

Abstract
Concussive brain injury results in neuronal degeneration, microglial activation and enhanced excitability in the hippocampal dentate gyrus, increasing the risk for epilepsy and memory dysfunction. Endogenous molecules released during injury can activate innate immune responses including toll-like receptor 4 (TLR4). Recent studies indicate that immune mediators can modulate neuronal excitability. Since non-specific agents that reduce TLR4 signaling can limit post-traumatic neuropathology, we examined whether TLR4 signaling contributes to early changes in dentate excitability after brain injury. Concussive brain injury caused a transient increase in hippocampal TLR4 expression within 4 hours, which peaked at 24 hours. Post-injury increase in TLR4 expression in the dentate gyrus was primarily neuronal and persisted for one week. Acute, in vitro treatment with TLR4 ligands caused bidirectional modulation of dentate excitability in control and brain-injured rats, with a reversal in the direction of modulation after brain injury. TLR4 antagonists decreased, and agonist increased, afferent-evoked dentate excitability one week after brain injury. NMDA receptor antagonist did not occlude the ability of LPS-RS, a TLR4 antagonist, to decrease post-traumatic dentate excitability. LPS-RS failed to modulate granule cell NMDA EPSCs but decreased perforant path-evoked non-NMDA EPSC peak amplitude and charge transfer in both granule cells and mossy cells. Our findings indicate an active role for TLR4 signaling in early post-traumatic dentate hyperexcitability. The novel TLR4 modulation of non-NMDA glutamatergic currents, identified herein, could represent a general mechanism by which immune activation influences neuronal excitability in neurological disorders that recruit sterile inflammatory responses.
Introduction
The rationale for independent analysis of immunological and electrophysiological consequences of neurological disease has been challenged by the recognition that inflammatory mediators can impact neuronal physiology (Stellwagen, 2011). Immune modulation of neuronal excitability is crucial to consider following brain trauma because mechanical injury to neurons is known to engage both inflammatory and neurophysiological responses (Goforth et al., 1999; Cohen et al., 2007; Dileonardi et al., 2012; Oliva et al., 2012; Ferrario et al., 2013). Concussive brain injury is characterized by neuronal damage and degeneration (Toth et al., 1997; Neuberger et al., 2014) and activation of sterile inflammatory responses (Kelley et al., 2007) within hours to days following impact. The early post-injury period is also marked by altered neuronal and network excitability in the hippocampal dentate gyrus (Ross and Soltesz, 2000; Santhakumar et al., 2003). However, whether immune signaling plays a role in post-traumatic alterations in network excitability has not yet been examined.
Endogenous molecules released from disrupted cells and extracellular matrix following brain injury can stimulate toll-like receptors (TLRs), a class of pattern-recognition receptors of the innate immune system (Okun et al., 2011) which are activated by damage-associated molecular patterns such as cellular injury products (Kielian, 2006). Certain TLR subtypes, including TLR4, are expressed in neurons and have been implicated in neuropathology following ischemic reperfusion injury and epilepsy (Hua et al., 2007; Gao et al., 2009; Maroso et al., 2010). Although contusive brain injury is known to augment TLR4 levels, the time course and cell-type specificity of post-traumatic changes in hippocampal TLR4 expression remains controversial (Chen et al., 2012; Mao et al., 2012). Interestingly, immunoactive drugs which improve neurological outcome after brain injury have been found to limit post-traumatic increases in TLR4 expression (Lu et al., 2007; Chen et al., 2008; Chen et al., 2009; Fan et al., 2009). However, the mechanistic link between TLR4 signaling and post-traumatic neurological dysfunction remains to be elucidated.
The early increases in dentate excitability after concussive brain injury have been proposed to augment the risk for epilepsy (Lowenstein et al., 1992; Toth et al., 1997; Santhakumar et al., 2001). TLR4 signaling has been shown to acutely influence excitability of central and peripheral neurons (Maroso et al., 2010; Diogenes et al., 2011; Pascual et al., 2012). Mechanistically, studies using hippocampal cultures have identified that activation of TLR4 enhances calcium entry through NMDA receptors (Balosso et al., 2014). Additionally, ifenprodil, an NMDA antagonist with non-specific adrenergic effects (Chenard et al., 1991), occludes pro-convulsive effects of TLR4 agonists (Maroso et al., 2010) indicating that TLR4 may influence neuronal excitability by activating NMDA receptors. Whether the underlying mechanism by which TLR4 contributes to network physiology is by modulating NMDA currents remains unclear. Given the proposed contribution of TLR4 to neuronal excitability, involvement in epilepsy and potential role in long-term neurodegenerative pathology (Bhaskaran and Smith, 2010; Maroso et al., 2010), we examined whether TLR4 signaling contributes to early increase in dentate excitability after concussive brain injury.
Materials and Methods
All procedures were performed under protocols approved by the Institutional Animal Care and Use Committee of the Rutgers New Jersey Medical School, Newark, New Jersey.
Fluid percussion injury
Juvenile male Wistar rats (25–27 days old) were subject to the moderate (2–2.2 atm) lateral fluid percussion injury (FPI) or sham-injury using standard methods (Santhakumar et al., 2003; Gupta et al., 2012). Briefly, the rats were placed in a stereotaxic frame under ketamine (80mg/kg)-xylazine (10mg/kg) anesthesia (i.p). A 3mm hole was drilled on the left side of the skull 3 mm antero-posterior and 3.5 mm lateral to the sagittal suture keeping the dura intact. Two steel screws were screwed into the skull and glued to support the cap. A Luer-Lock syringe hub was glued to the skull over the exposed dura and bonded to the skull with cyanoacrylate adhesive. One day later, animals were anesthetized with isoflurane and attached to the FPI device (Virginia Commonwealth University, VA). A pendulum was dropped to deliver a brief (20 ms) impact on the intact dura. For sham injury, the animals were anesthetized and attached to the FPI device, but the pendulum was not dropped. Only brain tissue and sections from the injured side were used in experiments.
Protein Isolation and Western Blotting
Western blots of hippocampal tissue from the injured side were performed as described previously (Tobon et al., 2012). Briefly, mice lacking the expression of TLR4 (tlr4−/−, Jackson Laboratory), and rats at various time points after FPI or sham-injury (4 hours, 24 hours, 3 days, 7 days and 1 month) were quickly perfused with cold artificial CSF (aCSF, 4°C) under isoflurane anesthesia, fresh hippocampal tissue was isolated under a dissecting microscope and stored at −80°C until use. Tissue was homogenized with CelLytic MT cell lysis reagent supplemented with phenylmethylsulfonyl fluoride 1mM, sodium fluoride 10 mM, sodium orthovanadate 1 mM, phosphatase inhibitor 10 μl/ml and protease inhibitor 10 μl/ml (Sigma). Samples were cooled on ice for 10 min, centrifuged (15,000 × g) for 20 min at 4°C and the supernatant was transferred to a new tube. Protein concentration of the sample lysates was measured using BCA assay (Santa Cruz). Equal amounts of protein samples were diluted at a ratio of 1:1 in Laemmli sample buffer (Sigma) and separated on pre-cast gel (4–12% Tris-glycine, Bio-Rad). Protein was electro-transferred onto nitrocellulose membrane (Thermo Scientific) in a Tris-Glycine-Methanol buffer. The membrane was blocked for 1 h in 5% nonfat dry milk in 0.05% Tween-20 Tris-buffer solution (TBS) at room temperature (RT) on a slow shaker. The membrane was washed in TBS, incubated with primary antibody (anti-TLR4: 1:500 in 5% BSA 0.05% Tween-20 TBS or anti-β-actin: 1:5000 in 2.5% nonfat milk 0.05% Tween-20 TBS) overnight at 4 °C. TLR4 antibodies were obtained from Cell Signaling (Cat. # 2219) and Santa Cruz (Cat. # H-80) and β-actin from Sigma-Aldrich (Cat. # A1978). This was followed by TBS wash and incubation in secondary antibody (HRP-conjugated goat anti-rabbit at 1:10000, Sigma or anti-mouse at 1:5000, Sigma) for 1 h at RT. Immunoreactivity was visualized and imaged on FluorChem using a chemiluminescent substrate, Westdura (Thermo Scientific). Densitometry analyses were performed to quantify signals generated by Western blotting using ImageJ software (NIH). The density of each sample was normalized to its own density of β–actin.
Immunohistology
Immunosatining was performed on hippocampal sections (50 μm) on the injured side obtained from rats and naïve adult wild-type (C57Bl6) and tlr4−/− mice perfused with 4% paraformaldehyde in phosphate buffered saline (PBS) with heparin (10 u/ml). Sections from rats were obtained 4 hours, 24 hours and 1week after FPI and from age-matched sham-operated controls. Sections were then washed in PBS and blocked in 10% normal goat serum in 0.3% Triton in PBS for 1 hour. For TLR4 staining, sections were incubated overnight in anti-TLR4 antibody (1:500, H-80 Rabbit polyclonal; Santa Cruz) in 0.3% Triton and 2% normal goat serum in PBS at RT and mounted on slides using vectashield with DAPI (Vector Laboratories). Double staining for TLR4 and markers for neurons (NeuN), reactive astrocytes (GFAP), microglia (Iba1) or mossy cells (CGRP) was performed by sequentially incubating sections in anti-TLR4 (1:500 rabbit polyclonal antibody H-80, Santa Cruz) primary antibody for one hour followed by addition of anti-NeuN (1:1000, Mouse monoclonal antibody MAB377, Millipore), anti-GFAP (1:500, Mouse monoclonal antibody MAB360, Millipore), anti-Iba1 (1:300, mouse monoclonal antibody MABN92, Millipore) or anti-CGRP (1:1000 mouse monoclonal antibody AB81887, Abcam) primary antibodies and overnight incubation at RT. Sections were washed in PBS and immunostained with fluorescent secondary antibodies: goat anti-rabbit Alexa 594 (A11037, 1:500; Invitrogen) or goat anti-rabbit Alexa 488 (A11001, 1:500; Life Technologies) to reveal TLR4, goat anti-mouse Alexa-488 (A11029, 1:800; Invitrogen) to reveal NeuN, GFAP or Iba1 and goat anti-mouse Alexa-594 (A11008, 1:500; Life Technologies) to reveal CGRP. Sections were mounted on slides using Vectashield (Vector labs). The following controls were implemented to ascertain specificity of TLR4 immunostaining: (1) Negative controls excluding the primary antibody confirmed absence of non-specific labeling by the secondary antibody (Supplementary Fig. 3B); (2) Specificity of the anti-TLR4 antibody (H-80 Rabbit polyclonal; Santa Cruz) was confirmed by the lack of immunostaining in sections from tlr4−/− mice (Fig. 2) although the pattern of TLR4 expression in wild-type mouse tissue was similar to that observed in sham-injured rats (Supplementary Fig. 3A).
Figure 2. Post-traumatic increases in TLR4 expression in dentate gyrus.

A–C. Single plane confocal images show fluorescence intensity of TLR4 labeling in dentate sections from a sham-operated rat (A), a rat 24 hours post FPI (B) and a TLR4 knockout mouse (C). Note the increase in TLR4 staining after FPI and absence of staining in the knockout (TLR4 KO). GCL: granule cell layer; ML: molecular layer. Scale bar: 25 μM. D–E Confocal images showing overlay of TLR4 positive cells with DAPI in the dentate hilus of sham-operated rat (D) and a rat 24 hours post FPI (E). Representative control and FPI sections illustrated here were stained in the same well and imaged using identical settings. Note the increase in background staining in the post-FPI section, indicating an increase in TLR4 labeling of the hilar neuropil after FPI. Arrows point to TLR4 positive cells co-localized with DAPI. Scale bar: 25 μM. (F) Summary data shows semi-quantitative analysis of immunostaining intensity in molecular layer and dentate hilus in sections obtained from rats 24 hours after FPI and the corresponding sham control. G–H. Single plane confocal images of cells from a sham (G) and FPI rat (H) shows immunostaining for TLR4 (top), NeuN (middle) and merged images (bottom). Scale bar: 25 μm. H. Summary plot shows the percentage of NeuN positive profiles in the hilus that are co-labelled with TLR4 at various time points. Error bars indicate s.e.m. * indicate p<0.05 by one way ANOVA followed by Tukey’s post hoc test. Or two way ANOVA followed by pairwise Tukey’s post hoc test.
Imaging and quantification was performed in the dentate hilus on every 13th section along the septo-temporal extent of the hippocampus. Cell counts were performed using the optical fractionator of Stereo Investigator V.10.02 (MBF Bioscience, Williston, VT) on an Olympus BX51 microscope with a 100X objective. In each section, the hilus was outlined by a contour traced using a 10X objective. Sampling parameters were set at 100X: counting frame=50 μm by 50 μm, dissector height =30 μm, and top guard zone=5 μm. Approximately 25 sites per contour were selected using randomized systematic sampling protocols in Stereo Investigator (West et al., 1991; Yu et al., 2013). In each section, an observer marked the outline of NeuN positive somata in the hilus under epifluorescence illumination and a 100X oil objective and switched filters to visually examine the expression of TLR4 in NeuN labeled soma. Neurons were deemed co-labeled if the staining for TLR4 shared the outline of the NeuN labeled profile and had a greater intensity than the hilar neuropil. For quantification of intensity of TLR4 staining, confocal images of the dentate gyrus were obtained using a Nikon A1R laser confocal microscope with a 0.75 NA 20X air objective with identical camera settings. Two regions of interest (ROI) of identical dimensions were drawn in both the hilus and the dentate molecular layer using NIS Elements (Nikon Instruments). The average gray scale intensity of TLR4 staining for each ROI was determined using standard routines in NIS Elements (Yu et al., 2013). An investigator blind to the treatment performed image analyses.
Fluoro-Jade C staining was performed on coronal sections from rats perfusion with 4% paraformaldehyde 4 h after sham or head injury. Sections (50 μm) were mounted on gelatinized slides and air dried. Slides were immersed in 100% alcohol, 70% ethanol, and water for 2 min each followed by a 15 min incubation in 0.06% potassium permanganate before being stained with 0.001% Fluoro-Jade C in 0.1% acetic acid in the dark for 30 min as described previously (Gupta et al., 2012; Neuberger et al., 2014).
Electrophysiology
Three to five days or one week (7–9 days) after FPI or sham-injury, rats were anesthetized with isoflurane and decapitated. Horizontal brain slices (400 μm for field recordings and 350 μm for whole cell recordings) were prepared in ice-cold sucrose-aCSF containing (in μM) 85 NaCl, 75 sucrose, 24 NaHCO3, 25 glucose, 4 MgCl2, 2.5 KCl, 1.25 NaH2PO4, and 0.5 CaCl2 using a Leica VT1200S Vibratome (Wetzlar, Germany) as described previously (Aradi et al., 2004; Proddutur et al., 2013). The slices were sagittally bisected and the slices from the left hemisphere (ipsilateral to the side of injury) were incubated at 32 ± 1°C for 30 minutes in a submerged holding chamber containing an equal volume of sucrose-aCSF and recording aCSF and subsequently held at RT. The recording aCSF contained (in mM) 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3 and 10 D-glucose. All solutions were saturated with 95% O2and 5% CO2 and maintained at a pH of 7.4 for 1–6 h. Slices were transferred to an interface field recording chamber (BSC2, Automate Scientific, Berkeley, CA) perfused and recorded in aCSF making note of the electrode positions. Slices were transferred to incubation chambers for treatment with either TLR4 antagonist (50 ng/ml LPS-RS, 1 to 2 hours), TLR4 antibody (2.5μg/ml, H-80 Rabbit polyclonal; Santa Cruz, 1 hour), TLR4 agonist HMGB1 (50 ng/ml, 1 hour) or vehicle (recording aCSF, 1–2 hours) before being transferred again to the interface field recording chamber perfused with aCSF for post-treatment recordings. The various TLR4 ligands used in the study and their concentrations are included in Table 1. Brain slices rested on a filter paper and were stabilized with platinum wire weights. The tissue was continuously superfused with humidified 95% O2-5% CO2 and the temperature of the perfusing solution was maintained at 34°C using a proportional control heating unit (PTC03, Automate Scientific). Field recordings of evoked population spikes in the granule cell layer of the dentate gyrus were obtained using patch pipettes filled with recording aCSF. To evoke the field responses, constant current stimuli (0.5–4 mA, 50 μs) were applied at 0.1 Hz through a bipolar 90 μm tungsten stimulating electrode placed in the perforant path, at the junction of the dorsal blade and the crest just outside the fissure where it was visualized as a fiber tract (Gupta et al., 2012; Neuberger et al., 2014) and coupled to a high voltage stimulus isolator (A365R, WPI, Sarasota, FL). Recordings were obtained using an AxoPatch200B amplifier, filtered at 4 kHz using a Bessel filter, and digitized at 10 kHz with a DigiData 1440A analog–digital interface (Molecular Devices, Sunnyvale, CA). Field responses in the granule cell layer were measured at five predetermined points in each slice (Santhakumar et al., 2000; Yu et al., 2013), including the tips of the dorsal and ventral blades, the middle of the dorsal and ventral blades and the middle of the crest, and the largest response was studied further. Population spike amplitude was measured as the amplitude of the first negative deflection overriding the field EPSP waveform as described previously (Neuberger et al., 2014).
Table 1.
Summary of effects of injury and TLR4 ligands on physiological measures
| Physiological measure | Effect of Injury: FPI vs. Sham | Effect of TLR4 Ligand | |||
|---|---|---|---|---|---|
| TLR4 Ligands | Sham | FPI | |||
| Dentate population spike amplitude | ⇧ | TLR4 Antagonists | LPS-RS (50 μg/ml) | ⇧ | ⇩ |
| TLR4 Antibody (2.5μg/ml) | ⇧a | ⇩ | |||
| LPS-RS (50 μg/ml) with APV (50 μM) | ⬄ | ⇩ | |||
| TLR4 Agonist | HMGB1 (10 ng/ml) | ⇩ | ⇧ | ||
| Granule Cell NMDA EPSC amplitude | ⬄ | TLR4 Antagonist | LPS-RS (50 μg/ml) | ⬄ | ⬄ |
| Granule cell non-NMDA EPSC amplitude | ⇧ | ⬄ | ⇩ | ||
| Mossy Cell non-NMDA EPSC amplitude | ⇧ | ⬄ | ⇩ | ||
⇧Indicates significant increase, ⇩indicates significant decrease and ⬄ indicates no change.
Trend did not reach statistical significance. Data in the shaded rows were obtained using whole-cell patch clamp recordings.
For whole cell recordings, slices (350 μm) were transferred to a submerged recording chamber and perfused with oxygenated aCSF at 34 °C. Slices were incubated either in aCSF or 50 ng/ml LPS-RS for 1 hour before being transferred to a submerged recording chamber. Whole-cell voltage-clamp recordings from dentate granule cells and hilar neurons were obtained under IR-DIC videomicroscopy using Axon Instruments MultiClamp 700A (Molecular Devices, Sunnyvale, CA, USA). The recording microelectrodes (5–7 MΩ) contained (in mM): 125 K-Gluconate, 5 KCl, 2 MgCl2, 10 HEPES, 0.2 EGTA, 2 MgATP, 0.5 NaGTP, and 10 phospho-creatine at a pH of 7.26. For hilar neuronal recordings either biocytin (0.2%) or Alexa Fluor 488 biocytin (50 μM) were included in the internal solution. Whole cell responses, were evoked using constant current stimuli (0.5 mA and 4 mA, 50μs) delivered at 0·1 Hz through a bipolar tungsten stimulating electrode (0.5 mΩ, World Precision Instruments, Inc., Sarasota, FL) placed in the perforant path at the junction of the dorsal blade and the crest(Santhakumar et al., 2000). Series resistance was monitored throughout the recordings and data were rejected if it increased by over 20%. NMDA currents were recorded in the presence of the GABAAR antagonist gabazine (SR95531, 20 μM) and AMPA/kainate antagonist DNQX (6,7-Dinitroquinoxaline-2,3-dione, 20 μM) at a holding potential of +40 mV. Non-NMDA currents were recorded in the presence of gabazine (20 μM) and the NMDA receptor antagonist APV (D-(−)-2-Amino-5-phosphonopentanoic acid, 50 μM). Following physiological recordings, slices were fixed in 0.1 M phosphate buffer containing 4% paraformaldehyde at 4°C for 2 d. For post hoc immunohistochemistry, thick slices (350 μm) were labeled with Alexa 488 were directly mounted while biocytin filled neurons were revealed with Alexa Fluor 488-conjugated streptavidin (1:1000). Sections were visualized and imaged with a Nikon A1R laser confocal microscope with a 1.2 NA X 60 water objective. All salts were purchased from Sigma-Aldrich (St. Louis, MO). Gabazine, APV and DNQX were obtained from Tocris (Minneapolis, MN).
Statistical analysis
Statistical analyses were performed using SigmaPlot 12.3. Quantitative data for Western blot analysis (8 groups) were not normally distributed. Thus Western blot data were compared using non-parametric tests (Kruskal-Wallis and Mann-Whitney U tests). Two way ANOVA was used for analysis of cell counts. Semi-quantitative data of TLR4 staining intensity was compared using Student’s t-test. All independent samples were tested for normality and homogeneity of variance in SPSS Levene’s test using descriptive statistic. Post-hoc Tukey’s test was used to assess statistical significance of between-group differences. Data from physiological experiments were compared using paired and unpaired Student’s t-test or two-way repeated measures ANOVA (RM-ANOVA) as appropriate. Significance was set to p < 0.05. Data are shown as mean ± s.e.m.
Results
Early and transient increase in TLR4 expression after brain injury
Fluid percussion brain injury leads to immediate mechanical damage to neurons in the hippocampal dentate gyrus (Toth et al., 1997) and dentate hilar cell death within hours of injury (Gupta et al., 2012). FluoroJade C labeling conducted 4 hours after FPI revealed that the moderate lateral FPI implemented in this study resulted in neuronal degeneration cortex, hippocampus and thalamus on the side of injury (Supplementary Fig. 1, based on n=3 rats). There was no evidence for early neuronal degeneration in the contralateral hemisphere of head-injured rats or in sham-injured controls (Supplementary Fig. 1–2, based on n=2 sham and n=3 FPI rats). However, contralateral hippocampal neuronal degeneration has been reported at later time points after FPI and cortical impact (Colicos et al., 1996; Neese et al., 2007; Pang et al., 2014), possibly due to secondary neuronal injury. Since neuronal injury and death can activate the innate immune responses, we used Western blot analysis to quantify hippocampal TLR4 protein levels at various time points after FPI. First, we tested the specificity of TLR4 antibodies from Cell Signaling and Santa Cruz used in earlier studies (Tang et al., 2007; Maroso et al., 2010). As illustrated in Figure 1A, the TLR4 antibody from Cell Signaling (Cat.# 2219) labeled a single band at 95 kD, the molecular weight of TLR4(Re and Strominger, 2002), in hippocampal samples from both sham-injured and FPI rats. In addition to the 95 kD band, the Cell Signaling antibody labeled a few bands with higher molecular weight which likely reflect glycosylation products. Antibody specificity was further confirmed by the complete absence of labeling in hippocampal tissue from mice lacking the TLR4 gene (Fig. 1A, lane labeled as TLR4 KO) even though the same antibody has been shown to reveal TLR4 in wild-type mouse tissue (Tang et al., 2007). Similarly, the TLR4 antibody from Santa Cruz (Cat# H-80) showed qualitatively similar results labeling a specific band at 95 kD and high molecular weight bands but failed to show the 95 kD band in mice lacking the TLR4 gene, confirming antibody specificity (data not shown). However, since the Cell Signaling antibody has been used for western blot analysis in earlier studies (Tang et al., 2007), the Cell Signaling antibody (Cat# 2219) was deemed to be better for Western analysis and used in all subsequent quantitative analysis. Compared to the sparse TLR4 expression in age-matched sham-controls, Western blots revealed a significant increase in TLR4 protein levels in hippocampal tissue from the injured side as early as 4 hours after FPI (Fig. 1A–B, TLR4 protein levels 4 hours after FPI was 121.83 ± 4.13% of protein levels in sham, n=6 rats each, p<0.05 by Kruskal-Wallis test followed by Mann-Whitney U tests). Increase in TLR4 expression peaked at 24 hours after FPI, reaching 191.17 ± 14.91 % of the expression levels in age-matched sham controls (Fig. 1A–B, n=6 rats each, p<0.05 Kruskal-Wallis test followed by Mann-Whitney U tests). A significant elevation in TLR4 expression was maintained 3 days after FPI (Fig. 1 A–B, TLR4 protein levels 3 days after FPI: 134.67 ± 6.44 % of age-matched sham, n=6 rats, p<0.05 by Kruskal-Wallis test followed by pairwise comparison by Mann-Whitney U tests). However, the small increase in TLR4 expression observed 7 days and 1 month after FPI failed to reach statistical significance (Fig. 1 A–B, TLR4 protein levels 7 days after FPI: 108.33 ± 3.72 % of the sham, 1 month after FPI: 105.65 ± 3.54 % of sham, n=6 rats all groups, p>0.05 by Mann-Whitney U tests). Remarkably, regardless of the time point after sham-injury, hippocampal tissue from sham-operated rats showed no difference in the levels of TLR4 expression from that observed at 4 hours (Fig. 1B) and in age-matched, naïve rats (95.67 ± 5.27 % of 4 hours post of sham, n=6 rats, p>0.05 by Mann-Whitney U tests) demonstrating that the surgical procedures did not increase TLR4 expression in the absence of brain injury. These data demonstrate that concussive brain injury results in early and transient up-regulation of TLR4 expression in hippocampus.
Figure 1. Early increase in hippocampal Toll-like Receptor 4 (TLR4) after fluid percussion injury (FPI).

A. Western blots of hippocampal tissue labeled with the Cell signaling (Cat # 2219) antibody against TLR4 show a single specific band at 95 KD (above). Note increase in intensity at various time points after FPI and absence of the band in TLR4 knockout (TLR4 KO). β-actin control for protein levels is shown below. B. Summary data shows percent of TLR4 protein levels (normalized to β-actin levels in the samples). Summary data demonstrate the TLR4 levels at various time points. Error bars indicate s.e.m. * indicate p<0.05 by Kriskal Wallis and Mann Whitney U tests.
In light of the Western blot data, we conducted immunostaining experiments to identify the regional distribution of post-injury alteration in TLR4 expression in the hippocampal dentate gyrus, a focus of cellular and physiological pathology after FPI (Lowenstein et al., 1992; Toth et al., 1997; Santhakumar et al., 2001). Since, consistent with vendor recommendations, the Cell Signaling TLR4 antibody was not compatible with immunohistology, the polyclonal Santa Cruz TLR4 antibody (Santa Cruz H-80) was used for immunostaining studies. In agreement with data from Western blots, we found an increase in the expression of TLR4 in the dentate gyrus of rats sacrificed 4 hours (not shown) and 24 hours after FPI compared to age-matched, sham-operated controls (Fig. 2A–B). The absence of cellular labeling in hippocampal sections from TLR4 knockout mice (Fig. 2C) despite labeling in wild-type mouse tissue (Supplementary Fig 3A) confirmed the specificity of the TLR4 antibody (Santa Cruz H-80) for immunostaining studies. TLR4 expression appeared enhanced in all dentate layers (Fig. 2A–B) and showed a prominent cellular pattern of expression in the dentate hilus (Fig. 2D–E). Semi-quantitative analysis of gray-scale images for fluorescence showed that the intensity of TLR4 labeling in both molecular layer and hilus was significantly elevated 24 hours after FPI (Fig. 2F, 43.01±6.48% increase in molecular layer; intensity in arbitrary units (A.U.), sham: 287.94±6.50 in 10 sections from 3 rats, FPI: 412.33±17.04 in 12 sections from 4 rats; 32.45±5.63% increase in the hilus; intensity in A.U., sham: 303.13±10.02 in 10 sections from 3 rats, FPI: 398.78±15.35 in 12 sections from 4 rats, p<0.01 by Student’s t-test). Similarly, as illustrated by confocal images of representative control and FPI sections stained in the same well and imaged using identical settings, the intensity of TLR4 labeling in the molecular layer and dentate hilus was enhanced 4 hours after FPI (36.96±8.62% increase in molecular layer; intensity in A.U., sham: 345.31±14.27 in 10 sections from 3 rats, FPI: 477.87±28.44 in 12 sections from 4 rats; 48.85±7.38% increase in the hilus; intensity in A.U., sham: 310.55±25.13 in 10 sections from 3 rats, FPI: 456.98±21.74 in 12 sections from 4 rats, p<0.01 by Student’s t-test). In addition to the cellular labeling in the hilus, TLR4 immunoreactivity in the hilar neuropil appeared elevated after FPI (Fig. 2D–E). While the increase in TLR4 levels in hippocampal tissue dissected en block failed to reach statistical significance 7 days post-injury (Fig. 1B), TLR4 labeling in the dentate molecular layer and hilus remained significantly elevated 1 week after FPI (66.85±9.40% increase in molecular layer; intensity in A.U., sham: 309.61±19.50 in 10 sections from 3 rats, FPI: 513.46±26.53 in 12 sections from 4 rats; 51.34±7.77% increase in the hilus; intensity in A.U., sham: 330.13±21.85 in 10 sections from 3 rats, FPI: 496.27±24.51 in 12 sections from 4 rats, p<0.01 by Student’s t-test). Thus, our data demonstrate that the post-traumatic increase in hippocampal TLR4 expression occurs as early as 4 hours after injury and persists up to 7 days.
Expression and post-injury increase in TLR4 is predominantly neuronal
Given the cellular expression pattern for TLR4 in the dentate hilus (Fig. 2D–E), we undertook double immunolabeling studies to determine the cell-type specific expression of TLR4. While TLR4 labeling in tissue from rats sacrificed 24 hours after sham injury was sparse, the cellular labeling of TLR4 was readily apparent in sections from rats 24 hours after FPI. As illustrated by representative maximum intensity projections of confocal image stacks, the intensity of TLR4 labeling in cellular profiles was greater in tissue from FPI rats (Supplementary Fig. 4A–B). Single plane confocal images (Fig. 2G–H) demonstrate that TLR4 positive profiles are positive for NeuN in hilar sections from both sham-operated controls and post-FPI rats sacrificed 24 hours after injury. However, not all NeuN positive profiles in the hilus were co-labeled with TLR4 (arrows in Supplementary Fig. 4A–B), indicating cell-type specific expression of TLR4 among the diverse neuronal populations in the hilus. Stereological quantification of the number of NeuN positive profiles that expressed TLR4 revealed a significant difference between sham-injured and FPI groups (F(1, 77) =58.66, p<0.001 by two-way ANOVA). The proportion of NeuN-positive hilar cells co-labeled for TLR4 showed no difference at 4 hours, 24 hours and 1 week after sham injury . Compared to sections from age-matched sham rats, sections from FPI rats showed a significant increase in the percentage of hilar neurons expressing TLR4 at all time-points tested (Fig. 2I, % of NeuN positive profiles expressing TLR4, 4 hours post-FPI: sham: 36.24±3.15%, based on 11 sections from 3 rats, FPI: 72.53±4.00% based on 13 sections from 4 rats; 24 hours post-FPI: sham: 33.88±2.57% based on 13 sections from 3 rats, FPI: 93.40±1.80 % based on 17 sections each from 4 rats; 1 week post-FPI: sham: 40.22±6.68% based on 13 sections from 3 rats, FPI: 69.67±6.68% based on 16 sections from 4 rats, p<0.05 by post-hoc Tukey’s test for all pairs). Thus, although the percentage of hilar neurons expressing TLR4 peaked 24 hours after FPI, there was a persistent increase in the number of hilar neurons labeled for TLR4 even 1 week after FPI.
Previous studies have reported that TLR4 may be expressed in microglia and reactive astrocytes (Kielian, 2006). Since brain injury can recruit microglia and contribute to reactive astrocytosis (Ogawa et al., 2005), we examined whether Iba1-positive microglia and GFAP-expressing reactive astrocytes express TLR4. Although we found cellular patterns of Iba1 and TLR4 expression in sections from the dentate hilus in both sham-controls and rats 24 hours after FPI, the expression patterns failed to overlap indicating the absence of TLR4 expression in microglia (Supplementary Fig. 5A–B, based on examination of 4 sections each from 3 sham rats and 4 post-FPI rats). Consistent with earlier studies (Fukuda et al., 2012), there was a striking increase in GFAP-expressing astrocytes in the hilus 24 hours after FPI (Supplementary Fig. 5C–D). However, there was little overlap between the GFAP and TLR4 in sections from both sham and FPI rats (Supplementary Fig. 5C–D), based on examination of 4 sections each from 3 sham and 4 post-FPI rats. Sections obtained from rats 4 hours and 7 days after FPI and age-matched, sham-controls revealed qualitatively similar results with no co-labeling of TLR4 with either Iba1 or GFAP (data not shown). Taken together, our data indicate that the post-traumatic increase in TLR4 expression in the dentate gyrus is primarily neuronal.
TLR4 antagonists decrease dentate excitability in brain injured rats
Since neuronal TLR4 expression in the dentate gyrus is increased up to 1 week after injury, and since recent studies have implicated TLR4 signaling in increased hippocampal excitability in epilepsy (Maroso et al., 2010), we examined if TLR4 signaling contributes to early post-traumatic increase in dentate excitability (Lowenstein et al., 1992; Toth et al., 1997; Gupta et al., 2012). First, we confirmed that the amplitude of the granule cell population response was substantially increased 1 week after FPI compared to age-matched sham controls (Fig. 3 A, C, E; population spike amplitude in mV in response to a 4mA stimulation, sham 0.14±0.06, n=9 slices from 4 rats, FPI: 1.61 ± 0.24, n=15 slices from 5 rats, p<0.05 by two-way RM-ANOVA followed by post-hoc Tukey’s test) as reported previously (Toth et al., 1997; Santhakumar et al., 2003). In field recordings from the granule cell layer of sham-injured controls, incubation in the TLR4 antagonist, LPS-RS (50 μg/ml for 1 to 2 hours), increased the amplitude of the population spike evoked by a 4 mA stimulus to the perforant path compared to responses in the same slice obtained prior to LPS-RS incubation (Fig. 3 A–B, population spike amplitude in mV, sham: before LPS-RS incubation 0.14±0.06; after LPS-RS incubation:0.856±0.36, n=9 slices from 4 rats, P<0.05 by two-way RM-ANOVA, followed by post-hoc Tukey’s test). Field potential response evoked by stimulation of the perforant path at increasing intensities showed a systematic increase in the amplitude of the granule cell population spike amplitude after LPS-RS treatment. These findings demonstrate a novel steady-state reduction of dentate excitability by TLR4 signaling (Fig.34E). In contrast to findings in slices from sham-control rats, incubation in LPS-RS reduced perforant path-evoked population spike amplitude in slices from FPI rats (Fig. 3C–E). Comparison of the population spike amplitudes in a slice from a FPI rat before (Fig. 3C) and after LPS-RS incubation (Fig. 3D) illustrates the LPS-RS-mediated suppression of population spike amplitude (Fig. 3C–D, population spike amplitude in mV at 4mA stimulation, FPI before LPS-RS: 1.61 ± 0.24, FPI after LPS-RS incubation: 0.55 ± 0.09, n=15 slices from 5 rats, p<0.05 by two-way RM-ANOVA followed by post-hoc Tukey’s test). One week after FPI, LPS-RS suppressed dentate population spike amplitude evoked by stimulation of the perforant path at increasing intensities (Fig. 3E). There was a significant interaction between the effects of injury and LPS-RS on the afferent-evoked granule cell population spike amplitude (F1,21=22.62, p<0.001 by two way RM-ANOVA). Additional control experiments in which slices were recorded in aCSF before and after a 1–2 hour incubation in aCSF ruled out the possibility that the observed effects were due to the process of recording slices before and after incubation (population spike amplitude at 4mA stimulation intensity in mV, sham pre-incubation : 0.40 ± 0.09, sham after aCSF incubation: 0.38 ± 0.11, n=6 slices from 3 rats, p>0.05; FPI pre-incubation: 1.56 ± 0.18, FPI after aCSF incubation: 1.58 ± 0.43, n=9 slices from 4 rats, p>0.05 by two-way RM-ANOVA followed by post-hoc Tukey’s test).
Figure 3. TLR4 antagonist decreases dentate excitability in injured rats.

A–B. Granule cell population responses evoked by a 4 mA stimulus to the perforant path in a slice from a sham rat, before (A) and after (B) incubation in the TLR4 antagonist, LPS-RS (50 ng/ml for 1–2 hr). C–D. Dentate field responses in a slice obtained from a rat 1 week after FPI before (C) and after (D) LPS-RS treatment. Arrows indicate truncated stimulus artifact. E. Summary data demonstrate the effect of LPS-RS on the perforant path-evoked granule cell population spike amplitude in slices from sham-operated and FPI rats at various stimulation intensities. Error bars indicate s.e.m. * indicates p<0.05 from control and # indicates p<0.05 for comparison of before and after drug treatment, by two-way RM-ANOVA followed by Tukey’s post hoc test.
To determine whether TLR4 signaling leads to bidirectional modulation of dentate excitability after FPI, we tested the effect of the TLR4 selective agonist HMGB1 (high mobility group box 1, 10ng/ml for 1 hour). HMGB1 reduced dentate population spike amplitude in sham injured rats (Fig. 4 A–B, E, population spike amplitude in mV, sham, before HMGB1 incubation 0.60±0.22; after HMGB1 incubation: 0.33±0.17, n=8 slices from 3 rats, P<0.05 by two-way RM-ANOVA, followed by post-hoc Tukey’s test) and increased dentate population spike amplitude in slices from rats 1 week after FPI, (Fig. 4C-E, population spike amplitude at 4mA stimulation in mV, FPI before HMGB1 incubation 1.65±0.20, FPI after HMGB1 incubation: 2.89±0.29, n=11 slices from 4 rats, p<0.05 by two-way RM-ANOVA followed by post-hoc Tukey’s test). Dentate population spike amplitude was significantly affected by both injury (F1,17 =29.15, p<0.001 by two-way RM-ANOVA) and HMGB1 (F1,17 =18.48, p<0.001 by two-way RM-ANOVA). Additionally, interaction between the effects of injury and HMGB1 (F1,17 =57.47, p<0.001 by two-way RM-ANOVA) was significant. Importantly, compared to LPS-RS, HMGB1 had opposite effects on granule cell population responses in slices from both sham- and brain-injured rats, suppressing excitability in sham- and increasing excitability in FPI rats. As an additional control for drug specificity, we examined whether the effects of TLR4 antibody (Santa Cruz H-80, 2.5μg/ml, 1 hour) on evoked dentate responses were opposite that of HMGB1 and similar to LPS-RS. Although TLR4 antibody tended to increase dentate population spike amplitude in slices from sham rats, the difference did not reach statistical significance (population spike amplitude in mV: sham in aCSF: 0.27±0.09, sham in TLR4 antibody: 0.72±0.29 n=10 slices from 4 rats p>0.05 by paired t-test). Crucially, like LPS-RS and in contrast to HMGB1, TLR4 antibody decreased dentate population spike amplitude 1 week after FPI (population spike amplitude in mV: FPI in aCSF: 1.85±0.26, FPI in TLR4 antibody: 0.57±0.27 n=12 slices from 4 rats p<0.05 by paired t-test). Similar results were also obtained with CLI-095 (Invivogen, 1μg/ml), a cyclohexene derivative that specifically suppresses TLR4 signaling (data not shown). In control experiments, incubation of slices from FPI rats in vehicle (aCSF, 1 hour) failed to alter dentate excitability (population spike amplitude in mV: FPI in aCSF: 1.67±0.11, FPI after aCSF incubation: 1.68±0.08, n=10 slices from 5 rats p>0.05 by paired t-test). Since LPS-RS and HMGB1 have both been suggested as non-specific agonists for TLR2 (Curtin et al., 2009) (also see http://www.invivogen.com/PDF/LPS_RS_TDS.pdf), the opposite effects of LPS-RS and HMGB1 and similar effects of LPS-RS, TLR4 antibody and CLI-095 eliminate the possibility that the observed effects of the TLR4 ligands result from non-specific effects on TLR2. These data validate the use of LPS-RS as an antagonist of the physiological effects of TLR4. Our findings summarized in Table 1 demonstrate that TLR4 signaling contributes to a tonic reduction in dentate excitability under control conditions and enhancement of dentate network excitability after FPI.
Figure 4. Bidirectional modulation of dentate excitability by TLR4 agonist HMGB1 after brain injury.
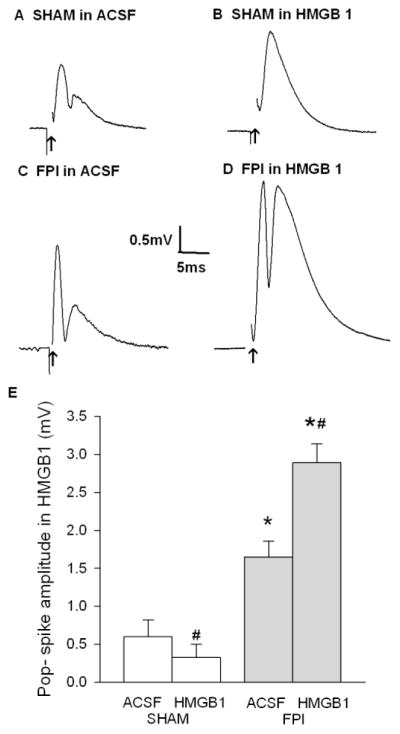
A–B. Granule cell population responses evoked by a 4 mA stimulus to the perforant path in a slice from a sham-control rat, before (A) and after (B) incubation in the TLR4 agonist, HMGB1 (10mg/ml for 30 min). C–D. Dentate field responses in a slice obtained from a rat 1 week after FPI before (C) and after (D) HMGB1 treatment. Arrows indicate truncated stimulus artifact. E. Summary data demonstrate the effect of HMGB1 on the perforant path-evoked granule cell population spike amplitude in slices from sham-operated and FPI rats. Error bars indicate s.e.m. * indicates p<0.05 from control and # indicates p<0.05 for comparison of before and after drug treatment, by two-way RM-ANOVA followed by Tukey’s post hoc test.
LPS-RS decreases dentate excitability in the presence of NMDA receptor antagonist
Recent studies have suggested that TLR4 signaling may impact neuronal excitability by modulating NMDA receptors (Maroso et al., 2010; Balosso et al., 2014). Therefore, we examined whether NMDA receptor antagonists occlude the LPS-RS-mediated suppression of post-traumatic dentate network excitability. Blocking NMDA receptors (APV, 50 μM) did not alter the amplitude of the afferent-evoked dentate population spike in slices from rats 3–9 days after sham- and FPI (population spike amplitude at 4 mA stimulation in mV, sham in aCSF: 0.55 ± 0.15, sham in APV: 0.49 ± 0.13, n=12 slices from 5 rats each, p>0.05,, FPI in aCSF: 1.73 ± 0.26, FPI in APV: 1.54 ± 0.20, n=12 slices from 5 rats each, p>0.05, by two-way RM-ANOVA). Thus the post-FPI increase in dentate population response appears to be independent of NMDA receptor activity. In slices from control rats, incubation in LPS-RS (50 μg/ml for 2 hours) did not alter the magnitude of the afferent-evoked population spike amplitude recorded in the presence of 50 μM APV (Fig. 5-B, E, population spike amplitude at 4mA stimulation in mV, sham before LPS-RS: 0.51±0.13, sham after LPS-RS incubation: 0.45 ± 0.16, n=12 slices from 5 rats, p>0.05, by two-way RM-ANOVA). Analysis of the perforant path-evoked population spike amplitudes measured at increasing stimulation intensities confirmed that LPS-RS failed to modulate dentate excitability in slices from sham-injured rats perfused with APV (Fig. 5E). However, LPS-RS incubation continued to reduce afferent-evoked granule cell population spike amplitude in slices from FPI rats even when the recordings were performed in the presence of APV (Fig. 5C–F, population spike amplitude in APV mV at 4mA stimulation, FPI before LPS-RS incubation 1.54 ± 0.20, FPI after LPS-RS incubation: 0.66 ± 0.18, n=12 slices from 5 rats, p<0.05 by two way RM-ANOVA followed by post-hoc Tukey’s test). In the presence of APV, there was a significant effect of both injury (F1,22=9.56, p<0.05 by two way RM-ANOVA) and LPS-RS (F1,22=18.67, p<0.05 by two way RM-ANOVA) and an interaction between effects of injury and LPS-RS (F1,22=8.36, p<0.05 by two way RM-ANOVA) on afferent-evoked granule cell population spike amplitude (Table 1). These data demonstrate that, while the tonic TLR4 modulation of dentate excitability is occluded in the presence of NMDA receptor antagonist, TLR4 mediated enhancement of dentate excitability after brain injury does not depend on increase in NMDA currents.
Figure 5. NMDA receptor antagonist does not occlude LPS-RS effects on post-traumatic dentate hyperexcitability.

A–D. Representative granule cell population responses evoked by a 4 mA stimulus to the perforant path. Recordings were obtained in slices from rats 3 days after sham- (A, B) or FPI (C, D) in the presence of the N-Methyl-D-aspartate (NMDA) receptor antagonist, (2R)-amino-5-phosphonovaleric acid; (2R)-amino-5-phosphonopentanoate (APV, 50 μm). Traces were obtained before (A, C) and after (B, D) incubation in LPS-Rs (50 ng/ml for 2 hr). Arrows indicate truncated stimulus artifact. E. Summary data demonstrate the effect of LPS-RS on the afferent-evoked dentate population spike amplitude at various stimulation intensities in sections obtained 3–9 days after FPI or sham -injury. F. Summary plots of the dentate population spike amplitude after incubation in LPS-RS normalized to the amplitude in the same slice before incubation in LPS-RS show that LPS-RS selectively decreases the amplitude of granule cell population responses both 3–5 days and 1 week after FPI but not after sham-injury. Error bars indicate s.e.m. * indicates p<0.05 by two-way RM-ANOVA followed by Tukey’s post hoc test.
To further assess if TLR4-modulation of post-traumatic dentate excitability was different between 3 days and 1 week after FPI, we separately analyzed LPS-RS modulation of granule cell population responses at the two time points (3–5 days and 7–9 days) after FPI. Similar to the pooled data (Fig. 5E), in the presence of APV, LPS-RS incubation selectively decreased perforant path-evoked population spike amplitude 3–5 days after FPI but not in sham-injured controls (Fig. 5F, population spike amplitude in LPS-RS normalized to aCSF, sham: 0.88 ± 0.17, FPI: 0.31 ±0.15 p<0.05 by paired t-test, response to a 4 mA stimulus). Similarly, LPS-RS incubation failed to modulate the population spike amplitude in APV 1 week after sham-injury (Fig. 5F, population spike amplitude in LPS-RS normalized to aCSF, sham: 0.92 ± 0.18 from 0.52 ± 0.26 before LPS-RS incubation to 0.48 ± 0.36 after LPS-RS incubation in n=6 slices from 2 rats p>0.05 by paired t-test, response to a 4 mA stimulus). Importantly, LPS-RS incubation continued to reduce afferent-evoked granule cell population spike amplitude in the presence of APV even 1 week after FPI (Fig. 5F, population spike amplitude in LPS-RS normalized to aCSF in mV, FPI: 0.51±0.19 from 1.25 ± 0.22 before LPS-RS to 0.62 ± 0.12 after LPS-RS incubation in n=6 slices from 2 rats p<0.05 by paired t-test, response to a 4 mA). Although there was an apparent reduction in the extent to which LPS-RS decreased the population spike amplitude in slices from rats 3–5 days and 1 week after injury, the difference did not reach statistical significance (Fig. 5F).
Next we directly examined whether LPS-RS modulates NMDA EPSCs in granule cells, the projection neurons of the dentate gyrus. Granule cells were held at +40 mV and NMDA EPSCs were recorded in response to a 4 mA stimulation of the perforant path in the presence of non-NMDA ionotropic glutamate and GABAA receptor antagonists (20 μM DNQX and 20 μM gabazine, respectively). Under these recording conditions, 50 μM APV completely eliminated perforant path-evoked granule cell EPSCs in 4 of 4 sham and FPI cells tested (not shown). Consistent with earlier findings (Santhakumar et al., 2000), granule cell NMDA EPSC peak amplitude and charge transfer were not altered 1 week after FPI (Fig. 6A, NMDA EPSC amplitude in pA, sham: 106.83 ± 8.38 in n=8 slices from 3 rats, FPI: 110.29 ± 11.84 in n=7 slices from 3 rats, p>0.05, NMDA EPSC charge transfer in pA.sec, sham: 5.29 ± 1.16, FPI: 6.40 ± 1.68, n=7 slices from 3 rats each, p>0.05 by two way ANOVA). Crucially, LPS-RS incubation failed to reduce NMDA EPSC peak amplitude in slices from both sham- and FPI rats (Fig. 6A, NMDA EPSC amplitude in pA, sham in aCSF: 106.83 ± 8.38, sham after LPS-RS treatment: 103.35 ± 7.46, n=8 slices from 3 rats each, p>0.05, FPI in aCSF: 110.29 ± 11.84, FPI after LPS-RS treatment: 101.53 ± 5.06, n=7 slices from 3 rats each, p>0.05 by post-hoc Tukey’s test). Similarly, LPS-RS incubation did not alter NMDA EPSC charge transfer (NMDA EPSC charge transfer in pA.sec, sham in aCSF: 5.29 ± 1.16, sham after LPS-RS treatment: 5.37 ± 0.52, n=8 slices from 3 rats each, p>0.05, FPI in aCSF: 6.40 ± 1.68, FPI after LPS-RS treatment: 5.41 ± 0.62, n=7 slices from 3 rats each, p>0.05 by post-hoc Tukey’s test). Neither injury nor LPS-RS had a significant effect on granule cell NMDA EPSC amplitude (effect of injury: F1,24 =0.01, p>0.05; effect of LPS-RS F1,24=0.77, p>0.05, by two way ANOVA) or charge transfer (effect of injury: F1,24 =0.28, p>0.05; effect of LPS-RS F1,24 =0.17, p>0.05, by two way ANOVA). Additionally, there was no interaction between effect of injury and LPS-RS on granule cell NMDA EPSC amplitude or charge transfer (NMDA EPSC amplitude: F1,24=0.16, p>0.05, NMDA EPSC charge transfer: F1,24=0.24, p>0.05 by two way ANOVA). These data confirm that LPS-RS mediated decrease in dentate excitability does not result from modulation of granule cell NMDA currents.
Figure 6. Lack of LPS-RS modulation of granule cell NMDA EPSCs.

A–D. Representative granule cell NMDA excitatory post-synaptic current (EPSC) traces in slices from sham-injured control rats in artificial cerebro spinal fluid (aCSF) (A) and after LPS-RS treatment (B) and in FPI rats in aCSF (C) and after LPS-RS treatment (D). Recordings were obtained at a holding potential of +40 mV in response to a 4 mA stimulation of the perforant path in the presence of gabazine (20 μM) and 6,7-dinitroquinoxaline-2,3-dione (DNQX, 20 μM). E–F. Summary data showing the lack of a post-FPI alteration or of LPS-RS treatment in the amplitude (E) and synaptic charge transfer (F) of NMDA EPSCs.
TLR4 antagonist modulates non-NMDA glutamatergic currents after brain injury
Earlier work has identified a post-traumatic increase in granule cell non-NMDA EPSC charge transfer, resulting from polysynaptic activity involving the dentate hilus (Santhakumar et al., 2000). Since TLR4 antagonists suppressed the post-traumatic increase in afferent evoked dentate excitability, we tested the ability of LPS-RS to reduce granule cell non-NMDA EPSC. Granule cells were held at −70 mV and non-NMDA EPSCs were isolated by recording responses to a 1 mA stimulus to the perforant path in the presence of the GABAA receptor antagonist gabazine (20 μM) and NMDA receptor antagonist APV (50 μM). Non-NMDA EPSCs were fully blocked by the non-NMDA ionotropic glutamatergic receptor antagonist DNQX (20 μM) in 3 of 3 cells tested from sham and FPI rats each (not shown). LPS-RS treatment had no effect on granule cell non-NMDA EPSC peak current amplitude or charge transfer in sham-injured rats (Fig. 7 A–B, E–F, non-NMDA EPSC amplitude in pA, sham in aCSF: 96.29 ± 11.48, sham after LPS-RS treatment: 75.67 ± 11.48, n=9 slices from 5 rats each, p>0.05 by Tukey’s test, non-NMDA EPSCs charge transfer in pA.sec, sham in aCSF: 4.57 ± 0.44, sham after LPS-RS treatment: 4.39 ± 0.34, n=9 slices from 5 rats each, p>0.05 by Tukey’s test). The lack of LPS-RS modulation of granule cell NMDA and non-NMDA EPSCs in sham rats (Fig. 6 and 7) suggests that effects on ionotropic glutamatergic currents do not underlie the tonic reduction of dentate network excitability by TLR4 in control rats. While earlier work revealed a small post-traumatic increase in granule cell non-NMDA EPSC amplitude that failed to reach statistical significance (Santhakumar et al., 2000), we find a significant increase in granule cell non-NMDA EPSC peak amplitude after FPI (Fig 7A, C E, non-NMDA EPSC amplitude in pA, sham in aCSF: 96.29 ± 11.48 in n=9 slices from 5 rats, FPI in aCSF: 139.69± 13.64, n=10 slices from 6 rats, p<0.05, by two way ANOVA followed by Tukey’s test). Moreover, consistent with earlier results (Santhakumar et al., 2000), granule cell non-NMDA EPSC charge transfer was also enhanced 1 week after FPI (Fig. 7 A, C, F, non-NMDA EPSC charge transfer in pA.sec, sham in aCSF: 4.57 ± 0.44 in n=9 slices from 5 rats, FPI in aCSF: 7.28 ± 0.85, n=10 slices from 6 rats, p<0.05 by two way ANOVA followed by Tukey’s test). LPS-RS incubation reduced both non-NMDA EPSC peak amplitude and charge transfer in slices from FPI rats (Fig. 7C–E, non-NMDA EPSCs amplitude in pA, FPI in aCSF: 139.69± 13.64, FPI after LPS-RS treatment: 92.44 ± 10.92, n=10 slices from 6 rats each, p<0.05,, non-NMDA EPSC charge transfer in pA.sec, FPI in aCSF: 7.28 ± 0.85, FPI after LPS-RS treatment: 4.69 ± 0.45, n=10 slices from 6 rats each, p<0.05 by two way ANOVA followed by post-hoc Tukey’s test). Statistical comparisons showed a significant effect of injury (F1,34 =6.532, p<0.05 by two way ANOVA) and LPS-RS (F1,34 =8.411, p<0.05 by two way ANOVA) on granule cell non-NMDA EPSC amplitude. However, the interaction between effect of injury and LPS-RS on EPSC amplitude was not statistically significant (F1,34=1.15, p>0.05 by two way ANOVA). The effect of injury (F1,34 =12.612, p<0.005 by two way ANOVA) and LPS-RS (F1,34 =9.985, p<0.005 by two way ANOVA) and the interaction between effects of injury and LPS-RS (F1,34=7.263, p<0.005 by two way ANOVA) on granule cell non-NMDA EPSC charge transfer were significant. Thus, consistent with the proposal that ongoing TLR4 modulation of non-NMDA EPSCs contributes to post-injury increase in dentate excitability, LPS-RS acutely decreased granule cell non-NMDA EPSC amplitude and charge transfer and dentate excitability after FPI.
Figure 7. LPS-RS reduces granule cell non-NMDA currents after brain injury.

A–D. Representative voltage clamp recordings of granule cell non-NMDA EPSCs from control rats in aCSF (A) and after LPS-RS incubation (B) and FPI rats in aCSF (C) and after LPS-RS incubation (D). LPS-RS does not alter the monosynaptic EPSC amplitude in control rats but reduces it FPI rats. FPI results in appearance of a late polysynaptic EPSC which is also reduced by LPS-RS treatment. Responses were evoked by a 1 mA stimulus to the perforant path and recorded at −70 mV in gabazine (20 μM) and APV (50 μM). E–F., Summary data of the effects of FPI and LPS-RS treatment on granule cell non-NMDA EPSC peak amplitude (E) and synaptic charge transfer (F). Error bars indicate s.e.m. * indicates p<0.05 by two-way ANOVA followed by Tukey’s post hoc test.
In addition to suppressing granule cell non-NMDA EPSC peak amplitude (Fig. 7), LPS-RS mediated reduction of granule cell charge transfer indicates that TLR4 signaling may increase dentate excitability after FPI, in part, by enhancing polysynaptic non-NMDA EPSCs. Previous studies have identified that excitatory hilar mossy cells, which form recurrent excitatory feedback synapses on granule cells (Buckmaster et al., 1996), are the most likely source of the polysynaptic non-NMDA EPSCs in granule cells after brain injury(Santhakumar et al., 2000; Ratzliff et al., 2002; Howard et al., 2007). Since over 90% of hilar neurons showed TLR4 expression 24 hours after FPI (Fig. 2) and mossy cells constitute approximately 50% of hilar neurons (Dyhrfjeld-Johnsen et al., 2007) we reasoned that hilar mossy cells express TLR4. Immunostaining for calcitonin gene-related peptide (CGRP), a marker for mossy cells(Freund et al., 1997), and TLR4 revealed that mossy cells express TLR4 24 hours (not shown) and 1 week after FPI and in age-matched controls (Fig. 8A, based on observations from 3 slices each from 2 sham and 2 FPI rats at each time point). Therefore, we directly examined the effect of brain injury and LPS-RS on perforant path evoked non-NMDA EPSCs in dentate hilar mossy cells. As illustrated in Figure 8 B, mossy cells were identified by their characteristic multipolar somata, dendrites in the hilus and the presence of thorny excrescences (Frotscher et al., 1991; Freund et al., 1997; Santhakumar et al., 2000). Mossy cells were held at −60 mV and non-NMDA EPSCs were recorded in responses to a 0.5 mA stimulus to the perforant path in the presence of gabazine (20 μM) and APV (50 μM). Perforant path evoked non-NMDA EPSCs in mossy cells had a longer latency than in granule cells (Latency in ms, granule cell: 1.88±0.26, n=30; mossy cell: 8.14±0.54, n=30, p<0.05 by Student’s t-test, control and FPI cells were pooled). This is consistent with polysynaptic activation of mossy cells following perforant path stimulation since only approximately 15% of mossy cells have molecular layer dendrites capable of receiving direct perforant path inputs (Scharfman, 1991; Buckmaster et al., 1992). Perforant path-evoked mossy cell non-NMDA EPSC amplitude and charge transfer were increased 1 week after FPI (Fig. 8 C–H, non-NMDA EPSC amplitude in pA, sham in aCSF: 89.90 ± 13.86 in n=5 slices from 4 rats, FPI in aCSF: 189.49 ± 11.88 in n=6 slices from 5 rats, p<0.05 by two way ANOVA followed by Tukey’s test, non-NMDA EPSC charge transfer in pA.sec, sham in aCSF: 3.88 ± 0.80, n=5 slices from 4 rats, FPI in aCSF: 7.44 ± 0.50, n=6 slices from 5 rats, p<0.05 by two way ANOVA followed by Tukey’s test). LPS-RS incubation failed to modulate non-NMDA EPSC peak amplitude in slices from control rats (Fig. 8 C, D and G, non-NMDA EPSC amplitude in pA, sham in aCSF: 89.90 ± 13.86, sham after LPS-RS treatment: 84.85 ± 14.43, n=5 slices from 4 rats each, p>0.05, Tukey’s test, non-NMDA EPSC charge transfer in pA.sec, sham in aCSF: 3.88 ± 0.80, sham after LPS-RS treatment: 3.56 ± 0.39, n=5 slices from 4 rats each, p>0.05 by Tukey’s test). However, LPS-RS significantly reduced both peak amplitude and charge transfer of mossy cell non-NMDA EPSCs from rats 1 week after FPI (Fig. 8 E–H, non-NMDA EPSC amplitude in pA, FPI in aCSF: 189.49 ± 11.88 in n=6 slices from 5 rats, FPI after LPS-RS treatment: 135.20 ± 5.89, n=5 slices from 5 rats, p<0.05 by two way ANOVA followed by post-hoc Tukey’s test, non-NMDA EPSC charge transfer in pA.sec, FPI in aCSF: 7.44 ± 1.22 in n=6 slices from 5 rats, FPI after LPS-RS treatment: 5.35 ± 0.20, n=5 slices from 5 rats, p<0.05 by two way ANOVA followed by post-hoc Tukey’s test). These findings are summarized in Table 1. Overall, there was a significant effect of both injury and LPS-RS on mossy cell non-NMDA EPSC amplitude (effect of injury: F1,17 =26.71, p<0.05; effect of LPS-RS F1,17 =5.43, p<0.005, by two way ANOVA) and charge transfer (effect of injury: F1,17 =38.55, p<0.005; effect of LPS-RS F1,17 =6.04, p<0.05, by two way ANOVA). However, the interaction between effect of injury and LPS-RS on mossy cell EPSC amplitude and charge transfer were not statistically significant (EPSC amplitude: F1,17=2.923, p>0.05 by two way ANOVA, charge transfer: F1,17=4.157, p>0.05 by two way ANOVA).
Figure 8. LPS-RS reduces post-traumatic increase in mossy cell non-NMDA currents.

A. Confocal images of sections obtained one week after sham (left) or FPI (right) show immunostaining for TLR4 (up), calcitonin gene-related peptide (CGRP, center) and merged images (bottom). Scale bar: 25 μm.B. Maximum intensity projections of confocal image stacks of biocytin-filled mossy cells in slices from sham- (left) and FPI rats (right) shows the typical morphology with large multipolar somata, dendrites in the hilus and axon in the inner molecular layer (arrows). Insets: images of the complex spines characteristic of mossy cells. Grey scale fluorescence images are shown in reverse color scheme for better visualization. Scale bar, 50 μm. C–F. Representative recordings of mossy cell non-NMDA EPSCs from control rats in aCSF (C) and after LPS-RS incubation (D) and FPI rats in aCSF (E) and after LPS-RS incubation (F), illustrate the post-traumatic increase in the monosynaptic EPSC peak amplitude and LPS-RS modulation EPSC amplitude in FPI rats. FPI results in appearance of a late polysynaptic EPSC which is also reduced by LPS-RS treatment. Responses were evoked by a 0.5 mA stimulus to the perforant path and recorded in voltage clamp at −60 mV in the presence of gabazine (20 μM) and APV (50 μM). G–H. Summary data of the effects of FPI and LPS-RS treatment on mossy cell non-NMDA EPSC peak amplitude (G) and synaptic charge transfer (H). Error bars indicate s.e.m. * indicates p<0.05 by two-way ANOVA followed by Tukey’s post hoc test.
Taken together, our findings demonstrate that TLR4 signaling contributes to early increase in dentate excitability after brain injury by NMDA receptor-independent mechanisms involving a novel enhancement of synaptic excitation in dentate granule cells and mossy cell through modulation of non-NMDA glutamate receptors.
Discussion
The potential for interaction between the brain’s electrical and immunological responses is particularly relevant to concussive brain injury, which elicits both acute neurophysiological (Ross and Soltesz, 2000) and sterile inflammatory responses (Redell et al., 2013). Our data demonstrate that TLR4 signaling contributes to an early increase in excitability of the hippocampal dentate gyrus after brain injury, which contrasts with a novel TLR4-dependent tonic attenuation of dentate excitability in controls. We report, for the first time, that TLR4 antagonists reduce excitatory non-NMDA glutamatergic currents in vitro after brain injury, indicating an active role for TLR4 signaling in post-traumatic increase in dentate excitability. The time course of the post-traumatic increase in TLR4 expression in the dentate molecular layer and hilar neurons is consistent with the early dentate hyperexcitability days to a week after FPI which resolves within a month after injury (Santhakumar et al., 2001; Santhakumar et al., 2003). While NMDA currents do not appear to contribute to TLR4 modulation of dentate excitability in the injured brain, our data reveal a novel TLR4-dependent enhancement of granule cell non-NMDA EPSCs after brain injury. The post-traumatic augmentation in mossy cells non-NMDA EPSC peak amplitude and charge transfer, identified herein, provide the long sought mechanistic basis for the increase in evoked mossy cell excitability after brain injury despite homeostatic regulation of intrinsic excitability (Santhakumar et al., 2000; Howard et al., 2007). Crucially, we demonstrate that TLR4-dependent signaling plays an active role in enhancing granule cell and mossy cell non-NMDA EPSCs after FPI. The apparent switch in the physiological effects of TLR4 signaling from reducing network excitability at control levels of expression to a pathological augmentation of non-NMDA EPSCs with increase in neuronal TLR4 expression is a potential mechanism by which recruitment of immune responses in the brain may modify neuronal excitability in certain diseases.
Enhanced TLR4 expression in the injured dentate gyrus
There is ample evidence for activation of inflammatory responses including early and transient elevation of TLR4 expression in other models of brain injury including cortical contusion (Chen et al., 2012; Mao et al., 2012). However, since most studies examined whole brain extracts, information regarding the regional changes in TLR4 after brain injury is limited (Chen et al., 2008; Chen et al., 2009). Additionally, there are discrepancies in the literature on expression of TLR4 in neurons and microglia after brain injury (Chen et al., 2012; Mao et al., 2012). Moreover, while TLR4 expression was reported in cortical but not hippocampal neurons after contusive brain injury (Chen et al., 2012), TLR4 co-localizes with hippocampal neurons in epilepsy (Maroso et al., 2010). Although regional and disease specific changes in TLR4 could have contributed to some of the conflicting observations, we find that several commercial TLR4 antibodies show non-specific immunostaining in tissue from mice lacking TLR4 (not shown). Using antibodies tested to eliminate non-specific labeling in TLR4 knockout mice (Figs. 1 and 2), we show that moderate concussive brain injury leads to a rapid increase in hippocampal TLR4 expression within 4 hours, earlier than previously reported (Mao et al., 2012). Hippocampal TLR4 expression reached a maximum at 24 hours after injury, consistent with results obtained from whole brain samples after focal brain injury (Mao et al., 2012). Although TLR4 levels in hippocampal tissue returned to control levels 7 days after injury, semi-quantitative analysis of sections immunolabeled for TLR4 identified significant post-injury increase in TLR4 levels in dentate molecular layer and hilus at this time point (Fig. 1,2), suggesting regional differences in injury-induced changes in TLR4 expression. While microglia, the resident immune cells of the central nervous system, are assumed to express TLR4, our finding that TLR4 in the dentate hilus is exclusively expressed in neurons not microglia is consistent with reports in the injured cortex and epileptic hippocampus (Maroso et al., 2010; Chen et al., 2012). Interestingly, TLR4 has been found to label microglia in multiple sclerosis but not epilepsy (Maroso et al., 2010), suggesting disease specific regulation of cellular expression of TLR4. Similarly, the focal nature of cortical contusion trauma could account for the lack of neuronal expression of TLR4 in the hippocampus after penetrating cortical trauma (Chen et al., 2012). Crucially, the post-traumatic increase in TLR4 expression in dentate neurons and not glia following concussive brain injury suggests a role for neuronal TLR4 signaling in post-traumatic dentate hyperexcitability. Additionally, TLR4 immunostaining in the molecular layer and surrounding unstained spaces in the granule cell layer (Fig. 2A–B) suggest that granule cell dendrites and possibly, interneuronal axons may express TLR4. Moreover, since TLR4 co-localized with over 70% of hilar neurons for up to 7 days after injury, GABAergic neuronal populations are also likely to express TLR4 early after brain injury.
Tonic bidirectional modulation of network excitability by TLR4
Our data summarized in Table 1 show that TLR4 ligands acutely modify network excitability in both sham- and brain-injured rats indicating a steady-state neurophysiological effect of TLR4 signaling. The ongoing basal neurophysiological effects of TLR4 could underlie the recently identified contribution of TLR4 to hippocampal long-term potentiation and memory function (Costello et al., 2011; Okun et al., 2012). The cellular mechanisms underlying TLR4 modulation of neurophysiology in the normal brain remain to be elucidated. However, in spite of the failure of LPS-RS to alter both NMDA and non-NMDA EPSCs in granule cells from control rats, the inability of LPS-RS to enhance dentate excitability following incubation of control slices in APV suggests that NMDA-dependent signaling may contribute to the tonic neurophysiological actions of TLR4. Alternatively, TLR4 signaling could influence dentate excitability in control rats by its effect on other receptor classes including metabotropic glutamate receptors and transient receptor potential channels which are regulated by TLR4 in certain central and peripheral neurons (Diogenes et al., 2011; Pascual et al., 2012). It is intriguing that TLR4 ligand modulation of network excitability in the injured dentate gyrus, the focus of the current study, was opposite to that observed in controls. It is possible that complementary downstream signaling pathways are engaged at different TLR4 expression levels or following trauma. Our finding that post-traumatic TLR4 signaling increases excitability after brain injury is consistent with the ability of TLR4 agonists to promote seizures (Rodgers et al., 2009; Maroso et al., 2010). However, unlike epilepsy where TLR4 acts through NMDA dependent pathways (Maroso et al., 2010), we find that NMDA currents are not involved in TLR4-mediated post-traumatic increase in dentate excitability. Since early increase in dentate excitability after brain injury is suggested to contribute to development of post-traumatic epilepsy (Lowenstein et al., 1992; Toth et al., 1997; Santhakumar et al., 2001), the ability of TLR4 antagonists to reduce dentate excitability after brain injury indicates that TLR4 may be targeted to reduce risk for epilepsy in brain injured patients. Indeed, several immunoactive drugs that reduce secondary neuronal degeneration and improve motor and cognitive outcome after brain injury have been suggested to reduce TLR4 signaling (Chen et al., 2009; Chen et al., 2012; Mao et al., 2012; Zhu et al., 2014). Our findings provide the first direct mechanistic link between TLR4 and post-traumatic neurological dysfunction and suggest a physiological mechanism by with TLR4 antagonism may reduce neuropathology.
TLR4 modulation of non-NMDA ionotropic glutamate currents contributes to post-traumatic dentate hyperexcitability
Apart from changes in inhibition (Lowenstein et al., 1992; Toth et al., 1997; Gupta et al., 2012) brain injury leads to increased excitation of granule cells (Santhakumar et al., 2000), we find that TLR4 antagonist reduces post-injury increases in granule cell monosynaptic and polysynaptic non-NMDA EPSCs to control levels. This suggests that blocking ongoing TLR4 modulation of non-NMDA EPSCs may underlie the ability of TLR4 antagonists to reduce dentate excitability after brain injury. Additionally, since mossy cells generate EPSCs in granule cells (Chancey et al., 2014) and contribute to dentate excitability (Ratzliff et al., 2004), and since granule cell polysynaptic activity is dependent on hilar excitatory inputs (Santhakumar et al., 2000), the post-injury increase in synaptic excitation of mossy cells likely underlies increases in granule cell polysynaptic activity and dentate excitability. Indeed, our demonstration of post-FPI increase in mossy cell non-NMDA currents is consistent with the larger spontaneous EPSC amplitude in mossy cells one week after FPI (Howard et al., 2007). Thus, it is possible that the TLR4-dependent increases in granule cell and mossy cell non-NMDA EPSCs (Fig. 7 and 8) identified in this study, contribute substantially to early post-traumatic dentate hyperexcitability. While LPS-RS reduced granule cell non-NMDA currents to control levels (Fig. 7), LPS-RS reversal of dentate network excitability, while significant, was not complete (Fig. 3E). Thus, although acute treatment with TLR4 antagonists significantly reduces post-traumatic dentate hyperexcitability, TLR4-independent mechanisms including network structural changes (Santhakumar et al., 2001) likely act in concert with the neurophysiological effects of TLR4 to enhance dentate excitability after brain injury. Further studies are needed to identify the molecular mechanisms by which TLR4 enhances non-NMDA EPSCs after FPI. An interesting candidate mechanism involves, tumor necrosis factor alpha (TNFα), a downstream effector of TLR4 signaling, which has been shown to regulate AMPA receptor trafficking (Beattie et al., 2002).
Our findings demonstrate a novel TLR4-dependent enhancement of non-NMDA glutamatergic synaptic currents in dentate mossy cells and granule cells after brain injury. Although TLR4 signaling may provide a valuable target for therapeutic interventions to prevent secondary neuronal degeneration and development of epilepsy after brain injury, the opposing physiological effects of tonic TLR4 signaling, identified herein, need to be considered before manipulating TLR4 signaling.
Supplementary Material
Highlights.
Early increase in dentate neuronal TLR4 expression after concussive brain injury
Acute and opposite effects of TLR4 on excitability of normal and injured brain
NMDA receptors do not underlie TLR4 effects on post-traumatic dentate excitability
Novel TLR4-mediated enhancement of mossy cell non-NMDA currents after brain injury
Continuing TLR4 signaling enhances dentate excitability one week after brain injury
Acknowledgments
We thank Drs. Ellen Townes-Anderson, Christine Rohowsky-Kochan and Eldo Kuzhikandathil, Rutgers NJMS for their extensive support with immunostaining and western blotting studies Dr. Robert F. Heary, Rutgers NJMS for equipment and Ms. Archana Proddutur for her helpful comments and technical assistance. The project was supported by CURE Foundation, F.M. Kirby Foundation, NJCBIR CBIR11PJT003, CBIR14IRG024 and NIH/NINDS R01 NS069861 to V.S.
Abbreviations
- aCSF
artificial Cerebrospinal Fluid
- AMPA
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- APV
(2R)-amino-5-phosphonovaleric acid
- A.U
Arbitrary Units
- CGRP
calcitonin gene-related peptide
- DNQX
6,7-dinitroquinoxaline-2,3-dione
- EPSC
Excitatory postsynaptic current
- FPI
Fluid Percussion Injury
- GFAP
Glial fibrillary acidic protein
- HMGB1
High-mobility group box 1
- Iba1
Ionized calcium binding adaptor molecule 1
- LPS-RS
Lipopolysaccharide from Rhodobacter sphaeroides
- NeuN
Neuronal Nuclei
- NMDA
N-Methyl-D-aspartate
- PBS
Phosphate buffered saline
- TLR4
toll-like receptor 4
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could a3ect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Aradi I, Santhakumar V, Soltesz I. Impact of heterogeneous perisomatic IPSC populations on pyramidal cell firing rates. J Neurophysiol. 2004;91:2849–2858. doi: 10.1152/jn.00916.2003. [DOI] [PubMed] [Google Scholar]
- Balosso S, Liu J, Bianchi ME, Vezzani A. Disulfide-Containing High Mobility Group Box-1 Promotes N-Methyl-d-Aspartate Receptor Function and Excitotoxicity by Activating Toll-Like Receptor 4-Dependent Signaling in Hippocampal Neurons. Antioxidants & redox signaling. 2014 doi: 10.1089/ars.2013.5349. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bhaskaran MD, Smith BN. Effects of TRPV1 activation on synaptic excitation in the dentate gyrus of a mouse model of temporal lobe epilepsy. Experimental neurology. 2010;223:529–536. doi: 10.1016/j.expneurol.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Wenzel HJ, Kunkel DD, Schwartzkroin PA. Axon arbors and synaptic connections of hippocampal mossy cells in the rat in vivo. Journal of Comparative Neurology. 1996;366:271–292. doi: 10.1002/(sici)1096-9861(19960304)366:2<270::aid-cne7>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Strowbridge BW, Kunkel DD, Schmiege DL, Schwartzkroin PA. Mossy cell axonal projections to the dentate gyrus molecular layer in the rat hippocampal slice. Hippocampus. 1992;2:349–362. doi: 10.1002/hipo.450020403. [DOI] [PubMed] [Google Scholar]
- Chancey JH, Poulsen DJ, Wadiche JI, Overstreet-Wadiche L. Hilar mossy cells provide the first glutamatergic synapses to adult-born dentate granule cells. J Neurosci. 2014;34:2349–2354. doi: 10.1523/JNEUROSCI.3620-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Hung TH, Wang YH, Lin CW, Wang PY, Lee CY, Chen SF. Wogonin improves histological and functional outcomes, and reduces activation of TLR4/NF-kappaB signaling after experimental traumatic brain injury. PLoS One. 2012;7:e30294. doi: 10.1371/journal.pone.0030294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Zhang S, Shi J, Ai J, Qi M, Hang C. Simvastatin reduces secondary brain injury caused by cortical contusion in rats: possible involvement of TLR4/NF-kappaB pathway. Exp Neurol. 2009;216:398–406. doi: 10.1016/j.expneurol.2008.12.019. [DOI] [PubMed] [Google Scholar]
- Chen G, Shi J, Jin W, Wang L, Xie W, Sun J, hang C. Progesterone Administration Modulates TLRs/NF-κ BSignaling Pathway in Rat Brain after Cortical Contusion. Annals of Clinical & Laboratory Science. 2008;38:65–74. [PubMed] [Google Scholar]
- Chenard BL, Shalaby IA, Koe BK, Ronau RT, Butler TW, Prochniak MA, Schmidt AW, Fox CB. Separation of alpha 1 adrenergic and N-methyl-D-aspartate antagonist activity in a series of ifenprodil compounds. J Med Chem. 1991;34:3085–3090. doi: 10.1021/jm00114a018. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Pfister BJ, Schwarzbach E, Grady MS, Goforth PB, Satin LS. Injury-induced alterations in CNS electrophysiology. Progress in Brain Research. 2007;161:143–169. doi: 10.1016/S0079-6123(06)61010-8. [DOI] [PubMed] [Google Scholar]
- Colicos MA, Dixon CE, Dash PK. Delayed, selective neuronal death following experimental cortical impact injury in rats: possible role in memory deficits. Brain Res. 1996;739:111–119. doi: 10.1016/s0006-8993(96)00819-0. [DOI] [PubMed] [Google Scholar]
- Costello DA, Watson MB, Cowley TR, Murphy N, Murphy Royal C, Garlanda C, Lynch MA. Interleukin-1alpha and HMGB1 mediate hippocampal dysfunction in SIGIRR-deficient mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:3871–3879. doi: 10.1523/JNEUROSCI.6676-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, Edwards MR, Michelsen KS, Kroeger KM, Liu C, Muhammad AK, Clark MC, Arditi M, Comin-Anduix B, Ribas A, Lowenstein PR, Castro MG. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS medicine. 2009;6:e10. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dileonardi AM, Huh JW, Raghupathi R. Differential effects of FK506 on structural and functional axonal deficits after diffuse brain injury in the immature rat. J Neuropathol Exp Neurol. 2012;71:959–972. doi: 10.1097/NEN.0b013e31826f5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diogenes A, Ferraz CC, Akopian AN, Henry MA, Hargreaves KM. LPS sensitizes TRPV1 via activation of TLR4 in trigeminal sensory neurons. J Dent Res. 2011;90:759–764. doi: 10.1177/0022034511400225. [DOI] [PubMed] [Google Scholar]
- Dyhrfjeld-Johnsen J, Santhakumar V, Morgan RJ, Huerta R, Tsimring L, Soltesz I. Topological determinants of epileptogenesis in large-scale structural and functional models of the dentate gyrus derived from experimental data. JNeurophysiol. 2007;97:1566–1587. doi: 10.1152/jn.00950.2006. [DOI] [PubMed] [Google Scholar]
- Fan H, Li L, Zhang X, Liu Y, Yang C, Yang Y, Yin J. Oxymatrine downregulates TLR4, TLR2, MyD88, and NF-kappaB and protects rat brains against focal ischemia. Mediators Inflamm. 2009;2009:704706. doi: 10.1155/2009/704706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario CR, Ndukwe BO, Ren J, Satin LS, Goforth PB. Stretch injury selectively enhances extrasynaptic, GluN2B-containing NMDA receptor function in cortical neurons. J Neurophysiol. 2013;110:131–140. doi: 10.1152/jn.01011.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Hajos N, Acsady L, Gorcs TJ, Katona I. Mossy cells of the rat dentate gyrus are immunoreactive for calcitonin gene-related peptide (CGRP) EurJ Neurosci. 1997;9:1815–1830. doi: 10.1111/j.1460-9568.1997.tb00748.x. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Seress L, Schwerdtfeger WK, Buhl E. The mossy cells of the fascia dentata: a comparative study of their fine structure and synaptic connections in rodents and primates. JComp Neurol. 1991;312:145–163. doi: 10.1002/cne.903120111. [DOI] [PubMed] [Google Scholar]
- Fukuda AM, Pop V, Spagnoli D, Ashwal S, Obenaus A, Badaut J. Delayed increase of astrocytic aquaporin 4 after juvenile traumatic brain injury: possible role in edema resolution? Neuroscience. 2012;222:366–378. doi: 10.1016/j.neuroscience.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Fang X, Sun H, Wang Y, Yao LJ, Li JP, Tong Y, Zhang B, Liu Y. Toll-like receptor 4-mediated myeloid differentiation factor 88-dependent signaling pathway is activated by cerebral ischemia-reperfusion in hippocampal CA1 region in mice. Biol Pharm Bull. 2009;32:1665–1671. doi: 10.1248/bpb.32.1665. [DOI] [PubMed] [Google Scholar]
- Goforth PB, Ellis EF, Satin LS. Enhancement of AMPA-mediated current after traumatic injury in cortical neurons. J Neurosci. 1999;19:7367–7374. doi: 10.1523/JNEUROSCI.19-17-07367.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Elgammal FS, Proddutur A, Shah S, Santhakumar V. Decrease in Tonic Inhibition Contributes to Increase in Dentate Semilunar Granule Cell. Journal of Neuroscience. 2012;32:2523–2537. doi: 10.1523/JNEUROSCI.4141-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard AL, Neu A, Morgan RJ, Echegoyen JC, Soltesz I. Opposing modifications in intrinsic currents and synaptic inputs in post-traumatic mossy cells: evidence for single-cell homeostasis in a hyperexcitable network. JNeurophysiol. 2007;97:2394–2409. doi: 10.1152/jn.00509.2006. [DOI] [PubMed] [Google Scholar]
- Hua F, Ma J, Ha T, Xia Y, Kelley J, Williams DL, Kao RL, Browder IW, Schweitzer JB, Kalbfleisch JH, Li C. Activation of Toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J Neuroimmunol. 2007;190:101–111. doi: 10.1016/j.jneuroim.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol. 2007;66:989–1001. doi: 10.1097/NEN.0b013e3181588245. [DOI] [PubMed] [Google Scholar]
- Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83:711–730. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Qu C, Goussev A, Jiang H, Lu C, Schallert T, Mahmood A, Chen J, Li Y, Chopp M. Statins increase neurogenesis in the dentate gyrus, reduce delayed neuronal death in the hippocampal CA3 region, and improve spatial learning in rat after traumatic brain injury. J Neurotrauma. 2007;24:1132–1146. doi: 10.1089/neu.2007.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao SS, Hua R, Zhao XP, Qin X, Sun ZQ, Zhang Y, Wu YQ, Jia MX, Cao JL, Zhang YM. Exogenous administration of PACAP alleviates traumatic brain injury in rats through a mechanism involving the TLR4/MyD88/NF-kappaB pathway. J Neurotrauma. 2012;29:1941–1959. doi: 10.1089/neu.2011.2244. [DOI] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, Bianchi ME, Vezzani A. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–419. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- Neese SL, Sherill LK, Tan AA, Roosevelt RW, Browning RA, Smith DC, Duke A, Clough RW. Vagus nerve stimulation may protect GABAergic neurons following traumatic brain injury in rats: An immunocytochemical study. Brain Res. 2007;1128:157–163. doi: 10.1016/j.brainres.2006.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuberger EJ, Abdul-Wahab R, Jayakumar A, Pfister BJ, Santhakumar V. Distinct effect of impact rise times on immediate and early neuropathology after brain injury in juvenile rats. Journal of Neuroscience Research. 2014 doi: 10.1002/jnr.23401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, Glass CK. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E, Griffioen KJ, Mattson MP. Toll-like receptor signaling in neural plasticity and disease. Trends in neurosciences. 2011;34:269–281. doi: 10.1016/j.tins.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E, Barak B, Saada-Madar R, Rothman SM, Griffioen KJ, Roberts N, Castro K, Mughal MR, Pita MA, Stranahan AM, Arumugam TV, Mattson MP. Evidence for a developmental role for TLR4 in learning and memory. PLoS One. 2012;7:e47522. doi: 10.1371/journal.pone.0047522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva AA, Jr, Kang Y, Sanchez-Molano J, Furones C, Atkins CM. STAT3 signaling after traumatic brain injury. J Neurochem. 2012;120:710–720. doi: 10.1111/j.1471-4159.2011.07610.x. [DOI] [PubMed] [Google Scholar]
- Pang KCH, Sinha S, Avcu P, Roland JJ, Nadpara N, Pfister BJ, Long M, Santhakumar V, Servatius RJ. Long-lasting suppression of acoustic startle response following mild traumatic brain injury. J Neurotrauma. 2014 doi: 10.1089/neu.2014.3451. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E197–205. doi: 10.1073/pnas.1111098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proddutur A, Yu J, Elgammal FS, Santhakumar V. Seizure-induced alterations in fast-spiking basket cell GABA currents modulate frequency and coherence of gamma oscillation in network simulations. Chaos. 2013;23:046109. doi: 10.1063/1.4830138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratzliff AH, Santhakumar V, Howard A, Soltesz I. Mossy cells in epilepsy: rigor mortis or vigor mortis? Trends Neurosci. 2002;25:140–144. doi: 10.1016/s0166-2236(00)02122-6. [DOI] [PubMed] [Google Scholar]
- Ratzliff AH, Howard AL, Santhakumar V, Osapay I, Soltesz I. Rapid deletion of mossy cells does not result in a hyperexcitable dentate gyrus: implications for epileptogenesis. J Neurosci. 2004;24:2259–2269. doi: 10.1523/JNEUROSCI.5191-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re F, Strominger JL. Monomeric recombinant MD-2 binds toll-like receptor 4 tightly and confers lipopolysaccharide responsiveness. J Biol Chem. 2002;277:23427–23432. doi: 10.1074/jbc.M202554200. [DOI] [PubMed] [Google Scholar]
- Redell JB, Moore AN, Grill RJ, Johnson D, Zhao J, Liu Y, Dash PK. Analysis of functional pathways altered after mild traumatic brain injury. J Neurotrauma. 2013;30:752–764. doi: 10.1089/neu.2012.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers KM, Hutchinson MR, Northcutt A, Maier SF, Watkins LR, Barth DS. The cortical innate immune response increases local neuronal excitability leading to seizures. Brain. 2009;132:2478–2486. doi: 10.1093/brain/awp177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross ST, Soltesz I. Selective depolarization of interneurons in the early posttraumatic dentate gyrus: involvement of the Na(+)/K(+)-ATPase. J Neurophysiol. 2000;83:2916–2930. doi: 10.1152/jn.2000.83.5.2916. [DOI] [PubMed] [Google Scholar]
- Santhakumar V, Voipio J, Kaila K, Soltesz I. Post-traumatic hyperexcitability is not caused by impaired buffering of extracellular potassium. J Neurosci. 2003;23:5865–5876. doi: 10.1523/JNEUROSCI.23-13-05865.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhakumar V, Ratzliff AD, Jeng J, Toth K, Soltesz I. Long-term hyperexcitability in the hippocampus after experimental head trauma. Ann Neurol. 2001;50:708–717. doi: 10.1002/ana.1230. [DOI] [PubMed] [Google Scholar]
- Santhakumar V, Bender R, Frotscher M, Ross ST, Hollrigel GS, Toth Z, Soltesz I. Granule cell hyperexcitability in the early post-traumatic rat dentate gyrus: the ‘irritable mossy cell’ hypothesis. J Physiol. 2000;524(Pt 1):117–134. doi: 10.1111/j.1469-7793.2000.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE. Dentate hilar cells with dendrites in the molecular layer have lower thresholds for synaptic activation by perforant path than granule cells. J Neurosci. 1991;11:1660–1673. doi: 10.1523/JNEUROSCI.11-06-01660.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D. The contribution of TNFalpha to synaptic plasticity and nervous system function. Adv Exp Med Biol. 2011;691:541–557. doi: 10.1007/978-1-4419-6612-4_57. [DOI] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci USA. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobon KE, Chang D, Kuzhikandathil EV. MicroRNA 142-3p mediates posttranscriptional regulation of D1 dopamine receptor expression. PLoS One. 2012;7:e49288. doi: 10.1371/journal.pone.0049288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth Z, Hollrigel GS, Gorcs T, Soltesz I. Instantaneous perturbation of dentate interneuronal networks by a pressure wave-transient delivered to the neocortex. J Neurosci. 1997;17:8106–8117. doi: 10.1523/JNEUROSCI.17-21-08106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJG. Unbiased Stereological Estimation of the Total Number of Neurons in the Subdivisions of the Rat Hippocampus Using the Optical Fractionator. Anatomical Record. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Yu J, Proddutur A, Elgammal FS, Ito T, Santhakumar V. Status epilepticus enhances tonic GABA currents and depolarizes GABA reversal potential in dentate fast-spiking basket cells. J Neurophysiol. 2013;109:1746–1763. doi: 10.1152/jn.00891.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu HT, Bian C, Yuan JC, Chu WH, Xiang X, Chen F, Wang CS, Feng H, Lin JK. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-kappaB signaling pathway in experimental traumatic brain injury. J Neuroinflammation. 2014;11:59. doi: 10.1186/1742-2094-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.