

Abstract
Impaired heparan sulfate (HS) synthesis in vertebrate development causes complex malformations due to the functional disruption of multiple HS-binding growth factors and morphogens. Here, we report developmental heart defects in mice bearing a targeted disruption of the HS-generating enzyme GlcNAc N-Deacetylase/GlcN N-Sulfotransferase 1 (NDST1), including ventricular septal defects (VSD), persistent truncus arteriosus (PTA), double outlet right ventricle (DORV), and retroesophageal right subclavian artery (RERSC). These defects closely resemble cardiac anomalies observed in mice made deficient in the cardiogenic regulator fibroblast growth factor 8 (FGF8). Consistent with this, we show that HS-dependent FGF8/FGF-receptor2C assembly and FGF8-dependent ERK-phosphorylation are strongly reduced in NDST1−/− embryonic cells and tissues. Moreover, WNT1-Cre/LoxP-mediated conditional targeting of NDST function in neural crest cells (NCCs) revealed that their impaired HS-dependent development contributes strongly to the observed cardiac defects. These findings raise the possibility that defects in HS biosynthesis may contribute to congenital heart defects in humans that represent the most common type of birth defect.
Keywords: fibroblast growth factor, heart development, heparan sulfate, neural crest, NDST1
1. Introduction
Neural crest cells (NCCs) are ecto-mesenchymal cells that migrate from the dorsal surface of the embryo to populate numerous structures along the dorsoventral axis. Cranial NCCs arise from the forebrain and hindbrain region to populate craniofacial structures as well as pharyngeal arches (PA) 1-3, giving rise to the maxilla, mandible and other structures of the neck and face. The cardiac neural crest (CNC) originates between the otic placode and the 3rd somite in the chick (Kuratani and Kirby, 1992) and populates the third, fourth, and sixth PA as well as the outflow tract (OFT) of the heart. Experimental models that ablate CNCs result in characteristic cardiovascular abnormalities such as failure of OFT septation and aortic arch artery defects (reviewed by (Kirby and Waldo, 1995)). Mouse models with abnormal NCC development also show these characteristic abnormalities (reviewed by (Maschhoff and Baldwin, 2000)), and several human birth defect syndromes, called 22q11 deletion syndromes, are thought to result from defective NCC development. 22q11 deletion syndromes result in failure of OFT septation (persistent truncus arteriosus (PTA), ventricular septal defects (VSDs), abnormal rotation and alignment of the OFT with the ventricles (double outlet right ventricle (DORV) and tetralogy of Fallot) or abnormal patterning of the aortic arch arteries. Craniofacial findings in these syndromes include cleft palate, abnormal facial features and external ear defects. An important family of factors involved in the formation of these syndromes and required for the survival, migration, proliferation and differentiation of NCCs are the fibroblast growth factors (FGFs) and their receptors (FGFRs). FGF1, FGF2, FGF9, FGF16, FGF20, FGFR1 and FGFR2 are expressed in mouse and chick ventricles during cardiac development (Colvin et al., 1999; Dell’Era et al., 2003; Hotta et al., 2008; Lavine et al., 2005; Pennisi et al., 2003) and activate cardiomyoblast proliferation in culture. Mouse mutants made deficient in FGF1 and FGF2 function, however, develop normally and have normal cardiac structure and mass (Miller et al., 2000; Schultz et al., 1999; Zhou et al., 1998). This indicates compensatory activities among FGF family members. FGF8, however, encodes a crucial member of the FGF family, providing survival, mitogenic, anti/-pro differentiation and patterning signals to adjacent tissues as well as autocrine activity (Park et al., 2008). Complete loss of FGF8 function in mice leads to early embryonic lethality (Meyers et al., 1998; Moon and Capecchi, 2000; Sun et al., 1999), and murine FGF8 hypomorphic mutants have a constellation of heart, OFT, great vessel and pharyngeal defects (Abu-Issa et al., 2002; Frank et al., 2002). Mesodermal FGF8 and FGF10 have overlapping roles in heart development (Watanabe et al., 2010). FGF9 is expressed in the midgestation mouse heart and regulates myocardial proliferation and differentiation in vivo (Lavine et al., 2005). FGF9 deficient mice die at birth with an enlarged dilated heart (Colvin et al., 1999).
FGF family members and their receptors require heparan sulfate (HS) for the formation of high affinity FGF- and FGFR-complexes and subsequent signaling (Rapraeger et al., 1991; Yayon et al., 1991). HS is produced by most mammalian cells as part of membrane and extracellular matrix proteoglycans (the HSPGs)(Esko and Lindahl, 2001). The polysaccharide chain grows by exostosin (Ext) copolymerization of GlcAβ1,4 and GlcNAcβ1,4 and is modified by one or more of the four NDST isozymes; the N-deacetylase activity of NDSTs removes acetyl groups from GlcNAc residues, which are then converted to GlcNS through the N-sulfotransferase activity. Subsequent modifications of the HS chain by most O-sulfotransferases and a GlcA C5-epimerase depend on the presence of GlcNS residues, making the NDSTs responsible for the generation of sulfated HS ligand binding sites (Lindahl et al., 1998). Mice deficient in EXT1, NDST1, 2-O-sulfotransferase and GlcA C5-epimerase show defective brain morphogenesis, axon guidance defects, craniofacial defects, defective formation of the lacrimal glands, skeletal defects, renal agenesis and eye defects due to simultaneous inhibition of multiple HS-binding factors (Bullock et al., 1998; Grobe et al., 2005; Inatani et al., 2003; Iwao et al., 2009; Li et al., 2003; McLaughlin et al., 2003; Pallerla et al., 2007; Pan et al., 2008; Pan et al., 2006). Mice deficient for the HSPG Glypican3 (GLP3) show defective heart development, as do mice lacking the HSPG Perlecan (Cano-Gauci et al., 1999; Costell et al., 2002; Ng et al., 2009). In humans, mutations in B3GAT3, the gene coding for glucuronosyltransferase-I (GlcAT-I), result in variable combinations of heart malformations, including mitral valve prolapse, VSD, and bicuspid aortic valve (Baasanjav et al., 2011). Importantly, craniofacial defects in NDST1-deficient mouse embryos are consistent with NCC deficiencies and resemble mutants deficient in Sonic hedgehog (SHH) and FGF8 function (Grobe et al., 2005). Therefore, we analyzed these mice for SHH/FGF- and NCC-related cardiac developmental defects, and found that NDST1 null mice indeed show multiple cardiovascular malformations, in large part due to impaired NCC function.
2. Results
2.1 Heart defects in NDST1 deficient embryos
FGF2 signaling and the development of NCC-derived facial and cranial structures are impaired in NDST1 null embryos (Grobe et al., 2005; Pallerla et al., 2007). Therefore, we analyzed E14.5 (n=4) and E18.5 (n=7) NDST1−/− embryos for potential FGF- and NCC-dependent developmental defects of the cardiovascular system. We detected membranous VSD in all E18.5 NDST1−/− mutants (Fig. 1B). Moreover, formation and remodeling of the fourth pharyngeal arch arteries to form the aortic arch and right subclavian artery are extremely sensitive to FGF8 dosage in the pharyngeal ectoderm (Macatee et al., 2003). Consistent with this, we detected retroesophageal right subclavian artery (RERSC) in one E18.5 NDST1 mutant (Fig. 1D), and double outlet right ventricle (DORV) was identified in one out of four E14.5 mutant embryos, indicating that proper alignment and rotation of the OFT were disrupted or delayed (Table 1). These findings provide an explanation for the perinatal lethality of NDST1 null mice, consistent with cyanosis and respiratory distress observed in NDST1−/− neonates (Fan et al., 2000; Ringvall et al., 2000).
Fig 1. Heart defects in NDST1 mutant E18.5 embryos.
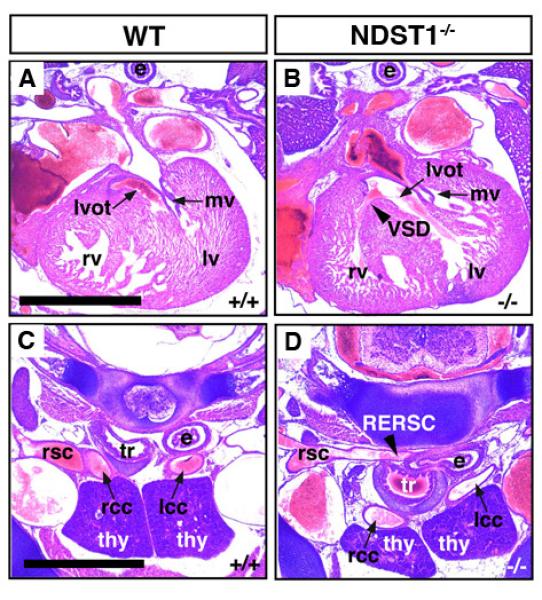
A-D) Ventricular septal defect (VSD, arrowhead in B) and retroesophageal right subclavian artery (RERSC, arrowhead in D) in transverse sections generated from E18.5 embryos. VSD was detected in all 7 mutant embryos investigated (Table 1). One embryo showed RERSC. Abbreviations used: lv: left ventricle, rv: right ventricle, mv: mitral valve, lvot: left ventricular outflow tract, rsc: right subclavian artery, rcc: right common carotid, lcc: left common carotid, e: esophagus, tr: trachea, thy: thymus.
Table 1.
Summary of phenotypes observed in systemic and conditional NDST mutant embryos. VSD: Ventricular Septal Defect, DORV: Double Outlet Right Ventricle, PTA: Persistent Truncus Arteriosus, RERSC: Retroesophageal right subclavian artery.
| VSD | DORV | PTA | RERSC | |
|---|---|---|---|---|
| WT (E18.5)(n=5) | 0/5 | 0/5 | 0/5 | 0/5 |
| NDST1−/− (E14.5)(n=4) | N/A | 1/4 | 0/4 | N/A |
| NDST1−/− (E18.5)(n=7) | 7/7 | 0/7 | 0/7 | 1/7 |
| NDST2−/− (E18.5)(n=8) | 0/8 | 0/8 | 0/8 | 0/8 |
| WNT1-Cre; NDST1f/f (E18.5)(n=4) | 3/4 | 0/4 | 0/4 | 0/4 |
| WNT1-Cre; NDST1f/f; NDST2−/− (E18.5)(n=4) | 4/4 | 0/4 | 4/4 | 1/4 |
2.2 NDST1 expression and HS composition in the mouse embryonic heart
Because all 4 NDST isoenzymes contribute to HS synthesis, and because multiple growth-promoting FGFs bind to HS during cardiogenesis, we first performed semiquantitative RT-PCR to determine FGF-, FGFR- and NDST expression in the E14.5-E18.5 embryonic heart (Table 2). NDST1 was strongly expressed at all stages investigated, whereas NDST3 and NDST4 mRNA expression was generally weaker or absent. The HS-binding fibroblast growth factors FGF1, FGF2, FGF8 and FGF9 as well as their receptors were also widely expressed. However, neither mRNA nor protein expression does strictly predict NDST activity. During biosynthesis, several HS-synthesizing enzymes form a multienzyme complex termed the GAGosome (Esko and Selleck, 2002). GAGosome composition of specific NDST isoforms can vary depending on their relative levels of expression or that of other proteins that may act as chaperones or scaffolding proteins. Therefore, as a functional readout for NDST1 enzyme activity, heart sections were stained with two different HS-specific antibodies, HepSS1 and 10E4. Both antibodies require NDST-generated GlcNS to bind HS. HepSS1 antibodies detected HS localized to various regions of the wildtype embryonic heart, whereas HS expressed in Ndst1 mutant tissues was not detected (Fig. 2). Likewise, we found that HS-staining by 10E4 specifically depended on NDST1 activity, consistent with previous findings (Pallerla et al., 2007; Pan et al., 2006). In E12.5 wildtype embryos, 10E4 stained the developing ventricles and ventricular septae (Fig. 2). 10E4 staining was undetectable in corresponding NDST1 null tissue sections.
Table 2.
Semiquantitative RT-PCR demonstrates ndst1-ndst4 expression and expression of the fgfs and their receptors in E14.5-E18.5 mouse embryonic hearts. Ndst1 and ndst2 are robustly expressed at all stages investigated, whereas ndst3 and ndst4 are not expressed or expressed at lower levels. Fgf1, fgf2, fgf8 and fgf9 as well as fgfrs1-4 were all detected during heart development. +: Strong expression, (+): weak expression, −: no detectable expression, NA: not assessed. RT-PCR was independently conducted 3-5 times on at least two independent cDNA preparations from whole embryonic hearts.
| E14.5 | E15.5 | E16.5 | E17.5 | E18.5 | |
|---|---|---|---|---|---|
| ndst1 | + | + | + | + | + |
| ndst2 | N/A | + | N/A | + | + |
| ndst3 | (+) | (+) | (+) | − | (+) |
| ndst4 | (+) | (+) | (+) | − | − |
| fgf1 | + | + | − | + | + |
| fgf 2 | + | + | + | + | + |
| fgf 8 | − | + | + | + | − |
| fgf 9 | + | + | − | + | + |
| fgfr1 | + | + | + | + | + |
| fgfr2 | + | + | + | + | + |
| fgfr3 | + | + | + | + | + |
| fgfr4 | + | − | − | + | + |
Fig 2. NDST expression in the developing heart.
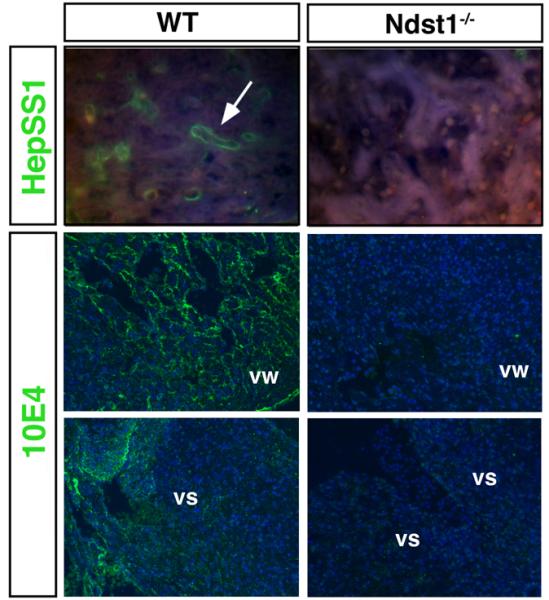
Top: HS detected by the monoclonal anti-HS antibody HepSS1. Bottom: HS detected by the monoclonal anti-HS antibody 10E4. HepSS1- and 10E4 reactive HS epitopes were widely expressed in developing heart tissues of wildtype embryos (E12.5), but not in NDST mutant embryos. vw: ventricular wall, vs: ventricular septum.
Next, HS was isolated from E18.5 wildtype and NDST1 mutant embryos for compositional analysis by quantitative mass spectrometry (Lawrence et al., 2008)(Table 3). Disaccharide analysis revealed strongly increased levels of unsulfated disaccharide D0A0 (68.6% relative abundance in NDST1−/− derived HS versus 51.5% in wildtype derived HS). In contrast, the relative abundance of N-sulfated, 2-O sulfated and 6-O sulfated disaccharides was reduced in the NDST1 null embryo (24.4% versus 40.7%, 13.7% versus 19.1% and 21.2% versus 23%, respectively). These findings confirm that deletion of NDST1 affects O-sulfation in addition to N-sulfation. 10E4 binding to HS requires specific N-sulfated and N-acetylated disaccharide units (van den Born et al., 2005); thus, complete loss of 10E4 binding to NDST1 null tissues despite only moderately reduced N-sulfation confirms specific, NDST1 dependent synthesis of the 10E4 HS epitope (Pallerla et al., 2007).
Table 3.
Quantitative disaccharide analysis revealed undersulfation of HS derived from E18.5 NDST1−/− embryos. Numbers denote the relative amounts of detected disaccharides.
| WT | NDST1−/− | |
|---|---|---|
| Unsulfated | 51.5 | 68.6 |
| N-sulfated | 40.7 | 24.4 |
| 2-O-sulfated | 19.1 | 13.7 |
| 6-O-sulfated | 23 | 21.2 |
2.3 NDST1 is a regulator of FGF function and proliferation in the embryonic heart
Cell proliferation depends on FGF function, and the NDST ortholog Sulfateless plays crucial roles in the generation of FGF binding sites in Drosophila melanogaster (Lin et al., 1999b) and in cell culture (Ishihara et al., 1993). FGFs bind and activate alternatively spliced forms of four tyrosine kinase FGF receptors (FGFRs 1–4)(Zhang et al., 2006). FGF activity is regulated by spatial and temporal expression patterns of FGFs and FGFRs, and by the regulated formation of specific ligand-receptor pairs. FGF ligand-receptor pairing is regulated by their binding specificity to HS chains of HSPGs, which in turn depends on specific HS sulfation motifs. These motifs are essential for the formation of signaling-active, trimeric FGF/FGFR/HS complexes. To test for HS-dependent FGF/FGFR assembly that regulates heart development starting at E10.5 (Ostrovsky et al., 2002), heart sections were incubated with FGF and FGFR fused with human IgG Fc domain (FGFR/Fc). Bound FGFR-Fc was then probed with a fluorescent anti-IgG antibody. In E10.5 wild type embryo sections, we observed FGFR1c, FGFR2b, FGFR2c and FGFR3b complex formation with FGF1 on ventricular and atrial tissue (Fig. 3 and not shown). As a control, no signal was detectable in the absence of FGF ligand under the same experimental conditions (Supplemental material), confirming that the observed FGF/FGFR binding in situ required cell surface HS. In E10.5 NDST1 null heart sections, FGF1/FGFR2c formation was reduced (Fig. 3), in contrast, FGF1/FGFR2b and FGF1/FGFR3b binding was only slightly affected or even unchanged (not shown). FGF2/FGFR2b and FGF2/FGFR4 binding was strongly reduced in the mutant, as was binding of FGF8/FGFR2c and FGF9/FGFR3b pairs. The observed differential binding of specific FGF/FGFR pairs thus suggests that NDST1 function is required for the assembly of some (but not all) FGF-FGFR pairs in heart tissue, including FGF8/FGFR2c. This is consistent with the observed comparable cardiovascular phenotypes in NDST1 null embryos and FGF8-deficient hypomorphs.
Fig 3. NDST1 mutation disrupted FGF/FGFR interaction.
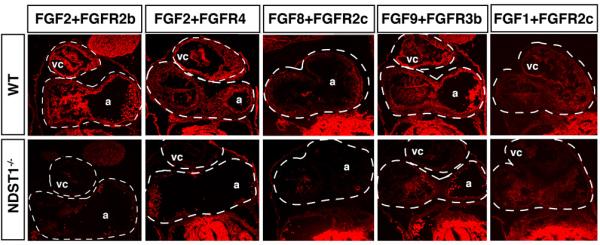
LACE-assay detection of FGF/FGFR binding to HS of E10.5 wildtype and NDST1−/− coronal heart sections. HS-mediated FGF2/FGFR2b, FGF2/FGFR4, FGF8/FGFR2c, FGF9/FGFR3b and FGF1/FGFR2c binding were observed in the wildtype heart, often specifically to endocardium, but were reduced in heart tissue of NDST1 mutant embryos. vc: ventricular chamber, a: atria. Comparable anatomical structures in wildtype and mutant tissue sections of given FGF/FGFR combinations are indicated by dashed lines.
The significant loss of FGF-FGFR binding to NDST1-null tissue suggested that FGF signaling was compromised during development. To test this possibility, we examined the expression of phospho-ERK1/2 (pERK1/2) acting as downstream effectors of the FGF-MAPK pathway. Using a phospho-specific antibody against ERK1/2, we detected reduced ERK1/2 phosphorylation in NDST1−/− E10.5 embryonic hearts (Fig. 4A,B, and insets), areas that also showed loss of FGF/FGFR pairing (Fig. 3). Total ERK protein levels were unchanged as demonstrated by an antibody directed against both, phosphorylated and unphosphorylated ERK (Fig. 4C,D). NDST1-dependent ERK-phosphorylation in the heart is consistent with its reported loss in NDST1 null eye lenses and lacrimal glands (Pan et al., 2008; Pan et al., 2006).
Fig 4. Loss of FGF signaling and impaired proliferation in NDST1−/− ventricular septa.
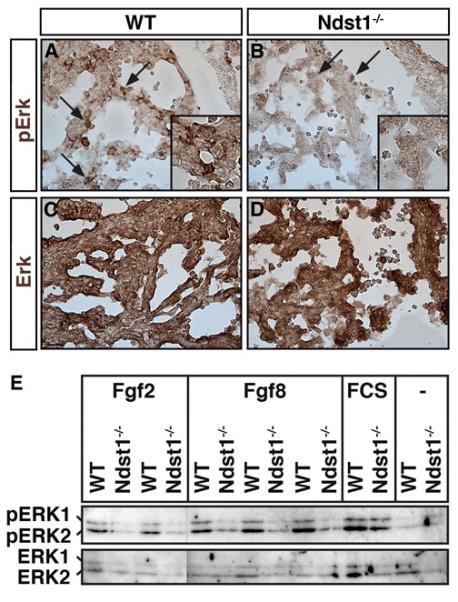
A,B) Specific pERK staining was detected in ventricle of E10.5 NDST1 mutant and wildtype embryos (brown stain, arrows). In NDST1 mutant littermates, pERK detection was reduced (arrows). Inset: magnification of pERK reactive cell clusters. Three embryos of each genotype were analyzed, one representative result is shown. C,D) Antibodies directed against phosphorylated and unphosphorylated (total) ERK proteins stained both tissue sections at comparable levels. Four embryos of each genotype were analyzed, one representative result is shown. E) FGF2- and FGF8-dependent ERK1/2 phosphorylation is significantly reduced in NDST1 mutant embryonic fibroblasts. Cultured cells derived from E14.5 wildtype and mutant littermates were stimulated with 10% serum in DMEM or FGF2/8 conditioned DMEM for 5 min before analysis. Phosphorylation of ERK1 and ERK2 (pERK1 and pERK2) were observed in the wildtype after addition of FGF2, FGF8 or serum, however, FGF addition failed to stimulate ERK1/2 phosphorylation in NDST1 mutant cells. n=2 (FGF2) and n=3 (FGF8). − refers to no stimulation.
To determine whether impaired FGF8 function contributes to reduced ERK-phosphorylation in NDST1 deficient cells, fibroblasts were isolated from wildtype and mutant E14.5 embryos. Cultured cells were starved from serum for 20 hrs and then stimulated for 5 min with FGF2 or FGF8 conditioned medium derived from FGF2/FGF8-transfected Bosc23 cells. Serum was added as a positive control, and DMEM from mock-transfected Bosc23-cells served as a negative control (Fig. 4E). ERK1/2 phosphorylation was strongly stimulated in wildtype fibroblasts in response to both FGF2 and FGF8. In contrast, ERK1/2 phosphorylation in NDST1−/− fibroblasts was unchanged in response to FGF2, consistent with previous findings (Grobe et al., 2005). Notably, FGF8 also failed to stimulate ERK1/2 phosphorylation. ERK1/2 in NDST1−/− cells was not affected per se, since serum efficiently stimulated ERK-phosphorylation. These findings indicate that FGF8 coreceptor function of HS expressed in NDST1−/− embryos is impaired, consistent with reduced HS-dependent FGF8/FGFR2c assembly observed in this work (Fig. 3).
FGF-induced MAPK-signaling stimulates cell proliferation, and conditional NDST1 mutant mice show impaired vascular smooth muscle cell proliferation (Adhikari et al., 2010). We thus tested next for possible changes in cell proliferation in NDST1 mutant heart tissue. Cell proliferation was determined by an antibody that labels cells in G1 and S-phase (Proliferating Cell Nuclear Antigen, PCNA) in E14.5 mutant animals and wild-type littermate controls (Supplemental materials). We found that cell proliferation was indeed significantly reduced in the NDST1−/− ventricular wall (~83% of wildtype levels (31%±3% versus 38%±3%, p=0.101, n=7)) and in the ventricular septum (~75% of wildtype levels (41%±2% versus 54%±2%, p=0.0028, n=4)), indicating that impaired growth factor activity may contribute to proliferation-related NDST1−/− cardiac phenotypes, such as VSD.
2.4 NDST1 expression and apoptosis in branchial arches
Ablation and genetic studies have shown that NCC production, delamination, directed migration and survival are critical for cardiovascular development (Gitler et al., 2002). Cardiac defects observed in NDST1 null embryos are consistent with impaired NCC development, such as failure of OFT septation and aortic arch artery defects. Importantly, NCCs require FGF8 function (Abu-Issa et al., 2002; Goddeeris et al., 2007), and NCCs in FGF8 mutant embryos undergo cell death in areas both adjacent and distal to the ectoderm and mesenchyme of the developing pharyngeal arches where FGF8 is normally expressed (Abu-Issa et al., 2002; Frank et al., 2002; Schneider et al., 2001; Song et al., 2010). We confirmed NDST1 protein expression (Fig. 5A) and 10E4 reactivity (Fig. 5B) in E10.5 pharyngeal arches, suggesting that its loss may result in increased NCC apoptosis due to reduced FGF8 activity in these tissues. To test this possibility, TUNEL staining was conducted in pharyngeal arches and maxillary prominences of E10.5 wildtype and mutant mouse embryos. We detected strongly increased apoptosis, particularly in pharyngeal pouches that express FGF8 (Song et al., 2010) and FGFR2 (Zhang et al., 2008)(Fig. 5D, arrows and inset). Increased apoptosis was also observed in NDST1−/− branchial arch mesenchyme (ba, Fig. 5F, arrows) and mesenchyme of the developing maxillary prominences (m, Fig. 5H, arrow) that both harbour migrating NCCs. Statistical analysis revealed that the observed increase of apoptosis in NDST1 null mesenchymal tissue was significant (16%±3% of TUNEL positive nuclei relative to total nuclei, versus 6.5%±1% in the wildtype (p<0.05, n=3 mutant and wildtype embryos)).
Fig 5. NDST expression, HS expression, and apoptosis in branchial arch tissue.
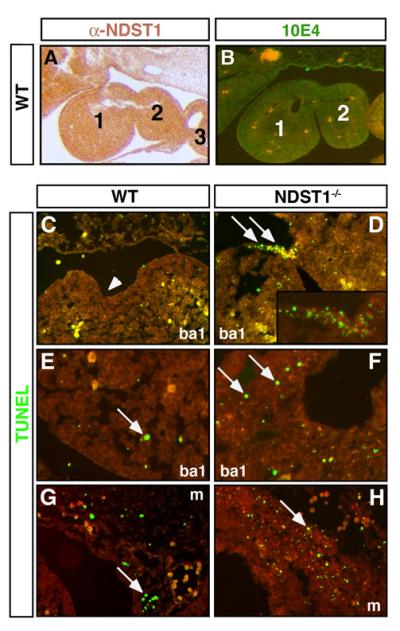
A) Immunohistochemical analysis using affinity purified rabbit-anti-mouse antiserum raised against murine NDST1 (left). NDST1 protein is strongly expressed (brown stain) and is also biosynthetically active, as demonstrated by 10E4 reactivity of pharyngeal arch HS (B, green stain). Preimmune sera or secondary antibodies alone did not result in detectable staining (not shown). 1-3: first to third pharyngeal arches. C-H: Apoptosis is significantly enhanced in branchial arches of NDST1 mutant embryos. (D) TUNEL staining reveals locally increased cell death (green nuclei) in the first branchial cleft (arrows) of E10.5 NDST1−/− embryos. Inset: Magnification of TUNEL positive clusters shown in D. ba1: First branchial arch. E,F) NDST1 deficient branchial arch mesenchyme also undergoes increased programmed cell death (arrows) if compared to wildtype branchial arch mesenchyme. G,H) Mesenchyme of the maxillary prominences likewise show increased programmed cell death in mutant embryos (H, arrow) if compared to wild type tissue (G, arrow). m: mesenchymal tissue. Five sections of each, three wildtype and three mutant embryos were analyzed.
2.5 SHH signaling in the embryonic heart depends on NDST1 expression
SHH mutant mice have atrial septum primum and septum secundum defects, membraneous VSD and defects in valve development (Washington Smoak et al., 2005) similar to those observed in NDST1 mutants. To understand whether SHH signaling is affected in the NDST1 mutant heart, we examined the expression of the SHH receptor Patched1 (PTC1) which reflects the extent of SHH signaling as well as signaling mediated by the other HH family members, Indian hedgehog and Desert hedgehog. Strong PTC1 expression was detected in all developing valves in E14.5 wildtype mice (Fig. 6). In the developing atrioventricular (A) and pulmonary (B) valves (arrows), PTC expression and thus SHH signaling was restricted to transdifferentiated cells populating the cardiac cushions, whereas the overlying endothelium strongly expressed 10E4 positive HS. On the other hand, in NDST1 mutant hearts, remodeling of the cardiac cushions into thinly tapered heart valves was impaired (Fig. 6C,D, asterisks). PTC expression was strongly reduced and remaining staining was patchy, and the endothelial expression of 10E4 reactive HS was absent. These data demonstrate that NDST1 contributes to HS sulfation in developing valve endothelium and also demonstrates that SHH signaling in valves, directly or indirectly, depends on NDST1 function.
Fig 6. Altered HS structure and loss of Ptc expression in NDST1 mutant valves.
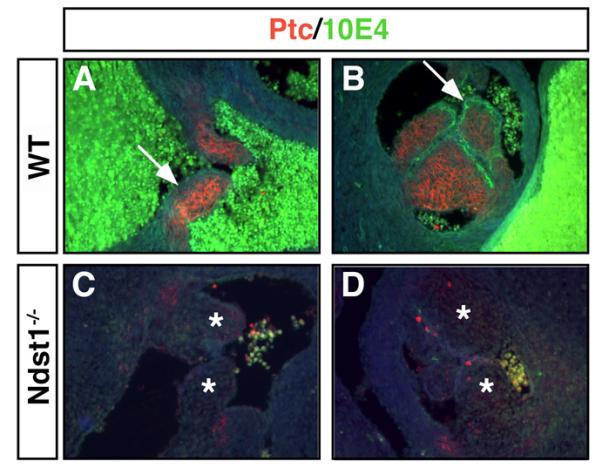
A-D) Sulfated HS expression recognized by 10E4 antibodies in wildtype valves (A,B, arrows, green fluorescence) is lost in E18.5 NDST1 mutant atrioventricular (C) and pulmonary (D) valve endothelia. PTC protein was expressed in wildtype valve cushion (A,B, red fluorescence) but undetectable in mutant valves (C,D, asterisks), indicating impaired SHH signaling in those structures. Also note malformation of the atrioventricular valve due to thickened cushions (asterisks). Representative results of three mutant and wildtype hearts are shown.
Notably, BMP, WNT and TGF-β functions also contribute to heart development and depend on HS-expression, raising the possibility that these pathways are impaired in NDST1 null mice. However, previous analysis demonstrated unaffected BMP/TGF-β and Wnt signaling in these mice (Pan et al., 2006). In contrast, FGF/FGFR and SHH signaling was strongly impaired in various developing NDST1−/− cells and tissues (Fuster et al., 2007; Grobe et al., 2005; Lanner et al., 2010; Pan et al., 2008; Pan et al., 2006; Qu et al., 2011; Qu et al., 2012). Thus, we suggest that FGF/FGFR function, and directly/indirectly HS-linked SHH signaling, most obviously depend on NDST1 expression during heart development.
2.6 Conditional NDST1 ablation in NCCs also results in developmental heart defects
To test for a possible role of NCC-autonomous HS expression, we employed WNT1-Cre recombinase expression (Brault et al., 2001) to specifically disrupt NDST1 function in these cells. It has previously been shown that WNT1-Cre driven deletion of EXT1 function resulted in striking craniofacial and cardiac defects (Iwao et al., 2009). Consistent with this, WNT1-Cre;NDST1f/f embryos survived to term with high incidence of VSD (n=3 out of 4)(Fig. 7B, compare to 7A)(Table 1). However, OFT defects were not detected in these embryos, possibly due to the compensatory activity of other NDST family members. NDST2 compensates for the loss of NDST1 function in various systems (Grobe et al., 2002), and HS N-sulfation is completely lost in cells lacking NDST1 and NDST2 gene function (Holmborn et al., 2004). Consistent with this, WNT1-Cre mediated disruption of NDST1 gene function in a NDST2 deficient background (WNT1-Cre;NDST1f/f;NDST2−/−) accentuates the observed defects, resulting in E14.5 hypoplastic hearts with thinned ventricular walls and large ventricular septal defects (n=4 out of 4)(Fig. 7C). Fully penetrant PTA and a complete failure of OFT septation into the aorta and pulmonary artery were also observed (Fig. 7F, compare to D,E). These defects disrupt the partitioning of blood flow required for adequate oxygenation and are thus lethal. RERSC was observed in one of four E14.5 compound mutant embryos (Fig. 7I). None of these defects were observed in NDST2 null (control) embryos (n=8), consistent with their postnatal survival (Forsberg et al., 1999). Thus, we conclude that cell autonomous NDST1 expression is absolutely required for CNC function, and demonstrate that heart defects observed in systemic NDST1−/− embryos can be explained, in large part, by impaired HS-dependent NCC development.
Fig 7. Neural crest cell specific knockout of NDST1 results in VSD and OFT defects.
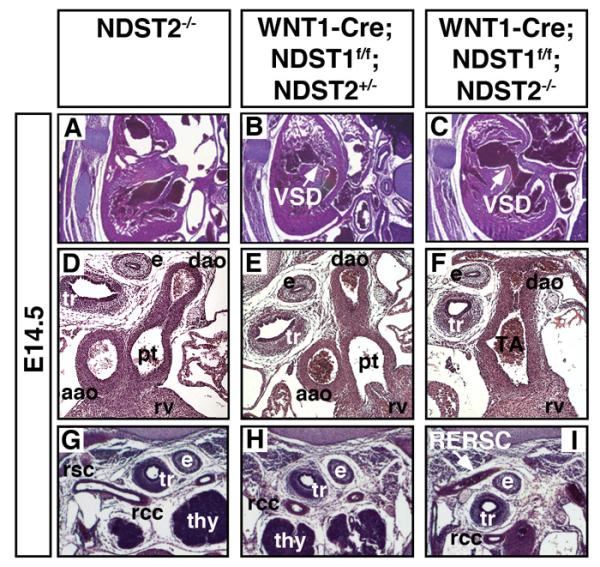
Tissue specific deletion of NDST1 in NCCs using E14.5 WNT1-Cre;NDST1flox/flox (n=4) and WNT1-Cre;NDST1flox/flox;NDST2−/− double mutants (n=4) results in severe heart defects. A-C) VSD was detected in single and double mutants, but defects were more severe in the compound mutant, indicating compensatory activity of NDST2. Of four WNT1-Cre;NDST1flox/flox and four WNT1-Cre;NDST1flox/flox;NDST2−/− compound mutants, all had large VSDs (arrow). D-F) In NDST1/2 compound mutants (WNT1-Cre;NDST1flox/flox;NDST2−/−), severe outflow tract defects such as PTA were also observed in four out of four embryos investigated. e: esophagus, tr: trachea, aao: ascending aorta, dao: descending aorta, rv: right ventricle, pt: pulmonary trunc, TA: truncus arteriosus. G-I) Of four WNT1-Cre;NDST1flox/flox;NDST2−/− embryos investigated, one had retroesophageal right subclavian artery (RERSC, arrow). thy: thymus, rsc: right subclavian artery, rcc: right common carotid.
2.7 Lack of NCC specific NDST1 expression results in decreased MAPK signaling and increased apoptosis
Comparable cardiac phenotypes in systemic and conditional NCC-specific NDST null embryos imply comparable underlying mechanisms, such as increased apoptosis and decreased MAPK signaling and proliferation in tissues populated by the NCC. Indeed, as shown in Fig. 8B, microscopic analysis of WNT1-Cre;NDST1f/f;NDST2−/− branchial arches showed strongly decreased pERK staining in 10E4-negative mesenchymal tissue (Fig. 8D,F, compare to 10E4-binding (red) in C,E). In contrast, epithelial cells in conditional mutant embryos showed unimpaired ERK-phosphorylation (Fig. 8A,B, arrows). Apoptosis in 10E4 negative, NDST1/NDST2 compound null mesenchyme increased from 2.4%±0.75% of total cells (NDST2−/−, n=14) to 11.45%±4% (WNT1-Cre;NDST1f/f;NDST2−/−, n=11, p<0.05) (Fig. 8G). This indicates that NDST1-dependent HS sulfation positively regulates cell survival. These data, taken together with the observed comparable cardiac phenotypes of WNT1-Cre;NDST1f/f;NDST2−/− and systemic NDST1−/− embryos, demonstrate that NCC-autonomous NDST1/HS expression is required for normal cardiac development. Undersulfation of NCC-expressed HS is associated with reduced MAPK signaling and increased cell death in branchial arch mesenchyme: both processes may contribute to the observed cardiac defects.
Fig 8. Reduced ERK-phosphorylation and increased apoptosis upon neural crest cell specific deletion of NDSTs.
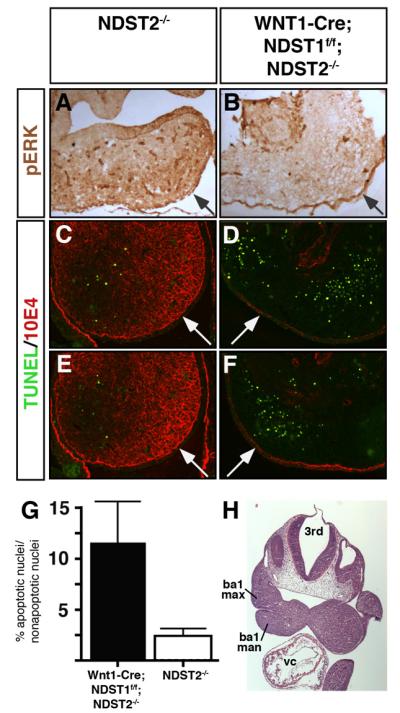
A,B) Tissue specific deletion of WNT1-Cre;NDST1flox/flox;NDST2−/− double mutants resulted in the loss of ERK phosphorylation in maxillary prominences, indicating strongly affected HS sulfation and FGF signaling in the mesenchyme. In contrast, ERK-phosphorylation was unaffected in epithelial cells (arrows). C-F) Loss of mesenchymal 10E4 staining (red) and increased apoptosis (green) in first branchial arch mesenchyme of compound mutant embryos. G) Quantification of cell death shown in C-F. Apoptosis is strongly increased in compound mutant tissue (NDST1−/−: 11.45%±4% versus 2.4%±0.75% in the wildtype (p<0.05, n=2 mutant and wildtype embryos, 11 (mutant) and 14 slides (wildtype) were analyzed, respectively). Numbers denote the percentage of TUNEL positive nuclei relative to total nuclei (DAPI-stained) in the entire branchial arch. Twenty sections of each, two wildtype and two mutant embryos were analyzed. H) H&E stained frontal section of a representative E10.5 embryo, showing maxillary (ba1 max) and mandibular (ba1 man) components of the first branchial arch, as analyzed in A-G. Abbreviations used: 3rd: third (midbrain) ventricle, vc: ventricular chamber.
3. Discussion
NDST1 null mice die perinatally/neonatally with a condition resembling respiratory distress syndrome in premature human infants (Ringvall et al., 2000) and show severe developmental defects of the skull, face and eyes due to impaired function of various HS-binding growth factors and morphogens (Fan et al., 2000; Grobe et al., 2005; Iwao et al., 2009; Pallerla et al., 2007; Pan et al., 2006; Ringvall et al., 2000). In this work, we characterized cardiovascular development in these mice, confirming previous studies implicating essential functions of glycosaminoglycans and proteoglycans in heart development (Adhikari et al., 2010; Baasanjav et al., 2011; Iwamoto et al., 2010; Ng et al., 2009). Strikingly, targeted disruption of NCC-autonomous HS synthesis resulted in the same malformations observed in systemic NDST1−/− embryos; this demonstrates an essential role of NDST1-modified HS for NCC function during cardiogenesis. This is consistent with experimental or genetic ablation of NCC development in other systems that also result in cardiovascular abnormalities. Moreover, human birth defect syndromes based on defective NCC development (Kirby and Waldo, 1995; Lindsay, 2001; Maschhoff and Baldwin, 2000) are often associated with craniofacial findings also seen in NDST1 mutant mice, such as agnathia (lack of lower jaw), formation of hypoplastic maxillary prominences and defects of the viscerocranium and neurocranium. SHH deficient mice have enhanced death of migrating NCCs that give rise to much of the skull and the face, resulting in severe craniofacial defects similar to those seen in NDST1 mutant embryos (Washington Smoak et al., 2005). SHH deficient embryos, and conditional Smoothened (a transmembrane protein that mediaties intracellular HH effects) mutants (Lin et al., 2006) also have ventricular septal defects and atrioventricular valve defects as found in NDST1 mutant embryos. These findings indicate that impaired SHH function may contribute not only to facial defects (Grobe et al., 2005) but also to defects in NDST1−/− embryonic heart and valve development. This possibility is supported by reduced SHH mRNA expression and signaling as a consequence of impaired GLP3 function as a co-receptor for FGF9 (Ng et al., 2009), a growth factor also affected in the NDST1−/− embryonic heart (Fig. 3). Notably, GLP3 loss-of-function mutations in humans cause Simpson-Golabi-Behmel syndrome, a complex overgrowth syndrome also associated with congenital heart disease (Lin et al., 1999a).
Multiple signaling molecules bind to HS and trigger cardiomyocyte differentiation or guide the formation of cardiac substructures, including WNT, BMP/TGF-β, HB-EGF (Andelfinger, 2008; Brand, 2003; Filmus et al., 2008; Iwamoto et al., 2010) and FGF family members (Abu-Issa et al., 2002; Armstrong and Bischoff, 2004; Delot et al., 2003; Frank et al., 2002; Liu et al., 2004; Park et al., 2008; Sanford et al., 1997). However, previous analysis demonstrated unaffected BMP/TGF-β and Wnt signaling in NDST1−/− embryos (Pallerla et al., 2007; Pan et al., 2006), suggesting that these proteins do not depend on NDST1 activity. In contrast, FGF/FGFR signaling was strongly impaired in numerous NDST1-deficient tissues or cells (Fuster et al., 2007; Grobe et al., 2005; Lanner et al., 2010; Pan et al., 2008; Pan et al., 2006; Qu et al., 2011; Qu et al., 2012). Consistent with this, we noticed that VSDs and OFT alignment defects in NDST1 mutant embryos closely resemble those commonly seen in FGF8 hypomorphs and tissue restricted conditional FGF8 mutants (Abu-Issa et al., 2002; Frank et al., 2002; Macatee et al., 2003; Moon et al., 2006; Park et al.; Park et al., 2006). In addition, FGF8 mutants share neural, limb and craniofacial defects similar to those found in NDST1 null embryos. This indicates that FGF8 function is impaired in NDST1 null mice, consistent with reduced ERK-phosphorylation upon FGF8-stimulation in NDST1−/− derived fibroblasts. Furthermore, RERSC detected in systemic NDST1−/− mutants and NCC-specific WNT1-Cre;NDST1f/f;NDST2−/− mutants is common in FGF8 hypomorphic mice and pharyngeal ectoderm conditional FGF8 mutants (Macatee et al., 2003). Consistent with the proposed role of HS in NCC function discussed above, and the proposed role of NDST1 function in FGF8 activity regulation, cardiovascular defects in FGF8 mutants have been linked to impaired development of NCCs (Abu-Issa et al., 2002; Macatee et al., 2003; Meyers et al., 1998). NDST1−/− phenotypes also resemble those found in FGFR mutant mice (Park et al., 2008). However, although ERK-phosphorylation is required in NCCs for normal heart and aortic arch development, NCCs are not direct targets of FGF8 signaling (Park et al., 2008).
NCC-specific deletion of NDST1 function in a systemic NDST2 background (WNT1-Cre;NDST1f/f;NDST2−/−) resulted in increased severity of all observed heart defects. WNT1-Cre;NDST1f/f;NDST2−/− mice showed large VSDs in addition to 100% penetrant PTA, whereas WNT1-Cre;NDST1f/f mice were less severely affected. Increased severity of heart defects in compound mutants corresponds with the reported effect of increased NCC ablation (Porras and Brown, 2008) that may be based on further reduced HS sulfation in compound mutants in our system (Grobe et al., 2002). Notably, in compound NDST1/NDST2 deficient NCCs, signaling by BMP/TGFβ (Iwao et al., 2009) may be affected as well. BMP/TGFβ depend on HS expression (Hacker et al., 2005; Hu et al., 2009) and contribute to cardiogenesis (Park et al., 2008). Thus, in addition to FGF signaling, we suggest that BMP and TGFβ function may also be affected in WNT1-Cre;NDST1f/f;NDST2−/− NCCs, resulting in more strongly affected OFT remodeling. In humans, this impaired ability of mutant HS to bind multiple soluble cardiogenic molecules, including but not restricted to FGFs, may thus contribute to congenital heart defect syndromes that affect nearly 1% of human newborns.
4. Experimental procedures
4.1 Mice
The generation of the NDST1− allele and of WNT1-Cre mice has been described previously (Brault et al., 2001; Grobe et al., 2005).
4.2 Histology and detection of mRNA expression
Embryos were fixed in 4% paraformaldehyde overnight, dehydrated, embedded in paraffin and sectioned at 8 μm. Sections were stained with haematoxylin and eosin for histological analysis. For immunohistochemical analysis, hearts were quick-frozen in prechilled isopentane and embedded in Tissue-Tec (Leica Instruments, Wetzlar, Germany). Hearts were cryotome-sectioned to 12μm slices. Sections were rinsed with PBS and permeabilized for 5-10 min in PBS supplemented with 0.1% Triton-X100 and blocked in 10% fetal calf serum/1% bovine serum albumin for 1-2 h.
4.3 Immunohistochemistry
For immunohistochemical HS detection, mouse monoclonal antibodies HepSS1 and 10E4 (Seikagaku, Tokyo, Japan) were used at a 1:100 dilution in PBS/5% milk powder at 4°C overnight. 10E4 recognizes a HS epitope specifically generated by NDST1 activity, serving as a readout for enzyme activity (Pallerla et al., 2007; Pan et al., 2006). Prior to antibody incubation, epitope retrieval was performed by boiling slides in 0.1M sodium citrate, pH6.0, for 20 minutes, followed by three washing steps in PBS. Control slides were heparinase I-III digested (Ibex, Montreal, Canada) (50mM Hepes, 100mM NaCl, 1mM CaCl2, 5 μg BSA/ml, pH 7.0) for 1h at 37°C to confirm antibody specificity. After antibody incubation, slides were washed three times for 10 minutes each in PBS prior to incubation with secondary Alexa- or FITC-labelled α-IgG/IgM H+L antibody (1:200, Jackson ImmunoResearch, West Grove, USA). Immunohistochemical analysis of NDST1 expression was performed using affinity-purified anti-NDST antiserum (Grobe and Esko, 2002) followed by detection with goat anti-rabbit HRP conjugated antibodies (Zymed, San Francisco, USA). Proliferation was quantified by using α-PCNA antibodies (1:1000, abcam). TUNEL assays were performed using the In Situ Cell Death Detection Kit (Roche, Mannheim, Germany) according to the manufacturer’s instructions. Total ERK and phospho-ERK (pERK) reactivity as a readout for MAPK activity in tissues and fibroblasts was assessed by α-phospho-ERK1/2 and α-ERK1/2 antibodies (1:1000, Anti-active MAPK family sampler, Promega) on frozen sections and Western blots of SDS-PAGE separated fibroblast cell lysates. Statistical analysis was performed in Prism using Students t-test (Two-tailed, unpaired). All values shown in text and figures are ±SD. FGF2 and FGF8 (Accession numbers AI158649 and BC048734) were cloned into pWIZ (Gene Therapy Systems, San Diego), expressed in Bosc23 cells and secreted into serum-free medium which was subsequently added to the fibroblasts. Heparin affinity purification was employed, showing elution at 0.8M salt and thus strong FGF/heparin interactions (not shown).
4.4 Preparation and analysis of tissue HS
Whole E18.5 embryos were digested overnight with 2mg/ml pronase in 320mM NaCl, 100mM sodium acetate (pH 5.5) at 40°C, diluted 1:3 in water and applied to 2.5ml DEAE sephacel columns. GAGs were applied to PD-10 (Sephadex G25) columns (Pharmacia). GAGs were lyophilized, ChondroitinaseABC-digested over night, purified on DEAE as described above, applied to PD-10 columns and again lyophilized. For disaccharide analysis, 10 μg GAG samples were digested using Heparin-lyases I, II and III (1.5mU of each in 100 μl reactions)(IBEX, Montreal, Canada) at 37°C for 1h and the resulting disaccharides were separated from undigested material using a 3kDa spin-column (Centricon, Bedford, USA). Compositional disaccharide analysis of cell-derived HS or HS derived from embryos was then carried out by liquid chromatography/mass spectrometry (LC/MS) (Lawrence et al., 2008). Analysis of the disaccharide composition by post-column derivatization with 2-cyanoacetamide or by the LC/MS method gave comparable results, and comparison of the two methods using 0.5 μg commercial porcine heparin showed an error of 2% for abundant disaccharides to 20% for minor species.
4.5 FGF ligand and carbohydrate engagement assay (LACE)
In situ binding of the FGF-FGFR complex with heparan sulfate was carried out using the LACE assay as previously described (Pan et al., 2006). Embryos (E10.5) were harvested and fixed in 4% PFA at 4°C overnight prior to paraffin embedding. Deparaffinized and rehydrated 5 μm sections were incubated with 0.5mg/ml NaBH4 for 10 minutes, in 0.1 M glycine for 30 minutes and blocked with 2% BSA. The slides were then incubated with 20 μM FGF and 20 μM human FGFR-Fc chimera (R&D Systems) in RPMI-1640 medium/15% fetal bovine serum at 4°C overnight, followed by 2 h incubation at room temperature with Cy-3 labelled anti-human Fc IgG secondary antibody. As a control, FGFR-Fc in the absence of ligand was added to the slides. Slides were washed in PBS before mounting and analysis. Images were taken on a Zeiss Axiophot microscope employing a 10×/0.3, a 20×/0.5 and a 63×/1.25 Zeiss objective and a Leica DFC280 camera. Also, a Leica inverted microscope (DMIRB) equipped with a cooled MicroMax CCD camera (Princeton Instruments, Stanford, USA) was used, employing 5×/0.15, 10×/0.3 and 20×/0.5 Leica objectives. Leica software was used for image capturing and Photoshop 7 software run on MacIntosh computers for the generation of figures. Contrast and brightness were adjusted for whole images during figure assembly.
4.6 RT-PCR analysis of mRNA expression
For RT-PCR analysis of mRNA derived from wildtype hearts, RNA was isolated using TriZol reagent (Invitrogen) according to the manufacturers instructions. cDNA was produced employing the First Strand cDNA Synthesis Kit (Fermentas, Burlington, Canada) according to the manufacturers instructions and used as a PCR template under the following conditions: 1/10 of cDNA (derived from 1 μg total RNA), hot start for 5min, 94°C for 20sec, 59°C for 30sec, 72°C for 2min, 30 cycles. For the specific amplification of mNDST1-4, eight specific, intron-spanning primers (Tm=62°C) were used: mNDST1-F(5′-cttgagccctcggcagatgc-3′) and -R(5′-ccagggtactcgttgtagaag-3′), mNDST2-F (5′-aggaacccttgcctctgccc-3′) and -R (5′-gatcgtgtgggtgaagaggc-3′), mNDST3-F (5′-gaaagtgaagtctctgggcgg-3′) and -R (5′-tccgtgaatactcttgtccag-3′), mNDST4-F (5′-aacaggaaatgacacttattgaaacg-3′) and -R (5′-aggtgtataagccgaggcgg-3′). For specific amplification of FGFs/FGFRs, eight intron spanning primers were used: mFGF1-F (5′-agatcacaaccttcgcagcc-3′) and -R (5′-caatttggtccctgtgttcc-3′), mFGF2-F (5′-atggctgccagcggcatca-3′) and -R (5′-tcagctcttagcagacattgg-3′), mFGF8-F (5′-cgagctgcctgctgttgca-3′) and -R (5′-tgtagagacctgtctctgcg-3′) and mFGF9-F (5′-agtgctcccttaggtgaagt-3′) and -R (5′-actttgtcagggtccactgg-3′). For specific amplification of FGFRs, eight intron spanning primers were used: mFGFR1-F (5′-tggagttcatgtgcaaggtg-3′) and -R (5′-atagagaggaccatcctgtg-3′), mFGFR2-F (5′-aaataccaaatctcccaacc-3′) and -R (5′-gccgcttctccatcttct-3′), mFGFR3-F (5′-actgtactcaagactgcagg-3′) and -R (5′-gtccttgtcagtcgcatcat-3′) and FGFR4-F (5′-ctgttgagcatctttcaggg-3′) and -R (5′-cgtggaaggcctgtccatcc-3′).
Supplementary Material
Acknowledgements
The authors would like to thank Andrea Powers (Indiana University, Indianapolis, USA) and Amy Talbot (University of Utah, Salt Lake City, USA) for histological work.
Abbreviations
- VSD
Ventricular Septal Defect
- DORV
Double Outlet Right Ventricle
- PTA
Persistent Truncus Arteriosus
- RERSC
Retroesophageal right subclavian artery
- NDST
GlcNAc N-Deacetylase/GlcN N-Sulfotransferase
- EXT1
Exostosin 1
- NCC
Neural crest cell
- FGF
Fibroblast growth factor
- FGFR
Fibroblast growth factor receptor
- PA
Pharyngeal arch
- CNC
Cardiac neural crest
- OFT
Outflow tract
- HS
Heparan sulfate
- HSPG
Heparan sulfate proteoglycan
- MAPK
Mitogen-activated protein kinase
- ERK
Extracellular signal-regulated kinase
- GlcNAc
N-acetyl glucosamine
- GlcNS
N-sulfo glucosamine
- GlcA
Glucuronic acid
- BMP
Bone morphogenetic protein
- TGFβ
Transforming growth factor beta
Footnotes
Conflict of interest statement
None declared
Author roles: Study design: AMM, XZ, KG; Study conduct: YP, CC, SK, UP, SO, MF, RS, SRR, RL; Data collection: AMM, XZ, KG; Data analysis: AMM, XZ, KG, Data interpretation: AMM, XZ, KG, Drafting manuscript AMM, JDE, XZ, KG. KG takes responsibility for the integrity of the data analysis.
References
- Abu-Issa R, Smyth G, Smoak I, Yamamura K, Meyers EN. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development. 2002;129:4613–4625. doi: 10.1242/dev.129.19.4613. [DOI] [PubMed] [Google Scholar]
- Adhikari N, Basi DL, Townsend D, Rusch M, Mariash A, Mullegama S, Watson A, Larson J, Tan S, Lerman B, Esko JD, Selleck SB, Hall JL. Heparan sulfate Ndst1 regulates vascular smooth muscle cell proliferation, vessel size and vascular remodeling. J Mol Cell Cardiol. 2010;49:287–293. doi: 10.1016/j.yjmcc.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andelfinger G. Genetic factors in congenital heart malformation. Clin Genet. 2008;73:516–527. doi: 10.1111/j.1399-0004.2008.01009.x. [DOI] [PubMed] [Google Scholar]
- Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circ Res. 2004;95:459–470. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baasanjav S, Al-Gazali L, Hashiguchi T, Mizumoto S, Fischer B, Horn D, Seelow D, Ali BR, Aziz SA, Langer R, Saleh AA, Becker C, Nurnberg G, Cantagrel V, Gleeson JG, Gomez D, Michel JB, Stricker S, Lindner TH, Nurnberg P, Sugahara K, Mundlos S, Hoffmann K. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am J Hum Genet. 2011;89:15–27. doi: 10.1016/j.ajhg.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand T. Heart development: molecular insights into cardiac specification and early morphogenesis. Dev Biol. 2003;258:1–19. doi: 10.1016/s0012-1606(03)00112-x. [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Bullock SL, Fletcher JM, Beddington RSP, Wilson VA. Renal agenesis in mice homozygous for a gene trap mutation in the gene encoding heparan sulfate 2-sulfotransferase. Genes Dev. 1998;12:1894–1906. doi: 10.1101/gad.12.12.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano-Gauci DF, Song HH, Yang H, McKerlie C, Choo B, Shi W, Pullano R, Piscione TD, Grisaru S, Soon S, Sedlackova L, Tanswell AK, Mak TW, Yeger H, Lockwood GA, Rosenblum ND, Filmus J. Glypican-3-deficient mice exhibit developmental overgrowth and some of the abnormalities typical of Simpson-Golabi-Behmel syndrome. J.Cell Biol. 1999;146:255–264. doi: 10.1083/jcb.146.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin JS, Feldman B, Nadeau JH, Goldfarb M, Ornitz DM. Genomic organization and embryonic expression of the mouse fibroblast growth factor 9 gene. Dev Dyn. 1999;216:72–88. doi: 10.1002/(SICI)1097-0177(199909)216:1<72::AID-DVDY9>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Costell M, Carmona R, Gustafsson E, Gonzalez-Iriarte M, Fassler R, Munoz-Chapuli R. Hyperplastic conotruncal endocardial cushions and transposition of great arteries in perlecan-null mice. Circ Res. 2002;91:158–164. doi: 10.1161/01.res.0000026056.81424.da. [DOI] [PubMed] [Google Scholar]
- Dell’Era P, Ronca R, Coco L, Nicoli S, Metra M, Presta M. Fibroblast growth factor receptor-1 is essential for in vitro cardiomyocyte development. Circ Res. 2003;93:414–420. doi: 10.1161/01.RES.0000089460.12061.E1. [DOI] [PubMed] [Google Scholar]
- Delot EC, Bahamonde ME, Zhao M, Lyons KM. BMP signaling is required for septation of the outflow tract of the mammalian heart. Development. 2003;130:209–220. doi: 10.1242/dev.00181. [DOI] [PubMed] [Google Scholar]
- Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J.Clin.Invest. 2001;108:169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko JD, Selleck SB. Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- Fan G, Xiao L, Cheng L, Wang X, Sun B, Hu G. Targeted disruption of NDST-1 gene leads to pulmonary hypoplasia and neonatal respiratory distress in mice. FEBS Lett. 2000;467:7–11. doi: 10.1016/s0014-5793(00)01111-x. [DOI] [PubMed] [Google Scholar]
- Filmus J, Capurro M, Rast J. Glypicans. Genome Biol. 2008;9:224. doi: 10.1186/gb-2008-9-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg E, Pejler G, Ringvall M, Lunderius C, Tomasini-Johansson B, Kusche-Gullberg M, Eriksson I, Ledin J, Hellman L, Kjellén L. Abnormal mast cells in mice deficient in a heparin-synthesizing enzyme. Nature. 1999;400:773–776. doi: 10.1038/23488. [DOI] [PubMed] [Google Scholar]
- Frank DU, Fotheringham LK, Brewer JA, Muglia LJ, Tristani-Firouzi M, Capecchi MR, Moon AM. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. 2002;129:4591–4603. doi: 10.1242/dev.129.19.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster MM, Wang L, Castagnola J, Sikora L, Reddi K, Lee PH, Radek KA, Schuksz M, Bishop JR, Gallo RL, Sriramarao P, Esko JD. Genetic alteration of endothelial heparan sulfate selectively inhibits tumor angiogenesis. J Cell Biol. 2007;177:539–549. doi: 10.1083/jcb.200610086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Brown CB, Kochilas L, Li J, Epstein JA. Neural crest migration and mouse models of congenital heart disease. Cold Spring Harb Symp Quant Biol. 2002;67:57–62. doi: 10.1101/sqb.2002.67.57. [DOI] [PubMed] [Google Scholar]
- Goddeeris MM, Schwartz R, Klingensmith J, Meyers EN. Independent requirements for Hedgehog signaling by both the anterior heart field and neural crest cells for outflow tract development. Development. 2007;134:1593–1604. doi: 10.1242/dev.02824. [DOI] [PubMed] [Google Scholar]
- Grobe K, Esko JD. Regulated translation of heparan sulfate N-acetylglucosamine N-deacetylase/N-sulfotransferase isozymes by structured 5′-untranslated regions and internal ribosome entry sites. J Biol Chem. 2002;277:30699–30706. doi: 10.1074/jbc.M111904200. [DOI] [PubMed] [Google Scholar]
- Grobe K, Inatani M, Pallerla SR, Castagnola J, Yamaguchi Y, Esko JD. Cerebral hypoplasia and craniofacial defects in mice lacking heparan sulfate Ndst1 gene function. Development. 2005;132:3777–3786. doi: 10.1242/dev.01935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobe K, Ledin J, Ringvall M, Holborn K, Forsberg E, Esko JD, Kjellen L. Heparan sulfate and development: Differential roles of the N-acetylglucosamine N-deacetylase/N-sulfotransferase (NDST) isozymes. Biochim. Biophys. Acta. 2002;1573:209–215. doi: 10.1016/s0304-4165(02)00386-0. [DOI] [PubMed] [Google Scholar]
- Hacker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005;6:530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- Holmborn K, Ledin J, Smeds E, Eriksson I, Kusche-Gullberg M, Kjellen L. Heparan sulfate synthesized by mouse embryonic stem cells deficient in NDST1 and NDST2 is 6-O-sulfated but contains no N-sulfate groups. J Biol Chem. 2004;279:42355–42358. doi: 10.1074/jbc.C400373200. [DOI] [PubMed] [Google Scholar]
- Hotta Y, Sasaki S, Konishi M, Kinoshita H, Kuwahara K, Nakao K, Itoh N. Fgf16 is required for cardiomyocyte proliferation in the mouse embryonic heart. Dev Dyn. 2008;237:2947–2954. doi: 10.1002/dvdy.21726. [DOI] [PubMed] [Google Scholar]
- Hu Z, Wang C, Xiao Y, Sheng N, Chen Y, Xu Y, Zhang L, Mo W, Jing N, Hu G. NDST1-dependent heparan sulfate regulates BMP signaling and internalization in lung development. J Cell Sci. 2009;122:1145–1154. doi: 10.1242/jcs.034736. [DOI] [PubMed] [Google Scholar]
- Inatani M, Irie F, Plump AS, Tessier-Lavigne M, Yamaguchi Y. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 2003;302:1044–1046. doi: 10.1126/science.1090497. [DOI] [PubMed] [Google Scholar]
- Ishihara M, Guo Y, Wei Z, Yang Z, Swiedler SJ, Orellana A, Hirschberg CB. Regulation of biosynthesis of the basic fibroblast growth factor binding domains of heparan sulfate by heparan sulfate-N-deacetylase/N-sulfotransferase expression. J.Biol.Chem. 1993;268:20091–20095. [PubMed] [Google Scholar]
- Iwamoto R, Mine N, Kawaguchi T, Minami S, Saeki K, Mekada E. HB-EGF function in cardiac valve development requires interaction with heparan sulfate proteoglycans. Development. 2010;137:2205–2214. doi: 10.1242/dev.048926. [DOI] [PubMed] [Google Scholar]
- Iwao K, Inatani M, Matsumoto Y, Ogata-Iwao M, Takihara Y, Irie F, Yamaguchi Y, Okinami S, Tanihara H. Heparan sulfate deficiency leads to Peters anomaly in mice by disturbing neural crest TGF-beta2 signaling. J Clin Invest. 2009;119:1997–2008. doi: 10.1172/JCI38519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby ML, Waldo KL. Neural crest and cardiovascular patterning. Circ Res. 1995;77:211–215. doi: 10.1161/01.res.77.2.211. [DOI] [PubMed] [Google Scholar]
- Kuratani SC, Kirby ML. Migration and distribution of circumpharyngeal crest cells in the chick embryo. Formation of the circumpharyngeal ridge and E/C8+ crest cells in the vertebrate head region. Anat Rec. 1992;234:263–280. doi: 10.1002/ar.1092340213. [DOI] [PubMed] [Google Scholar]
- Lanner F, Lee KL, Sohl M, Holmborn K, Yang H, Wilbertz J, Poellinger L, Rossant J, Farnebo F. Heparan sulfation-dependent fibroblast growth factor signaling maintains embryonic stem cells primed for differentiation in a heterogeneous state. Stem Cells. 2010;28:191–200. doi: 10.1002/stem.265. [DOI] [PubMed] [Google Scholar]
- Lavine KJ, Yu K, White AC, Zhang X, Smith C, Partanen J, Ornitz DM. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev Cell. 2005;8:85–95. doi: 10.1016/j.devcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Lawrence R, Olson SK, Steele RE, Wang L, Warrior R, Cummings RD, Esko JD. Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J Biol Chem. 2008;283:33674–33684. doi: 10.1074/jbc.M804288200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JP, Gong F, Hagner-McWhirter A, Forsberg E, Abrink M, Kisilevsky R, Zhang X, Lindahl U. Targeted disruption of a murine glucuronyl C5-epimerase gene results in heparan sulfate lacking L-iduronic acid and in neonatal lethality. J Biol Chem. 2003;278:28363–28366. doi: 10.1074/jbc.C300219200. [DOI] [PubMed] [Google Scholar]
- Lin AE, Neri G, Hughes-Benzie R, Weksberg R. Cardiac anomalies in the Simpson-Golabi-Behmel syndrome. Am J Med Genet. 1999a;83:378–381. [PubMed] [Google Scholar]
- Lin L, Bu L, Cai CL, Zhang X, Evans S. Isl1 is upstream of sonic hedgehog in a pathway required for cardiac morphogenesis. Dev Biol. 2006;296:756–763. doi: 10.1016/j.ydbio.2006.03.053. [DOI] [PubMed] [Google Scholar]
- Lin X, Buff EM, Perrimon N, Michelson AM. Heparan sulfate proteoglycans are essential for FGF receptor signaling during Drosophila embryonic development. Development. 1999b;126:3715–3723. doi: 10.1242/dev.126.17.3715. [DOI] [PubMed] [Google Scholar]
- Lindahl U, Kusche-Gullberg M, Kjellén L. Regulated diversity of heparan sulfate. J.Biol.Chem. 1998;273:24979–24982. doi: 10.1074/jbc.273.39.24979. [DOI] [PubMed] [Google Scholar]
- Lindsay EA. Chromosomal microdeletions: dissecting del22q11 syndrome. Nat Rev Genets. 2001;2:858–868. doi: 10.1038/35098574. [DOI] [PubMed] [Google Scholar]
- Liu W, Selever J, Wang D, Lu MF, Moses KA, Schwartz RJ, Martin JF. Bmp4 signaling is required for outflow-tract septation and branchial-arch artery remodeling. Proc Natl Acad Sci U S A. 2004;101:4489–4494. doi: 10.1073/pnas.0308466101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macatee TL, Hammond BP, Arenkiel BR, Francis L, Frank DU, Moon AM. Ablation of specific expression domains reveals discrete functions of ectoderm- and endoderm-derived FGF8 during cardiovascular and pharyngeal development. Development. 2003;130:6361–6374. doi: 10.1242/dev.00850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschhoff KL, Baldwin HS. Molecular determinants of neural crest migration. Am J Med Genet. 2000;97:280–288. doi: 10.1002/1096-8628(200024)97:4<280::aid-ajmg1278>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- McLaughlin D, Karlsson F, Tian N, Pratt T, Bullock SL, Wilson VA, Price DJ, Mason JO. Specific modification of heparan sulphate is required for normal cerebral cortical development. Mech Dev. 2003;120:1481–1488. doi: 10.1016/j.mod.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- Miller DL, Ortega S, Bashayan O, Basch R, Basilico C. Compensation by fibroblast growth factor 1 (FGF1) does not account for the mild phenotypic defects observed in FGF2 null mice. Mol Cell Biol. 2000;20:2260–2268. doi: 10.1128/mcb.20.6.2260-2268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon AM, Capecchi MR. Fgf8 is required for outgrowth and patterning of the limbs. Nat Genet. 2000;26:455–459. doi: 10.1038/82601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon AM, Guris DL, Seo JH, Li L, Hammond J, Talbot A, Imamoto A. Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev Cell. 2006;10:71–80. doi: 10.1016/j.devcel.2005.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng A, Wong M, Viviano B, Erlich JM, Alba G, Pflederer C, Jay PY, Saunders S. Loss of glypican-3 function causes growth factor-dependent defects in cardiac and coronary vascular development. Dev Biol. 2009;335:208–215. doi: 10.1016/j.ydbio.2009.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrovsky O, Berman B, Gallagher J, Mulloy B, Fernig DG, Delehedde M, Ron D. Differential effects of heparin saccharides on the formation of specific fibroblast growth factor (FGF) and FGF receptor complexes. J.Biol.Chem. 2002;277:2444–2453. doi: 10.1074/jbc.M108540200. [DOI] [PubMed] [Google Scholar]
- Pallerla SR, Pan Y, Zhang X, Esko JD, Grobe K. Heparan sulfate Ndst1 gene function variably regulates multiple signaling pathways during mouse development. Dev Dyn. 2007;236:556–563. doi: 10.1002/dvdy.21038. [DOI] [PubMed] [Google Scholar]
- Pan Y, Carbe C, Powers A, Zhang EE, Esko JD, Grobe K, Feng GS, Zhang X. Bud specific N-sulfation of heparan sulfate regulates Shp2-dependent FGF signaling during lacrimal gland induction. Development. 2008;135:301–310. doi: 10.1242/dev.014829. [DOI] [PubMed] [Google Scholar]
- Pan Y, Woodbury A, Esko JD, Grobe K, Zhang X. Heparan sulfate biosynthetic gene Ndst1 is required for FGF signaling in early lens development. Development. 2006;133:4933–4944. doi: 10.1242/dev.02679. [DOI] [PubMed] [Google Scholar]
- Park EJ, Ogden LA, Talbot A, Evans S, Cai CL, Black BL, Frank DU, Moon AM. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development. 2006;133:2419–2433. doi: 10.1242/dev.02367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EJ, Watanabe Y, Smyth G, Miyagawa-Tomita S, Meyers E, Klingensmith J, Camenisch T, Buckingham M, Moon AM. An FGF autocrine loop initiated in second heart field mesoderm regulates morphogenesis at the arterial pole of the heart. Development. 2008;135:3599–3610. doi: 10.1242/dev.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi DJ, Ballard VL, Mikawa T. Epicardium is required for the full rate of myocyte proliferation and levels of expression of myocyte mitogenic factors FGF2 and its receptor, FGFR-1, but not for transmural myocardial patterning in the embryonic chick heart. Dev Dyn. 2003;228:161–172. doi: 10.1002/dvdy.10360. [DOI] [PubMed] [Google Scholar]
- Porras D, Brown CB. Temporal-spatial ablation of neural crest in the mouse results in cardiovascular defects. Dev Dyn. 2008;237:153–162. doi: 10.1002/dvdy.21382. [DOI] [PubMed] [Google Scholar]
- Qu X, Hertzler K, Pan Y, Grobe K, Robinson ML, Zhang X. Genetic epistasis between heparan sulfate and FGF-Ras signaling controls lens development. Dev Biol. 2011;355:12–20. doi: 10.1016/j.ydbio.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Pan Y, Carbe C, Powers A, Grobe K, Zhang X. Glycosaminoglycan-dependent restriction of FGF diffusion is necessary for lacrimal gland development. Development. 2012;139:2730–2739. doi: 10.1242/dev.079236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapraeger AC, Krufka A, Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252:1705–1708. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- Ringvall M, Ledin J, Holmborn K, Kuppevelt T, Ellin F, Eriksson I, Olofsson AM, Kjellén L, Forsberg E. Defective heparan sulfate biosynthesis and neonatal lethality in mice lacking N-deacetylase/N-sulfotransferase-1. J.Biol.Chem. 2000;275:25926–25930. doi: 10.1074/jbc.C000359200. [DOI] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider RA, Hu D, Rubenstein JL, Maden M, Helms JA. Local retinoid signaling coordinates forebrain and facial morphogenesis by maintaining FGF8 and SHH. Development. 2001;128:2755–2767. doi: 10.1242/dev.128.14.2755. [DOI] [PubMed] [Google Scholar]
- Schultz JE, Witt SA, Nieman ML, Reiser PJ, Engle SJ, Zhou M, Pawlowski SA, Lorenz JN, Kimball TR, Doetschman T. Fibroblast growth factor-2 mediates pressure-induced hypertrophic response. J Clin Invest. 1999;104:709–719. doi: 10.1172/JCI7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Li Y, Wang K, Zhou CJ. Cardiac neural crest and outflow tract defects in Lrp6 mutant mice. Dev Dyn. 2010;239:200–210. doi: 10.1002/dvdy.22079. [DOI] [PubMed] [Google Scholar]
- Sun X, Meyers EN, Lewandoski M, Martin GR. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev. 1999;13:1834–1846. doi: 10.1101/gad.13.14.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Born J, Salmivirta K, Henttinen T, Ostman N, Ishimaru T, Miyaura S, Yoshida K, Salmivirta M. Novel heparan sulfate structures revealed by monoclonal antibodies. J Biol Chem. 2005;280:20516–20523. doi: 10.1074/jbc.M502065200. [DOI] [PubMed] [Google Scholar]
- Washington Smoak I, Byrd NA, Abu-Issa R, Goddeeris MM, Anderson R, Morris J, Yamamura K, Klingensmith J, Meyers EN. Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev Biol. 2005;283:357–372. doi: 10.1016/j.ydbio.2005.04.029. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Miyagawa-Tomita S, Vincent SD, Kelly RG, Moon AM, Buckingham ME. Role of mesodermal FGF8 and FGF10 overlaps in the development of the arterial pole of the heart and pharyngeal arch arteries. Circ Res. 2010;106:495–503. doi: 10.1161/CIRCRESAHA.109.201665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64:841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]
- Zhang J, Lin Y, Zhang Y, Lan Y, Lin C, Moon AM, Schwartz RJ, Martin JF, Wang F. Frs2alpha-deficiency in cardiac progenitors disrupts a subset of FGF signals required for outflow tract morphogenesis. Development. 2008;135:3611–3622. doi: 10.1242/dev.025361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Sutliff RL, Paul RJ, Lorenz JN, Hoying JB, Haudenschild CC, Yin M, Coffin JD, Kong L, Kranias EG, Luo W, Boivin GP, Duffy JJ, Pawlowski SA, Doetschman T. Fibroblast growth factor 2 control of vascular tone. Nat Med. 1998;4:201–207. doi: 10.1038/nm0298-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.