs) from the peripheral blood mononuclear cells (PBMCs) of healthy human volunteers and exposed them to purified toxins A and B as well as culture filtrates from C. difficile strain r20291. We evaluated IL-23a gene expression by quantitative reverse transcription-PCR, as human IL-23p19 ELISAs suffer from relatively low sensitivity. Purified toxins A and B did not induce IL-23a gene expression. However, wild-type R20291 supernatant did induce significant IL-23a expression, and this was lost in the presence of nontoxigenic filtrate from R20291 AB-. The addition of purified toxins A and B restored some level of IL-23a gene expression (Fig. 2), leading us to conclude that human cells respond similarly to the presence of C. difficile toxins and PAMPs.
FIG 2 .
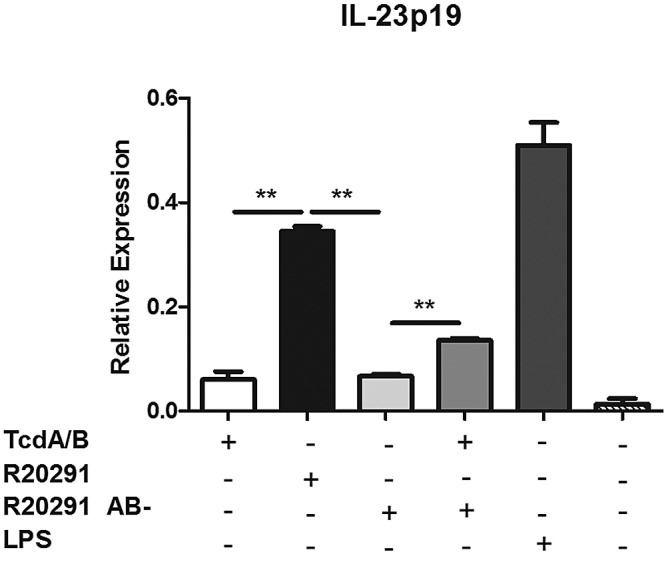
C. difficile toxins and PAMPs induce IL-23 expression in human cells. MoDCs were generated from monocytes isolated from healthy human peripheral blood samples and were treated for 24 h with purified toxins A and B (2 ng/ml) or with filter-sterilized culture supernatants from C. difficile strain r20291 or an isogenic toxin mutant, R20291 AB-. RNA was isolated and assayed by quantitative reverse transcription-PCR for IL-23 gene expression. Student’s t test was used to determine statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data shown are representative of three independent experiments.
A priming signal significantly enhances IL-1β production in response to toxins A and B.
Toxins A and B have been shown to activate an ASC-containing inflammasome to secrete IL-1β. Both the NLRP3 inflammasome and a pyrin-dependent inflammasome have been implicated in this process (10–12). Two signals are a well-established requirement for activation of the inflammasome. The first “priming” signal induces NF-κB-dependent expression of pro-IL-1β, and the second “activation” signal induces assembly of the inflammasome complex and processing of IL-1β into its mature form (24). Because IL-1β is thought to play a role in the induction of IL-23 (25), we assayed BMDCs for IL-1β production (Fig. 3). We found that toxins A and B alone induced low levels of IL-1β production, but treating BMDCs with LPS in combination with toxins A and B significantly increased levels of IL-1β (Fig. 3A). Additionally, we found that treating BMDCs with supernatant from strain R20291 also induced significant levels of IL-1β, indicating that this supernatant may be sufficient to provide both priming and activation signals. IL-1β production was lost when BMDCs were exposed to R20291 AB- filtrate but restored when purified toxins A and B were added back (Fig. 3A). We also found that SAA was sufficient to serve as a priming signal to induce increased IL-1β production in the presence of toxins A and B (Fig. 3B). Because many IL-1β ELISAs can also detect pro-IL-1β released from dying cells, we confirmed by Western blotting that the IL-1β produced in response to toxins A and B was indeed processed and secreted rather than simply released from intoxicated, nonviable cells (Fig. 3C). We concluded that danger signals and toxins A and B synergistically increase IL-1β production by BMDCs.
FIG 3 .

IL-1β secretion by BMDCs in response to C. difficile toxins is enhanced by a priming signal. (A) BMDCs were treated for 24 h with purified toxins A and B (2 ng/ml), LPS (100 ng/ml), or filter-sterilized culture supernatants from C. difficile strain R20291 or an isogenic toxin mutant, R20291 AB-. BMDC medium was assayed for IL-1β via an ELISA. (B) BMDCs were incubated with serum amyloid A (500 ng/ml) with or without toxins A and B (2 ng/ml). (C) Western blotting results for supernatants (Sup) and whole-cell lysate (Lys) from treated BMDCs. Blots were probed with antibodies directed against IL-1β p17 (supernatant) and β-actin (lysate). Lane 1 contains recombinant IL-1β as a positive control. Student’s t test was used to determine statsitical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data shown were combined from at least 3 experiments.
MyD88 signaling is required for IL-23 production.
Multiple TLRs as well as the IL-1 receptor require the adaptor protein MyD88 in order to successfully activate NF-κB to induce downstream cytokine production (26). In order to determine whether these pathways were required for the induction of IL-23 by C. difficile, we utilized BMDCs from MyD88−/− mice. MyD88−/− BMDCs showed significantly reduced IL-23 production in response to R20291 and R20291 AB- supernatant with purified toxins A and B added (Fig. 4A). To further delineate the contribution of TLR signaling to this process, we next utilized TLR2−/−, TLR4−/−, and TLR5−/− BMDCs. TLR4−/− BMDCs, but not TLR2−/− or TLR5−/− BMDCs, showed significantly decreased IL-23 production in response to R20291. These data implicate the SLPs of C. difficile, known to signal through TLR4 (13, 26), as a major PAMP involved in IL-23 induction (Fig. 4B). TLR4−/− BMDCs also demonstrated significantly decreased IL-23 production in the presence of LPS and toxins A and B, further demonstrating the necessity for a danger signal in addition to the toxins for robust IL-23 production.
FIG 4 .

TLR4 and MyD88 signaling contribute to IL-23 production in response to C. difficile. BMDCs from C57BL6/J or MyD88−/− mice (A) or C57BL6/J, TLR2−/−, TLR4−/−, or TLR5−/− mice (B) were treated for 24 h with either LPS or filter-sterilized culture supernatant from C. difficile strain r20291 or the nontoxigenic strain R20291 AB-, in the presence or absence of purified toxins A and B (2 ng/ml). IL-23 protein levels were quantified with an ELISA; data in panel B are relative to 100% of C57BL6 levels under each condition. Student’s t test was used to determine statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001. #, result below the detection level. Data shown were combined from 3 independent experiments.
Toxin-induced IL-1β signaling enhances IL-23 production.
Because IL-1β has been shown to contribute to IL-23 production in other models (25) and MyD88-deficient cells did not show an increase in IL-23 in the presence of toxins A and B, we hypothesized that IL-1β produced in response to toxins A and B enhances IL-23 production. To test this hypothesis, we treated BMDCs with filtrate from strain R20291 in the presence of an anti-IL-1β neutralizing antibody or an IL-1 receptor antagonist (Fig. 5). Both the neutralizing antibody and the receptor antagonist significantly decreased the amount of IL-23 produced, compared to an isotype control antibody (Fig. 5A). As expected, when cells were exposed to R20291 AB-, IL-23 production was minimal. However, IL-23 could be significantly enhanced by adding recombinant IL-1β to R20291 AB-, suggesting that IL-1 receptor signaling could account for the increase in IL-23 observed in the presence of toxins A and B. Next, we utilized BMDCs from IL-1R−/− mice to further assess the contribution of this receptor to IL-23 production. We found that IL-1R−/− BMDCs displayed significantly reduced IL-23 production in response to R20291 as well as R20291 AB- with added toxins A and B (Fig. 5B). As expected, no significant difference was observed for R20291 AB- in the absence of toxins A and B. These results suggest that, following a priming signal, toxins A and B may activate the inflammasome to secrete IL-1β, which in turn signals through the IL-1 receptor to enhance IL-23 production.
FIG 5 .
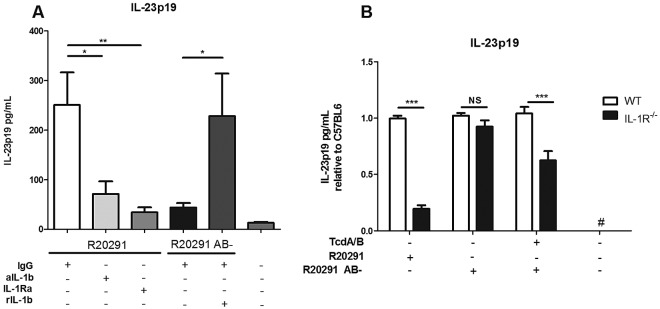
IL-1β signaling contributes to IL-23 production. BMDCs were treated for 24 h with filter-sterilized culture supernatant from C. difficile strain R20291 or R20291 AB-. (A) Recombinant murine IL-1β (rIL-1β; 50 ng/ml), IL-1β neutralizing antibody (aIL-1β; 1 µg/ml), IL-1 receptor antagonist (IL-1RA; 500 ng/ml), or IgG isotype control antibody (1 µg/ml) was added, as indicated. (B) BMDCs from C57BL6 (wild-type [WT]) or IL-1R−/− mice were treated for 24 h with filtrates from C. difficile strain R20291 or R20291 AB-, in the presence or absence of purified toxins A and B. Data are shown relative to 100% of the C57BL6 results under each condition. Data were combined from at least 3 independent experiments. Student’s t test was used to determine statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Reducing inflammasome activation decreases the production of IL-23 in the presence of toxins A and B.
Although multiple diverse stimuli have been shown to activate the NLRP3 inflammasome, these stimuli share the downstream requirement for efflux of potassium (27). Thus, the addition of excess extracellular potassium can inhibit NLRP3 inflammasome activation, although this is not specific to NLRP3 and can also inhibit NLRP1 oligomerization (24). In order to confirm the role of inflammasome-produced IL-1β in enhanced IL-23 production, BMDCs were incubated with culture filtrate from R20291 in the presence or absence of 40 mM KCl and results were compared to those with 40 mM NaCl, an osmotic control. We found that the addition of extracellular K+ to BMDCs significantly inhibited the secretion of IL-1β as well as IL-23 (Fig. 6A and B). Similarly, the addition of YVAD-FMK, a caspase-1/4 inhibitor, or glybenclamide, an inhibitor of K+ efflux, decreased both the amount of IL-1β and the amount of IL-23 secreted by BMDCs in the presence of R20291 culture filtrate (Fig. 6C and D). These data provided further evidence that inflammasome activation and the resulting IL-1 receptor signaling contribute to IL-23 production.
FIG 6 .

Inflammasome inhibition reduces IL-23 secretion. BMDCs were treated with filter-sterilized culture supernatant from strain R20291 in the presence of 40 mM KCl, 40 mM NaCl (A and B) or 20 µM YVAD-FMK or 25 µg/ml glybenclamide (C and D) for 24 h. IL-1β (A and C) and IL-23 (B and D) in the cell supernatant were measured in an ELISA. Student’s t test was used to determine statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data were combined from at least 3 independent experiments.
Patients with C. difficile infection display elevated serum IL-1β.
Finally, in order to confirm the presence of IL-1β during human infection, we analyzed sera from CDI-positive and CDI-negative patients with diarrhea, as well as from healthy outpatient controls. Utilizing a high-sensitivity IL-1β ELISA specifically designed for cytokine detection in serum (limit of detection, 0.16 pg/ml), we found that patients with CDI had significantly higher serum IL-1β than healthy outpatient controls (Fig. 7). IL-23 in serum was not present at detectable levels (data not shown). Interestingly, patients with diarrhea from other causes also displayed increased IL-1β, demonstrating that this cytokine is not specific to CDI. These results suggest that inflammasome activation occurs in vivo during CDI and may potentiate the IL-23 production in the lamina propria that we previously reported (19). Additionally, the presence of IL-1β in patients with non-CDI diarrhea suggests that this cytokine is present and could be involved in IL-23 production in other models of colitis.
FIG 7 .
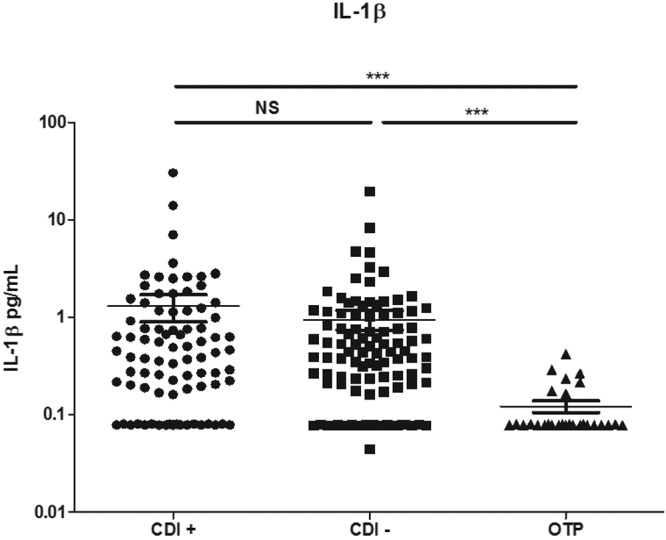
IL-1β is increased in the sera of patients with CDI. Sera from C. difficile-positive patients with diarrhea (CDI+), C. difficile-negative patients with diarrhea (CDI−), and healthy outpatients (OTP) were assayed for IL-1β via a high-sensitivity ELISA. Each dot represents one patient sample. The Mann-Whitney test was used to determine statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
Toxins A and B (TcdA and TcdB) are the major virulence factors of C. difficile and act via intoxication of Rho family GTPases, including RhoA, Rac1, and Cdc42 (6). Both toxins are capable of causing epithelial cell death, as well as apoptosis of neurons, endothelial cells, and monocytes (28). Toxins A and B alone can also induce the secretion of IL-8/CXCL1, TNF-α, and IL-6 (8). Toxin-induced activation of the inflammasome and IL-1β secretion in primed cells have also been demonstrated (10–12). It is well appreciated that C. difficile toxins and other factors contribute to disease severity, as nontoxigenic strains do not cause symptomatic infection (29, 30) and genetic manipulation of C. difficile sporulation and motility can influence disease outcome (31).
Despite the importance of toxins A and B during infection, recent findings implicate host-derived inflammation as a contributing factor to the severity of CDI. Therefore, a thorough understanding of C. difficile pathogenesis requires examination of both host- and pathogen-derived mediators. We previously demonstrated that the cytokine IL-23 is pathogenic during CDI, as mice deficient in IL-23 were significantly protected from mortality (19). We have also demonstrated that IL-23 is expressed during human infection with C. difficile. Although the role of toxins A and B in the expression of various cytokines has been extensively studied, the induction of IL-23 by C. difficile has thus far been incompletely characterized. Thus, the goal of this work was to determine the role of C. difficile toxins A and B in the induction of IL-23.
Here, we found that, surprisingly, toxins A and B alone do not induce expression of measurable amounts of IL-23 from BMDCs (Fig. 1). However, supernatant from the ribotype 027 strain R20291 was capable of inducing IL-23, and IL-23 production was synergistically increased in the presence of both toxins A and B and a danger signal (Fig. 1 and 2). Interestingly, we also found that robust secretion of IL-1β likewise required a priming signal, and C. difficile PAMPs were sufficient to prime BMDCs for IL-1β secretion (Fig. 3). IL-1 receptor signaling following inflammasome activation contributed to the production of IL-23, accounting for the increase in IL-23 observed in the presence of toxins A and B (Fig. 4 to 6).
Induction of IL-23 by C. difficile strain R20291.
Previously, purified SLPs of C. difficile strain R13537 have been shown to induce IL-23 expression in a TLR4-dependent manner (13), demonstrating that C. difficile PAMPs can induce IL-23. Additionally, C. difficile strains 630 and R20291 also induced IL-23 in vitro, and mutants of strain 630 that do not produce toxins A and B showed significantly decreased IL-23 production (11). However, mutants of strain R20291 lacking toxin A and B production were not investigated, although R20291 was shown to induce significantly more IL-23 than strain 630. Additionally, it was unknown whether purified toxins A and B could induce IL-23. Our data confirmed that toxins A and B enhance IL-23 production by ribotype 027 strains, and we demonstrated that the toxins alone are not sufficient for IL-23 production (Fig. 1). The ability of toxins A and B alone to induce production of IL-8 suggests an underlying difference between this cytokine and IL-23, possibly due to differences in expression level or cell source. Additionally, our results suggest that C. difficile PAMPs in combination with toxins A and B are capable of inducing IL-23 (Fig. 1) and that both TLR4 and MyD88 signaling are involved in PAMP recognition (Fig. 4).
Although bacterial danger signals were sufficient to allow IL-23 production in response to toxins A and B, we also examined the possibility that host-derived signals could serve this function. Serum amyloid A, an endogenous danger signal released from damaged epithelial cells (23), was also sufficient to allow IL-23 production from BMDCs (Fig. 1). Therefore, the production of IL-23 is not dependent on the presence of bacterial PAMPs. These data provide insight into the inflammatory response to C. difficile in germ-free mice, which are notably susceptible to CDI (32). Under germ-free conditions, C. difficile PAMPs or host-derived danger signals in combination with toxins A and B could be sufficient to invoke a robust inflammatory response involving both IL-1β and IL-23.
Inflammasome activation by toxins A and B.
The abilities of C. difficile toxins to activate the inflammasome are well known; however, the specific inflammasome components involved are still in question. Originally, toxins A and B were thought to activate the NLRP3 inflammasome in an ASC-dependent manner (12). However, recent work has demonstrated that inflammasome activation by these toxins may be NLRP3 independent and instead involve activation of the pyrin inflammasome (10, 11). Although we have not examined a necessity for specific inflammasome components, we have found that BMDCs are capable of robust IL-1β production in response to toxins A and B when a priming signal is provided (Fig. 3). Interestingly, potassium efflux appears to be involved in this process, as excess extracellular potassium decreased both IL-1β and, consequently, IL-23 production (Fig. 6). However, potassium efflux is not specific to NLRP3 inflammasome activation and does not rule out involvement of other inflammasomes.
Contribution of IL-1β signaling to IL-23 production.
The ability of IL-1β to influence IL-23 signaling has been demonstrated previously in the context of autophagy, where inhibition of the autophagosome increased IL-23 production in response to TLR agonists, and this increase was mediated by IL-1 receptor signaling (25). Similarly, we found that IL-1 receptor signaling contributes to IL-23 production in the presence of C. difficile toxins and bacterial products (Fig. 4 and 5). The use of anti-IL-1β neutralizing antibody or an IL-1 receptor antagonist decreased the levels of IL-23 produced under inflammasome-activating conditions (Fig. 5). Recombinant IL-1β increased the amount of IL-23 produced, demonstrating that IL-1 receptor signaling can account for much of the increase in IL-23 found in the presence of toxins A and B. However, we cannot rule out additional signaling pathways activated by the toxins which may also contribute to IL-23 production.
We also found that IL-1β was elevated in the serum of patients with CDI (Fig. 7); this had been previously shown in stools of such patients (33). Detection of serum IL-1β required the use of a high-sensitivity ELISA. Although we have not examined the presence of toxins A and B in patient serum samples, the toxins have been shown to be present in the serum of infected animals, suggesting that inflammasome activation may be a systemic response to CDI (22). Increased serum IL-1β was also noted in patients with diarrhea from other causes, suggesting that elevation of this cytokine is a common response to diarrheal illness. Indeed, IL-1β release is triggered in response to other bacterial infections, and it is increased in cells from patients with inflammatory bowel disease (34, 35). IL-1β may therefore play a role in the induction of IL-23 in other diarrheal illnesses, an interesting possibility for which more investigation is warranted.
The role of IL-1β during CDI is complex, as reduced IL-1β production in ASC−/− mice has been demonstrated to be protective during challenge with toxins A and B (12) but pathogenic in an infection model (17). IL-1β is thought to play an essential role in the induction of CXCL1 and recruitment of neutrophils, which may be pathogenic during challenge with toxins alone but play an essential role in controlling infection with live C. difficile (16, 36). Our data further demonstrated that IL-1β likely plays a multifaceted role during CDI by inducing the pathogenic cytokine IL-23.
In conclusion, we suggest that C. difficile is initially recognized in a TLR4- and MyD88-dependent manner, resulting in low-level expression of IL-23. Intoxication of primed host cells by toxins A and B leads to robust IL-1β secretion, likely due to inflammasome activation. IL-1β then signals through the IL-1 receptor to further enhance the production of IL-23. This model therefore suggests that two signals are required for a robust inflammatory response to C. difficile: detection of bacterium- or host-derived danger signals and also intoxication of host cells. Our work demonstrates that although toxins A and B are essential virulence factors of C. difficile, additional signals play a role in shaping the inflammatory response during CDI. Therefore, targeting innate signaling pathways could present novel therapies to disrupt pathogenic host signaling during disease.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
C. difficile strain r20291 AB- was generated using the ClosTron system, and functional inactivation of the targeted genes was confirmed by Western blotting as previously described (29). Strains were inoculated onto brain heart infusion agar and incubated at 37°C overnight in an anaerobic work station (SHEL Lab). Single colonies were inoculated into tryptone-yeast extract medium (37) and grown anaerobically overnight at 37°C. C. difficile cultures were prepared for cell stimulation by resuspending each culture to an optical density corresponding to 2 × 107 CFU/ml. Cultures were spun in a centrifuge, and the supernatant was removed and sterilized through a 0.2-µm filter.
Mice and cells.
C57BL6, MyD88−/−, TLR2−/−, TLR4−/−, TLR5−/−, and IL-1R−/− mice were purchased from The Jackson Laboratory. Mice were males aged 8 to 12 weeks for all experiments. All animals were housed under specific-pathogen-free conditions at the University of Virginia’s animal facility. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Virginia. BMDCs were generated as previously described with minor modifications (38). Briefly, femurs and tibia were removed and bone marrow was flushed with phosphate-buffered saline (PBS). Cells were counted, viability was assessed via trypan blue staining, and cells were resuspended in RPMI 1640 medium (Life Technologies) containing 10% fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin, and 100 U/ml streptomycin. Medium was supplemented with 10 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF; PeproTech) and 55 µM β-mercaptoethanol (Gibco), and 3 × 106 cells were seeded into a T75 vent cap tissue culture flask. Cells were cultured for 7 days and supplemented with fresh medium on days 2 and 4. On day 7, cells were harvested for stimulation. Human MoDCs were generated as described previously (39). Briefly, peripheral blood mononuclear cells were isolated from the blood of healthy human volunteers by Ficoll-Paque density gradient centrifugation. Mononuclear lymphocytes were collected and resuspended in RPMI 1640 containing 10% fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin, and 100 U/ml streptomycin. PBMCs were adjusted to 2 × 106 cells/ml, and 10 ml of cell suspension was added to each T75 tissue culture flask. PBMCs were incubated at 37°C for 3 h, and suspension cells were aspirated. Adherent monocytes were washed thoroughly and detached with gentle scraping. Monocytes were counted and assessed for viability by trypan blue exclusion. Monocytes were adjusted to 1 × 106 cells/ml, and 50 ng/ml recombinant human GM-CSF (PeproTech), 50 ng/ml recombinant human IL-4 (PeproTech), and 55 µM β-mercaptoethanol were added. Monocytes were differentiated for 6 days, with addition of fresh medium on days 2 and 4.
Cell stimulation.
BMDCs or MoDCs were harvested and resuspended in complete RPMI 1640. A total of 2 × 105 cells per well were seeded into 48-well tissue culture plates. C. difficile filtrate was diluted 1:20 into the BMDC suspension, and the remaining volume was made up with complete culture medium (containing toxins A and B where indicated). Purified toxins A and B were a kind gift from Techlab, Inc. (Blacksburg, VA). Recombinant serum amyloid A (PeproTech), TLR ligand-tested lipopolysaccharide (Sigma), anti-mouse IL-1β monoclonal antibody (clone B122; eBioscience), IgG isotype control (eBioscience), recombinant IL-1 receptor antagonist (R&D Systems), recombinant murine IL-1β (R&D Systems), YVAD-FMK (Enzo Life Sciences), and glybenclamide (InvivoGen) were diluted in RPMI 1640 and added to cells as indicated. For incubation of BMDCs with excess potassium, 0.8 M stock solutions of KCl and NaCl were prepared, filter sterilized, and diluted 1:20 into the BMDC suspension. Stimulated cells were incubated for 24 h at 37°C with 5% CO2. Cells were spun at 300 × g for 5 min, and the supernatant was harvested and frozen at − 80°C. Remaining cells were washed in 1× PBS once and lysed in an appropriate volume of RLT buffer (lysis buffer of the RNeasy kit; Qiagen). RNA-containing lysates were frozen at −80°C until RNA isolation.
Detection of cytokines.
IL-1β and IL-23 were detected in protein supernatants from BMDCs by using the mouse IL-1β Ready-Set-Go! ELISA kit (eBioscience) and the mouse IL-23p19 DuoSet kit (R&D Systems) according to the manufacturers’ instructions. IL-1β processing was confirmed by Western blotting. To generate supernatant samples, protein supernatant from stimulated BMDCs was harvested and concentrated by methanol precipitation. Whole-cell lysate was generated from the same BMDCs by washing cells once in ice-cold PBS and adding 100 µl lysis buffer to each well (50 mM Tris-HCl, 5 mM EDTA, 150 mM NaCl, 1% Triton X-100, 1 ml HALT protease inhibitor [Pierce]). Lysates for each condition were pooled and shaken at 4°C for 30 min before spinning at 12,000 rpm at 4°C. Supernatant and whole-cell lysate protein samples were run on BioRad Mini-Protean TGX gradient gels (4 to 15%) and transferred to Amersham Hybond-C nitrocellulose. Blots were probed with either rabbit polyclonal anti-IL-1β antibody (supernatant samples; ab9722; Abcam) or rabbit polyclonal anti-β-actin loading control (whole-cell lysates; ab75186; Abcam) followed by LI-COR anti-rabbit 800 secondary antibody. Fluorescent protein bands were detected using an Odyssey infrared imaging system (LI-COR Biosciences). Image manipulation was limited to adjustment of brightness. MoDC IL-23 production was assessed by quantitative reverse transcription-PCR. RNA was isolated using the RNeasy isolation kit (Qiagen). Contaminating genomic DNA was digested by using the Turbo DNA-free kit (Ambion), and RNA was reverse transcribed with the Tetro cDNA synthesis kit (Bioline) according to the manufacturer’s instructions. The resulting cDNA was purified using Qiagen’s PCR purification kit. IL-23a gene expression was quantified via a QuantiTect primer assay (Qiagen) using Sensifast SYBR and fluorescein mix (Bioline) in the QuantiTect 2-step amplification protocol. Gene expression was normalized to that of β-actin (forward primer, ATTGCCGACAGGATGCAGAA; reverse primer, GCTGATCCACATCTGCTGGAA).
Detection of human serum IL-1β.
Serum samples collected at the time of diagnosis of C. difficile diarrhea from 85 patients at the University of Virginia, as well as samples from 104 patients with diarrhea but who were negative for C. difficile, and also samples from 28 healthy outpatients were stored at −80°C until assayed. IL-1β in the serum samples was quantified using the eBioscience high-sensitivity ELISA kit according to the manufacturer’s instructions. The study was approved by the University of Virginia Institutional Review Board for Health Sciences Research.
SUPPLEMENTAL MATERIAL
Toxins A and B cause cell death and activate NF-κB, and toxin A induces CXCL1 secretion. RAW-Blue NF-κB reporter macrophages (InvivoGen) were exposed to purified toxins A and B as indicated. (A) Cell supernatants were harvested at 8 h and assayed for secreted embryonic alkaline phosphatase activity in a QUANTI-Blue assay. (B) RAW-Blue cells were assayed for cytotoxicity in a lactate dehydrogenase (LDH) release assay (Promega). Cell death is represented as a percentage of total lysis for the positive control. (C) BMDCS were exposed to purified toxins A and B as indicated. CXCL1/KC production was determined by using the R&D Systems Quantikine ELISA kit. Student’s t test was used to determine statistical significance (compared to the untreated control): *, P < 0.05; **, P < 0.01; ***, P < 0.001. Download
ACKNOWLEDGMENTS
We thank Alison Criss for assistance with monocyte generation, and TechLab, Inc. for providing purified toxins A and B.
This work was supported by grants from the National Institutes of Health (5R01AI026649-25 and 5T32AI07046-38).
Footnotes
Citation Cowardin CA, Kuehne SA, Buonomo EL, Marie CS, Minton NP, Petri WA, Jr. 2015. Inflammasome activation contributes to interleukin-23 production in response to Clostridium difficile. mBio 6(1):e02386-14. doi:10.1128/mBio.02386-14.
REFERENCES
- 1.Miller BA, Chen LF, Sexton DJ, Anderson DJ. 2011. Comparison of the burdens of hospital-onset, healthcare facility-associated Clostridium difficile infection and of healthcare-associated infection due to methicillin-resistant Staphylococcus aureus in community hospitals. Infect Control Hosp Epidemiol 32:387–390. doi: 10.1086/659156. [DOI] [PubMed] [Google Scholar]
- 2.Ananthakrishnan AN. 2011. Clostridium difficile infection: epidemiology, risk factors and management. Nat Rev Gastroenterol Hepatol 8:17–26. doi: 10.1038/nrgastro.2010.190. [DOI] [PubMed] [Google Scholar]
- 3.Bacci S, Mølbak K, Kjeldsen MK, Olsen KE. 2011. Binary toxin and death after Clostridium difficile infection. Emerg Infect Dis 17:976–982. doi: 10.3201/eid/1706.101483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartlett JG, Chang TW, Gurwith M, Gorbach SL, Onderdonk AB. 1978. Antibiotic-associated pseudomembranous colitis due to toxin-producing clostridia. N Engl J Med 298:531–534. doi: 10.1056/NEJM197803092981003. [DOI] [PubMed] [Google Scholar]
- 5.Buffie CG, Pamer EG. 2013. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol 13:790–801. doi: 10.1038/nri3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. 1995. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375:500–503. doi: 10.1038/375500a0. [DOI] [PubMed] [Google Scholar]
- 7.Chaves-Olarte E, Weidmann M, Eichel-Streiber C, Thelestam M. 1997. Toxins A and B from Clostridium difficile differ with respect to enzymatic potencies, cellular substrate specificities, and surface binding to cultured cells. J Clin Invest 100:1734–1741. doi: 10.1172/JCI119698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jefferson KK, Smith MF Jr, Bobak DA. 1999. Roles of intracellular calcium and NF-κB in the Clostridium difficile toxin A-induced up-regulation and secretion of IL-8 from human monocytes. J Immunol 163:5183–5191. [PubMed] [Google Scholar]
- 9.Lee JY, Kim H, Cha MY, Park HG, Kim Y-J, Kim IY, Kim JM. 2009. Clostridium difficile toxin A promotes dendritic cell maturation and chemokine CXCL2 expression through p38, IKK, and the NF-kappaB signaling pathway. J Mol Med 87:169–180. doi: 10.1007/s00109-008-0415-2. [DOI] [PubMed] [Google Scholar]
- 10.Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong Y-N, Peng X, Xi JJ, Chen S, Wang F, Shao F. 2014. Innate immune sensing of bacterial modifications of Rho GTPases by the pyrin inflammasome. Nature 513:237–241. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 11.Jafari NV, Kuehne SA, Bryant CE, Elawad M, Wren BW, Minton NP, Allan E, Bajaj-Elliott M. 2013. Clostridium difficile modulates host innate immunity via toxin-independent and dependent mechanism(s). PLoS One 8:e69846. doi: 10.1371/journal.pone.0069846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng J, Hirota SA, Gross O, Li Y, Ulke-Lemee A, Potentier MS, Schenck LP, Vilaysane A, Seamone ME, Feng H, Armstrong GD, Tschopp J, Macdonald JA, Muruve DA, Beck PL. 2010. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 139:542–552. doi: 10.1053/j.gastro.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Ryan A, Lynch M, Smith SM, Amu S, Nel HJ, McCoy CE, Dowling JK, Draper E, O’Reilly V, McCarthy C, O’Brien J, Ní Eidhin D, O’Connell MJ, Keogh B, Morton CO, Rogers TR, Fallon PG, O’Neill LA, Kelleher D, Loscher CE. 2011. A role for TLR4 in Clostridium difficile infection and the recognition of surface layer proteins. PLoS Pathog 7:e1002076. doi: 10.1371/journal.ppat.1002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshino Y, Kitazawa T, Ikeda M, Tatsuno K, Yanagimoto S, Okugawa S, Yotsuyanagi H, Ota Y. 2013. Clostridium difficile flagellin stimulates Toll-like receptor 5, and toxin B promotes flagellin-induced chemokine production via TLR5. Life Sci 92:211–217. doi: 10.1016/j.lfs.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 15.Hasegawa M, Yamazaki T, Kamada N, Tawaratsumida K, Kim Y-G, Núñez G, Inohara N. 2011. Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol 186:4872–4880. doi: 10.4049/jimmunol.1003761. [DOI] [PubMed] [Google Scholar]
- 16.Jarchum I, Liu M, Shi C, Equinda M, Pamer EG. 2012. Critical role for MyD88-mediated neutrophil recruitment during Clostridium difficile colitis. Infect Immun 80:2989–2996. doi: 10.1128/IAI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasegawa M, Kamada N, Jiao Y, Liu MZ, Núñez G, Inohara N. 2012. Protective role of commensals against Clostridium difficile infection via an IL-1β-mediated positive-feedback loop. J Immunol 189:3085–3091. doi: 10.4049/jimmunol.1200821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El Feghaly R, Stauber JL, Deych E, Gonzalez C, Tarr PI, Haslam DB. 2013. Markers of intestinal inflammation, not bacterial burden, correlate with clinical outcomes in Clostridium difficile infection. Clin Infect Dis 56:1713–1721. doi: 10.1093/cid/cit147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buonomo EL, Madan R, Pramoonjago P, Li L, Okusa MD, Petri WA. 2013. Role of IL-23 signaling in Clostridium difficile colitis. J infect Dis 208:917–920. doi: 10.1093/infdis/jit277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu W, Ouyang X, Yang J, Liu J, Li Q, Gu Y, Fukata M, Lin T, He JC, Abreu M, Unkeless JC, Mayer L, Xiong H. 2009. AP-1 activated by Toll-like receptors regulates expression of IL-23 p19. J Biol Chem 284:24006–24016. doi: 10.1074/jbc.M109.025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, Lawley TD, Sebaihia M, Quail MA, Rose G, Gerding DN, Gibert M, Popoff MR, Parkhill J, Dougan G, Wren BW. 2009. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol 10:R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steele J, Chen K, Sun X, Zhang Y, Wang H, Tzipori S, Feng H. 2012. Systemic dissemination of Clostridium difficile toxins A and B is associated with severe, fatal disease in animal models. J Infect Dis 205:384–391. doi: 10.1093/infdis/jir748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckhardt ER, Witta J, Zhong J, Arsenescu R, Arsenescu V, Wang Y, Ghoshal S, de Beer MC, de Beer FC, de Villiers WJ. 2010. Intestinal epithelial serum amyloid A modulates bacterial growth in Vitro and pro-inflammatory responses in mouse experimental colitis. BMC Gastroenterol 10:133. doi: 10.1186/1471-230X-10-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Von Moltke J, Ayres JS, Kofoed EM, Chavarría-Smith J, Vance RE. 2013. Recognition of bacteria by inflammasomes. Annu Rev Immunol 31:73–106. doi: 10.1146/annurev-immunol-032712-095944. [DOI] [PubMed] [Google Scholar]
- 25.Peral de Castro C, Jones SA, Ní Cheallaigh C, Hearnden CA, Williams L, Winter J, Lavelle EC, Mills KH, Harris J. 2012. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J Immunol 189:4144–4153. doi: 10.4049/jimmunol.1201946. [DOI] [PubMed] [Google Scholar]
- 26.Akira S, Takeda K. 2004. Toll-like receptor signalling. Nat Rev Immunol 4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 27.Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. 2013. K+ efflux is the common trigger of Nlrp3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chumbler NM, Farrow MA, Lapierre LA, Franklin JL, Haslam DB, Goldenring JR, Haslam D, Lacy DB. 2012. Clostridium difficile toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog 8:e1003072. doi: 10.1371/journal.ppat.1003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuehne SA, Collery MM, Kelly ML, Cartman ST, Cockayne A, Minton NP. 2013. The importance of toxin A, toxin B and CDT in virulence of an epidemic Clostridium difficile strain. J Infect Dis 209:83–86. doi: 10.1093/infdis/jlt426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 31.Deakin LJ, Clare S, Fagan RP, Dawson LF, Pickard DJ, West MR, Wren BW, Fairweather NF, Dougan G, Lawley TD. 2012. The Clostridium difficile spo0a gene is a persistence and transmission factor. Infect Immun 80:2704–2711. doi: 10.1128/IAI.00147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pawlowski SW, Calabrese G, Kolling GL, Freire R, AlcantaraWarren C, Liu B, Sartor RB, Guerrant RL. 2010. Murine model of Clostridium difficile infection with aged gnotobiotic C57BL/6 mice and a BI/NAP1 strain. J Infect Dis 202:1708–1712. doi: 10.1086/657086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steiner TS, Flores CA, Pizarro TT, Guerrant RL. 1997. Fecal lactoferrin, interleukin-1beta, and interleukin-8 are elevated in patients with severe Clostridium difficile colitis. Clin Diagn Lab Immunol 4:719–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X, Cheng Y, Xiong Y, Ye C, Zheng H, Sun H, Zhao H, Ren Z, Xu J. 2012. Enterohemorrhagic Escherichia coli specific enterohemolysin induced IL-1β in human macrophages and EHEC-induced IL-1β required activation of Nlrp3 inflammasome. PLoS One 7:e50288. doi: 10.1371/journal.pone.0050288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stevens C, Walz G, Singaram C, Lipman ML, Zanker B, Muggia A, Antonioli D, Peppercorn MA, Strom TB. 1992. Tumor necrosis factor-alpha, interleukin-1beta, and interleukin-6 expression in inflammatory bowel disease. Dig Dis Sci 37:818–826. doi: 10.1007/BF01300378. [DOI] [PubMed] [Google Scholar]
- 36.Kelly CP, Becker S, Linevsky JK, Joshi MA, O’Keane JC, Dickey BF, LaMont JT, Pothoulakis C. 1994. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J Clin Invest 93:1257–1265. doi: 10.1172/JCI117080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cartman ST, Minton NP. 2010. A Mariner-based transposon system for in vivo random mutagenesis of Clostridium difficile. Appl Environ Microbiol 76:1103–1109. doi: 10.1128/AEM.02525-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gross O. 2012. Measuring the inflammasome. Methods Mol Biol 844:199–222. doi: 10.1007/978-1-61779-527-5_15. [DOI] [PubMed] [Google Scholar]
- 39.Tedder TF, Jansen PJ. 2001. Isolation and generation of human dendritic cells. Curr Protoc Immunol Chepter 7:Unit 7.32. doi: 10.1002/0471142735.im0732s23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Toxins A and B cause cell death and activate NF-κB, and toxin A induces CXCL1 secretion. RAW-Blue NF-κB reporter macrophages (InvivoGen) were exposed to purified toxins A and B as indicated. (A) Cell supernatants were harvested at 8 h and assayed for secreted embryonic alkaline phosphatase activity in a QUANTI-Blue assay. (B) RAW-Blue cells were assayed for cytotoxicity in a lactate dehydrogenase (LDH) release assay (Promega). Cell death is represented as a percentage of total lysis for the positive control. (C) BMDCS were exposed to purified toxins A and B as indicated. CXCL1/KC production was determined by using the R&D Systems Quantikine ELISA kit. Student’s t test was used to determine statistical significance (compared to the untreated control): *, P < 0.05; **, P < 0.01; ***, P < 0.001. Download