

Abstract
Biosensors are valuable tools used to monitor many different protein behaviors in vivo. Demand for new biosensors is high, but their development and characterization can be difficult. During biosensor design, it is necessary to evaluate the effects of different biosensor structures on specificity, brightness, and fluorescence responses. By co-expressing the biosensor with upstream proteins that either stimulate or inhibit the activity reported by the biosensor, one can determine the difference between the biosensor’s maximally activated and inactivated state, and examine response to specific proteins. This involves considerable labor and expense, as expression conditions must be optimized to saturate the biosensor with the regulator, and multiple replicates and controls are required. We describe here a protocol for biosensor validation in a 96-well plate format using an automated microscope. This protocol produces dose-response curves, enables efficient examination of many parameters, and unlike cell suspension assays allows visual inspection (eg for cell health and biosensor or regulator localization). Optimization of single chain and dual chain Rho GTPase biosensors is addressed, but the assay is applicable to any biosensor that can be expressed or otherwise loaded in adherent cells. The assay can also be used for purposes other than biosensor validation, using a well-characterized biosensor as a readout for variations in upstream molecules.
Keywords: biosensor, HC screening, microplate assay, fluorescence, FRET
Unit Introduction
Biosensors have become valuable tools in cell biology. They can report a wide range of protein activities, including conformational change, post-translational modification, enzymatic activity, protein-protein interaction and small molecule concentration. The design and validation of biosensors requires time consuming and laborious measurements of biosensor response, specificity, dynamic range etc. In a common approach to this, biosensors and upstream regulators are transfected into the same cells, and regulator effects on biosensor fluorescence are determined by suspending these cells in a cuvette or well plate and measuring fluorescence. By keeping the biosensor DNA fixed and adding increasing amounts of regulator DNA it is possible to obtain a titration of effects on the biosensor and thus determine when a plateau in regulator effects has been reached. Only by finding a plateau can one gauge whether the biosensor has been saturated by the regulatory molecule. These assays are complicated by the fact that high levels of expression, of either the biosensor or the regulator, can be toxic to cells or reduce biosensor response, generating spurious fluorescence results. Multiple controls and multiple replicates of each condition are also important.
This unit describes a method for performing such titration analysis in a 96-well microplate format using automated microscopy. The procedure enables extensive and informative titration that would be prohibitively laborious when performed ‘by hand’. Validation is greatly facilitated through increased throughput of transfection, detection, and analysis, with simultaneous visual confirmation of cell health. Imaging adherent cells with an automated microscope means that there is no need to detach and resuspend cells, which can affect both biosensor response and cell health. Furthermore, the image data that is obtained may be stored and examined to assess biosensor or regulator localization, transfection efficiency, cell health or other aspects of cell behavior.
As an example, we present validation of Rho family GTPase biosensors that use fluorescence resonance energy transfer (FRET). We discuss designs based on a single protein chain (intramolecular FRET biosensor) or on two separate, interacting chains (intermolecular FRET biosensor). When developing new FRET biosensors, it is desirable to maximize the level of FRET output, the extent to which FRET efficiency changes when the biosensor responds to the targeted biological activity, and to maximize selectivity for the desired biological activity. These parameters are optimized by varying linkers within the biosensors, choosing the best fluorescent proteins, and using fluorescent protein circular permutants to affect fluorophore dipole orientation. The automated procedure we describe simplifies performing titrations for multiple biosensor variants.
Most biosensors are based upon genetically encoded fluorescent proteins, but recent advances in labeling proteins in vivo indicate that even biosensors based on dyes can be readily ‘loaded’ into cells without using microinjection, electroporation or other mechanical loading procedure. The assay described here is useful for any fluorescent molecule that can be readily introduced into populations of cells adhered to the bottom of well plates.
The assay can also be valuable for biochemical investigations of protein activity. Once a biosensor is validated, that biosensor can be used as a readout, using the assay to examine interactions between upstream molecules and the protein activity reported by the biosensor.
Strategic Planning
Biosensor design
The Rac1 FLARE.dc biosensor (Figure 1) will be used as an example throughout this article (Kraynov, 2000; Machacek et al., 2009). This biosensor consists of two separate proteins: a fusion of Rac1 with the donor fluorescent protein, and another chain consisting of the ‘affinity reagent’ (a small protein that binds selectively to the activated conformation of Rac1) fused to the acceptor fluorophore. In this biosensor, the affinity reagent is the p21 binding domain of PAK1, a Rac1 effector. Rac1 is fused at its N-terminus to CyPet. This configuration retains the Rac1 C-terminal CAAX box, which is required to localize Rac1 to the membrane and for interaction with one of Rac’s negative regulators, RhoGDI-1. The p21 binding domain is tagged with the acceptor fluorescent protein YPet. When Rac1 is in the inactive, GDP-bound state, it does not interact with the affinity reagent, and FRET efficiency is low. When Rac1 is in the active, GTP-bound state, it binds to the affinity reagent, bringing the donor and acceptor into proximity, which increases FRET efficiency.
Figure 1.

The Rac1 FLARE.dc biosensor. When Rac1 is in the GDP-bound state, it has low affinity for the p21 binding domain of PAK1 (PBD). The two peptides are unassociated and there is negligible FRET from the CyPet on Rac1 to the YPet on the PBD. When in the GTP-bound state, Rac1 has increased affinity for the PBD and interacts with it, bringing the CyPet and YPet tags into proximity and allowing FRET to take place. Rac1 can be converted from the GDP-bound to the GTP-bound state by GEFs, and back to the GDP-bound state by GAPs. The GDP-bound GTPase can also be complexed with GDI, which sequesters Rac1 in the cytoplasm and prohibits interaction with the PBD.
Regulators
To validate Rho family GTPase biosensors they are co-expressed with both positive and negative regulators. Three major types of regulators directly bind to Rac1 to control its GTP/GDP binding status: (1) guanine nucleotide exchange factors (GEFs) stimulate the release of GDP and subsequent binding of GTP (Rossman et al., 2005); (2) GTPase activating proteins (GAPs) stimulate the hydrolysis of GTP into GDP (Moon and Zheng, 2003); and, (3) guanine nucleotide dissociation inhibitors bind to GDP-associated Rac1 and sequester it in the cytoplasm in an inactive state (Garcia-Mata et al., 2011). When different families of positive or negative regulators regulate the targeted activity by different mechanisms, as in the case of GAPs and GDIs for Rac1, a representative of each family would ideally be tested. In many cases, the regulator molecules are themselves regulated, so it is necessary to overcome their negative regulation to generate strong effects on the biosensor. For example, many GEFs and GAPs are autoinhibited through intramolecular interaction of an autoinhibitory domain with the GTPase-interacting domain. Expression of a truncated GEF or GAP that lacks the autoinhibitory domain is used to produce uninhibited stimulation of the downstream Rho GTPases.
For quantitation of regulator expression, it is possible to tag the regulator with a fluorescent protein that has excitation and emission wavelengths orthogonal to those of the biosensor. For example, a biosensor employing a cyan fluorescent protein (CFP) donor and a yellow fluorescent protein (YFP) acceptor could be combined with an mCherry-tagged regulator. This fluorophore combination enables imaging of the regulator with little bleedthrough from the biosensor fluorophores into the regulator imaging channel, and vice versa. Directly tagging the regulator with a fluorescent protein has been helpful when interactions between the regulator and the biosensor are transient, as with GEFs and GAPs. However, regulators that stably bind the GTPases, such as GDIs, may undergo FRET that is detectable in the assay (e.g., YFP-> mCherry FRET in the example above.) We have observed such effects when examining Rho family biosensors’ response to mCherry-tagged GDI. Data obtained using mCherry-tagged regulators indicated that expression level is linearly related to the mass of DNA transfected over two to three orders of magnitude (Figure 2). Therefore, it is frequently preferable to omit the fluorescent tag on the regulator to avoid distortions of the FRET index. This approach is presented in this unit.
Figure 2.

Regulator expression increases nearly linearly with the mass of DNA transfected over a wide range of DNA mass. Samples transfected with varying dosage of expression plasmid encoding mCherry-tagged regulator (filled circles: RhoGDI1; empty circles: active Vav2 GEF) were imaged and total mCherry emission, after background subtraction, was plotted against transfected regulator plasmid mass.
Controls
Various controls can be employed to alert the user to artifacts and guard against misinterpretation of the results:
Donor-only and acceptor-only controls
In donor-only and acceptor-only controls, the biosensor is replaced with a biosensor that lacks either the acceptor or the donor, but, when feasible, contains all other elements of the biosensor. (When that proves difficult, simple expression of a single fluorophore alone can serve). These controls can be incorporated into each experiment as single samples without regulators, to calculate bleedthrough coefficients that are used to correct bleedthrough into the FRET channel from donor and acceptor emission that is not sensitized emission (see below). Additionally, the FRET:donor ratio of a donor-only sample, or the FRET:acceptor ratio of an acceptor-only sample, can be used as a basis for normalization of data to allow comparison across experiments imaged with different exposure times. Finally, control biosensors with donor or acceptor fluorophore only can be subjected to the full range of regulator co-expressions. Since these control biosensors should have a FRET: Donor ratio that is dependent only upon the physics of the fluorophore, this control serves to confirm that the image analysis is correctly performed, that the best-case variance of replicates is acceptably low, that the transfection of regulator does not affect the emission spectrum of the donor, and that the imaging conditions are consistent over time, over different regions of well plates, and from plate to plate.
Donor-Acceptor controls
Overcrowding of biosensors or their aggregation or partial degradation can lead to artifactual increases or decreases in FRET efficiency or in the sensitivity of the biosensor. To control for this type of artifact, one can use a control biosensor that contains both of the fluorescent proteins, but is missing one or more biologically active components to render it non-functional (e.g., a biosensor without an affinity reagent, or with mutations abrogating GTPase-affinity reagent interactions). The FRET efficiency of such a construct should be independent of biosensor or regulator expression level, unless one of the aforementioned artifacts is present.
Non-specific regulator controls
A “regulator” protein known not to regulate the activity of interest may be transfected with the biosensor to demonstrate specificity of the response. For example, there are a large number of GEFs and GAPs that target members of the Rho family of GTPases, but do not target Rac1. Alternatively, a specific regulator that is mutated to a non-functional form could serve this purpose.
Biosensor mutant controls
In some cases, there are well-described mutants of your protein that produce permanent activation or knock out activation. These are good positive and negative controls. Examples for the Rac1 biosensor include: 1) the Rac1(Q61L) mutant, constitutively active due to an inability to catalyze hydrolysis of GTP to GDP; 2) the Rac1(T17N) mutant, a dominant negative because it is unable to release GEFs and bind to GTP after the GEF has catalyzed release of GDP; 3) the PAK1(H83,86D) affinity reagent mutant is unable to bind to the active form of Rac1. Such mutants often do not exactly replicate the active and inactive states of the wild-type protein. For example, Rac1(Q61L) is reported to have a higher affinity for the PBD affinity reagent than GTP-bound wild-type Rac1 (Thompson et al., 1998), and in our hands, the FRET efficiency for the Rac1(T17N) mutant is higher than for wild-type Rac1 down-regulated by RhoGDI (data not shown).
Basic Protocol #1
Title: Transfection of LinXE or HEK 293T cells with gradients of biosensor and/or regulators in 96-well microplates
-
Basic protocol introduction
In this protocol, we will describe how to transfect a biosensor together with regulators, and all of the necessary controls, into cells in a 96-well microplate. This protocol was optimized for transfection of intramolecular and intermolecular Rho family GTPase biosensors developed in the Hahn lab, and their GEF, GAP, and GDI regulators, in LinXE cells using Lipofectamine transfection reagent. This should be generalizable to other biosensors, regulators, and cell types, but some steps may require re-optimization.
-
Basic protocol materials list
Black walled, tissue culture treated MicroClear 96-well plate (Greiner Bio-one catalog #655090)
Clear 96-well V-bottom sterile polystyrene microplates (Greiner Bio-one catalog #651161)
DMEM complete (10% FBS, no antibiotics)
DMEM, serum-free and antibiotic free
LinXE or HEK 293T cells, T75 flask near confluency
Biosensor expression construct
Donor only fluorescent protein expression construct
Acceptor only fluorescent protein expression construct
12-channel pipettor for 20–200 μL
Multichannel aspirator (8 or 12 channels)
Single well v-bottom liquid reservoir that fits multichannel pipettor
Trypsin/EDTA
DPBS without calcium or magnesium
100μg/mL Poly-L-Lysine (PLL; mol. wt. 150,000 to 300,000, Sigma Aldrich, cat. no. P4832
-
Basic protocol steps and annotations
-
We will demonstrate the protocol using transfection of a fixed amount of biosensor with 20 two-fold dilutions of a regulator. The following figures illustrate how this experiment is designed, and can serve as a template for variations on the experiment. Each figure is also provided as an Excel template file.
Figure 3 illustrates the layout of transfected cells within a 96-well microplate. Each well is labeled with the identity and amount (when variable) of expression plasmids transfected into the cells. Note that column 1 contains mock-transfected cells. These samples are used later for background subtraction. The second column contains alternating donor only and acceptor only samples. These are used for bleedthrough correction in the calculation of sensitized emission (they may be omitted if bleedthrough correction is not required). The remaining columns are transfected with biosensor, and with varying amounts of a regulator plasmid. The biosensor is transfected at 4.25 ng per well, and the maximum amount of regulator plasmid transfected in this example is 42.5 ng per well. These are approximately equivalent to 100 and 1000 ng per well, respectively, in a 6-well tray, based on ng plasmid/well surface area. Note that there are four replicate wells for each transfection type. Rows 1 and 2 are replicated in rows 3 and 4, rows 5 and 6, and rows 7 and 8.
Figure 4 illustrates the layout of transfection reactions as they are prepared in a 96-well microplate prior to transfer onto cells as outlined in Figure 3. A single reaction well can transfect up to four wells of cells, and therefore replicate wells of the transfected cell map in Figure 3 are supplied by a single well within the transfection reaction map in Figure 4. In the example, the transfection reaction map in Figure 4 is equivalent to the first two rows of the transfected cell map in Figure 3, with the amount of each DNA simply increased to 4.7 fold the amount that goes into each cell well.
Figure 5 is a table of DNA/medium solutions that will be prepared in microfuge tubes as a first step of preparing the transfection reactions. This table is also supplied as an Excel file (DNA solutions table form.xlsx) so that the reader can fill it out for their own experiment. Solution #1 consists of only empty vector DNA for a mock transfection. Solutions #2 and #3 consist of donor-only and acceptor-only expression plasmids, respectively. Solution #4 consists of Biosensor expression plasmid. Solution #5 consists of Biosensor and Regulator expression plasmids. Variable amounts of regulator transfection will be achieved by serially diluting Solution #5 into Solution #4. Figure 6 shows how the DNA solutions will be distributed into a 96-well microplate as a first step towards preparation of individual transfection reactions. Note that 20 μL of DNA solution is required for each well into which it will be distributed, except for DNA solution #5, which contains the regulator. It requires 40 μL for a single well, because half of that volume will be transferred into the well to the left for serial dilution. Figure 5 has a column for total number of wells, which can be counted from Figure 6, and a column for μL per well. Total volume for each DNA solution will be equal to (total number of wells) X (volume per well) X (1.1). The factor 1.1 ensures that excess volume is produced and guards against running short of volume when distributing into wells. This total volume consists of the volumes of DNA listed in Figure 5 (a total of 220 ng of plasmid per 20 μL of total solution volume), and the remainder is serum-free, antibiotic-free medium.
Dilute PLL to 1 μg/mL in DPBS for a total volume of 10 mL. Transfer the solution to a reservoir and use a multichannel pipettor to dispense 100 μL into each well of a 96-well plate.
Incubate the plate overnight in a tissue culture incubator at 37 °C. Alternativley, you can reduce the incubation time to one hour by increasing the PLL concentration 5- fold.
Using a multi-channel aspirator, aspirate the PLL solution from the wells and wash with 100 μL DPBS per well two times. Store the plate with the second wash in the incubator until you are ready to seed cells into the wells in a later step.
Aspirate the media from your flask of cells and rinse once or twice with DPBS without calcium or magnesium. Aspirate off all DPBS and add 0.5 ml Trypsin/EDTA. Evenly spread the solution and incubate for 5 minutes at 37 °C.
Resuspend trypsinized cells in 25 mL complete DMEM. Pipette up and down several times to ensure a single-cell solution.
Determine cell count per volume using a hemacytometer and phase contrast microscope.
-
Seed 40,000 cells in 100 uL complete per well. To seed all wells of one plate, use the following numbers:
First create a cell suspension by adding 4,400,000 cells into complete DMEM for a final volume of 11mL.
Next, transfer the cell solution to a single well v-bottom reservoir. Use an 8 or 12 channel pipettor to distribute 100 μL of cell solution into each well. Incubate overnight at 37 °C. Inserting the pipette tips nearly to the bottom of the wells and placing the ends of the tips against the well wall will ensure that all of the solution reaches the bottom, rather than sticking to the well wall or the pipette tip. Air bubbles are likely to form when the dispensed solution contains serum, and they may interfere with even dispersal of cells on the growing surface. To avoid the formation of air bubbles, pipette the cell suspension up and down several times in the source reservoir to expel any air bubbles before beginning the transfer to the microplate. Additionally, when dispensing into the microplate, only depress the plunger to the first stop.
The day after seeding the cells, pre-warm serum-free DMEM to 37 °C in a water bath.
Prepare for the transfection reaction by diluting all DNAs that are to be transfected to 50 ng/μL. This simplifies making the transfection reactions by allowing you to use the same volumes per microgram across all plasmids.
In microfuge tubes, create the DNA/medium solutions outlined in Figure 5.
-
Following the DNA solution distribution map in Figure 6, distribute DNA solutions into a sterile v-bottom polystyrene 96-well microplate as follows:
Add 20 uL DNA solution #1 to wells A1 and B1.
Add 20 μL DNA solution #2 to well A2.
Add 20 μL DNA solution #3 to well B2.
Distribute 20 μL of DNA solution #4 into wells in row A, columns 3–11, and in row B, columns 3–12.
Add 40 μL DNA solution #5 into well A12.
Serially dilute DNA solution #5 into DNA solution #4 1:2. To do this, pipette 20 μL from well A12 into A11. Insert pipette tips fully into the bottom of the well, and pipette up and down slowly eight times to thoroughly mix the contents. Then pipette up 20 uL and transfer it to well A10. Repeat this process from right to left until reaching well A3. Then, proceed to dilute from A3 into B12, and again from right to left until you reach well B4. Leave well B3 without any regulator. If you want to prepare reactions for more than one regulator at a time, simply repeat the layout of DNA solutions in lower wells of the same wellplate, and use a multichannel pipettor with a tip on every other nozzle to perform serial dilutions of all regulators simultaneously. We usually perform 20 two-fold dilutions to capture the full dose-response curve for both GEFs (which have a very low EC50) and GDIs (which have a 100-fold higher EC50 than GEFs). However, you may be able to capture the peaks of the response curve of your biosensor of interest with only 10 two-fold dilutions. This would only require a single row of dilutions for each regulator.
In a microfuge tube, combine 23.23 μL Lipofectamine with 504.8 μl serum-free medium. This is equivalent to .88 μL Lipofectamine in a total volume of 20 μL per well of transfection reaction, multiplied by an excess volume factor of 1.1.
To a second, empty v-bottom polystyrene 96-well tray, add 260 uL diluted Lipofectamine solution to rows wells A1 and B1.
Using a multichannel pipettor with two pipette tips, transfer 20 μL of the diluted Lipofectamine solution from wells A1 and B1 of the second 96-well v-bottom microplate to the DNA solutions in the wells of rows A and B of the first 96-well v-bottom microplate, working from left to right, and pipette up and down several times to mix. Change tips once after the addition to column 2. By making additions to the DNA solutions from left to right, you work up the regulator concentration gradient. In this way, inadvertent transfer of minute amounts of reaction from one well to the next will have negligible effect on the “contaminated” reaction. It is important to change tips after column 2, however, because contamination of a biosensor reaction with donor or acceptor control expression plasmid could affect FRET measurements. This pipetting strategy will be repeated throughout this protocol, and we will simply refer to it as the “column-wise left to right pipetting strategy”. It minimizes the use of pipette tips, and it becomes especially useful if the experiment is scaled up to assay multiple regulators and the transfection reaction are arrayed in rows of a single v-bottom microplate.
Incubate DNA/Lipofectamine reactions for 15 minutes at room temperature.
Fill a v-bottom reservoir with 22 mL of serum-free medium, then use a 12-channel pipettor to distribute 195 uL of the medium to each well of the first v-bottom polystyrene 96-well microplate in rows A and B containing a DNA/Lipofectamine solution. This dilutes the reaction to a final concentration that can be applied directly to cells. This can be done quickly by using a “row-wise pipetting strategy”, using all 12 channels of the pipettor to add to all wells in one row at a time, working from top to bottom. To avoid contamination of reactions with other reactions, dispense the medium from above the row of wells without touching the contents of the wells. Dispensing the medium with some reasonable force will help to mix the diluted reaction.
Using a multichannel aspirator, gently aspirate medium from the cells in the black-walled 96-well microplate. To minimize cell loss and confine that loss to the unimaged edge of the well, use the lowest aspiration rate that efficiently removes the medium, and always slide the aspirator tips down one wall of the well until contacting the growth surface.
Using the row-wise pipetting strategy, pipette the diluted reactions in rows G and H of the v-bottom 96-well microplate up and down several times and then add 50 μL of each diluted reaction to the corresponding four replicate wells of cells in the black-walled 96-well microplate, according to the map in Figure 3.
Discard the clear v-bottom microplates with residual transfection reaction, and incubate the black-walled microplate with transfected cells for 3 hours in a 37 °C tissue culture incubator.
Meanwhile, pre-warm DMEM with 20% serum to 37 °C in a water bath.
After the 3 hour incubation, fill a v-bottom reservoir with 6 mL of pre-warmed DMEM with 20% serum. Then use the multichannel pipettor to add 50 μL DMEM plus 20% serum to each well of the transfected cell microplate. When expelling the medium, just touch the pipette tips to the well wall above the surface of the medium already in the well and depress the plunger only to the first stop to avoid introducing bubbles. Additions may be column-wise or row-wise. Changing tips between is not necessary if care is taken not to insert the tips into the well contents.
Return the transfected cells to the incubator overnight.
Perform imaging according to Basic Protocol #2 approximately 24 hours after addition of the transfection reaction to the cells.
-
Figure 3.

Layout of cell transfections in black-walled, flat-bottom imaging 96-well microplate. Column 1 contains 8 replicates of mock transfected cells. Column 2 contains four replicate wells each of cells transfected with a donor-only or acceptor-only control. The remainder of the columns contain cells transfected with biosensor plus varying amounts of regulator. Regulator amount halves serially from right to left across row A, and then again across row B, to cover a dosage range of greater than 200,000 fold. Subsequent rows contain replicates of rows A and B, for a total of four replicate wells for each biosensor/regulator combination. M = mock. D = donor only. A = Acceptor only. X = 4.26. B = biosensor. Y = 42.56. R = regulator.
Figure 4.

Layout of Lipofectamine/DNA transfection reactions in v-bottom 96-well microplates. M = mock. D = donor only. A = Acceptor only. X = 20. B = biosensor. Y = 200. R = regulator.
Figure 5.

DNA solutions prepared for transfection reactions. Volumes that are added to the solutions are highlighted in gray.
Figure 6.
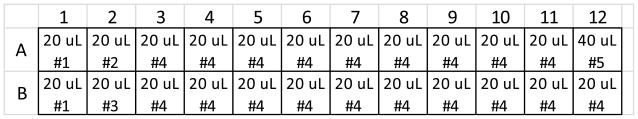
Distribution of Figure 5 DNA solutions into v-bottom microplate prior to serial dilution.
Basic Protocol #2
Title: Automated microscopy of cells transfected with biosensor in a 96-well microplate
-
Introduction
This protocol describes how to use a high content analysis (HCA) microscope to efficiently acquire images of all wells of the wellplate transfected in Basic Protocol #1. Automated microscopy enables fast acquisition of images in all wells of the plate in an objective manner. Ultimately, the images will be used to quantitate fluorescence emission intensities, as described in Basic Protocol #3. Automated microscopes are produced by several different companies, and are usually sold as high content analysis (HCA) or high content screening (HCS) microscopes. Alternately, they can be assembled by a lab, and each has unique capabilities and software used to operate the microscope. Therefore, in this protocol, we will provide general guidelines that should be applicable to most systems, rather than a specific step-by-step protocol.
-
Materials list
-
Imaging medium
HBSS without phenol red, with calcium and magnesium
2% FBS
10 mM HEPES
-
Automated microscope with the following features:
a motorized stage that is capable of precise movements in x, y, and z, and that accepts microplates
laser-based or image-based autofocus.
-
motorized filter and dichroic mirror wheels containing filters and dichroic mirrors capable of capturing images in the following channels:
Donor channel: excitation of the donor and collection of the donor emission without collection of any acceptor emission.
FRET channel: excitation of the donor with collection of the acceptor emission.
Acceptor channel: excitation of the acceptor and collection of the acceptor emission, without any excitation of the donor.
Software that allows programming the automated imaging of an entire wellplate
-
-
Steps and annotations
-
Set up the microscope for imaging before preparing samples.
Warm up the microscope until the lamp reaches a constant intensity.
If temperature control is available, warm the incubation chamber to 37 °C.
Choose the objective. A dry objective must be used to image across a 96-well plate. We typically use an Olympus 10X, 0.4 NA plan apochromat objective and acquire four fields of view per well in a 2 × 2 grid.
Moderate binning (e.g., 2 × 2) can be performed to speed imaging if exposure times are long or to reduce file size. Neutral density filters may be used if the signal is too bright or to avoid photobleaching or cytotoxicity when setting exposure times.
Use a multichannel aspirator to gently remove all culture medium from the microplate to be imaged and replace with 100 μL of imaging medium using a multichannel pipettor. Transfer the plate onto the microscope and manually focus on cells in any well using transmitted light or epifluorescence.
Set the exposure times for each channel to maximize use of the camera dynamic range while avoiding saturation of pixels. First, navigate to a well that is expected to produce the highest level of signal for the given channel. For each channel, set exposure times that produce images with maximum intensities of 50% - 75% of the grayscale maximum for the camera. The donor channel may be brightest in a donor-only well or in a well containing biosensor in the lowest FRET state. The acceptor channel may be brightest in the acceptor-only channel if present, or in a well expressing biosensor. The FRET channel will likely be brightest in the well in which the biosensor is expected to undergo maximal FRET. Check multiple wells for each channel using the chosen exposure times to ensure that they are set properly. Saturated pixels under-report the true intensity value for the pixel in that channel, and can lead to incorrect calculations of the FRET:Donor ratio if included. Therefore, avoid pixel saturation.
Set up autofocus. Autofocus may be image- or laser-based. Image-based autofocus acquires images in one selected channel through a z-stack and considers the optimally focused z-position to be that which maximizes a measured value representative of the degree of focus. If image-based autofocus is used, focus using the FRET channel, because there will be an image in the FRET channel in the donor only, acceptor only, and biosensor samples. Image-based autofocus may fail on mock-transfected wells, because there is no fluorescent protein on which to focus. If so, you may acquire images of mock-transfected wells by programming the microscope to skip autofocus on these wells and use instead the current focal position from the previously imaged well. Laser-based autofocus uses reflection of laser light from the glass/media interface, and then focuses at a plane at a user-determined offset from the interface. For laser-based autofocus, set the offset such that your first imaged channel becomes optimally focused.
If necessary, set offsets between channels. The optimal focal plane will likely not be identical from one channel to the next. Determine the offsets necessary to maintain optimal focus from one channel to the next. One way to do this is to acquire a z-stack in all channels of a field containing fluorescence in all channels. Determine from the image stacks the z-distance between optimal focal planes in each pair of successively imaged channels. Program the microscope to adjust z-position by those distances between each pair of channels. Once determined for a set of channels on a microscope, these values could be expected to remain constant between experiments.
The acquired images should be saved with information about the image within the pathname of the file in a consistent pattern. This information will be extracted and associated with the measurements taken from the images in the analysis phase. The information can all be within the filename itself, or within the hierarchy of directory names containing the file. The minimal information that must be contained within the path includes the row and column of the well and the channel name. Depending on the extent of the experiment, it may be necessary to also include a plate ID (if more than one plate is run in an experiment), or a field ID (if multiple fields are imaged per well). Additional information may be included in and extracted from the filenames if desired. Images should be saved as tiff files or some other file format that does not perform lossy compression. An example filename, which was produced when acquiring images using Metamorph control software, is “140128_plate1_A01_s1_w1.TIF”. This image is part of the experiment “140128”, within the plate 1, at well position A01. It is field 1 of the well and the channel is channel 1.
Scan the wells in a back-and-forth mode, as opposed to a raster mode, if possible. This makes autofocus easier because it minimizes large changes in focus between wells.
-
Basic Protocol #3
Title: Analysis algorithm for biosensor response in microplate image data
-
Introduction
-
In this protocol, we will describe the basic steps in extracting data from the images acquired in Basic Protocol #2, and computing dose-response curves that plot various FRET indices versus regulator expression levels. Because of the large number of images that need to be processed, it is essential that the analysis be performed automatically by software. Several different software programs can be used to perform automated image analysis, including software that comes with many HCA microscopes, freely available software such as CellProfiler, and commercially available software such as PipelinePilot. We use a custom written Matlab script. In this basic protocol, we will describe the analysis steps that should be performed in general language. We also supply our own Matlab script for use, and as one example of an implementation of the algorithm. Instructions for using the Matlab script are provided as Support Protocol #1, and data analysis using Support Protocol #1 can be performed without reference to this basic protocol.
The steps listed here describe the calculation of several different readout measurements and FRET indices. Some of these readouts are only relevant to certain biosensor designs and experimental designs. Each readout is listed and explained in Figure 7. Calculation of irrelevant readouts may be omitted from the algorithm implementation, unless they are a prerequisite to calculating another readout.
-
Materials list
A desktop computer running any software capable of batch processing of images, such as Matlab, and a spreadsheet program.
-
Steps and annotations
-
Since images are large files, and these experiments require hundreds of images, each image is loaded into memory only as needed. Therefore, in order to find the appropriate image when needed, the image path needs to be associated with information characterizing the image. As described in basic protocol #2, each image pathname contains certain pieces of information describing the image. Therefore, the first step in analysis is to load the image pathnames into memory, extract this data from the pathnames, and record the data in a database table or a similar data structure (the “Image Data Table”), where each image is represented as a record in the data table, and each piece of data an attribute. Depending on the software used, this may be done by splitting the filename or pathname into multiple substrings using one or more delimiting characters, or by using a regular expression to identify substrings, and then assigning those substrings to variables. The Image Data Table should contain at least the following attributes:
path: The path for accessing the original image file.
filename: The filename of the original image file.
experiment: The name or ID number of the experiment.
plate: The name or ID number of the plate.
row: The row letter of the well within the plate.
column: The column number of the well within the plate.
channel: The spectral channel in which the image was acquired.
field: A number representing the field of view within the well.
-
Data associated with each well must also be loaded into memory. This data will be loaded into the Well Data Table, where each record represents a single well. One way to do this is to create a spreadsheet file that contains the data. The spreadsheet file would have multiple worksheets, each labeled with the name of an attribute. Within each attribute’s worksheet, the values for that attribute are laid out within the cells as they are spatially arranged within the analyzed microplates (as in Figure 3). Exactly how the microplates are arranged within the spreadsheet space is unimportant. The important thing is that a given cell represents the same well within each attribute’s worksheet. Blank cells can be ignored. The analysis software should then be programmed to read the spreadsheet file (this can be done in Matlab for Excel files using the “xlsread” function), and extract the value for each attribute of each well and assign the values to the appropriate well records within the Well Data Table. The critical attributes that should be loaded into the Well Data Table are the following:
experiment: The name or ID number of the experiment.
plate: The name or ID number of the plate.
row: The row letter of the well within the plate.
column: The column number of the well within the plate.
channel: The spectral channel in which the image was acquired.
field: A number representing the field of view within the well.
replicate: A unique number for each replicate well of a given sample.
biosensor: The name of the biosensor expressed in each well. May be a functional biosensor, a control biosensor, donor only, or acceptor only, or none, in the case of background images.
biosensor_amount: This is the mass of biosensor DNA transfected.
regulator: The name of the regulatory protein expressed in each well. This can be set to “none” for background, donor only, acceptor only, and biosensor only wells.
regulator_amount: This is the mass of regulator DNA transfected.
-
Calculate sum of pixel intensities for all acquired images.
For each record in the Image Data Table, first load the image into memory using the path attribute of the record.
Next, sum all pixels in the image to produce a scalar value.
Store the sum intensity value as the attribute sumIntensity.
Delete the image from memory and proceed to the next record.
-
The records of the Image Data Table are then grouped into a new ChannelWell Data Table, where each record in the ChannelWell Data Table comprises records from the Image Data Table with common values for the experiment, plate, row, column, and channel attributes, i.e., all fields imaged for a given well and channel are grouped. The multiple grouped values for the sumIntensity attribute of the Image Data Table are summed to produce a sumIntensity attribute for each record of the ChannelWell Data Table.
-
ChannelWell(i).sumIntensity = sum(Image(j).sumIntensity), where:
Image(j).channel = ChannelWell(i).channel
Image(j).experiment = ChannelWell(i).experiment
Image(j).plate = ChannelWell(i).plate
Image(j).row = ChannelWell(i).row
Image(j).column = ChannelWell(i).column
-
-
The sumIntensity values from the ChannelWell Data Table are then assigned to records within the Well Data Table based on common values of the attributes experiment, plate, row, and column. Within the appropriate record, each value is assigned to an attribute that is named after the channel, suffixed by Channel_sumIntensity, to create the donorChannel_sumIntensity, FRETChannel_sumIntensity, and acceptorChannel_sumIntensity (generalized here as the Channel_sumIntensity attributes).
-
Well(i).donorChannel_sumIntensity = ChannelWell(j).sumIntensity, where:
Well(i).experiment = ChannelWell(j).experiment
Well(i).plate = ChannelWell(j).plate
Well(i).row = ChannelWell(j).row
Well(i).column = ChannelWell(j).column
ChannelWell(j).channel = “donor”
-
Well(i).FRETChannel_sumIntensity = ChannelWell(j).sumIntensity, where:
Well(i).experiment = ChannelWell(j).experiment
Well(i).plate = ChannelWell(j).plate
Well(i).row = ChannelWell(j).row
Well(i).column = ChannelWell(j).column
ChannelWell(j).channel = “FRET”
-
Well(i).acceptorChannel_sumIntensity =
ChannelWell(j).sumIntensity, where
Well(i).experiment = ChannelWell(j).experiment
Well(i).plate = ChannelWell(j).plate
Well(i).row = ChannelWell(j).row
Well(i).column = ChannelWell(j).column
ChannelWell(j).channel = “acceptor”
-
-
Group Well Data Table records by common values for the experiment, biosensor, regulator, and regulator_amount attributes. Find the median of the multiple grouped values for each of the three Channel_sumIntensity attributes of the Well Data Table to produce three corresponding Channel_sumIntensity_median attributes for each record of the Sample Data Table.
-
Sample(i).donorChannel_sumIntensity_median =
median(Well(j).donorChannel_sumIntensity), where
Well(j).experiment = Sample(i).experiment
Well(j).biosensor = Sample(i).biosensor
Well(j).regulator = Sample(i).regulator
Well(j).regulator_amount = Sample(i).regulator_amount
-
-
Set background values for each channel equal to the median sum intensity values of each channel in the background wells.
Create the Experiment Data Table (this data table will only have a single record unless multiple experiments are being simultaneously analyzed).
-
Experiment(i).donorChannel_background_median=
Sample(j).donorChannel_sumIntensity_median, where
Experiment(i).experiment = Sample(j).experiment
Sample(j).biosensor = “none”
Sample(j).regulator = “none”
-
Experiment(i).FRETChannel_background_median =
Sample(j).FRETChannel_sumIntensity_median, where
Experiment(i).experiment = Sample(j).experiment
Sample(j).biosensor = “none”
Sample(j).regulator = “none”
-
Experiment(i).acceptorChannel_background_median =
Sample(j).acceptorChannel_sumIntensity_median, where
Experiment(i).experiment = Sample(j).experiment
Sample(j).biosensor = “none”
Sample(j).regulator = “none”
-
Background subtract all Channel_sumIntensity values in the Well Data Table.
-
Well(i).donorChannel_sumIntensity_bs =
Well(i).donorChannel_sumIntensity –
Experiment(j).donorChannel_background_median, where
Well(i).experiment = Experiment(j).experiment
-
Well(i).FRETChannel_sumIntensity_bs =
Well(i).FRETChannel_sumIntensity –
Experiment(j).FRETChannel_background_median, where
Well(i).experiment = Experiment(j).experiment
-
Well(i).acceptorChannel_sumIntensity_bs =
Well(i).acceptorChannel_sumIntensity –
Experiment(j).acceptorChannel_background_median, where
Well(i).experiment = Experiment(j).experiment
-
-
Calculate FRET:Donor ratios for each well, and then calculate the medians for all wells in each sample.
-
Well(i). FRET_donor_ratio = Well(i). FRETChannel_sumIntensity_bs/
Well(i). donorChannel_sumIntensity_bs
-
Sample(i). FRET_donor_ratio_median = median (Well(j).FRET_donor_ratio), where
Sample(i).experiment = Well(j).experiment
Sample(i).biosensor = Well(j).biosensor
Sample(i).regulator = Well(j).regulator
Sample(i).regulator_amount = Well(j).regulator_amount
-
-
Calculate FRET:Acceptor ratios for each well, and then calculate the medians for all wells in each sample.
-
Well(i). FRET_acceptor_ratio = Well(i).
FRETChannel_sumIntensity_bs/
Well(i).acceptorChannel_sumIntensity_bs
-
Sample(i). FRET_acceptor_ration_median = median (Well(j).
FRET_acceptor_ratio), where
Sample(i).experiment = Well(j).experiment
Sample(i).biosensor = Well(j).biosensor
Sample(i).regulator = Well(j).regulator
Sample(i).regulator_amount = Well(j).regulator_amount
-
-
Calculate Donor:Acceptor ratios for each well.
-
Well(i). donor_acceptor_ratio =
Well(i).donorChannel_sumIntensity_bs/
Well(i).acceptorChannel_sumIntensity_bs
-
-
Determine donor-to-FRET and acceptor-to-FRET bleedthrough coefficients for each channel.
-
Experiment(i). donor2FRET_bleedthrough_coefficient =
Sample(j).FRET_donor_ratio_median, where
Sample(j).experiment = Experiment(i).experiment,
Sample(j).biosensor = “donor-only”,
Sample(j).regulator = “none”
-
Experiment(i). acceptor2FRET_bleedthrough_coefficient =
Sample(j).FRET_acceptor_ratio_median, where
Sample(j).experiment = Experiment(i).experiment,
Sample(j).biosensor = “acceptor-only”,
Sample(j).regulator = “none”
-
-
Calculate bleedthrough into the FRET channel in all wells
-
Well(i).acceptor_bleedthrough =
Well(i).acceptorChannel_sumIntensity_bs *
Experiment(j).acceptor2FRET_bleedthrough_coefficient, where
Experiment(j).experiment = Well(i).experiment.
-
Well(i).donor_bleedthrough =
Well(i).donorChannel_sumIntensity_bs *
Experiment(j).donor2FRET_bleedthrough_coefficient, where
Experiment(j).experiment = Well(i).experiment.
-
-
Perform bleedthrough correction on the FRET channel in all wells
-
Well(i).FRETChannel_sumIntensity_bs_btca =
Well(i).FRETChannel_sumIntensity_bs -
Well(i).acceptor_bleed through
-
Well(i).FRETChannel_sumIntensity_bs_btcd =
Well(i).FRETChannel_sumIntensity_bs - Well(i).donor_bleedthrough
-
Well(i).FRETChannel_sumIntensity_bs_btcad =
Well(i).FRETChannel_sumIntensity_bs_btca -
Well(i).donor_bleedthrough
-
-
Calculate bleedthrough-corrected FRET:donor and FRET:acceptor ratios for all wells
-
Well(i).FRET_donor_ratio_btca =
Well(i).FRETChannel_sumIntensity_bs_btca /
Well(i).donorChannel_sumIntensity_bs
-
Well(i).FRET_donor_ratio_btcad =
Well(i).FRETChannel_sumIntensity_bs_btcad /
Well(i).donorChannel_sumIntensity_bs
-
Well(i).FRET_acceptor_ratio_btcd =
Well(i).FRETChannel_sumIntensity_bs_btcd /
Well(i).acceptorChannel_sumIntensity_bs
-
Well(i).FRET_acceptor_ratio_btcad =
Well(i).FRETChannel_sumIntensity_bs_btcad /
Well(i).acceptorChannel_sumIntensity_bs
-
-
Normalize the FRET:donor and bleedthrough-corrected FRET:donor ratios to the donor bleedthrough coefficient, and normalize the FRET:acceptor and bleedthrough-corrected FRET:acceptor ratios to those of the acceptor bleedthrough coefficient.
-
Well(i).FRET_donor_ratio_norm2dbtc = Well(i).FRET_donor_ratio /
Experiment(j).donor2FRET_bleedthrough_coefficient, where
Well(i).experiment = Experiment(j).experiment
-
Well(i).FRET_donor_ratio_btca_norm2dbtc =
Well(i).FRET_donor_ratio_btca /
Experiment(j).donor2FRET_bleedthrough_coefficient, where
Well(i).experiment = Experiment(j).experiment
-
Well(i).FRET_donor_ratio_btcad_norm2dbtc =
Well(i).FRET_donor_ratio_btcad /
Experiment(j).donor2FRET_bleedthrough_coefficient, where
Well(i).experiment = Experiment(j).experiment
-
Well(i).FRET_acceptor_ratio_norm2abtc =
Well(i).FRET_acceptor_ratio /
Experiment(j).acceptor2FRET_bleedthrough_coefficient, where
Well(i).experiment = Experiment(j).experiment
-
Well(i).FRET_acceptor_ratio_btcd_norm2abtc =
Well(i).FRET_acceptor_ratio_btcd /
Experiment(j).acceptor2FRET_bleedthrough_coefficient, where
Well(i).experiment = Experiment(j).experiment
-
Well(i).FRET_acceptor_ratio_btcad_norm2abtc =
Well(i).FRET_acceptor_ratio_btcad /
Experiment(j).acceptor2FRET_bleedthrough_coefficient, where
Well(i).experiment = Experiment(j).experiment
-
-
Average all Well Data Table attributes Well(i).attribute over all replicates of each sample, and assign each mean to an attribute of the Sample Data Table, Sample(j).attribute_mean. Use a similar approach to calculate any additional desired statistics across all wells of a sample record.
-
Sample(j).attribute_mean = mean(Well(i).attribute), where
Well(i).experiment = Sample(j).experiment
Well(i).biosensor = Sample(j).biosensor
Well(i).regulator = Sample(j).regulator
Well(i).regulator_amount = Sample(j).regulator_amount
-
-
-
Figure 7.

Ratiometric FRET indices. A = acceptor bleedthrough correction, acceptor bleedthrough coefficient, acceptor-only sample, or acceptor channel, depending on the column heading. D = donor bleedthrough correction, donor bleedthrough coefficient, donor-only sample, or donor channel, depending on the column heading. F = FRET channel. Inter = intermolecular biosensor design. Intra = intramolecular biosensor design. An equal sign means “equivalent”. M = monotonic. L = linear. P = proportional. IL = inversely linear. NA = not applicable.
Support Protocol #1
Title: Matlab scripts for analysis of biosensor response in microplate image data
-
Introduction
This section provides specific instructions regarding how to use the Matlab functions provided with this unit for analyzing biosensor image data in a 96-well microplate format. Three files are supplied. The first is the Matlab function Biosensor4b.m. This function processes images, performs calculations of FRET indexes per well, and calculates statistics over well replicates. The results are saved as tables in Excel and as Matlab .mat files. The second file is an Excel file called map.xlsx. This serves as a template for creating a wellmap for each experiment that will provide input data to the Biosensor4b function. The third file is the Matlab function chartfromtable.m. This function makes it easy to visualize the data produced by the Biosensor4b function as scatterplots. Use of these tools requires only a minimal knowledge of how to run and operate Matlab.
-
Materials list
A personal computer equipped with Matlab software. We have tested the functions using Matlab R2013a (8.1.0.604). The functions may also work on earlier or later versions.
-
Steps and annotations
On the analysis computer, create a directory of any name that will hold the experimental images and results.
Make a copy of the Biosensor4b.m, map.xlsx, and chartfromtable.m files and paste them into the new directory
Transfer the images for the experiment into the same directory. The images may be immediately within this directory, or may be within subfolders in the directory. The Biosensor4b.m function will search recursively through the hierarchy of directories within the active Matlab directory for image files that match a regular expression string. Also, additional files irrelevant to the analysis will be ignored, so may be included within the directory without causing problems.
Open the Matlab software
Within the Current Folder panel, navigate to the directory created above.
Open the map.xlsx file from the Current Folder panel by right clicking the filename and selecting ‘Open Outside Matlab’.
-
In the workbook that opens in Excel, you will find nine worksheets. The name of each worksheet, shown on the tab at the bottom, represents the information that will be input by the user on that worksheet for each well imaged in the experiment. By clicking on each tab, you bring up a worksheet that contains an outline of a 96-well microplate, with values within each well for the variable represented by the tab. The values are prefilled, but should be changed if necessary to match the particular experiment. Note that the values for experiment, plate, row, and column are used to match image files to wells, and therefore the values entered for each well must exactly match portions of the associated image file paths. Values for biosensor, biosensor_amount, regulator, and regulator_amount, are additional values supplied by the user, that are not found in the image file paths, but which are required for proper functioning of the Biosensor4b.m function. Finally, the replicate value can be entered by the user to aid in analysis, but is not required and may be omitted. Any wells that were not imaged should be left blank within each workbook of the wellmap file. Blank wells are ignored. Otherwise, the Biosensor4b function will attempt to pair the well with image files and will encounter an error. The variables entered in the wellmap are described below:
Experiment: a character string that names the experiment. The experiment string will be the same for all wells analyzed at the same time. The same character string must be found in the image file paths.
Plate: an identification number for each plate that will be analyzed. The number will be the same for all wells of the plate. The same plate identification number must be found in the associated image file paths.
Row: a letter representing the row for each well. The same letter must be found in the associated image file paths.
Column: a number representing the column for each well. The same number must be found in the associated image file paths.
Biosensor: a string that represents the name of the biosensor, or biosensor control, expressed in each well. For example, “Rac1 FLARE.sc(Cer3/YPet)”, and “donor-only”, are both valid biosensor names. If no biosensor is expressed, as in mock-transfected samples, enter “none”. The string “none” will be recognized by the analysis software to indicate that the sample is to be treated as a mock transfection.
Biosensor_amount: a number representing the amount of biosensor transfected. The units are the choice of the user.
Regulator: a string that represents the name of the regulator expressed in each well, e.g., “GEF”. If no regulator is expressed, as in mock-transfected samples, enter “none”.
Regulator_amount: a number representing the amount of regulator transfected. The units are the choice of the user.
Replicate: a number identifying each replicate of otherwise identical wells. For example, if there are eight mock-transfected wells, then here they can be labeled as 1–8.
If more than one plate will be analyzed in one experiment, simply copy the microplate outline within each worksheet and paste it anywhere else in the worksheet. Where it is pasted does not matter, but it is critical that the second plate occupies the same cells in each worksheet. Update the values in the second microplate as necessary. Repeat for each additional microplate.
Save and close the map.xlsx file.
-
Open the Biosensor4b.m file by double clicking its filename in the Current Folder panel of the Matlab window. The code for the file will open in an editor window. At the top of the progam code is a section clearly delineated by rows of % symbols and labeled “User input here: ”. This section assigns values to input variables that may be changed by the user. Do not make changes to any other section, unless you intend to alter the operation of the function. Make changes to each input value as necessary. Following is a description of each variable value:
-
Experiment.imageFilenameOrPath:
Description: a string with the value ‘filename’ if the regular expression described below matches the files’ filename, or ‘path’ if it matches the files’ entire path.
-
Experiment.imageFileRegularExpression:
-
Description: a regular expression that will match your image files, including named tokens for each of the following elements: experiment, plate, row, column, field, channel. A description of regular expressions can be found within the Matlab help documentation. In brief, to obtain a regular expression with named tokens, first copy an actual image filename or path. For each of the variables that will be extracted from the image filename/path (experiment, plate, row, column, channel, field (optional)), replace the characters representing that variable’s value with the following construct: (?<variableName>).
Next, after the ‘>’ insert a general representation of all possible values that could exist for that variable. Some useful representations are:
\d = a single digit
\d{2} = two successive digits
\d+ = one or more successive digits
[A-P] = a single capital letter ranging from A to P
-
\w+ = one or more characters including any alphabetic, numeric, or underscore character
Next, replace those literal characters that are outside of the named tokens if the character has a non-literal interpretation. For example, the period has a non-literal interpretation in regular expressions, so you must replace ‘.’ with ‘\.’ to represent a literal period. Finally, enclose the whole string in single quote marks.
-
Example:
-
To match the following filename string:
“140128_plate1_A01_s1_w1.TIF”
-
use the following regular expression:
‘(?<experiment>\w+)_plate(?<plate>\d+)_(?<row>[A-P])(?<column>\d{2})_s(?<field>\d+)_w(?<channel>\d+)\ .TIF’
-
This will extract the following values:
experiment = ‘140128’
plate = ‘1’
row = ‘A’
column = ‘01’
field = ‘1’
channel = ‘1’
-
-
-
Experiment.imageAttributeValueSwitching
Description: If you desire to change the values of the data extracted from the filenames, you can do that using this cell array. Leave the cell array as an empty array if you do not wish to change any values. For each attribute to be changed, create a row in the cell array. The first cell in each row should be a string that equals the attribute name to be changed. The following cell values of the row alternate between initial values and new values.
-
Example:
-
To change channel attribute values from
‘1’ to ‘YFP’,
‘2’ to ‘FRET’, and
‘3’ to ‘CFP’,
-
and to change experiment attribute values from
‘140220’ to ‘ImportantExperiment’
-
input the following cell array
{‘channel’ ‘1’ ‘YFP’ ‘2’ ‘FRET’ ‘3’ ‘CFP’; ‘experiment’ ‘140220’ ‘ImportantExperiment’}
-
-
Experiment.wellMapPath
Description: input a string containing the relative or absolute path of the wellmap Excel file. If the map is in the currently active Matlab directory, then only a filename is required.
Example: ‘map.xlsx’
-
Experiment.donorChannel
Description: input the channel name for the donor channel as it appears in the image paths (or as modified by the Experiment.imageAttributeValueSwitching input). Input ‘none’ if the channel image was not acquired.
Example: ‘CFP’
-
Experiment.acceptorChannel
Description: input the channel name for the acceptor channel, if it exists, as it appears in the image paths (or as modified by the Experiment.imageAttributeValueSwitching input). Input ‘none’ if the channel image was not acquired.
Example: ‘YFP’
-
Experiment.FRETChannel
Description: input the channel name for the FRET channel as it appears in the image paths (or as modified by the Experiment.imageAttributeValueSwitching input). Input ‘none’ if the channel image was not acquired.
Example: ‘FRET’
-
Experiment.donorOnlyBiosensor
Description: input the biosensor value assigned to the donor only wells, if they exist, in the wellplate map excel file. Enter ‘none’ if it does not exist. The software will recognize the string ‘none’ to indicate that donor bleedthrough correction and normalization to the donor bleedthrough coefficient should not be performed.
Example: ‘donor’
-
Experiment.acceptorOnlyBiosensor =
Description: input the biosensor value assigned to the acceptor only wells, if they exist, in the wellplate map excel file. Enter ‘none’ if it does not exist. The software will recognize the string ‘none’ to indicate that acceptor bleedthrough correction and normalization to the acceptor bleedthrough coefficient should not be performed.
Example: ‘acceptor’
-
Experiment.bitdepth
Description: input the bitdepth of the camera used to acquire the images. This is used to identify saturated pixels.
Example: 12
-
Experiment.groupImagesIntoChannelWellsBy
Description: a cell array containing the names of Image attributes that must have common values to group multiple image records into a single ChannelWell record. The default value, shown in the example, should be sufficient for most experiments.
Example: {‘experiment’, ‘plate’, ‘row’, ‘column’, ‘channel’}
-
Experiment.groupWellsIntoSamplesBy
Description: a cell array containing the names of Well attributes that must have common values to group multiple Well records into a single Sample record. The default value, shown in the example, should be sufficient for most experiments.
Example: {‘experiment’, ‘biosensor’, ‘biosensor_amount’, ‘regulator’, ‘regulator_amount’}
-
When all inputs have been updated as necessary, save the Biosensor4b.m file.
In the Matlab command window, type Biosensor4b and press Enter to run the function. The Matlab Command Window will display user input values for verification by the user. To continue, press any key. When the function has completed, the message ‘Complete!’ will be displayed in the Command Window. If all inputs have been correctly entered into the map.xlsx file and the Biosensor4b.m file input section, then the function should run with no errors in less than one minute per plate. Actual processing time will depend on the number of wells and channels acquired, the image file size, and the capabilities of the analysis computer. Processing speed may be adversely affected by additional open applications or processes running in the background. See the troubleshooting section for dealing with errors encountered when the analysis is run.
As output, Biosensor4b creates an analysis folder, called ‘Analysis#’ within the current directory, where # is incremented so that each run of the function creates a unique analysis folder. Within the analysis folder, Biosensor4b saves Experiment, Sample, and Well data tables as Excel files called ExperimentData.xls, SampleData.xls, and WellData.xls, respectively. It also saves each data table in a Matlab “structure” variable within the Analysis_variable.mat file.
To examine the results in scatterplots using the chartfromtable.m function, first, open the newest analysis folder, and then double click Analysis_variable.mat to load the contained variables into the Matlab workspace.
An example will be used to illustrate the use of the chartfromtable function. In the example, mean FRET:Donor ratios of all wells in each sample will be plotted as a function of regulator amount, with vertical error bars showing standard deviations across the wells. Each unique biosensor/regulator combination will be displayed as a data series with a unique color.
In the Command Window, type chartfromtable(Sample). For other plots, Experiment, Well, ChannelWell, or Image datatables could be plotted in place of Sample.
A dialog box appears, asking what type of chart to produce. Choose ‘scatterplot’ from the popup list.
Select the “Series” radio button to reveal a listbox of Sample attributes that may be chosen to group records into color-coded series. From the series listbox, select “biosensor” and “regulator” (hold down the control button to make multiple selections).
Select the “X axis” radio button to reveal a listbox of numerical Sample attributes that may be chosen as the x axis attribute. From the x axis listbox, select “regulator_amount“.
Select the “Y axis” radio button to reveal a listbox of numerical Sample attributes that may be chosen as the y axis attribute. From the y axis listbox, select “FRET_donor_ratio_Well_mean”.
Select the “Error bars” radio button to make a listbox of numerical Sample attributes visible that may be chosen as the error bar attribute then select “FRET_donor_ratio_Well_stdDev”.
After series, x axis, and y axis attributes have been selected, the inactive Graph pushbutton becomes activated. Click the Graph pushbutton to create the scatterplot in a separate figure window. A possible result is shown in Figure 8. Properties of the chart may be modified, and the graph may be saved, using the features available in Matlab and described in the Matlab help documentation.
Figure 8.

Biosensor response to regulator expression. The Rac1 FLARE.dc biosensor or a donor-only control biosensor were transfected into LinXE cells along with varying amounts of active Vav2 GEF or RhoGDI1 regulators. For each well, the sensitized emission:donor emission ratio was calculated and normalized by dividing by the donor bleedthrough coefficient. Plotted are mean ratio values for two replicate wells of each transfection condition with error bars representing standard deviations, as a function of regulator plasmid mass transfected.
Commentary
Background Information
Förster resonance energy transfer (FRET) is the non-radiative transfer of energy from a donor fluorescent molecule to an acceptor (Lakowicz, 1999). The FRET efficiency, E, is the percentage of donor energy transferred. FRET efficiency is a function of the the interfluorophore distance, r, according to the equation,
where, R0 is the Förster distance. R0 is a function of the donor fluorophore’s quantum yield and unit emission spectrum, the acceptor fluorophore’s molar extinction coefficient as a function of wavelength, the refractive index of the medium, and the relative orientations of the donor emission transition dipole and acceptor absorption transition dipole. Thus, for a given FRET pair in a given medium, E depends on interfluorophore distance and inter-dipole relative orientation. For a given orientation (which is often assumed to average to a constant), when r = R0, then E = 0.5, and E is most sensitive to changes in r. For fluorescent protein pairs commonly used in FRET-based biosensors, R0 is typically in the range of 4–6 nm, which makes the FRET efficiency very sensitive to r in the distance range of interacting proteins.
By fusing donor and acceptor fluorescent proteins to biologically active molecules, FRET can be harnessed to produce reporters of protein-protein interaction or protein activity. These FRET-based biosensors can take two general designs, intramolecular or intermolecular. In the intramolecular design, the entire biosensor, including both fluorescent proteins, is encoded as a single polypeptide chain. Changes in status of the biologically active components affect their conformation, which alters the interfluorophore distance or angle, resulting in changes in FRET efficiency (Miyawaki and Tsien, 2000; Itoh et al., 2002; Pertz et al, 2006). In the intermolecular design, the biosensor is expressed as two separate polypeptides, with each containing either the donor or the acceptor fused to a biologically active component. FRET is negligible when the two halves are not interacting, and it increases when interaction between the biological components brings the donor and acceptor into proximity (Miyawaki et al, 2000; Kraynov et al, 2000; Machacek et al, 2009).
There are several methods for directly measuring the apparent FRET efficiency of a sample. However, it is not necessary to directly measure FRET efficiency to assess relative degrees of donor-acceptor interaction. It can be simpler to instead compute one of various FRET indices from measurements of donor emission after direct donor excitation (referred to here as the donor channel emission), acceptor emission after direct acceptor excitation (acceptor channel emission), and acceptor emission after direct donor excitation (FRET channel emission). FRET channel emission arises from three sources (after background subtraction): 1) donor emission, here called donor bleedthrough, 2) acceptor emission due to direct acceptor excitation, here called acceptor bleedthrough, and 3) acceptor emission that arises from energy transferred from the donor by FRET, which is called sensitized emission. When the number of molecules involved is fixed, increases in FRET efficiency result in decreased donor channel emission and increased sensitized emission. Acceptor emission is unaffected. If the number of biosensor molecules in each sample is known or fixed, then the donor channel or sensitized emission can act as a FRET index, but in most experimental designs, the number of biosensor molecules cannot be controlled. Therefore, it is necessary to normalize these measurements by taking a ratio, using either the donor emission or the acceptor emission as the denominator. Many ratios can be employed as a FRET index, and some possibilities are described in Figure 7. Which index is best depends on many factors. Some indices are only applicable to certain biosensor designs. Some are linearly related to donor-acceptor interaction status, some only monotonically. The error present in an index will depend upon the signal-to-noise ratios of the contributing channels and how the noise is propagated through the calculation of the index.
To assess a biosensor’s function, a FRET index must be measured when the biosensor is in the high FRET and low FRET states. Sometimes, mutant versions of the biosensor may mimic these states, but it is often the case that they do not perfectly replicate the on and off states of the WT biosensor. Obtaining a population of wild-type biosensor that is fully in the on or off state can usually be accomplished by saturating it with a positive or negative regulator. Different biosensor/regulator combinations will require different levels of expression to reach saturation, depending upon their mode of action. For example, GEF expression induces a saturated level of Rho family signaling activity at an expression level approximately 100-fold lower than the level of RhoGDI-1 necessary to induce saturated inactivity. Presumably, this is because GEFs act catalytically, while RhoGDI-1 acts through stoichiometric binding. Some regulators can also cause cytotoxicity or unexpected regulatory effects when expressed at high levels. This may cause poor quality measurements of FRET index, or a loss of the saturating effect at high levels. Testing a wide range of regulator expression levels and construction of a dose-response curve allows an investigator to identify the point of saturation. Construction of these curves is greatly simplified by the assay described here.
A biosensor’s expression level can also affect its function. At very low expression levels, biosensor brightness may not be sufficient to achieve a good signal-to-noise ratio. However, at high expression levels, the biosensor may lose sensitivity for any of several reasons. It could aggregate, misfold, or become mislocalized, leading to aberrantly high or low FRET and/or inaccessibility to regulators. It could also overwhelm endogenous regulators, such that it is no longer reporting the activity level that would exist in its absence. Testing a range of biosensor expression levels using fixed amounts of positive or negative regulators, or no regulators, will reveal the expression level that optimizes sensitivity.
Critical Parameters and Troubleshooting
Since values determined in certain well sets are applied to all wells across the plate, all wells must receive identical treatment, aside from the intentionally varied biosensor and regulator. This is aided by careful, accurate pipetting practices and mindful planning of each pipetting step. The best results will be obtained in these experiments if, at the time of imaging, cells are evenly dispersed, nearly confluent, have a high proportion of transfected cells, and exhibit reasonably low heterogeneity of expression level. These conditions will produce the least variability in measured FRET indices for replicate wells. Again, careful pipetting will be helpful here. Adjusting the number of cells seeded may be necessary to achieve the desired confluency without too much crowding. Overcrowding and piling up of cells can lead to unfocused cells in the image. If there is a high degree of variability across the population, imaging more fields of view may help to average out the contributions of extreme cells.
Good signal-to-noise ratio in these experiments is aided by a higher level of cellular confluence, a higher proportion of transfected cells, and lower background fluorescence. Background can make up 50–90% of the sum of pixel intensities of an image, so anything that decreases signal or increases background may erode the signal-to-noise ratio. Using HBSS as an imaging buffer has been found to produce less background than growth medium. Phenol red, serum, and certain vitamins can contribute to background fluorescence.
When setting up the microscope for imaging, it is advisable to avoid saturation of pixels. Saturated pixels under-report their true value and may lead to inaccurate values for FRET index measurements. It is a good practice to set exposure times so that the brightest pixels are about half the maximal possible value for the camera. This will help ensure that even brighter pixels will not saturate the detector. Sometimes, biosensors may aggregate or accumulate in bright spots. This can force the investigator to set very short exposure times, which produce otherwise dim images with low signal-to-noise ratios. In some cases, lowering the biosensor expression level can reduce aggregation and allow longer exposure times. Performing a biosensor titration is useful for recognizing this situation. Another problem that may be found at the time of imaging is that different wells have very large differences in expression level. For example, if an unfused fluorescent protein is used as the donor-only or acceptor-only bleedthrough control biosensor, it may express much more highly than the true biosensor. In this case, the investigator would have to set a short exposure time to avoid saturation of the bleedthrough control wells. If the results are unsatisfactory, then either the transfection can be repeated with less of the brighter expression construct, or a different bleedthrough control can be designed that contains those components of the biosensor that limit expression. Poor image focus can also affect the measurements. It is advisable to watch the images as they are acquired through at least one plate to ensure that the autofocus is not failing frequently.
Determining accurate values for background and bleedthrough coefficients is critical to accurate calculation of FRET indices. Background values and bleedthrough coefficents are set to the median values of control wells. As opposed to mean values, the median should be robust to outlier values. Nevertheless, it is a good practice check a plot of the sum of pixel intensities of each channel versus replicate for the mock-transfected background wells, to verify that the values are consistent from well to well (Figure 9). Likewise, one should plot the FRET:donor ratio for donor-only control wells (Figure 10), or the FRET:acceptor ratio for acceptor-only control wells, versus replicate to detect large variances that could lead to errors in calculated bleedthrough coefficients. The user can also get a feeling for the signal:noise ratio of experimental samples in each emission channel by plotting the background-subtracted emission intensity of those wells and comparing their values to the standard deviation of the background-subtracted emission intensities of the mock-transfected wells.
Figure 9.

Checking for consistency in the background measurements. Mock transfected wells, which are used to determine background values for each channel, were imaged, and the sum of pixel intensities were computed in all channels. The resulting values for the Donor channel are shown plotted against the well replicate number. One well appears to have an outlying value, but this will not affect the median value, which will be calculated and used for background subtraction of other wells.
Figure 10.
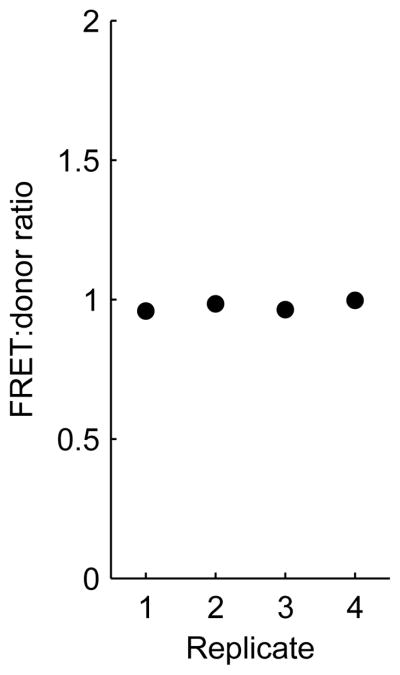
Checking for consistency in bleedthrough coefficient measurements. Donor-only transfected wells, which are used to calculate donor bleedthrough coefficients, were imaged, and the FRET:donor ratio (equivalent to the donor bleedthrough coefficient in the case of donor-only control wells) was plotted versus replicate number. There is very little variance across the replicates.
We have optimized the transfection of cells by adjusting the amount of Lipofectamine, the order of reagent addition, and by adding Plus Reagent to the reaction (Life Technologies -- an additive that increases Lipofectamine transfection efficiency in some cell lines) (Figure 11). We found that using Lipofectamine at a 4:1 ratio (μL Lipofectamine: μg DNA) produced much better transfection efficiencies than a 2:1 or 1:1 ratio. Addition of Lipofectamine to DNA solutions prior to titration, rather than after titration, did not negatively affect the transfection efficiency, but had a large negative effect on biosensor response to regulators. One explanation is that the biosensor and regulator are not efficiently co-transfected into the same cells when the titration is performed after Lipofectamine:DNA complex formation. Plus Reagent at 6:1 or 1:1 ratios (μL Plus: μg DNA) did not provide any substantial benefit, so we leave out this costly reagent.
Figure 11.

A Rac1 biosensor was transfected into LinXE cells with a gradient of Vav2 GEF, using different transfection conditions as listed in the legend. The amount of Lipofectamine, the amount of Plus Reagent, and the order of addition (Lipofectamine added either before or after DNA titration) were varied. Mean values of a FRET index (error bars are standard deviations of four replicate wells) were plotted against transfected Vav2 plasmid mass.
Calculated FRET indices for wells could be affected by such factors as position on the wellplate or time on the microscope (particularly if the plate is imaged without a controlled temperature or atmosphere). Replicating the same biosensor transfection across all wells of a plate could enable the detection or dismissal of such complicating factors.
The Biosensor4b function can output error messages that should guide the user in troubleshooting. The most common cause of an error in running the Biosensor4b function is a mistake in filling out the wellmap Excel file. If an error is encountered, be sure to examine the wellmap thoroughly for empty cells that should be filled, filled cells that should be empty, or mismatches between strings entered in the wellmap and the strings extracted from the filenames. Another common problem is mismatches between the user-input parameter values at the beginning of the Biosensor4b code and values in the image filenames or in the wellmap. Carefully examine the input values to find mistakes in entry. In our hands, on certain machines or versions of Matlab, the final step in the function, saving data to a Microsoft Excel file, inexplicably fails and produces an error. The code is arranged such that the data is saved as a Matlab .mat file before attempting to save as an Excel file, Analysis_variables.mat, so that even if this error is encountered, the analysis is still completed and the data may still be examined via the chartfromtable function.
Anticipated Results
Cell density and transfection efficiency should be high, such that the acquired images resemble those in Figure 12. A functional biosensor should exhibit a dose-dependent change in each FRET index in response to a positive regulator, and the opposite dose-dependent change in response to a negative regulator (Figure 8). The magnitude and reproducibility of the response may be different for different FRET indexes, depending on the fluorescent proteins used and the design of the biosensor. If the response is specific, then control biosensors, such as a donor-only (Figure 8) or acceptor-only control, or a fused donor-acceptor control, should not exhibit a dose-dependent change in the FRET index in response to the regulator.
Figure 12.

A typical image of Rac1 FLARE.dc biosensor expressed in LinXE cells (contrast enhanced).
Time Considerations
To prepare and image one 96-well microplate will take four days (or only three if PLL treatment is reduced to one hour rather than overnight). PLL treatment requires about 10 minutes plus incubation time. On the second day, cell seeding requires approximately 30–60 minutes. On the third day, a total of four hours is required, including approximately one hour to prepare and distribute transfection reactions, three hours of incubation, and about 10 minutes to add serum to transfected cells. On the fourth day, allow about 1 hour to pre-warm imaging medium and set up the microscope. Imaging time depends upon the capabilities of the microscope, the number of channels and fields acquired, and the exposure times, but may run from 15–60 minutes per plate. After transfer of data to an analysis computer, the images from one plate can be analyzed by the provided Matlab function in less than 5 minutes. With practice, four to eight plates can easily be prepared, imaged, and analyzed in one four day period. If multiple plates are assayed simultaneously, assume each active step will require proportionally more time.
Acknowledgments
We thank Ellen O’Shaughnessy, Dan Marston, Onur Dagliyan, and Mihai Azoitei for testing the assay and suggesting technical improvements. We are grateful to Ellen O’Shaughnessy for critical reading of the manuscript, and thank the American Heart Association (12POST10950000 to SDS) and NIH for support of this work (GM103723 and GM094663 to KMH).
Literature Cited
- Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nature Reviews: Molecular Cell Biology. 2011;12:493–504. doi: 10.1038/nrm3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz RD, Letzelter M, Reimann A, Mertin K, Fusco L, Ritsma L, Ponsioen B, Fluri E, Schulte-Merker S, van Rheenen J, Pertz O. A versatile toolkit to produce sensitive FRET biosensors to visualize signaling in time and space. Science Signaling. 2013;6:rs12. doi: 10.1126/scisignal.2004135. [DOI] [PubMed] [Google Scholar]
- Itoh RE, Kurokawa K, Ohba Y, Yoshizaki H, Mochizuki N, Matsuda M. Activation of Rac and Cdc42 video imaged by fluorescent resonance energy transfer-based single-molecule probes in the membrane of living cells. Molecular and Cellular Biology. 2002;22:6582–6591. doi: 10.1128/MCB.22.18.6582-6591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. Kluwer Academic/Plenum Publishers; New York, N.Y: 1999. [Google Scholar]
- Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM. Localized Rac Activation dynamics visualized in living cells. Science. 2000;290:333–337. doi: 10.1126/science.290.5490.333. [DOI] [PubMed] [Google Scholar]
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461:99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A, Tsien RY. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. In: Thorner J, Emr S, Abelson J, Simon M, editors. Methods in Enzymology, Vol. 327: Application of Chimeras in Cell Physiology. Academic Press; New York: 2000. pp. 472–500. [DOI] [PubMed] [Google Scholar]
- Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. TRENDS in Cell Biology. 2003;13:13–22. doi: 10.1016/s0962-8924(02)00004-1. [DOI] [PubMed] [Google Scholar]
- Pertz O, Hodgson L, Klemke R, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440:1069–1072. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on Rho GTPases with guanine nucleotide-exchange factors. Nature Reviews: Molecular Cell Biology. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Thompson G, Owen D, Chalk PA, Lowe PN. Delineation of the Cdc42/Rac1-binding domain of p21-activated kinase. Biochemistry. 1998;37:7885–7891. doi: 10.1021/bi980140+. [DOI] [PubMed] [Google Scholar]