

Abstract
MiR-181 has deleterious effects on stroke outcome, and reducing miR-181a levels prior to middle cerebral artery occlusion (MCAO) was shown previously to be protective. Here we tested the effect of post-ischemic treatment with miR-181a antagomir by intracerebroventricular and intravenous routes of administration on infarct size, neurological outcome, inflammatory response and long term behavioral outcome. Post-treatment with miR-181a antagomir significantly reduced infarction size, improved neurological deficits and reduced NF-κB activation, numbers of infiltrating leukocytes and levels of Ibal. Targets affected by miR-181a antagomir administered after stroke onset include BCL2 and X-linked inhibitor of apoptosis protein (XIAP). Post-treatment with miR-181a antagomir significantly improved behavioral outcome assessed by rotarod at one month. These findings indicate that post-treatment with miR-181a antagomir has neuroprotective effects against ischemic neuronal damage and neurological impairment in mice, and the protection is long lasting including recovery of motor function and coordination over one month. The ability to protect brain with post-treatment with miR-181a antagomir with long lasting effect makes this a promising therapeutic target and may be an innovative and effective new approach for stroke therapy.
Keywords: microRNA, miR-181a, stroke, BCL2, inflammation, XIAP, rotarod, focal ischemia
Introduction
Stroke is one of the leading causes of death worldwide and the leading cause of long-term neurological disability. Although many clinical stroke trials have been completed, the only clinically efficacious treatment to date is thrombolysis (Blakeley and Llinas, 2007). Suggested reasons for the many failures include the complex interplay among multiple signaling pathways and intracellular organelles, interaction between different cell types, and the potentially short therapeutic window for neuroprotection after stroke.
MicroRNAs (miRs) are a class of small, non-coding RNAs. Mature miRs are generated from primary miR transcripts by sequential endonucleolytic processing and act as posttranscriptional regulators of gene expression including in the setting of cerebral ischemia (for review see Ouyang et al. (2013)). Many miRs exist in families. The miR-181 family contains four highly conserved members, miR-181a, miR-181b, miR-181c and miR-181d, which are derived independently from 6 precursors located on 3 chromosomes as identified using Target Scan (http://targetscan.org).
Our previous study reported increased injury with increased levels of miR-181 and a protective effect of reducing miR-181a levels using antagomir when administered the day prior to middle cerebral artery occlusion (MCAO) in male mice (Ouyang et al., 2012b). Reduced levels of miR-181 were associated with reduced oxidative stress in in vitro ischemia (Ouyang et al., 2012a). Previously validated targets of miR-181 include the ER stress protein GRP78 (Ouyang et al., 2012b) and three antiapoptotic proteins, BCL2, MCL1 (Ouyang et al., 2012a) and XIAP (Hutchison et al., 2013). While treatment before stroke provides evidence that anti-miR-181 can protect in acute stroke, treatment after stroke onset will be needed in most cases, as patients often present hours after stroke onset. Thus to assess potential translational relevance we tested the effect of post-ischemic treatment with miR-181a antagomir administered by intracerebroventricular (ICV) or intravenous (IV) injection in a transient focal cerebral ischemia model. We assessed both short term and long term outcome with post-treatment, including rotarod neurobehavioral assessment.
Materials and Methods
miRNA-181a antagomir
miRNA-181a antagomir and a negative control (mismatched (MM)-miR-181a antagomir) were from Thermo Scientific (Hudson, New Hampshire, USA) and the sequences are:
Antagomir miR-181a (MAGWA-000005)
mA.*.mC.*.mU.mC.mA.mC.mC.mG.mA.mC.mA.mG.mC.mG.mU.mU.mG.mA.mA.mU.*.mG.*.mU.*.mU.*.3′-Chl
MM - Antagomir miR-181a (MAGWA-00006)
mA.*.mG.*.mU.mC.mA.mG.mC.mG.mA.mG.mA.mG.mC.mC.mU.mU.mG.mA.m
U.mU.*.mG.*.mU.*.mU.*.3′-Chl
mN = 2′-O-Methyl nucleotide (N = A or C or G or U); * = phosphorothioate linkage
Reverse Transcription Quantitative Real-time Polymerase Chain Reaction (RT-qPCR)
RT-qPCR for miRNA quantitation in brain tissue was as reported previously (Ouyang et al., 2012b). All materials were from Applied Biosystems (Foster City, CA). Total RNA was isolated with TRIzol®, then reverse transcription of equal amounts of RNA (200 ng) was performed using the TaqMan MicroRNA Reverse Transcription Kit and 1.3 mM dNTPs (with dTTP), 50 U reverse transcriptase, 10 U RNase inhibitor, and specific miRNA reverse transcriptase primers at 16°C for 30 min, 42°C for 30 min, and 85°C for 5 min. PCR reactions were then conducted using the TaqMan® MicroRNA Assay Kit at 95°C for 10 min, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 min. Each reaction contained 0.75 μl of the RT reaction product, 5 μl TaqMan 2×Universal PCR Master Mix in a total volume of 10 μl using the 7900HT Fast Real-Time PCR System (Life Technologies, South San Francisco, CA, USA). Predesigned primer/probes for miRNAs and mouse U6 were from Applied Biosystems. The expression of miR-181a was normalized using U6 as the internal control. Measurements were normalized to U6 (ΔCt) and comparisons calculated as the inverse log of the ΔΔCT to give the relative fold change for all miRNA levels (Livak and Schmittgen, 2001). Liu et al have validated U6 as not changing in cerebral ischemia (Liu et al., 2010). The PCR experiments were repeated 3 times, each using separate sets of samples.
Transient Focal Cerebral Ischemia
All experimental protocols using animals were performed according to protocols approved by the Stanford University Animal Care and Use Committee and in accordance with the NIH guide for the care and use of laboratory animals. Adult male CB57/B6 mice (25–30 g from Charles River) were anesthetized with 2% isoflurane in balance O2 by facemask and focal cerebral ischemia was produced by 1 hour of middle cerebral artery occlusion (MCAO) with a silicone-coated 6-monofilament (Doccol Co, Redlands, CA, USA) followed by reperfusion as described before (Ouyang et al., 2012b). Sham-operated mice underwent an identical procedure, without inserting the suture but tying off the ipsilateral external carotid artery. Rectal temperature was maintained at 37±0.5°C controlled by a Homeothermic blanket control unit (Harvard Apparatus, Holliston, MA, USA). Temperature and respiratory rate were monitored continuously. Mice were randomized to surgery or sham, and mice with no evidence of acute neurological deficit or with evidence of hemorrhage were excluded from analysis. A total of 220 mice were subjected to sham or MCAO surgery, 18 were excluded from analysis, 12 animals died prior to day 7, 4 had evidence of hemorrhage, and 2 had no evidence of neurological deficit acutely. After different durations of reperfusion, mice were deeply anesthetized, and brains were removed after transcardiac perfusion with ice cold phosphate buffered saline (PBS) for RT-qPCR, Western blot analysis, or first with PBS and then 4% paraformaldehyde in PBS for immunohistochemistry.
Intracerebroventricular Infusion (ICV) and Intravenous Injection (IV) of 181a Antagomir
Two hours after MCAO miR-181a antagomir or control mismatch antagomir (MM) was injected intracerebroventricularly as previously (Ouyang et al., 2012b). Mice were anesthetized and placed in a stereotaxic frame with a mouse head holder. The brain infusion cannula was stereotaxically placed into the left lateral ventricle (bregma: −0.58 mm; dorsoventral: 2.1 mm; lateral: 1.2 mm) via a burr hole (Xiong et al., 2011) and affixed to the skull. Antagomir (3 pmol/gram in 2μl final volume) or MM (same amount) was mixed with the cationic lipid DOTAP (4 μl; Roche Applied Science, San Francisco, CA). After mixing for 5 seconds and incubating at 37°C for 15 min, the mixture (total 6 μl) was infused into the left lateral cerebral ventricle over 20 min. After that the bone wound was closed with bone wax. For intravenous infusion, 1 hour after MCAO miR-181a antagomir (30 pmol/gram) or control (MM) in sterile saline (100μl) was administered into the internal jugular vein. The schematic timecourse is shown in Figure 1A.
Figure 1.
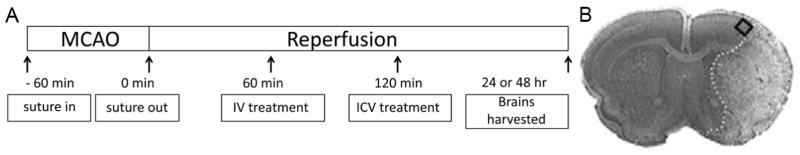
A. Timeline of post-treatment with miR-181a antagomir in mouse stroke. miR-181a antagomir was injected 60 min (IV) or 120 min (ICV) after 60 min MCAO. After 24 or 48 hours of reperfusion brains were collected to determine infarction size, protein levels, or fixed for immunofluorescence staining. B. An example of a post-stroke cresyl violet stained brain section shows the ischemic infarction and the square indicates the approximate area within the penumbra used for cell counting and immunohistochemistry.
Determination of Infarct Volume and Neurological Status
Cerebral infarction volume was determined after 2,3,5 triphenyltetrazolium chloride (TTC, Sigma, T8877, Louis, MO 63103, USA) staining of brains harvested at 48 hours reperfusion. Four sections/mouse were analyzed by a blinded observer, and corrected for edema using Adobe Photoshop CS3 as described previously (Han et al., 2009). Neurological status was assessed by neurologic deficit score at 24 hours reperfusion (Xiong et al., 2011). Neurological deficit score: 0 - no observable neurological deficits, 1 - failure to extend right forepaw, 2 - circling to the right, 3 - falling to the right, 4 - cannot walk spontaneously.
Rotarod test
Motor coordination and learning were assessed with the accelerating rotarod (SD Instruments, San Diego, CA). Beginning on the first day of training and continuing every other day for a total of 9 training days each mouse was placed on the 2.75cm diameter rod with rotation speed increasing from 5 to 10 rpm over 5 min. The time in which the mouse was able to stay on the rotating rod before falling was determined up to a maximum duration of 300 seconds. The test was repeated three times each day for each mouse, and the scores were averaged for each day. Any mouse not able to achieve at least a 200 seconds average on the rotarod after 9 days of training was excluded from the study. Rotarod testing was performed 3 days, and 1–4 weeks after surgery.
Immunohistochemistry
Ischemic or sham-operated mice were deeply anesthetized and perfused with cold 0.9% saline, followed by 4% paraformaldehyde in phosphate-buffered saline (PBS) (pH 7.4) 24 or 48 hours after MCAO. The brains were kept in 4% paraformaldehyde in PBS (pH 7.4) for 3 days then cut into 50 μm sections with a vibratome (VT1000S, Leica Microsystems, Wetzlar, Germany). Sections were immunostained using Polyclonal Rabbit Anti-Human Myeloperoxidase (MPO dilution 1:600, Cat # A03978 Dako, Carpinteria, CA, USA) to detect infiltrating neutrophils, polyclonal antibody to Iba1 (Iba1, dilution1:200, Cat # 019-19741 Wako Chemicals, Richmond, VA, USA) to identify activated microglia/macrophages, and X-linked inhibitor of apoptosis (XIAP) antibody (1:500 dilution, catalog #3331, ProSci, Poway, CA, USA).
After staining, evaluation was done in a fixed 1 mm × 1 mm area of the penumbra. The penumbra was defined as the region of generally morphologically normal cells surrounding the core (see Figure 1B). To quantify levels of fluorescence intensity 4 brain sections for each of five animals per group were evaluated using ImageJ (National Institutes of Health, Bethesda, MD, USA). The number of MPO positive cells was counted in an area of 220 uM × 200 uM in the penumbra on 4 slides/mouse for 5 mice/group.
Immunoblotting
Protein samples from ipsilateral brain tissue were harvested 24 hours after MCAO. Immunoblotting was performed as previously (Ouyang et al., 2012b). Equal amounts (50μg) of protein were separated on 12.5% polyacrylamide gels, and electrotransferred to Immobilon polyvinylidene fluoride membrane. Membranes were blocked and incubated overnight with primary antibody against GRP78 (1:10, PA1-37806, Affinity BioReagents), BCL2 (1:1000, #2870, Cell Signaling, Danvers, MA, USA), XIAP (1:1000, Catalog #3331, ProSci) and β-actin (1:1000, #926–42,210, LiCOR Bioscience), washed and incubated with secondary antibodies. Immunoreactive bands were visualized using the LICOR Odyssey infrared imaging system according to the manufacturer’s protocol. Densitometric analysis of bands was performed using ImageJ software (NIH) and the intensity was normalized to β-actin.
Measurement of NF-κB (P65) Activation by ELISA
At 5 hours and 24 hours following MCAO or sham treatment mice were transcardially perfused with cold saline, and the ipsilateral cerebral cortices were dissected and placed in liquid nitrogen. Complete lysis buffer AM2 from the TransAM NF-κB p65 Activity Assay kit (Active Motif, Carlsbad, CA 92008, USA) was added to tissue at a 1:5 ratio and the mixture was sonicated for 5 seconds. In total 20 μl of the mixture was added to each well and measured according to manufacturer’s instructions. All samples were run in duplicate. Data are presented as the fold change in absorbance value from sham to MCAO.
Statistical Analysis
Numbers of animals are indicated in figure legends. Data reported are means±SE. Statistical difference was determined using T-test for comparison of two groups or ANOVA followed by Bonferroni correction for experiments with N>2 groups using Sigmaplot (Systat Software, San Jose, CA, USA). P<0.05 was considered significant.
Results
Post-stroke ICV Treatment with miR-181a Antagomir Reduces Infarct Volume and Reduces Acute Neurological Deficit
Antagomir treatment reduced miR-181a in the brain to very low levels 24 hours later by quantitative RT-PCR analysis (Figure 2A). Using the same dose of miR-181a antagomir as used previously for pretreatment (Ouyang et al., 2012b), antagomir or MM was administered ICV 2 hours after MCAO. The infarction size after miR-181a antagomir treatment (31.9±3.6%) compared to MM control group (49.4±2.3%) was reduced about 35% (p<0.05) at 48 hours reperfusion (Figure 2B) and the neurologic deficit at 24 hours was significantly improved (Figure 2C).
Figure 2.

Post-stroke treatment with ICV administration of miR-181a antagomir 2 hours after MCAO protects brain. A. ICV injection of antagomir results in significantly reduced levels of miR-181a 24 hours later, N=5 mice/group. B. Infarct volume assessed at 48 hours reperfusion was reduced and C. the neurologic deficit score at 24 hours was improved, N=9. * indicates p<0.05 compared to MM control. D. ICV injection of antagomir results in better recovery of rotarod performance after MCAO. Sham treated mice in miR-181a antagomir group (n = 6) and MM group (n = 6) and MCAO mice in miR-181a antagomoir group (n = 21) and MM group (n = 22) learned the task and by the 9 times training performance exceeded 270 seconds. * p<0.05 vs sham control; # p<0.05 vs MM control. MM: mismatched control, con: control; antag: antagomir.
Post-stroke-treatment with miR-181a antagomir improves long-term recovery
Both miR-181a antagomir and MM groups of mice were able to learn the rotarod task and by the 7th training day performance reached a plateau (Figure 2D). Sham mice had similar rotarod performance immediately before and after sham surgery and through 4 weeks of post-testing, demonstrating that the mice achieved maximum performance prior to surgery and sham surgery did not affect their ability to perform the rotarod task when assessed beginning 3 days after anesthesia and sham surgery. At 3 days after MCAO surgery there was a marked drop in performance in both groups with MM group dropping more than miR-181a antagomir group. With increasing time, the miR-181a antagomir mice had greater recovery and reached that of sham by 21 days, while the MM mice still had a significant deficit in performance which appeared stable by 21 days. Treatment with antagomir also improved survival (Table 1). For the MM control group 5/22 animals died by the end of 7 days, whereas in the antagomir treated group only 2/21 mice died. No mice died between 7 and 28 days.
Table 1.
| Number died on post-stroke day | 28 day | |||||
|---|---|---|---|---|---|---|
| Groups | Total mice | Day 1 | Day 2 | Day 3 | Day 7 | survival % |
| 181a antagomir | 21 | 0 | 0 | 0 | 2 | 90% |
| MM control | 22 | 1 | 1 | 2 | 1 | 72% |
Post-stroke IV Treatment with miR-181a Antagomir Reduces Brain miR-181a Level and protects from MCAO
For clinical relevance we next tested IV administration to see if this route could also provide protection. The time-course of change in brain miR-181a expression level following IV injection of miR-181a antagomir is shown in Figure 3A. MiR-181a levels were significantly reduced as early as 2 hours following IV injection. Levels were still significantly reduced 24 hours after IV treatment with antagomir (Figure 3A). IV injection of antagomir 1 hour after MCAO significantly reduced infarct volume at 48 hours by TTC staining (Figure 3B and C). Neurologic deficit score was also significantly improved following IV injection of miR-181a antagomir (Figure 3D).
Figure 3.

Post-stroke treatment with IV administration of miR-181a antagomir protects brain. A. Time-course of relative expression levels of miR-181a following IV injection of miR-181a antagomir. N=4–5 mice/group. B. One hour following MCAO miR-181a antagomir was injected IV. Infarct volume 48 hours following MCAO was assessed with TTC staining, representative brains are shown. C. Bar graph shows post-treatment with IV injected miR-181a antagomir significantly reduced infarct volume. N=10 mice/group. D. Neurologic deficit score was significantly improved following IV injection of antagomir. MM: mismatched control. N=10/group, * indicates p<0.05 vs MM control for panels C and D and p<0.01 for panel A.
Post-stroke Treatment with miR-181a Antagomir Reduces Inflammation
The significant increase in activation of the inflammatory mediator NF-κB at 24 hours following MCAO was reduced by post-stroke IV injection of antagomir relative to control injection (MM), though early activation at 5 hours reperfusion was not blocked (Figure 4A). MCAO causes significant microglial/macrophage activation (increased IBa1 staining) in MM treated brains 48 hours after MCAO compared to sham (Figure 4B, left panel). IV injection of miR-181a antagomir 1 hour following MCAO significantly reduced this activation by about 60% compared to control (MM MCAO; Figure 4, right panel). In sham-operated brain, there are almost no myeloperoxidase (MPO) positive cells identified (left panel Figure 4C). A marked increase in MPO positive cells was observed following MCAO, indicating infiltration of inflammatory neutrophils (Figure 4, left panel). MPO positive cell number was significantly reduced by post-stroke IV injection of 181a antagomir compared to control (MM) (Figure 4C, right panel).
Figure 4.

Post-stroke treatment with IV injection of miR-181a antagomir reduces inflammation. A. The significant increase in activation of the transcription factor NF-κB at 24 hours following MCAO was prevented by post-stroke IV injection of antagomir relative to control injection (N=6–8 mice/group). * p< 0.01 compared to MM, # p<0.05 compared to sham. B. Representative images show activated microglia/monocytes visualized with IBa1 staining (green, left panel). IV injection of miR-181a antagomir significantly reduces microglial/monocyte activation (Iba1 fluorescence intensity) relative to MM control assessed 48 hours after MCAO (right panel). C. Representative images show infiltrating neutrophils which stain for MPO (green, left panel) 48 hours after injury. Scale bar 50uM. IV injection of miR-181a antagomir significantly reduces the number of infiltrating neutrophils (MPO stained cells) in the penumbra, right panel. N=5/group, * p<0.001 vs MM control. MM: mismatched control.
miR-181a Antagomir treatment increases BCL2 and XIAP
To further investigate the effects of miR-181a antagomir by IV post-treatment we examined levels of 3 known targets. GRP78 (Ouyang et al., 2012b), BCL2 (Ouyang et al., 2012a), and X-linked inhibitor of apoptosis (XIAP; Hutchison et al. (2013)). Using Western blot we found no significant difference in GRP78 protein level between miR-181a antagomir treated and MM treated brains (Figure 5A). In contrast protein levels of BCL2 and XIAP were modestly but significantly higher in miR-181a antagomir treated brains compared with the MM control, 24 hours after cerebral ischemia (Figure 5A). To further confirm the increased XIAP Western results we immunostained sections from post stroke brain (Figure 5B). The brightness of the images was increased representing increased XIAP expression in the miR-181a antagomir group after MCAO compared with MM group.
Figure 5.
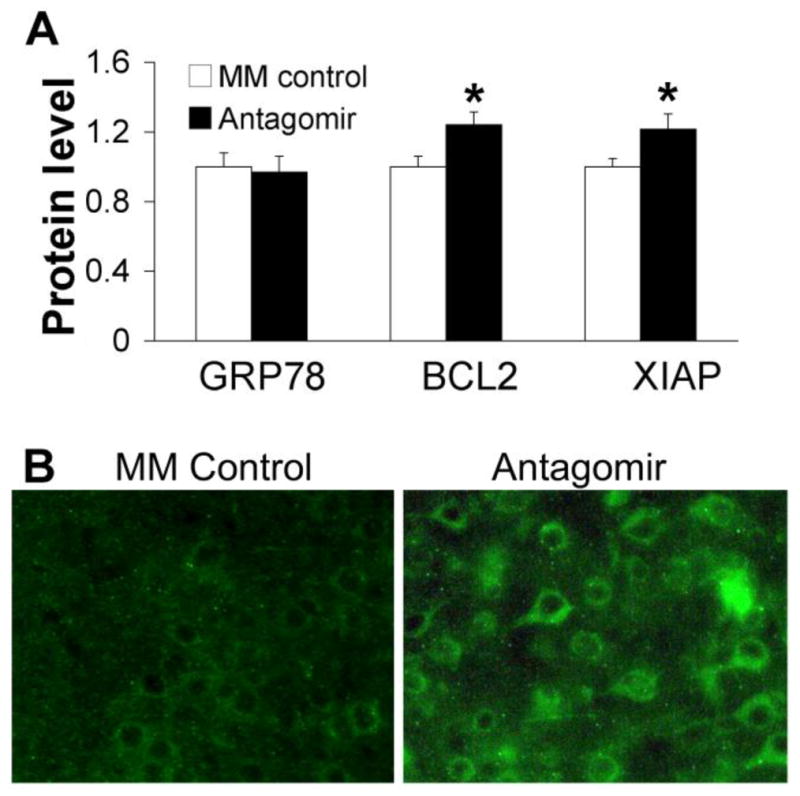
miR-181a antagomir increases anti-apoptotic protein BCL2 and apoptosis inhibitor XIAP with IV post-stroke treatment. A. Western blots showing no significant change in GRP78 protein level, but increased BCL2 and XIAP protein levels 24 hours after MCAO with post-treatment. For comparison the immunofluorescence intensities of Western blots were normalized to MM control in each group in the bar graphs. N= 7 (GRP78), N=6 (BCL-2), N=10 (XIAP), * p<0.05 vs MM control. B. Representative images showing increased XIAP staining (green). MM: mismatched control.
Discussion
The main findings in this study are (1) post-stroke treatment with miR-181a antagomir has neuroprotective effects against ischemic neuronal damage, and improves long-term neurobehavioral recovery in mice; (2) both ICV and IV administration were effective; (3) miR-181a antagomir administered shortly after stroke reduces evidence of inflammation and (4) miR-181a antagomir increases levels of BCL2 and XIAP but not GRP78 when given during reperfusion.
Previous studies have reported that miR-181a has anti-tumor effects (Fei et al., 2012; Shi et al., 2008; Shin et al., 2011). In agreement with those findings our recent work demonstrated that reducing miR-181a prior to stroke protects brain cells from ischemic injury in vivo and in vitro (Ouyang et al., 2012a; Ouyang et al., 2012b). To further assess translational potential we tested post-stroke treatment with both ICV and IV administration. We found that post-treatment with miR-181a antagomir by either route significantly reduced infarction size and improved neurological deficits. There are several reports from different labs of the effect of pre-stroke treatment with individual miRNAs, but this is the first report, to our knowledge, testing post-stroke treatment or IV administration.
Several neuroprotective strategies have been shown to have transient effects. Dietrich et al. demonstrated that strong protection by mild post-ischemic hypothermia seen at 3 days was reduced at 7 days and absent at 2 months (Dietrich et al., 1993). Intraischemic isoflurane treatment reduced infarct area acutely, but the effect was lost by 14 days (Kawaguchi et al., 2000). Thus assessment of long-term outcome is essential. Here we show that when animals are allowed to survive for 4 weeks after focal ischemia neurobehavioral performance is significantly improved in miR-181a antagomir treated mice.
The immune response is an important element contributing to the fate of the ischemic brain (Harari and Liao, 2010; Iadecola and Anrather, 2011) and the miR-181 family is implicated in immune regulation in several studies. An earlier report found miR-181 increased in B cells and overexpression in precursors increased the number of B lineage cells (Chen et al., 2004). T cell sensitivity to peptide antigens was found to be regulated by miR-181a (Li et al., 2007). A key element of inflammation is activation of the master inflammatory transcription factor NF-κB, which also plays a role in control of apoptosis (Harari and Liao, 2010; Hoffmann and Baltimore, 2006).
A recent report demonstrated an anti-inflammatory effect of miR-181a, direct down-regulation of IL1a, in monocyte and macrophage cell lines (Xie et al., 2013). Other work showed an anti-inflammatory effect of miR-181c in astrocytes exposed to LPS, and also validated XIAP as a target of miR-181c (Hutchison et al., 2013). Although anti-inflammatory effects of miR-181 family members have been reported, these were in response to a pure pro-inflammatory stimulus, and differ from our observations in the complex setting of stroke.
While our and others’ results consistently show that miR-181 aggravates cell injury after stress (Hutchison et al., 2013; Ouyang et al., 2012a; Ouyang et al., 2012b) and miR-181 was shown to sensitize glioblastoma cells to apoptosis by reducing BCL2 (Chen et al., 2010) this family clearly regulates both cell death regulatory pathways and proinflammatory pathways, and these pathways may overlap in signaling. The reduced inflammation observed in our study with miR-181a likely reflects several factors. First, the strong protective effect likely leads to a significant reduction in the induction of inflammation. Second, since miR-181 targets proteins that affect mitochondria, improved mitochondrial function may contribute directly to an anti-inflammatory effect (Voloboueva et al., 2013) just as impaired mitochondria can contribute to inflammation (López-Armada et al., 2013).
Mitochondria are centrally involved in both ischemic injury and recovery of brain tissue after cerebral ischemia due to their major roles in ATP generation, ROS production and regulation of cell death and inflammation. Both the heat shock protein 70 family (HSP70) and the antiapoptotic BCL2 protein family influence mitochondrial function and protect the brain from ischemia when overexpressed. MiR-181 targets both families, HSP70 family member GRP78, and anti-apoptotic BCL2 family members BCL2 and MCL1. Protection from ischemia with pretreatment with miR-181a inhibitor was associated with improved mitochondrial function (Ouyang et al., 2012a). The significant increase of BCL2 protein level in the miR-181a antagomir group suggests it is likely one of the mechanisms reducing injury and improving mitochondrial function with possibly a secondary reduction in inflammatory response.
It should also be mentioned that miR-181a affects different cell types differently. Reducing miR-181a increased BCL2 as one of the targets and increased survival of primary astrocytes (Ouyang et al., 2012a) and N2a cells (Moon et al., 2013) while it failed to significantly change levels of BCL2 and did not change outcome in primary neurons after ischemia-like injury (Moon et al., 2013). Combined with the fact that expression of the miR-181 family is strongly enriched in cultured astrocytes compared to neurons (Hutchison et al., 2013) the protective effect of anti-miR-181a in the present and earlier studies (Moon et al., 2013; Ouyang et al., 2012b) may result significantly from protection of astrocytes, in addition to possible direct protection of neurons.
Members of the miR-181 family are directly involved in regulation of both apoptosis regulatory proteins (BCL2 family and XIAP) and inflammatory mediators. Apoptosis regulatory proteins, especially the inhibitors of apoptosis family and caspases are also involved in the regulation of inflammation independent of cell death regulation, so it is clear that the effects of altered miR-181 levels on survival and inflammation will depend on the cell type and the presence or absence of other mediators, such as pro-inflammatory signaling. There is already evidence that the functions of the miR-181 family members are not redundant, and disparate effects are observed in different cells types. This work thus adds to current knowledge of the effects of modulating miR-181a on brain injury.
The ability to reduce brain damage in both focal (Ouyang et al., 2012b) and forebrain ischemia (Moon et al., 2013) as well as with either pre- or post-stroke treatment, makes miR-181a antagomir a promising target. Defining the effective post-treatment window remains an important next step to investigate. The ability to reduce brain injury with intravenous post-stroke treatment is encouraging for potential translation to clinical use.
Highlights.
Post-stroke treatment with miR-181a antagomir reduces infarction size.
Post-stroke treatment with miR-181a antagomir reduces long term neurobehavioral deficits.
Both ICV and IV administration are effective.
Post-stroke treatment with miR-181a antagomir reduces inflammation.
miR-181a antagomir administered after stroke onset targets BCL2 and XIAP.
Acknowledgments
The authors thank Siwei Liu for help with Westerns, Jeong-Mi Moon for help with RT-PCR, and Elysa Hall and William Magruder for help preparing the manuscript.
Sources of Funding:
Grant sponsor: NIH; Grant numbers: NS053898, and NS084396.
Footnotes
Disclosures:
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blakeley JO, Llinas RH. Thrombolytic therapy for acute ischemic stroke. J Neurol Sci. 2007;261:55–62. doi: 10.1016/j.jns.2007.04.031. [DOI] [PubMed] [Google Scholar]
- Chen CZ, et al. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–6. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- Chen G, et al. MicroRNA-181a sensitizes human malignant glioma U87MG cells to radiation by targeting Bcl-2. Oncology Reports. 2010;23:997–1003. doi: 10.3892/or_00000725. [DOI] [PubMed] [Google Scholar]
- Dietrich WD, et al. Intraischemic but Not Postischemic Brain Hypothermia Protects Chronically Following Global Forebrain Ischemia in Rats. J Cereb Blood Flow Metab. 1993;13:541–549. doi: 10.1038/jcbfm.1993.71. [DOI] [PubMed] [Google Scholar]
- Fei J, et al. miR-181a post-transcriptionally downregulates oncogenic RalA and contributes to growth inhibition and apoptosis in chronic myelogenous leukemia (CML) PLoS One. 2012;7:e32834. doi: 10.1371/journal.pone.0032834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han RQ, et al. Postischemic brain injury is attenuated in mice lacking the beta2-adrenergic receptor. Anesth Analg. 2009;108:280–7. doi: 10.1213/ane.0b013e318187ba6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harari OA, Liao JK. NF-kappaB and innate immunity in ischemic stroke. Ann N Y Acad Sci. 2010;1207:32–40. doi: 10.1111/j.1749-6632.2010.05735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–86. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- Hutchison ER, et al. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia. 2013;61:1018–28. doi: 10.1002/glia.22483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi M, et al. Isoflurane Delays but Does Not Prevent Cerebral Infarction in Rats Subjected to Focal Ischemia. Anesthesiology. 2000;92:1335–1342. doi: 10.1097/00000542-200005000-00023. [DOI] [PubMed] [Google Scholar]
- Li QJ, et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–61. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Liu DZ, et al. Brain and blood microRNA expression profiling of ischemic stroke, intracerebral hemorrhage, and kainate seizures. J Cereb Blood Flow Metab. 2010;30:92–101. doi: 10.1038/jcbfm.2009.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- López-Armada MJ, et al. Mitochondrial dysfunction and the inflammatory response. Mitochondrion. 2013;13:106–118. doi: 10.1016/j.mito.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Moon J-m, et al. Inhibition of microRNA-181 reduces forebrain ischemia-induced neuronal loss. J Cereb Blood Flow Metab. 2013 doi: 10.1038/jcbfm.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, et al. miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion. 2012a;12:213–9. doi: 10.1016/j.mito.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, et al. miR-181 regulates GRP78 and influences outcome from cerebral ischemia in vitro and in vivo. Neurobiol Dis. 2012b;45:555–63. doi: 10.1016/j.nbd.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, et al. microRNAs: innovative targets for cerebral ischemia and stroke. Curr Drug Targets. 2013;14:90–101. doi: 10.2174/138945013804806424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, et al. hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain Res. 2008;1236:185–93. doi: 10.1016/j.brainres.2008.07.085. [DOI] [PubMed] [Google Scholar]
- Shin KH, et al. miR-181a shows tumor suppressive effect against oral squamous cell carcinoma cells by downregulating K-ras. Biochem Biophys Res Commun. 2011;404:896–902. doi: 10.1016/j.bbrc.2010.12.055. [DOI] [PubMed] [Google Scholar]
- Voloboueva LA, et al. Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Letters. 2013;587:756–762. doi: 10.1016/j.febslet.2013.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, et al. miR-181a Regulates Inflammation Responses in Monocytes and Macrophages. PLoS ONE. 2013;8:e58639. doi: 10.1371/journal.pone.0058639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, et al. Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke. 2011;42:2026–32. doi: 10.1161/STROKEAHA.110.593772. [DOI] [PMC free article] [PubMed] [Google Scholar]