

Abstract
A series of self-associating foldamers have been designed as heparin reversal agents, as antidotes to prevent bleeding due to this potent antithrombotic agent. The foldamers have a repeating sequence of Lys-Sal, in which Sal is 5-amino-2-methoxy-benzoic acid. These foldamers are designed to self-associate along one face of an extended chain in a β-sheet-like interaction. The methoxy groups were included to form intramolecular hydrogen bonds that preclude the formation of very large amyloid-like aggregates, while the positively charged Lys side chains were introduced to interact electrostatically with the highly anionic heparin polymer. The prototype compound (Lys-Sal)4 carboxamide weakly associates in aqueous solution at physiological salt concentration in a monomer-dimer-hexamer equilibrium. The association is greatly enhanced at either high ionic strength or in the presence of a heparin derivative, which is bound tightly. Variants of this foldamer are active in an antithrombin III–factor Xa assay, showing their potential as heparin reversal agents.
Foldamers are nonbiological sequence-specific polymers of defined-length oligomers that adopt well-defined secondary and tertiary structures.1−4 Not only are they used to test hypotheses concerning biomacromolecular folding and function, but also they can be used to design molecules for practical applications, including mimics of antimicrobial peptides and inhibitors of protein–protein interactions.5−9 Previously, the design of biologically active foldamers has focused on molecules that act as monomolecularly folded units. However, a number of foldamers are also known to self-assemble into more complex architectures than might be easily achieved using a monomeric unit,10−16 because a large cooperatively formed unit can be assembled from shorter pieces that individually are less well structured.17−20 Here, we design self-assembling foldamers that recognize and antagonize the antithrombotic action of heparin.
Heparin, also known as unfractionated heparin (UFH), is a complex and highly sulphated glycosaminoglycan. This oligosaccharide is recognized for its ability to prevent the coagulation of blood. Heparin inhibits blood clotting by binding to the enzyme inhibitor antithrombin III (ATIII), consequently inactivating factor Xa (FXa) and other proteolytic enzymes involved in forming fibrin-rich clots.21−25 Because of its antithrombotic function, heparin is widely used clinically as an injectable anticoagulant for the prevention and treatment of thrombosis.26 For example, heparin is commonly used for the prevention of clot formation during cardiothoracic and vascular surgical procedures. However, following surgery, heparin’s activity needs to be reversed. The only agent presently approved to reverse the action of UFH is the arginine-rich small heterogeneous protein preparation protamine. Protamine restores clotting by binding UFH,27 thereby reducing its ability to bind to ATIII. Because UFH is highly negatively charged, protamine and related basic peptides are able to neutralize its action through electrostatic interactions.28 However, protamine and related peptides29−31 have a number of serious adverse side effects,32,33 and there are no approved agents for reversal of low molecular weight heparins (LMWHs) such as fondaparinux (Arixtra), a synthetic pentasaccharide heparin analogue.34
Previous studies of heparin–protein interactions with Arg-rich peptides indicated that the minimal requirement for activity is the presentation of a high density of cationic groups along one face of a secondary structure.29−31 Here, we design self-associating foldamers that similarly interact with heparin using an electrostatic mechanism.
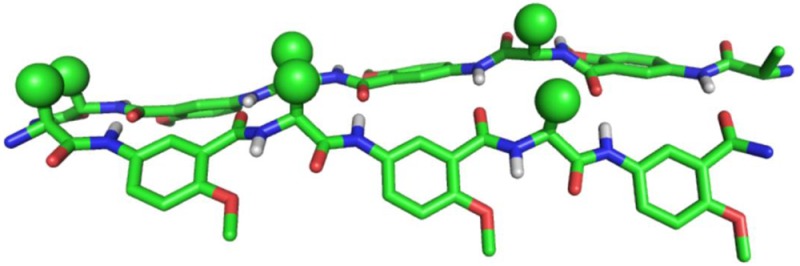
As a model for the foldamer backbone, we used principles previously developed in the pioneering and encompassing contributions of the groups of Huc,12−14 Nowick,10,15 and Gong.11,16 Specifically, we chose a previously characterized backbone in which an α-amino acid and a salicylamide alternate in sequence, which we refer to as SalAA foldamers (Figure 1).11,16,34−37 A short prototype for this family of foldamers, designated (Sal-Gly-Sal) (Figure 1A), has been crystallized from organic solvents and shown to form an antiparallel, two-stranded sheet-like dimer, stabilized by intermolecular hydrogen bonds between the main chain amides.11 Aryl-methoxy groups form intramolecular hydrogen bonds with amides not involved in the dimer interface (Figure 1A), thereby further stabilizing the structure and preventing the formation of long fibrils. To use this backbone as a reversibly associating scaffold, we positioned cationic side chains along the backbone to attract heparin via electrostatic interactions as in previous heparin-binding peptides and proteins. This led to the design of (Sal-Lys)n foldamers (Figure 1B, C, and D). Intermolecular electrostatic repulsion between the Lys side chains would lead to weak association in solution at physiological salt concentrations. However, we expected that the SalAA foldamers would associate under conditions in which the electrostatic repulsion was minimized (e.g., when they bind to heparin).
Figure 1.
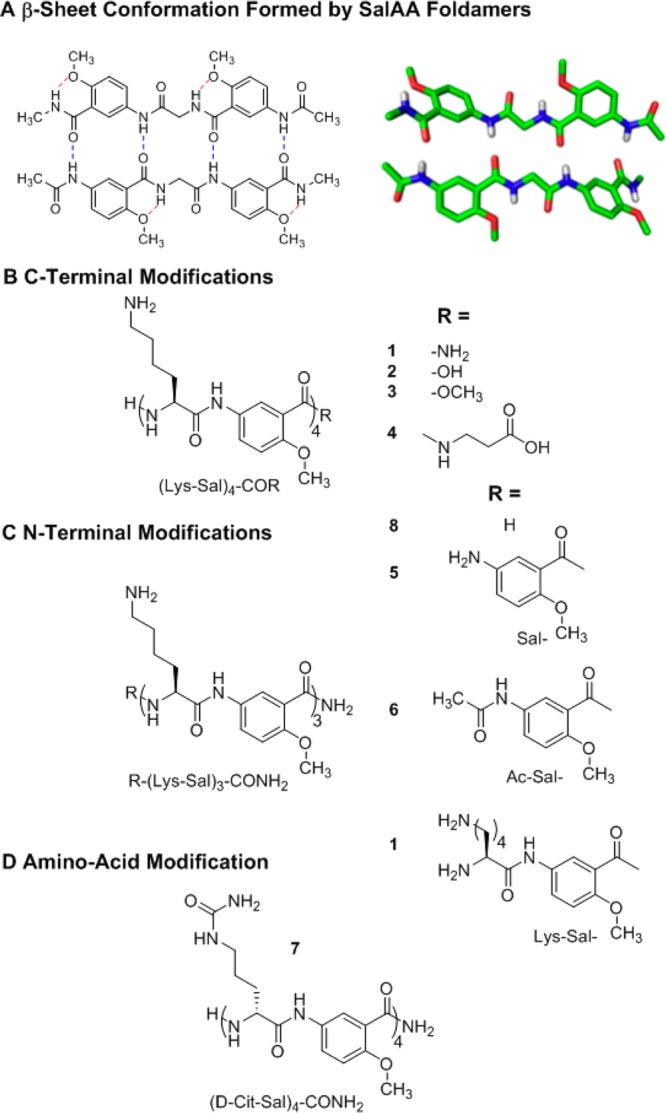
Chemical structures of SalAA foldamers explored in this study. (A) Schematic (left) and crystal structure (right) of a dimeric foldamer based on the salicylamide backbone, Ac-Sal-Gly-Sal-NMe amide, in which Ac is acetyl, Sal is the indicated amino-salicylic acid methyl ether unit, Gly is glycine, and NMe amide is an N-methyl amide. (B) Chemical structures of salicylamide-α-amino acid peptides (SalAA foldamers) for C-terminal and N-terminal (C) modified series. (D) Chemical structure of compound 7, (d-Cit-Sal)4-CONH2; in this variant of (Lys-Sal)4-CONH2, compound 1, cationic lysine is replaced with a neutral citrulline amino acid.
Here, we examine the association of the (Lys-Sal)n foldamer by using the techniques of analytical ultracentrifugation (AUC), concentration-dependent UV–vis absorption, circular dichroism (CD) spectroscopy, and isothermal calorimetry (ITC). The foldamers exist in a monomer-dimer-hexamer (trimer of dimers) equilibrium. The hexameric structure, although not anticipated in the initial design, can be rationalized in terms of the predicted structure of the dimer, due to the hydrophobicity of the salicylamide backbone. The salt-dependence of the interaction demonstrates that the association is opposed by electrostatic repulsion in the absence of heparin. Importantly, the pentasaccharide heparin analogue fondaparinux binds tightly to the associated form of the foldamer, as anticipated in the design.
Results and Discussion
Design Considerations
The key to heparin’s activity is a specific pentasaccharide sulfation sequence, which is important for antithrombin III binding.34,38−41 The foldamers were designed to target this pentasaccharide region of heparin, based on the overall length and shape of the saccharide. However, in this first round of design, we did not strive to specifically recognize the fine-grained geometry of the target. Instead, based on earlier studies with poly-Arg peptides30,42−44 and heparin-binding foldamers,5 we expected that the creation of a positively charged patch of appropriate dimensions would be sufficient to bind the pentasaccharide.
Based on the structure of (Sal-Gly-Sal) (Figure 1A), we designed a series of (Sal-Lys)n foldamers (Figure 1B, C, and D), which should form two-stranded β-sheet-like structures in which the basic Lys side chains project from one face of the structure. Molecular models suggested that four Sal-Lys units (as in compound 1, Figure 1) would be sufficient to mediate binding to the pentasaccharide. This also turned out to be the minimal length required to allow association into the active oligomeric form in aqueous solution. To probe the requirements for self-association and heparin binding, we varied the chain length and the nature of the N-and C-terminal functional groups (compounds 2–6; Figures 1B, C). Furthermore, to determine the effect of charge, we replaced the Lys residues with citrulline (d-Cit) in compound 7 (Figure 1D).
Anti-heparin Activity Is Dependent on Chain Length and Charge
The ability of the foldamers to interact with heparin was evaluated using the pentasaccharide fondaparinux. This pentasaccharide interacts with anti-thrombin III, triggering the inhibition of factor Xa (FXa). The foldamers were able to reverse this interaction in a concentration-dependent manner. We first examined the chain-length dependence of the potency of the foldamers, as assessed by their EC50 values in this assay (Table 1, top). A foldamer with only three (Lys-Sal) units was inactive, but extending its length by one Sal unit resulted in a compound, 5, with a 6.8 μM EC50. Further extension of an Ac-Sal unit, compound 6, increased activity to an EC50 of 2.3 μM; addition of a Lys-Sal unit, compound 1, failed to further increase potency but rather decreased potency slightly. The activity did not depend on chirality, as the enantiomer of 1, 1a, was nearly equipotent to 1. To test whether the electrostatic interactions between Lys and the foldamer were essential for activity, we synthesized compound 7, in which neutral citrulline replaced the lysine residues. As expected, this compound was fully inactive.
Table 1. Anti-fondaparinux Activities of N-Terminal Truncations, Side Chain Modifications, and C-Terminal Truncations of SalAA Foldamersa.
| compound | sequence | FXa EC50 (μM) |
|---|---|---|
| N-Terminal Modifications | ||
| 8 | H2N-(Lys-Sal)3-CONH2 | inactive |
| 5 | Sal-(Lys-Sal)3-CONH2 | 6.8 |
| 6 | Ac-Sal-(Lys-Sal)3-CONH2 | 2.3 |
| 1 | Lys-Sal-(Lys-Sal)3-CONH2 | 3.6 |
| 1a | H2N-(d-Lys-Sal)4CONH2 | 3.0 |
| 7 | H2N-(d-Cit-Sal)4-CONH2 | inactive |
| C-Terminal Modifications | ||
| 1 | H2N-(Lys-Sal)4-CONH2 | 3.6 |
| 2 | H2N-(Lys-Sal)4-COOH | inactive |
| 3 | H2N-(Lys-Sal)4-COOCH3 | 11 |
| 4 | H2N-(Lys-Sal)4-CONH-(CH2)2-COOH | 6.0 |
Average standard deviation was ±9%.
We next tested the effect of changes to the C-terminus of compound 1, (Lys-Sal)4-CONH2 (Table 1, bottom). The C-terminal carboxamide was expected to be important for self-association, so we explored replacing it with a carboxylate in compound 2, (Lys-Sal)4-COOH, which resulted in an inactive analogue. The corresponding methyl ester (Lys-Sal)4-COOCH3, 3, showed weak potency. By contrast, the extension of the C-terminal residue to a β-Ala unit (Lys-Sal)4-CO-β-Ala-carboxylate, 4, had an EC50 of 6.0 μM. Thus, a carboxylate is not necessarily detrimental if it is moved sufficiently away from the (Lys-Sal)n core. In summary, the C-terminal Sal monomer should optimally be an amide, but the substituent from the amide group can be either a carboxamide (CONH2) or a β-Ala residue.
Self-Association of Foldamers
We expected that the Lys-Sal foldamers would self-associate weakly at physiological pH, because of electrostatic repulsion between the positively charged Lys residues. Association of the highly anionic pentasaccharide would alleviate this repulsion, thereby driving the self-association of the foldamer upon heparin complexation. If this model is correct, then we would expect that compound 1, (Lys-Sal)4-CONH2, would associate only weakly in the absence of fondaparinux. Also, its self-association would depend on ionic strength, becoming more favorable at high salt concentrations that could electrostatically shield the interactions between neighboring Lys side chains. Similarly, the folding and assembly of (d-Cit-Sal)4-CONH2 (compound 7) would be highly favorable, because it lacks the destabilizing electrostatic interactions between the charged Lys side chains. Furthermore, as the chain length is shortened from four to three Lys-Sal units (compounds 1 and 8, respectively), we expect that the free energy of self-association of the foldamer would become less favorable, explaining this compound’s lack of activity. Finally, if the C-terminal carboxamide of 1 stabilized folding through the formation of an intermolecular hydrogen bond, then the C-terminal carboxyl-containing 2, (Lys-Sal)4-COOH, and the corresponding methyl ester (3) might weakly self-associate.
To address these questions, we first extensively examined the assembly and folding of compound 1, as a prototype of the series. Next we examined the ability of the variants of compound 1 to assemble in aqueous solution. Finally, we examined the direct interaction of compound 1 and selected variants with fondaparinux.
Analytical Ultracentrifugation (AUC) of Compound 1 Reveals a Monomer-Dimer-Hexameric Assembly
Compound 1 was examined by sedimentation equilibrium AUC. The compound was dissolved in TBS buffer at physiological salt concentration (0.050 M Tris, pH 7.4; 0.15 M NaCl). Curves derived from a single species of a fixed association state failed to fit the data, as did fully cooperative monomer-nmer equilibrium schemes. We therefore examined multistep assemblies. The best fit was obtained with a dimer-hexamer model (Supplementary Figure S1).
CD and UV–vis Spectroscopy of Compound 1 Reveals a Concentration-Dependent Monomer-Dimer-Hexamer Equilibrium
Compound 1 showed marked changes in its electronic spectra for the range of concentrations it was shown to associate by AUC. The CD spectrum of 1 (Figure 2A, B) shows large changes in mean residue ellipticity with increasing concentration, consistent with the formation of a stable organized structure. The aromatic absorption from 300 to 350 nm (Figure 2A, C) shows a single peak (Figure 2A) at low concentrations in the CD spectrum, which closely matches that of the corresponding absorbance spectrum (Figure 2C). However, as the concentration and degree of association increase, this band resolves into a pair of bands of opposite sign. This splitting is indicative of excitonic coupling between the aromatic groups, as anticipated from the designed conformation. In parallel, the absorbance spectrum shows a shift to longer wavelengths, from about 310 to 320 nm, as the concentration is increased.
Figure 2.

Concentration-dependent CD spectra of compound 1 in the near (A) and far (B) UV regions. Extinction coefficient as a function of wavelength (C), determined from near UV–vis absorption spectra. CD mean residue ellipticity (MRE) at 333 nm and UV–vis absorption shifts in λmax are overlaid (D) with their respective fits; Khex equals 10–18.4 (or 4.0 × 10–19) M5 for CD MRE333 and 10–19.3 (or 5 × 10–20) M5 for UV–vis λmax.
The change in the concentration-dependent spectra of both the CD (mean residue ellipticity at 333 nm, MRE333 nm) and UV–vis (λmax) values (Figure 2D) can be fit into a monomer-dimer-hexamer equilibrium. However, due to the lower degree of sensitivity and greater scatter of the data in this experiment, we were unable to precisely fit both monomer-dimer (K(mon-dim)) and dimer-hexamer (K(dim-hex)) equilibrium constants. The product of the two equilibrium constants for the overall assembly (K(mon-hex) = K(mon-dim)3 × K(dim-hex)) was well-defined, but the two equilibrium constants covaried when fit individually. Therefore, we fit the curves to a cooperative monomer-hexamer equilibria (K(mon-hex)). The values of the derived dissociation constants were similar (4.0 × 10–19 M5 and 5 × 10–20 M5, from CD and UV–vis respectively). These dissociation constants correspond to midpoints of association ([1]mid) of 0.29 and 0.19 mM, respectively. Thus, under physiological salt concentration, 1 weakly associates into a well-structured oligomer with a midpoint of approximately 250 μM, and the agreement of the fit from both CD and absorbance spectroscopy indicates that the two techniques are comparable.
ITC Investigation of the Dissociation of Compound 1 Confirms Concentration-Dependent Self-Assembly
We used isothermal calorimetry to measure the heat of dissociation of compound 1 (Figure 3). The compound was dissolved in TBS at 1 mM concentration where it is predominantly in the associated form and then serially diluted into buffer. Initially, a large heat change is observed as the compound dissociates, but with each additional increment the concentration of 1 in the cell increases, leading to decreased net association (Figure 3A, B).
Figure 3.
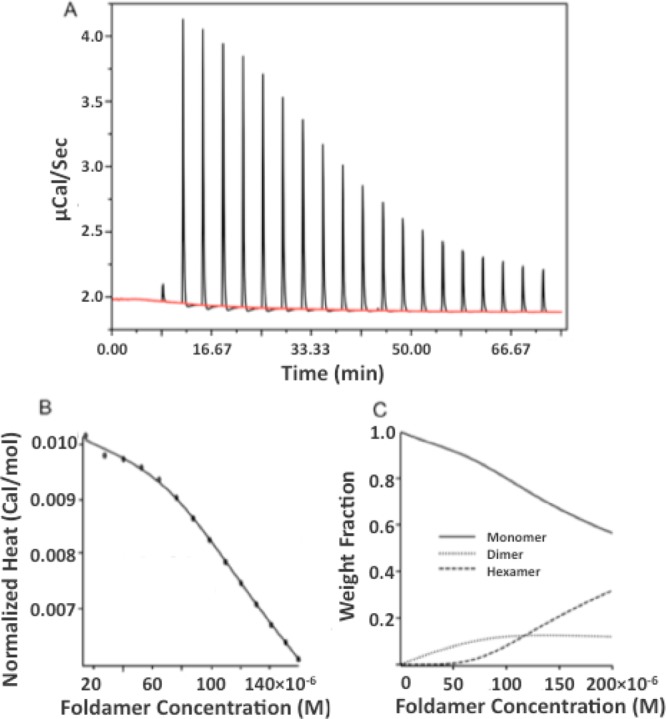
Concentration-dependent ITC of 1. A stock solution of 1.0 mM 1 in TBS was serially diluted into the same buffer (A). The heat associated with each injection was determined by integrating the peaks. The heat generated for a given final concentration is then summed and expressed in cal/mol vs final concentration (B). The smooth line shows the best fit of a monomer-dimer-hexamer equilibrium to the data (see Methods). The weight fraction distribution for hexamer, dimer, and monomer species are plotted vs concentration (C).
Association of Compound 1 Variants Measured by UV–vis Spectroscopy
We next used UV–vis spectroscopy to determine how C-terminal modifications affected the assembly of the foldamers. As for 1, the changes in λmax were measured as a function of each variant’s concentration (Figure 4A, B). Compound 4, which has an additional β-Ala residue compared to 1, showed a similar curve, but the midpoint was shifted to lower concentrations. Fitting a monomer-hexamer equilibrium to the data gave values of λmax for the monomer and hexamer that were the same within experimental error for the two compounds, but the midpoint was shifted from 0.19 mM for 1 to 0.06 mM for 4. By contrast, the C-terminal methyl ester, 3, showed a much weaker association as compared to 1 (midpoint = 0.37 mM), and (Lys-Sal)4-COOH, 2, did not appear to associate at all over this concentration range.
Figure 4.

UV–vis absorption λmax and normalized ITC heats of dilution as a function of SalAA foldamer concentration. (A) UV–vis absorption λmax as a function of SalAA foldamer concentration for C-terminal modifications. (B) UV–vis absorption λmax as a function of SalAA foldamer concentration for N-terminal modifications. Normalized ITC heats of dilution (pK(hex-mon)) values are summarized in Table 2 as a function of SalAA foldamer concentration for C-terminal (C) and N-terminal (D) modified series, and their respective fits are illustrated as lines.
Compound 7 (Cit-Sal)4-CONH2, in which the Lys side chains are converted to neutral citrulline residues, did not appear to show significant dissociation over the concentration range where 1 showed a concentration-dependent spectrum. Compound 7 showed a λmax value of 316 ± 1 nm, as seen for the hexameric form of 1. However, its association appeared to be so strong that it failed to dissociate over this concentration range, presumably because it lacked the electrostatic repulsive interactions between closely opposing Lys side chains seen in 1.
The UV–vis spectra for the N-terminally truncated foldamers showed that the N-terminal Lys residue in (Lys-Sal)4-CONH2 was not essential for association, but further truncation led to a loss of association. Compounds 5 and 6 associated more tightly than 1, with midpoints of 0.08 and 0.07 mM for 5 and 6 respectively. The foldamer with an unacylated salicylamide monomer (5) had a slightly greater λmax in the hexamer, which we attribute to the fact that only this foldamer had a free aniline group. The tighter association of compound 5 and 6, which lack the N-terminal Lys of 1, is consistent with the idea that electrostatic repulsion disfavors association of 1, as removal of a Lys from the N-terminus would decrease the net charge. However, further truncation of the N-terminal Lys-Sal dipeptide unit gave a compound that did not associate over the measured concentration range, as assessed from the fact that compound 8, (Lys-Sal)3-CONH2, did not show any spectral changes at concentrations as high as 500 μM (Supplementary Figure S2; Table 2).
Table 2. Effects of C-Terminal Modifications and N-Terminal Modifications on the Association of Compound 1, Determined from the Concentration Dependence of the UV–vis Absorption and ITCa.
| pK(hex-mon) |
|||||||
|---|---|---|---|---|---|---|---|
| compound | sequence | ITC | UV–vis | λmax(hex) (nm) | λmax(mon) (nm) | Δλmax (nm) | ΔH(hex-mon) (cal/mol) |
| C-Terminal Modifications | |||||||
| 1 | H2N-(Lys-Sal)4-CONH2 | 18.8 | 19.3 | 318.1 | 303.5 | 14.6 | 14100 |
| 2 | H2N-(Lys-Sal)4-COOH | 302 | |||||
| 3 | H2N-(Lys-Sal)4-COOCH3 | 17.9 | 315.1 | 304.4 | 10.7 | ||
| 4 | H2N-(Lys-Sal)4-CONH-(CH2)2-COOH | 21.0 | 21.8 | 317.6 | 303.1 | 14.5 | 13800 |
| N-Terminal Modifications | |||||||
| 8 | (Lys-Sal)3-CONH2 | <15 | 304.0 | ||||
| 5 | Sal-(Lys-Sal)3-CONH2 | 20.3 | 21.1 | 323.3 | 305.7 | 17.6 | 15700 |
| 6 | Ac-Sal-(Lys-Sal)3-CONH2 | 21.7 | 21.5 | 319.8 | 305.1 | 14.7 | 15600 |
| 1 | Lys-Sal-(Lys-Sal)3-CONH2 | 18.8 | 19.3 | 318.1 | 303.5 | 14.6 | 14100 |
Δλmax is the difference between the λmax values of the hexameric and monomeric species, determined from the fits. The association of compound 2 was too weak for UV–vis data fitting. ITC heats of formation were too small to accurately determine dissociation constants for compounds 2 and 3.
Dissociation of Variants of Compound 1 Measured by ITC
For concentration-dependent ITC, variants of 1 in TBS buffer were diluted in a calorimetric cell at 25 °C in the same manner described for compound 1 (Figure 4C, D). Because the dissociation of 1 was highly cooperative, the data were fit to a cooperative monomer-dimer-hexamer equilibrium (Table 2). As before, the UV–vis and ITC results are in good agreement and share the same trends with tightest self-assembly for the N-terminally and C-terminally modified variants (5, 6, and 4, respectively). Weaker association was seen for the methyl ester variant (3 relative to compound 1), and the degree of heat formation was too small to allow accurate fitting of the dissociation constant to the curve. Also, as seen by UV–vis spectroscopy, very weak to no self-assembly was observed for the carboxylate variant, 2. UV–vis and ITC data are summarized together, for comparison, in Table 2.
Ionic Strength Dependence of the Association of Compound 1
The above data on variants of 1 strongly suggest that its association is electrostatically destabilized by close opposition of the Lys side chains upon association. If this is the case, then decreasing the ionic strength should weaken the association of 1, while increasing ionic strength should have the opposite effect. Figure 5 shows the concentration-dependent UV–vis spectra for 1 at NaCl concentrations ranging from 0 to 1.0 M NaCl. Global fitting of the curves indicated that the midpoint of the transition shifts from less than 4 μM to greater than 1 mM as the [NaCl] is varied from 0 to 1.0 M.
Figure 5.
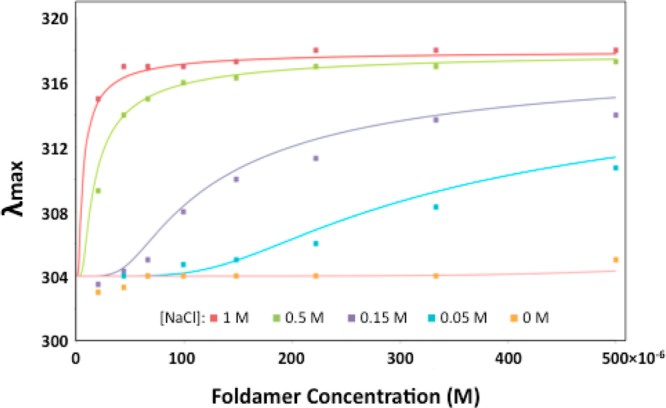
Concentration-dependent shifts in λmax for 1 at varying [NaCl] in 0.05 M Tris, pH 7.4. The smooth curves were generated by global fitting of the data to a monomer-hexamer equilibrium. The value of λmax for the monomer (304 nm) and hexamer (318 nm) were globally fit, while the value of pK(hex-mon) was allowed to vary for each curve. The fit pK(hex-mon) values were <15 at 0 M NaCl, 17.4 at 0.05 M, 19.7 at 0.15 M, 24.1 at 0.5 M, and 26.5 at 1.0 M.
Binding of Fondaparinux Induces Association and Folding of Compound 1: CD and ITC
Next, we sought to determine how fondaparinux influenced the association of the SalAA foldamer. The ability of compound 1 to bind to fondaparinux was measured by CD titrations (Figure 6A) in which increasing equivalents of fondaparinux were titrated into 25 μM compound 1. At this low concentration compound 1 gave a CD spectrum typical of the monomeric form, but a spectrum that was identical in shape to that of the hexamer was observed at high fondaparinux concentrations (Figure 2A). The titration curve is linear up to 6.25 μM fondaparinux; beyond this point no further spectral changes were observed. This titration behavior is typical of a tight-binding isotherm. An increase in light-scattering was also observed as the concentration of fondaparinux was increased, which made it difficult to obtain quality spectra at higher fondaparinux concentrations.
Figure 6.
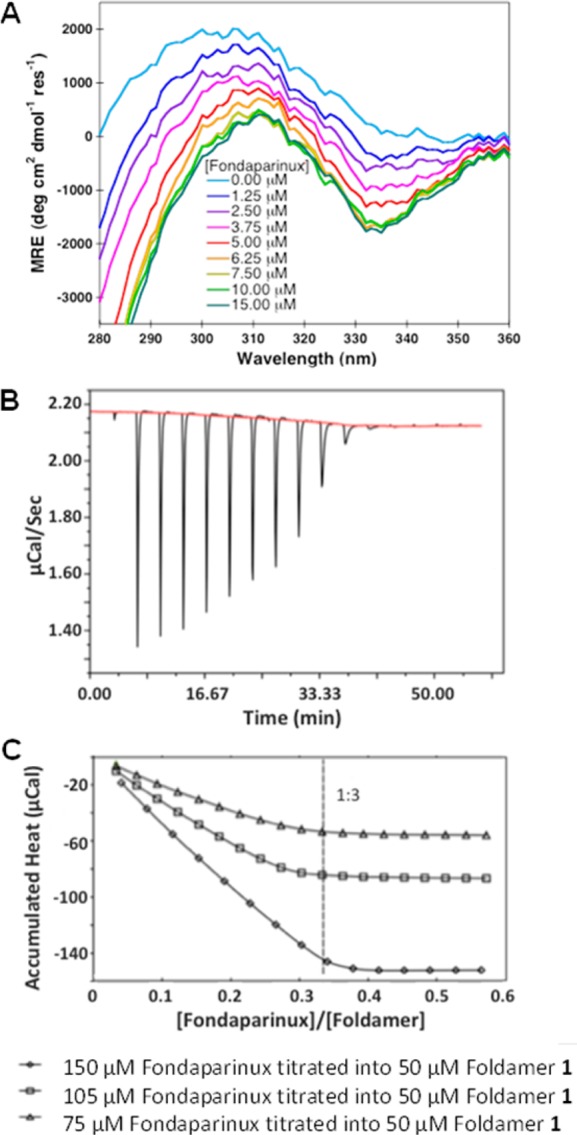
Near UV CD and ITC characterizations of SalAA compound 1 binding to fondaparinux. (A) Near UV CD spectra of the fondaparinux titration of compound 1, (Lys-Sal)4-CONH2, where increasing concentrations of fondaparinux were titrated into 25 μM compound 1. (B) Concentration-dependent ITC of fondaparinux titrated into compound 1, (Lys-Sal)4-CONH2. Shown are the raw data of 150 μM fondaparinux (stock solution) titrated into 50 μM compound 1. (C) Accumulated integrated heat changes as a function of fondaparinux:foldamer molar ratio for three experiments at the listed concentrations.
In order to confirm the association of fondaparinux with 1, the titration was also monitored using ITC. Compound 1 was dissolved in TBS at concentrations between 25 μM and 50 μM, where it is largely monomeric (Figure 6B). This solution was titrated with fondaparinux; the concentration of the stock solution of fondaparinux was adjusted to ensure equal equivalents per injection for each titration. In each case linear binding curves were observed (Figure 6B). Interestingly, while the heat change was positive for the dissociation of 1 (Figure 3A) the heparin-binding curves showed negative enthalpy changes (Figure 6B). This finding is consistent with the finding that the low molecular weight heparin analogue induces association of 1, although the association of fondaparinux complicates a quantitative analysis of the energetics. At each concentration of 1 examined, the fondaparinux-promoted SalAA foldamer self-association saturates at a 1:3 molar ratio. Given the stoichiometry and similarity of the CD spectrum of the bound form to that of the hexameric form of 1, it appears that two fondaparinux molecules bound per tetramer. The titration behavior suggests that the dissociation constant for the interaction is significantly lower than the concentration of 1.
Conclusions
Here,
we design a series of foldamers that
use the principle of self-assembly to create a heparin-interactive
surface. Our data confirm this design principle and suggest that the
stable and active form of SalAA foldamers occurs in the presence of
fondaparinux; in the absence of fondaparinux the foldamer favors a
monomer. The association of foldamers is best described below in a
monomer-dimer-hexamer equilibrium in the absence and presence of fondaparinux
as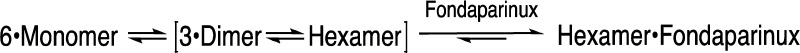
While our original design called for a dimeric association, we experimentally discovered that 1 further associated to a hexamer. We have not yet solved the structure for the hexamer but expect that it is driven by the amphiphilic conformation of the dimer. The Lys side chains project from one face of the structure while the other face of the dimer is rich in aromatic groups and the methyl ethers of the salicylamide. Thus, it is likely that this hydrophobic surface leads to further self-association. The association into the hexameric form also requires close association of the Lys side chains, which suggested that the association should be more favorable in the presence of increasing ionic strength that would screen the electrostatically repulsive interaction. This was found to be the case and further supports the idea that the hexameric form associates with fondaparinux by electrostatic interactions. Also supporting this idea, the neutral citrulline-containing foldamer (7) self-associated but did not bind fondaparinux.
The detailed SAR that emerged from this work is also consistent
with the linked equilibrium in which the binding of fondaparinux is
dependent on oligomerization of the foldamer. The data in Table 2 show that there is a minimal core that is required
for association of the Lys-Sal foldamers into a hexamer, shown in
blue in the chemical structures shown below. 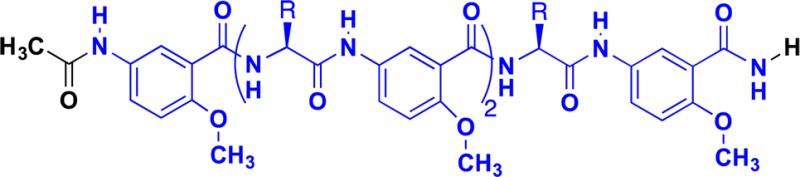
Compounds in which the N-terminus was elongated beyond this core (e.g., with a Lys- in 1 or Ac- in 6) retained the ability to associate. Also, the C-terminus could be extended with a β-Ala with retention of the ability to self-associate. Moreover, deletions or replacements to the core led to molecules that failed to self-associate. For example, deletion of the N-terminal Sal residue or substitution of the C-terminal amide with either a carboxylate or a methyl ester strongly decreased self-association.
According to the self-assembly scheme, binding of fondaparinux requires both self-association into hexamers as well as interaction with the pentasaccharide, so both steps contribute to the potency of a given compound. Compounds that failed to show appreciable self-association (e.g., 2, 8) were completely inactive in the FXa assay. Similarly, the methyl ester of 1 showed reduced affinity for self-association as well as reduced potency in the factor Xa assay. On the other hand, self-association per se was not sufficient to ensure interaction with fondaparinux. For example, compound 7, (Cit-Sal)4-CONH2, which has neutral side chains was inactive in the FXa assay, and compounds with reduced positive charge, such as 4, self-associated but was somewhat less potent in the factor Xa assay. The interrelation between self-association and fondaparinux activity can be further seen in a comparison of compound 5, Sal-(Lys-Sal)3CONH2, and 6, Ac-Sal-(Lys-Sal)3CONH2. Both compounds have similar charge (we assume the N-terminal Sal aniline group of 5 would be neutral at the assay pH of 8.4), but compound 6 is more potent at reversing the effects of fondaparanux, likely because of its greater tendency to self-associate (Table 2).
In summary, this work extends the use of the SalAA backbone to the design of a self-assembling foldamer that inhibits fondaparinux in a clinically relevant assay. Also compound 1 was highly active in a standard activated partial thromboplastin time aPTT assay (IC50 = 1.1 μM versus unfractionated heparin, as described in the Supporting Information). Future papers will describe further modifications of this backbone to increase affinity and specificity for this target.
Methods
Synthesis of SalAA Foldamers
Foldamers 1–3 were prepared by solution phase methods that are described in detail in the Supporting Information. Foldamers 5–8 were synthesized by solid phase methods as described in the Supporting Information. Foldamer 4 was similarly synthesized on preloaded Fmoc-β-alanine Wang resin (Novabiochem) (Supporting Information).
Heparin Reversal Assay
Neutralization of antifactor Xa activity was determined in a kinetic in vitro amidolytic assay for factor Xa activity. Human antithrombin III and bovine factor Xa were obtained from Diapharma; 0.036 IU/mL antithrombin was combined with 0.636 nkat/mL bovine factor Xa in a NaCl Tris pH 8.4 buffer. Activity was determined by kinetically monitoring cleavage of a chromogenic substrate, S-2765 (Diapharma) in a SpectraMax 250 plate reader at OD405 nm. Fondaparinux was added at a final concentration of 0.02 μg/mL to inhibit factor Xa activity. Reversal of fondaparinux was measured following addition of serially diluted compound. EC50 was calculated as the amount of compound needed to return factor Xa activity to 50% that of normal.
Sedimentation Equilibrium Analytical Ultracentrifugation (AUC)
The oligomerization state and affinity for compound 1, (Lys-Sal)4-CONH2, was characterized by sedimentation equilibrium AUC, using a Beckman XL-I analytical ultracentrifuge at 25 °C in Tris-buffered saline (TBS, 0.050 M Tris, 0.150 M NaCl, pH 7.4). Here, 250 μM (Lys-Sal)4-CONH2 was centrifuged at 35, 40, 45, 48 KRPM, respectively, and the concentration distributions vs rotor diameter were collected at 280 nm. The data were analyzed by globally fitting using nonlinear least-squares (IGOR Pro Wavemetrics). The data are not well described by a single molecular species, which prompted the examination of several different association schemes. The simplest that was able to fit the data adequately was a dimer-hexamer scheme. The fitting procedures and experimental method are provided in more detail in the Supporting Information.
Isothermal Titration Calorimetry (ITC)
Each foldamer was diluted directly into Tris-buffered saline (TBS, 0.050 M Tris, 0.150 M NaCl, pH 7.4). The foldamers were prepared at final concentrations of 1 mM in TBS buffer and titrated into TBS buffer in the calorimetric cell at 25 °C. The heat exchange evolved in each injection was obtained from the integral area of the calorimetric signal and has been normalized upon each accumulated addition. The normalized heat of compound 1 was plotted as a function of foldamer concentration and analyzed using a monomer-dimer-hexamer model, while a monomer-hexamer fitting model was applied to the other foldamers.
Detailed procedures for data fitting can be found in the Supporting Information.
Circular Dichrosim (CD) Spectroscopy
Concentration-dependent CD data were collected on a Jasco CD spectropolarimeter (J-810) using either a 10.0, 5.0, 1.0, 0.5, or 0.2 mm sample cuvette, and the ellipticity was expressed as mean residue ellipticity. Concentration-dependent CD experiments of foldamers were obtained in TBS buffer at 25 °C in step scan mode with a 1 nm data pitch, 4 s response time, 1 nm bandwidth, and 3 accumulations. CD experiments for fondaparinux-foldamer titrations were conducted in a 10 mm sample cuvette as described above with the addition of 5 min mixing upon each addition of fondaparinux.
The observed MRE (MREobs) at 333 nm as a function of foldamer concentration was analyzed by nonlinear least-squares fitting to an equilibrium hexamer-monomer disassociation by IGOR Pro (Wavemetrics). The experimental observables are MREobs at 333 nm and the total foldamer concentration [T], which is given by eqs 2A and 2B (Supporting Information). The MREobs was expressed as the sum of the MRE of the hexamer and the monomer (eq 3). MREhex and MREmon are dependent variables, which are obtained by nonlinear least-squares fitting to eq 3:
| 3 |
in which C is a constant, which was found to be close to zero.
UV–vis Absorption Spectroscopy
UV–vis concentration-dependent and fondaparinux-foldamer titration data were collected on a HP diode array or Varian CARY 100 (used for dilution series) spectrophotometer using either a 10 or 1 mm sample cuvette. λmax was plotted as a function of peptide concentration; the data were analyzed according to eq 3, except that the value of λmax for the hexamer and monomer were the fitting variables.
Acknowledgments
This work was primarily supported by GM54616 (NIH), with additional support from Phase 2 SBIR 5R44HL090113 and a grant from the MRSEC program of NSF to the University of Pennsylvania (grant DMR-1120901).
Supporting Information Available
Further description of AUC, ITC, UV–vis fits, UV absorption spectra of compound 1 with and without fondaparinux, and other details. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ University of California, San Francisco, San Francisco, CA 94158.
Author Contributions
¶ These authors contributed equally to this work.
The authors declare the following competing financial interest(s): Trevor M. Young, Michael J. Costanzo, Katie B. Freeman, Dylan J. Clements, Robert W. Kavash, Richard W. Scott, Dahui Liu were employees of PolyMedix, a company that was working to commercialize this type of foldamer. William F. DeGrado was the scientific advisor of Polymedix. PolyMedix has been acquired by Cellceutix, and the authors are no longer employed by Cellceutix.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gellman S. H. (1998) Foldamers: a manifesto. Acc. Chem. Res. 31, 173–180. [Google Scholar]
- Goodman C. M.; Choi S.; Shandler S.; DeGrado W. F. (2007) Foldamers as versatile frameworks for the design and evolution of function. Nat. Chem. Biol. 3, 252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichard G.; Huc I. (2011) Synthetic foldamers. Chem. Commun. 47, 5933–5941. [DOI] [PubMed] [Google Scholar]
- Hill D. J.; Mio M. J.; Prince R. B.; Hughes T. S.; Moore J. S. (2001) A field guide to foldamers. Chem. Rev. 101, 3893–4012. [DOI] [PubMed] [Google Scholar]
- Choi S.; Clements D. J.; Pophristic V.; Ivanov I.; Vemparala S.; Bennett J. S.; Klein M. L.; Winkler J. D.; DeGrado W. F. (2005) The design and evaluation of heparin-binding foldamers. Angew. Chem., Int. Ed. 44, 6685–6689. [DOI] [PubMed] [Google Scholar]
- Choi S.; Isaacs A.; Clements D.; Liu D.; Kim H.; Scott R. W.; Winkler J. D.; DeGrado W. F. (2009) De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc. Natl. Acad. Sci. U.S.A. 106, 6968–6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritzer J. A.; Hodsdon M. E.; Schepartz A. (2005) Solution structure of a β-peptide ligand for hDM2. J. Am. Chem. Soc. 127, 4118–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. F.; Sadowsky J. D.; Smith B. J.; Czabotar P. E.; Peterson-Kaufman K. J.; Colman P. M.; Gellman S. H.; Fairlie W. D. (2009) High-resolution structural characterization of a helical α/β-peptide foldamer bound to the anti-apoptotic protein Bcl-xL. Angew. Chem., Int. Ed. 48, 4318–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Choi S.; Chen B.; Doerksen R. J.; Clements D. J.; Winkler J. D.; Klein M. L.; DeGrado W. F. (2004) Nontoxic membrane-active antimicrobial arylamide oligomers. Angew. Chem., Int. Ed. 43, 1158–1162. [DOI] [PubMed] [Google Scholar]
- Cheng P. N.; Liu C.; Zhao M.; Eisenberg D.; Nowick J. S. (2012) Amyloid β-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nat. Chem. 4, 927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B.; Yan Y.; Zeng H.; Skrzypczak-Jankunn E.; Kim Y. W.; Zhu J.; Ickes H. (1999) A new approach for the design of supramolecular recognition units: hydrogen-bonded molecular duplexes. J. Am. Chem. Soc. 121, 5607–5608. [Google Scholar]
- Haldar D.; Jiang H.; Leger J. M.; Huc I. (2006) Interstrand interactions between side chains in a double-helical foldamer. Angew. Chem., Int. Ed. 45, 5483–5486. [DOI] [PubMed] [Google Scholar]
- Huc I. (2004) Aromatic oligoamide foldamers. Eur. J. Org. Chem. 2004, 17–29. [Google Scholar]
- Maurizot V.; Dolain C.; Huc I. (2005) Intramolecular versus intermolecular induction of helical handedness in pyridinedicarboxamide oligomers. Eur. J. Org. Chem. 1293–1301. [Google Scholar]
- Nowick J. S. (2008) Exploring β-sheet structure and interactions with chemical model systems. Acc. Chem. Res. 41, 1319–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford A. R.; Yamato K.; Yang X.; Yuan L.; Han Y.; Gong B. (2004) Well-defined secondary structures. Eur. J. Biochem. 271, 1416–1425. [DOI] [PubMed] [Google Scholar]
- Price J. L.; Horne W. S.; Gellman S. H. (2010) Structural consequences of β-amino acid preorganization in a self-assembling α/β-peptide: fundamental studies of foldameric helix bundles. J. Am. Chem. Soc. 132, 12378–12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels D. S.; Petersson E. J.; Qiu J. X.; Schepartz A. (2007) High-resolution structure of a β-peptide bundle. J. Am. Chem. Soc. 129, 1532–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliano M. W.; Horne W. S.; Gellman S. H. (2009) An α/β-peptide helix bundle with a pure β3-amino acid core and a distinctive quaternary structure. J. Am. Chem. Soc. 131, 9860–9861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersson E. J.; Schepartz A. (2008) Toward β-amino acid proteins: design, synthesis, and characterization of a fifteen kilodalton β-peptide tetramer. J. Am. Chem. Soc. 130, 821–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capila I.; Linhardt R. J. (2002) Heparin-Protein Interactions. Angew. Chem., Int. Ed. 41, 391–412. [DOI] [PubMed] [Google Scholar]
- Jin L.; Abrahams J. P.; Skinner R.; Petitou M.; Pike R. N.; Carrell R. W. (1997) The anticoagulant activation of antithrombin by heparin. Proc. Natl. Acad. Sci. U.S.A. 94, 14683–14688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D. J.; Huntington J. A. (2003) Crystal structure of antithrombin in a heparin-bound intermediate state. Biochemistry (Moscow) 42, 8712–8719. [DOI] [PubMed] [Google Scholar]
- Olson S. T.; Chuang Y. J. (2002) Heparin activates antithrombin anticoagulant function by generating new interaction sites (exosites) for blood clotting proteinases. Trends Cardiovasc. Med. 12, 331–338. [DOI] [PubMed] [Google Scholar]
- Rosenberg R. D.; Damus P. S. (1973) The purification and mechanism of action of human antithrombin-heparin cofactor. J. Biol. Chem. 248, 6490–6505. [PubMed] [Google Scholar]
- Lever R.; Page C. P. (2002) Novel drug development opportunities for heparin. Nat. Rev. Drug Discovery 1, 140–148. [DOI] [PubMed] [Google Scholar]
- Jones G. R.; Hashim R.; Power D. M. (1986) A comparison of the strength of binding of antithrombin-III, protamine and poly(L-lysine) to heparin samples of different anticoagulant activities. Biochim. Biophys. Acta 883, 69–76. [DOI] [PubMed] [Google Scholar]
- Mecca T.; Consoli G. M. L.; Geraci C.; La Spina R.; Cunsolo F. (2006) Polycationic calix[8]arenes able to recognize and neutralize heparin. Org. Biomol. Chem. 4, 3763–3768. [DOI] [PubMed] [Google Scholar]
- Cardin A. D.; Weintraub H. J. (1989) Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 9, 21–32. [DOI] [PubMed] [Google Scholar]
- Fromm J. R.; Hileman R. E.; Caldwell E. E.; Weiler J. M.; Linhardt R. J. (1997) Pattern and spacing of basic amino acids in heparin binding sites. Arch. Biochem. Biophys. 343, 92–100. [DOI] [PubMed] [Google Scholar]
- Margalit H.; Fischer N.; Ben-Sasson S. A. (1993) Comparative analysis of structurally defined heparin binding sequences reveals a distinct spatial distribution of basic residues. J. Biol. Chem. 268, 19228–19231. [PubMed] [Google Scholar]
- Chang L. C.; Liang J. F.; Lee H. F.; Lee L. M.; Yang V. C. (2001) Low molecular weight protamine (LMWP) as nontoxic heparin/low molecular weight heparin antidote (II): in vitro evaluation of efficacy and toxicity. AAPS PharmSci. 3, E18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L. M.; Chang L. C.; Wrobleski S.; Wakefield T. W.; Yang V. C. (2001) Low molecular weight protamine as nontoxic heparin/low molecular weight heparin antidote (III): preliminary in vivo evaluation of efficacy and toxicity using a canine model. AAPS PharmSci. 3, E19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choay J.; Petitou M.; Lormeau J. C.; Sinay P.; Casu B.; Gatti G. (1983) Structure-activity relationship in heparin: a synthetic pentasaccharide with high affinity for antithrombin III and eliciting high anti-factor Xa activity. Biochem. Biophys. Res. Commun. 116, 492–499. [DOI] [PubMed] [Google Scholar]
- Zeng H.; Miller R. S.; Flowers R. A.; Gong B. (2000) A highly stable, six-hydrogen-bonded molecular duplex. J. Am. Chem. Soc. 122, 2635–2644. [Google Scholar]
- Zeng H.; Yang X.; Flowers R. A.; Gong B. (2002) A noncovalent approach to antiparallel β-sheet formation. J. Am. Chem. Soc. 124, 2903–2910. [DOI] [PubMed] [Google Scholar]
- Lindahl U.; Backstrom G.; Hook M.; Thunberg L.; Fransson L. A.; Linker A. (1979) Structure of the antithrombin-binding site in heparin. Proc. Natl. Acad. Sci. U.S.A. 76, 3198–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl U.; Backstrom G.; Thunberg L.; Leder I. G. (1980) Evidence for a 3-O-sulfated D-glucosamine residue in the antithrombin-binding sequence of heparin. Proc. Natl. Acad. Sci. U.S.A. 77, 6551–6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl U.; Thunberg L.; Backstrom G.; Riesenfeld J.; Nordling K.; Bjork I. (1984) Extension and structural variability of the antithrombin-binding sequence in heparin. J. Biol. Chem. 259, 12368–12376. [PubMed] [Google Scholar]
- Thunberg L.; Backstrom G.; Lindahl U. (1982) Further characterization of the antithrombin-binding sequence in heparin. Carbohydr. Res. 100, 393–410. [DOI] [PubMed] [Google Scholar]
- Fromm J. R.; Hileman R. E.; Caldwell E. E.; Weiler J. M.; Linhardt R. J. (1995) Differences in the interaction of heparin with arginine and lysine and the importance of these basic amino acids in the binding of heparin to acidic fibroblast growth factor. Arch. Biochem. Biophys. 323, 279–287. [DOI] [PubMed] [Google Scholar]
- Schedin-Weiss S.; Arocas V.; Bock S. C.; Olson S. T.; Bjork I. (2002) Specificity of the basic side chains of Lys114, Lys125, and Arg129 of antithrombin in heparin binding. Biochemistry (Moscow) 41, 12369–12376. [DOI] [PubMed] [Google Scholar]
- Udit A. K.; Everett C.; Gale A. J.; Reiber Kyle J.; Ozkan M.; Finn M. G. (2009) Heparin antagonism by polyvalent display of cationic motifs on virus-like particles. ChemBioChem 10, 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.