

Abstract
B-cell malignancies are a common type of cancer. One approach to cancer therapy is to either increase oxidative stress or inhibit the stress response systems on which cancer cells rely. In this study, we combined non-toxic concentrations of Auranofin (AUR), an inhibitor of the thioredoxin (Trx) system, with non-toxic concentrations of buthionine-sulfoximine (BSO), a compound that reduces intracellular glutathione (GSH) levels, and investigated the effect of this drug combination on multiple pathways critical for malignant B-cell survival.
AUR interacted synergistically with BSO at low concentrations to trigger death in multiple malignant B-cell lines and primary mantle cell lymphoma (MCL) cells. Additionally, there was less toxicity toward normal B-cells. Low AUR concentrations inhibited Trx reductase (TrxR) activity, an effect significantly increased by BSO co-treatment. TrxR over-expression partially reversed AUR+BSO toxicity. Interestingly, the combination of AUR+BSO inhibited NF-κB signaling. Moreover, synergistic cell death induced by this regimen was attenuated in cells over-expressing NF-κB proteins, arguing for a functional role for NF-κB inhibition in AUR+BSO-mediated cell death.
Together, these findings suggest that AUR+BSO synergistically induce malignant B-cell death, a process mediated by dual inhibition of TrxR and NF-κB, and such an approach warrants further investigation in B-cell malignancies.
Keywords: B-cell malignancies, Auranofin, BSO, NF-κB, Thioredoxin Reductase
Introduction
Although the survival of patients suffering from B-cell lymphoproliferative disorders has improved in recent years, many ultimately succumb to their disease. Consequently, newer and more effective therapeutic approaches are urgently needed. One such strategy is based on the concept that neoplastic cells require a higher level of oxidative stress than their normal cellular counterparts to maintain their neoplastic phenotype. Such cells have been described as being “non-oncogene addicted” to oxidative stress [1, 2]. To survive such greater degrees of oxidative stress, cancer cells are more dependent than their normal counterparts on stress response systems, such as the thioredoxin (Trx) and glutathione (GSH) systems. Therefore, one approach to cancer therapy involves targeting this “addiction” by either increasing oxidative stress or inhibiting the stress response systems [1]. Given that the neoplastic cells are closer to the oxidative stress “lethal threshold” than their normal counterparts, such an approach has the potential for cancer-selective lethality.
Auranofin (AUR), a sulfur-containing, oral, gold(I)-based agent, has an excellent safety profile and was approved by the US Food and Drug Administration (FDA) in 1985 for the treatment of patients with rheumatoid arthritis (RA) [3–8]. AUR inhibits thioredoxin reductase (TrxR), a major cellular anti-oxidant protein that is a component of the cellular Trx system [9]. AUR has been shown to induce apoptotic death of multiple human tumors including breast cancer [10], ovarian cancer [11], multiple myeloma (MM) [12], and chronic lymphocytic leukemia (CLL) [13].
Homeostasis of the cellular redox state is regulated not only by the Trx-dependent system, but also by the glutathione (GSH) system. Recent studies demonstrate that genetic ablation of TrxR1 results in a compensatory increase in the activity of the GSH system [14], supporting the notion of redundancy in antioxidant pathways. Given this redundancy, it is not surprising that inhibition of the GSH system in conjunction with genetic inhibition of either TrxR1 or TrxR2 results in the elicitation of pronounced neoplastic cell death [14]. A pharmacological approach to inhibit GSH is to use buthionine-sulfoximine (BSO), an inhibitor of the rate-limiting enzyme in GSH biosynthesis, γ-glutamylcysteine synthetase [15]. Therefore, combining AUR with agents that inhibit or deplete GSH is a rational therapeutic combination, and in fact, combinations of AUR and BSO have recently been shown to selectively kill human head and neck and lung cancer cells by inducing oxidative stress [16, 17]. Whether this strategy would be effective in malignant hematopoietic cells, and more specifically, malignant B-cells, has not yet been investigated.
Increasing cellular oxidative stress can induce cancer cell death directly as a result of the irreversible damage that reactive oxygen species (ROS) elicit on cellular lipids, DNA, and/or proteins. Oxidative stress can also induce cancer cell death indirectly by modulating numerous redox-dependent cellular pathways that mediate cell survival. One such redox-dependent regulatory mechanism involves the post-translational modification of reduced cysteines (or thiols) on proteins involved in diverse cellular functions, a process known as thiol-based redox switching [18]. The nuclear factor-kappa B (NF-κB) signaling pathway, a pivotal cellular prosurvival pathway that is often dysregulated in cancer, is regulated in part by thiol-based redox switching. Notably, AUR inhibits the nuclear translocation, and thus activation, of NF-κB by inhibiting the activation of the inhibitory kappa B kinase (IKK) complex through its modification of cysteine 779 on IKK [19, 20].
In addition to inhibiting NF-κB activation, AUR is thought to inhibit TrxR activation via modification of cysteine and selonocysteine residues (thiols) in the redox centers of TrxR. Thus, given that BSO results in GSH depletion, and GSH reduces free thiols, we hypothesized that BSO would augment the inhibitory effect of AUR on both TrxR activity and NF-κB activation. Further, we hypothesized that the cytotoxicity elicited by the combination of AUR and BSO was due, in part, to inhibition of both TrxR and NF-κB.
Here we show that mantle cell lymphoma (MCL) cells exhibit dramatically reduced viability following combined exposure to AUR and BSO, even at low concentrations of both agents. Similar effects were observed for both diffuse large B-cell lymphoma (DLBCL) and MM cells; however, normal B-cell viability was less affected. Notably, BSO augmented AUR-induced inhibition of both TrxR and NF-κB signaling. Finally, the cytotoxic effects of AUR and BSO were attenuated by over-expressing either TrxR or NF-κB proteins, supporting a significant functional role for inhibition of these signaling pathways in the AUR/BSO synergistic toxicity. Collectively, these findings support the notion that dual targeting of cellular antioxidant systems may exert selective toxicity toward malignant B-cells.
Materials and Methods
Cell culture and primary cells
The human DLBCL cell lines, SUD-HL6 and OCI-LY10, the MCL cell lines, Rec-1 and Granta, and the MM cell lines, U266 and KMS-12-PE, were cultured as described [21–24].
B-cells were isolated from fresh peripheral blood mononuclear cells (PBMC) from one healthy donor by negative magnetic bead selection of non-B-cells according to the manufacturer’s protocol (Miltenyi Biotec, Auburn, CA, USA). This donor provided signed written informed consent. All procedures and methods were approved by the Research Subjects Review Board at the University of Rochester Medical Center (URMC).
Primary patient derived MCL cells were obtained from the URMC Tissue Core under an Institutional Review Board approved protocol. These tissue specimens were fully anonymous, hence patient demographics are not available.
Chemical reagents
All chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated.
Cell viability assays
Cells were treated with varying concentrations of AUR or BSO, alone or together, for 24 h, and a MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was performed as described previously [23]. The alamarBlue® cell viability assay (Life Technologies, Grand Island, NY, USA) was used according to the manufacturer’s protocol to assess the viability of primary cells. The Annexin V-FITC Apoptosis Detection kit (BD Biosciences, San Jose, CA, USA) was also used according to the manufacturer’s instructions.
Thioredoxin reductase activity
To measure TrxR activity, Granta cells were incubated with 100 nM AUR or 5 μM BSO, alone or in combination, for 18 h. Subsequently, TrxR activity was measured spectrophotometrically using a kit, according to the manufacturer’s protocol (Sigma-Aldrich, St. Louis, MO, USA).
Electrophoretic mobility shift assay
Granta cells were treated with the indicated concentrations of AUR and BSO, alone or in combination, for 6 h. Following these treatments, nuclear extracts were prepared as described [25–27]. Electrophoretic mobility shift assays (EMSA) were performed by incubating nuclear extracts with IR-Dye-700 conjugated, double-stranded DNA probe at room temperature for 10 minutes, or an additional 10 minute incubation with NF-κB-specific antibodies for supershift analysis, followed by resolution of the complexes on native 4% polyacrylamide gels. The bands were then visualized using images acquired on an Odyssey infrared imager (LI-COR Biosciences, Lincoln, NE, USA). The double stranded oligonucleotide probe is as follows (upper strand): 5′/5IRD700/CAACGGCAGGGGAATTCCCCTCTCCTT-3′.
Luciferase assay
Luciferase reporter plasmids containing either the NF-κB or OCT-1 responsive elements upstream of a firefly luciferase gene were transfected into Granta cells, together with a vector expressing IκBαS1/2 or an empty vector, using Nucleofector (Amaxa/Lonza, Basel, Switzerland). The total amount of plasmid DNA was kept constant at 5 μg for each transfection. Thirty minutes after the transfection, cells were plated into single wells and were left untreated or incubated for 6 h with the indicated doses of AUR or BSO, alone or in combination. Cell lysates were prepared using reporter lysis buffer and luciferase activity was measured with a SpectraMax M3 plate reader (Molecular Devices, Sunnyvale, CA, USA). In these assays, total protein amount as determined by Bradford assay was used to normalize the samples as a single transfection was divided into multiple wells for subsequent treatments.
Immunoblot analyses
Following the indicated manipulations, whole cell lysates were prepared in ELB buffer [28]. Lysates were fractionated on 10% SDS-PAGE gels and protein was electrophoretically transferred to Hybond ECL nitrocellulose membrane (GE Healthcare Bio-Sciences Corporation, Piscataway, NJ, USA). The membranes were analyzed for immunoreactivity with primary antibodies raised against RelA (1:1000), RelB (1:1000), IκBα (1:1000), or α-Tubulin (1:1000; all from Santa Cruz Biotechnologies Inc., Santa Cruz, CA, USA), thioredoxin reductase, p100/p52, or actin (1:1000; from Cell Signaling Technology, Danvers, MA, USA). Bound antibodies were detected by species-specific, IRDye-conjugated secondary antibodies (1:20,000; LI-COR Biosciences, Lincoln, NE, USA). The membranes were then subjected to Odyssey infrared imaging (LI-COR Biosciences, Lincoln, NE, USA).
Quantitative Real Time PCR (qRT-PCR) analysis
Bcl-xL mRNA levels were determined via qRT-PCR using the following primers: Bcl-xL F 5′-GATCCCCATGGCAGCAGTAAAGCAAG-3′, R 5′-CCCCATCCCGGAAGAGTTCATTCACT-3′; GAPDH F 5′-AGGTGAAGGTCGGAGTCA-3′, R 5′-GGTCATTGATGGCAACAA-3′. mRNA levels were compared using the ΔΔCt method.
Adenovirus transduction
U266 cells were transduced with recombinant adenovirus vectors that express green fluorescent protein (GFP) and RelA [26], RelB (Cell Biolabs Inc., San Diego, CA, USA), or an empty control vector (Cell Biolabs Inc.) (biosafety level 2). Vectors were constructed and propagated using previously described methods [29], and were handled following standard biosafety level 2 procedures. The viral vectors were used to transduce U266 cells at a multiplicity of infection (MOI) of 100. Eighteen-24 h post-transduction, cells were treated with AUR or BSO, alone or in combination, for an additional 24 h.
Statistical analyses
Synergy was tested at each testable dose combination using Laska et al’s model-free test of synergy [30], using unequal variance t-tests (in R software) as the building blocks. Adjustment for multiple comparisons over dose combinations via control of the false discovery rate (FDR) was considered but unnecessary, given that the majority of the synergy p-values were significant and they always clustered in contiguous synergistic dose combination regions.
Mean data values and the standard error of the mean (SEM) were calculated for each variable. One-way ANOVA followed by Bonferroni’s test for multiple comparisons was used to analyze data involving the analysis of multiple sample groups. A value of p<0.05 was designated as statistically significant (Prism 4.0 software; GraphPad Software, La Jolla, CA, USA).
Results
Combined toxicity of Auranofin and BSO
The effect of AUR in combination with BSO on the viability of various malignant B-cell lines was determined using an MTT assay following 24 h exposure to these drugs, either alone or together. At low nanomolar concentrations, AUR alone caused minimal levels of cell death, and BSO alone was non-toxic in the MCL cell lines, Granta and Rec-1. However, simultaneous exposure of the cells to both drugs dramatically enhanced the cytotoxicity, even at the lowest tested AUR concentration of 100 nM (Figure 1A). Similarly, BSO enhanced AUR-mediated toxicity in the DLBCL cell lines, OCI-LY10 and SUDHL6 (Figure 1B), and the MM cell lines, U266 and KMS-12-PE (Figure 1C). Laska et al’s model-free test of synergy [30] confirmed the synergistic action of these two drugs together on cancer cell death, at multiple dose combinations that were tested. This method has previously been used to test the effects of drug combinations on cancer cell growth [31–33]. The most profound combined toxicity of these drugs occurred in Granta cells and U266 cells, hence these two cell lines were used in subsequent experiments investigating potential mechanisms underlying this synergism.
Figure 1. Toxicity of Auranofin and BSO in malignant B-cell lines.
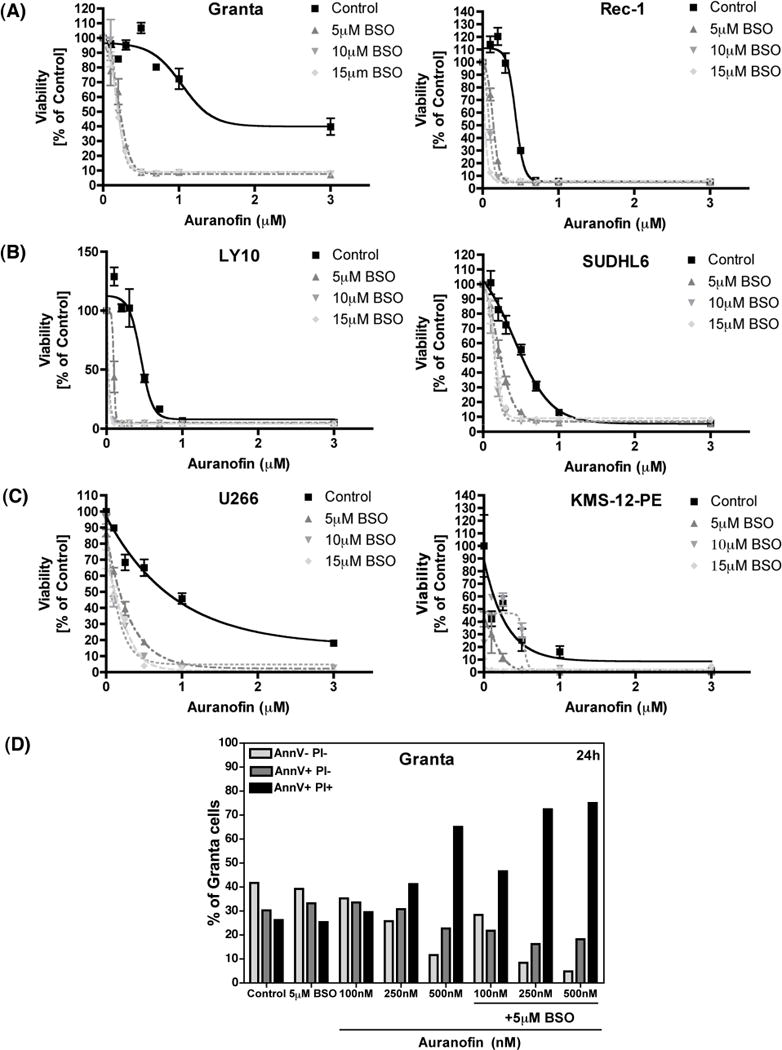
(A) Granta cells and Rec-1 cells (human mantle cell lymphoma cell lines) were treated with Auranofin or BSO, alone or together, at the indicated concentrations for 24 h. Cell viability was measured using the MTT assay. Percent survival was calculated as compared to DMSO-treated control cells. (B) LY10 and SUDHL6 (human diffuse large B-cell lymphoma cell lines) were treated as indicated for 24 h and cell viability was measured as described above. (C) U266 and KMS-12-PE cells (human multiple myeloma cell lines) were treated as indicated for 24 h and cell viability was measured as described above. (D) Granta cells were treated as indicated for 24 h and cell death was analyzed using Annexin V/PI staining followed by flow cytometry. AnnexinV− PI− indicates viable cells, AnnexinV+ PI− indicates early apoptotic cells, and AnnexinV+ PI+ indicates late apoptotic/dead cells.
We performed Annexin V/Propidium Iodide (PI) staining in Granta cells at multiple time points following treatment to determine the mechanism of AUR + BSO-induced cell death. At 6 h post-treatment there were no changes in levels of Annexin V single positive (early apoptotic) or Annexin V/PI double positive (late apoptotic/dead) cell populations (data not shown). However, at both 18 h (data not shown) and 24 h (Figure 1D) post-treatment there was a large increase in the Annexin V/PI double positive (late apoptotic/dead) cell population, thus suggesting that AUR + BSO may be directly inducing necrotic cell death in Granta cells. This is consistent with the report by Sobhakumari et al. [16] where they suggest that AUR + BSO induce necrosis in human head and neck cancer cells.
Enhanced lethality involves inhibition of thioredoxin reductase activity
AUR is a well-known inhibitor of the Trx antioxidant system, through inhibition of mitochondrial TrxR [34–38]. As expected, we observed a significant reduction in TrxR activity after 18 h treatment with AUR in Granta cells. Interestingly, TrxR activity was further reduced following exposure to AUR + BSO in combination (Figure 2A). The effect of TrxR over-expression on AUR + BSO-mediated cell death was next determined using an MTT assay in U266 cells stably over-expressing either wild-type TrxR or mutated TrxR (mutTrxR), which was unable to reduce oxidized thioredoxin. The untransfected, parent cell line, U266, was used as a control. A higher level of TrxR expression in both the TrxR and mutTrxR stable cell lines was confirmed by immunoblot analysis (Figure 2B). AUR alone caused minimal cell death, and this was not significantly altered with TrxR over-expression (Figure 2B). When combined with 5 μM BSO, AUR drastically decreased cell viability. Interestingly, at the two lowest AUR concentrations, TrxR over-expression significantly protected against AUR + BSO-mediated toxicity (Figure 2C), thus confirming that AUR’s inhibition of TrxR contributed to the cell death induced by this drug combination.
Figure 2. Effect of Auranofin and BSO on thioredoxin reductase activity.
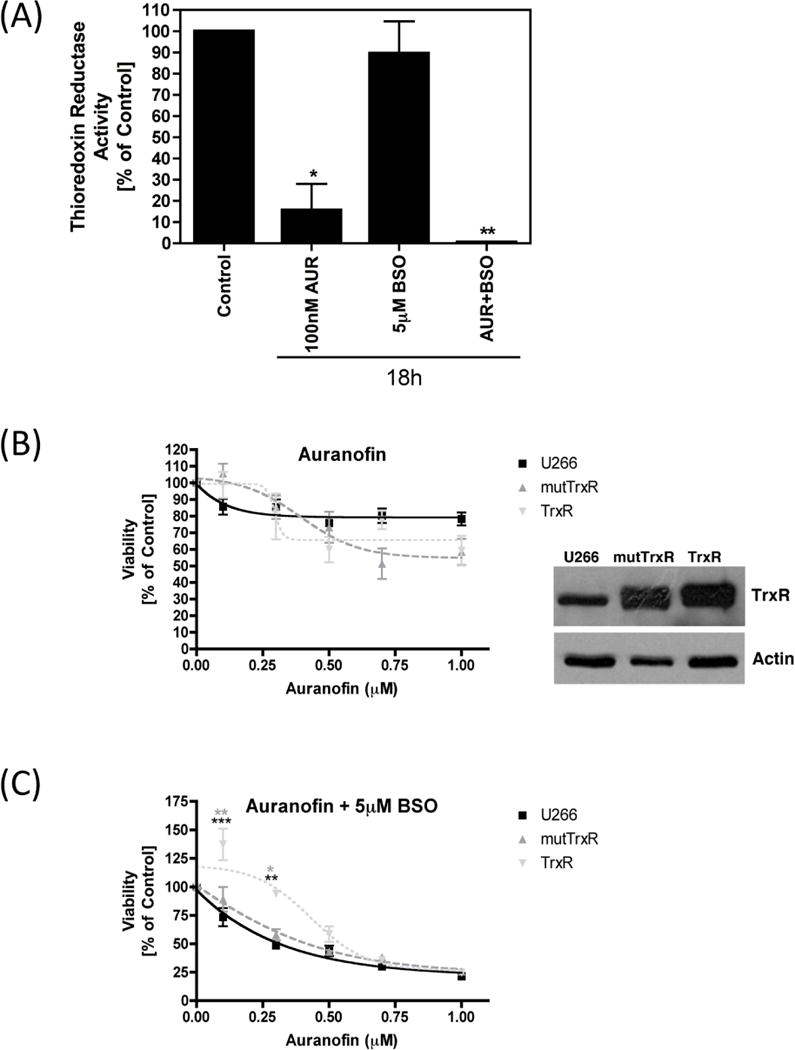
(A) Granta cells were treated with Auranofin or BSO, alone or together, at the indicated concentrations for 18 h. Thioredoxin reducatase activity was measured using the Sigma Thioredoxin Reductase assay kit. Results are shown as percent activity compared to DMSO-treated control cells. (B, C) U266 cells either untransfected (control), or stably over-expressing mutant thioredoxin reductase (mutTrxR) or wild-type thioredoxin reductase (TrxR) were treated with the indicated concentrations of Auranofin either alone (B) or together with 5 μM BSO (C) for 24 h. Cell viability was measured using the MTT assay. Percent survival was calculated as compared to DMSO-treated control cells. Immunoblot analysis was performed on whole cell lysates from U266 cells either untransfected (control), or stably over-expressing mutTR or wild-type TrxR. Over-expression of TrxR was confirmed using an anti-TrxR antibody. Actin was used as a loading control. Statistical significance is indicated, as compared to untransfected control cells (***p<0.001, **p<0.01) or as compared to mutTrxR expressing cells (**p<0.01, *p<0.05).
BSO augments AUR inhibition of NF-κB signaling
The survival of malignant cells is often linked to constitutive activation of the NF-κB signaling pathway [39]. As AUR has been previously shown to inhibit NF-κB signaling [19, 20], we sought to determine the effect of AUR + BSO on NF-κB activation in Granta cells, having hypothesized that BSO would augment the AUR-induced inhibition of NF-κB. As shown in Figure 3A, following 6 h treatment with AUR and BSO, alone or together, levels of NF-κB DNA binding activity were dramatically reduced, particularly with the drug combination. To analyze the NF-κB DNA binding activity further, we performed supershift analyses. As shown in Figure 3B, the antibodies against RelA and RelB altered the mobility of bands, suggesting the presence of these two molecules in NF-κB/DNA complexes. Together our data suggest that combined treatment of Granta cells with AUR and BSO was able to block RelA- and RelB-specific activities. We also used luciferase assays as an independent measure of NF-κB activation status in Granta cells following AUR + BSO treatment. AUR alone significantly reduced NF-κB activity, to a level similar to that observed following over-expression of the NF-κB super-repressor, IκBαS1/2, and BSO further augmented AUR’s inhibition of NF-κB in these cells (Figure 3C). A parallel transfection with a luciferase reporter plasmid in which the luciferase gene is downstream of OCT-1 transcription factor response elements was used as a control for the specificity of AUR + BSO’s inhibition of NF-κB (Figure 3C). These findings support the notion that BSO enhanced the inhibition of NF-κB activity by AUR.
Figure 3. Effect of Auranofin and BSO on NF-κB activity.
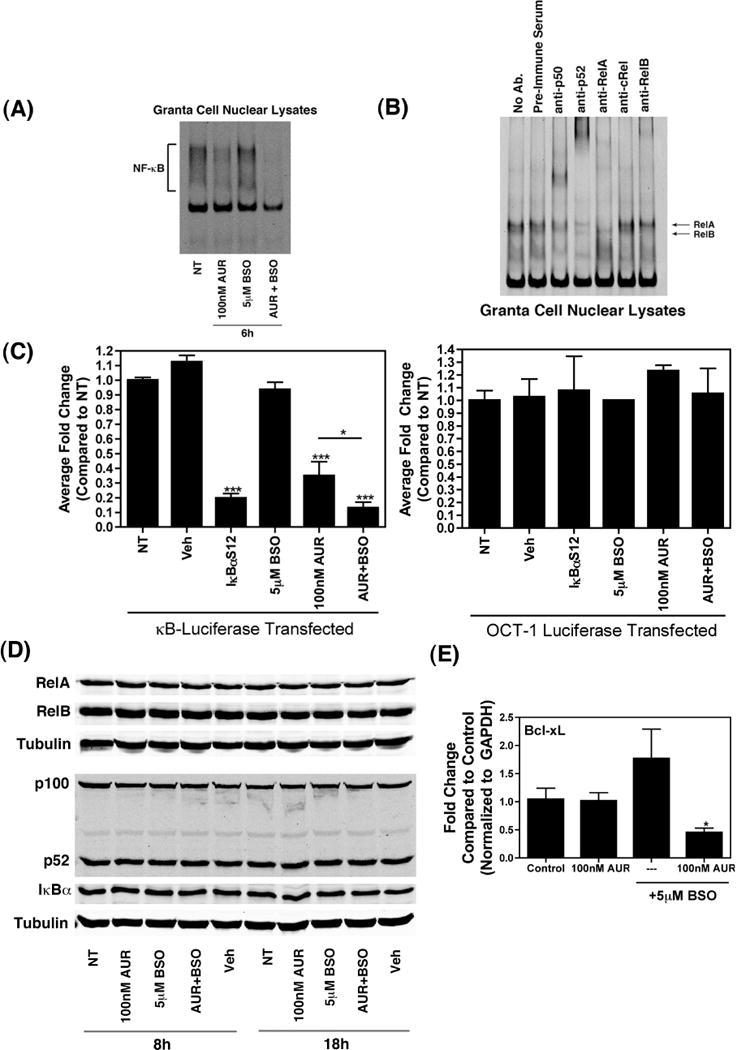
(A) Granta cells (1×106) were treated with the indicated concentrations of Auranofin and BSO, either alone or together, for 6 h. Following these treatments, nuclear extracts were prepared and EMSA was performed by incubating nuclear extracts with IR-Dye-700 conjugated, double-stranded DNA probe at room temperature for 10 minutes, followed by resolution of the protein-DNA complexes on native 4 % polyacrylamide gels. The bands were then visualized on an Odyssey infrared imager (LI-COR Biosciences). (B) Nuclear extracts were prepared from untreated Granta cells and super-shift assays were performed by incubating nuclear extracts with IR-Dye-700 conjugated, double-stranded DNA probe at room temperature for 10 minutes, followed by incubation with the indicated antibodies for 5 minutes. The protein-DNA-antibody complexes were resolved on native 4 % polyacrylamide gels. The bands were then visualized as described above. (C) Luciferase reporter plasmids containing NF-κB or OCT-1 responsive elements upstream of a firefly luciferase gene was transfected into Granta cells (5×106), together with either a vector expressing IκBαS1/2 or an empty vector, using Nucleofector (Amaxa/Lonza). The total amount of plasmid DNA was kept constant at 5 μg for each transfection. Thirty minutes after the transfection, cells were plated into single wells and were either left untreated or incubated for 6 h with the indicated doses of Auranofin or BSO, alone or together. Treatment with DMSO (Veh) was used as a control. Cell lysates were prepared using reporter lysis buffer and luciferase activity was measured with a SpectraMax M3 plate reader (Molecular Devices). Data are presented as fold change compared to non-treated cells and are shown as mean ± SEM of values derived from three replicates from each of four combined experiments for NF-κB or from two combined experiments for OCT-1. ***, p<0.001, **, p<0.01 as compared to cells transfected with the luciferase reporter and left untreated. (D) Whole cell lysates from Granta cells treated as indicated were subjected to immunoblot analysis with antibodies against RelA (first panel), RelB (second panel), p100/p52 (fourth panel), IκBα (fifth panel), or α-Tubulin (third and sixth panels). (E) Bcl-xL mRNA levels were determined via qRT-PCR in Granta cells treated as indicated for 24 h. mRNA levels were compared using the ΔΔCt method. *, p<0.05 as compared to DMSO-treated cells (control).
In addition, we used immunoblot analysis and qRT-PCR to examine NF-κB protein levels and NF-κB target gene expression respectively. As shown in Figure 3D, there was no significant change in RelA, RelB, p100/p52, or IκBα protein levels following 8 h and 18 h treatment with AUR alone or together with BSO, however, the intracellular mobilization of NF-κB dimers, rather than changes in NF-κB protein levels, determines NF-κB activity. Alternatively, Bcl-xL mRNA levels were significantly reduced following 24 h treatment with AUR + BSO (Figure 3E). This was expected considering that Bcl-xL is a pro-survival Bcl-2 homologue that is an NF-κB target gene [40].
Genetic evidence for the role of NF-κB inhibition in AUR + BSO-mediated toxicity
In order to further investigate the involvement of NF-κB signaling in AUR + BSO-induced cell death, we over-expressed the NF-κB proteins, RelA and RelB, using adenoviral vectors, and assessed cell viability via MTT assay following AUR + BSO treatment for 24 h. Under these conditions the over-expressed NF-κB molecules are expected to overcome the inhibitory actions of IκBα via stoichiometric changes, thereby assuming a functional presence in the nucleus [41]. Transduction with an empty adenoviral vector followed by AUR + BSO treatment for 24 h was used as a control. U266 cells are highly amenable to adenovirus transduction, as shown by fluorescent microscopy and flow cytometry to measure GFP expression (Figure 4A), hence, we used this cell type for these analyses. Over-expression of both RelA and RelB was confirmed by immunoblot analysis (Figure 4B). A longer exposure of the blot containing RelB bands suggests the constitutive presence of this molecule in the cells (data not shown). Enforced RelA and RelB expression completely protected against the cell death caused by AUR alone. Additionally, over-expression of both of these NF-κB proteins significantly protected cells from AUR + BSO-induced toxicity, an effect particularly apparent at the lowest AUR concentrations (Figure 4B). Collectively, these data suggest that the combined effect of these two drugs on NF-κB signaling plays an important role in AUR + BSO-mediated cell death.
Figure 4. Over-expression of NF-κB molecules rescues U266 cells from Auranofin + BSO-induced toxicity.
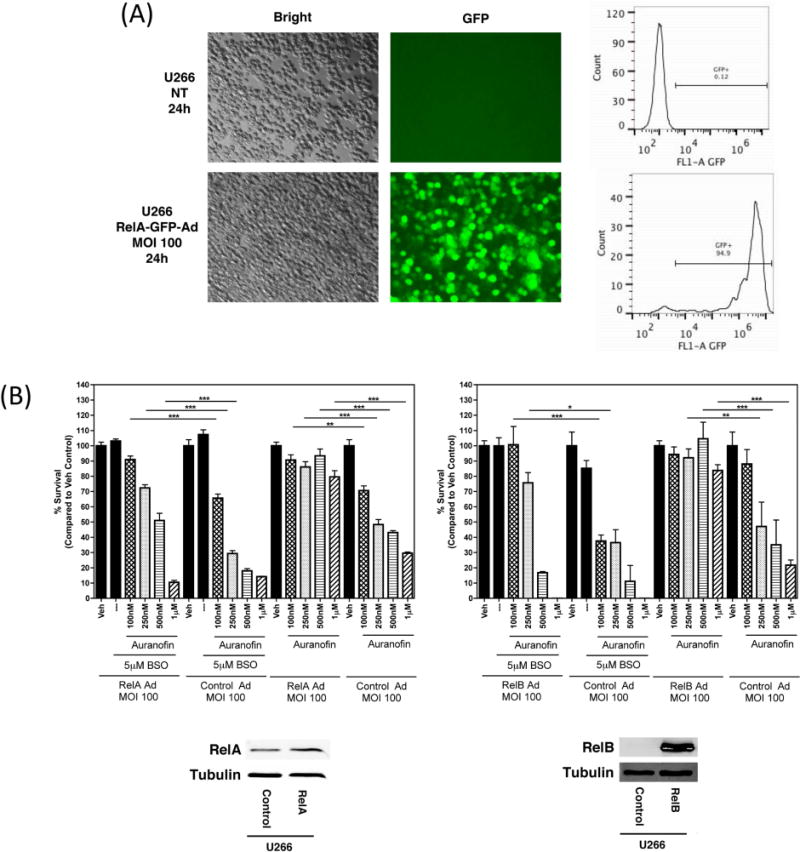
(A) U266 cells were either left untransduced, or were transduced with an adenoviral vector expressing RelA and GFP at a multiplicity of infection (MOI) of 100 for the indicated time. GFP expression was captured by fluorescence microscopy. Cells were then fixed with 4 % paraformaldehyde and were subjected to flow cytometric analysis. The percentage of GFP-positive cells is indicated. (B) U266 cells were transduced with a control adenoviral vector or with an adenoviral vector expressing RelB (first panel) or RelA (second panel) for 24 h. Cells were then treated with the indicated concentrations of Auranofin and/or BSO for an additional 24 h. Cell viability was measured using the MTT assay. Percent survival was calculated as compared to DMSO-treated (Veh) control cells. Data are presented as mean ± SEM of values derived from a single experiment that was performed in triplicate. ***, p<0.001, **, p<0.01, *, p<0.05 indicates statistical significance.
Combined toxicity in primary MCL-derived cells
Finally, we determined the effect of AUR + BSO on primary human cells, derived from MCL patients, as well as a healthy volunteer. Following AUR + BSO treatment for 24 h, alamarBlue® reagent was used to assess cell viability. Because of the sensitivity of alamarBlue® reagent to as few as 50 cells, we chose this assay due to the small number of cells available in our MCL patient samples. In two out of the three primary MCL cell samples that were tested, there was decreased cell viability with AUR + BSO, compared to AUR alone (Figures 5A, B, C). With the normal B-cells, there was a 40 % reduction in cell viability, however there was not any combined effect of AUR + BSO in these cells (Figure 5D). Together these data suggest that this drug combination, particularly at low concentrations, may be preferentially toxic to malignant cells, as compared to healthy, normal B-cells.
Figure 5. Toxicity of Auranofin and BSO in primary cells.
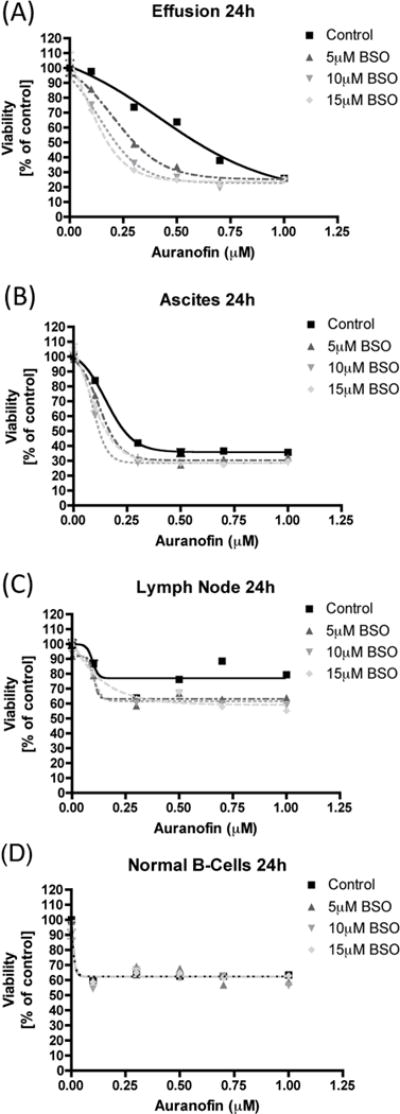
(A, B, C) Unseparated, single cell suspensions derived from the indicated sources were treated with Auranofin or BSO, alone or together, at the indicated concentrations for 24 h. Cell viability was measured using the alamarBlue® assay. Percent survival was calculated as compared to DMSO-treated control cells. (D) Normal B-cells isolated from a healthy individual were treated as indicated for 24 h and cell viability was measured via the alamarBlue® assay. Percent survival was calculated as compared to DMSO-treated control cells.
Discussion
There has been great interest in repurposing existing and approved drugs for the treatment of cancer [42], as this would shorten the time and cost required to provide new and effective cancer therapeutics to patients in need. These agents are already approved for human use, and therefore ample information is available regarding formulation dosing, pharmacology, and toxicity. Consequently, their use as anti-cancer agents can be more rapidly advanced to patients, in contrast to traditional drug discovery pathways. Indeed, AUR represents a prototype of a repurposed agent, as it is currently in clinical trials for patients with CLL [42].
AUR is a gold-based drug that is FDA approved for the treatment of patients with rheumatoid arthritis. The pharmacological activity of AUR is due primarily to its ability to react with physiological thiols and form stable thiol gold adducts. As described, such post-translational modification of protein thiols is a major mechanism for the regulation of multiple cellular pathways. One important target of AUR is TrxR, a component of the cellular thioredoxin system and major regulator of the cellular thiol state. Additionally, through its regulation of ribonucleotide reductase, TrxR regulates deoxyribonucleotide production and thus regulates DNA synthesis and cellular proliferation [43]. That inhibiting TrxR is an attractive target for cancer therapeutics is supported by the finding that TrxR is over-expressed in a number of cancers compared to its normal cellular counterpart.
Another proposed mechanism of action of AUR is inhibition of NF-κB. NF-κB represents a family of dimeric transcription factors including five members, cRel, RelB, RelA (p65), p50/p105, and p52/p100. NF-κB target genes play central roles in a multitude of cellular processes including immune responses, inflammation, proliferation, and survival [44]. In addition, inhibition of NF-κB has been shown to be an effective therapeutic strategy for B-cell lymphoproliferative disorders such as DLCL, MCL, and MM [45–48]. Redox regulation of NF-κB activation has been shown to occur both in the nucleus as well as in the cytoplasm [49, 50]. In this regard, AUR inhibits nuclear translocation of NF-κB by inhibiting IKK activation through its modification of cysteine 779 on IKK [19, 20].
Over-expression of both RelA and RelB attenuated the cytotoxicity of both AUR alone, as well as that of the AUR/BSO combination. The RelA/p50 combination is most commonly involved with activation of the canonical NF-κB pathway, whereas the RelB/p52 combination is mainly involved in the non-canonical NF-κB pathway [51]. Although there is a significant amount of overlap between these two pathways, the different heterodimers differentially regulate gene expression [51, 52]. Whereas the canonical NF-κB pathway has been extensively studied in the context of cancer, the same is not true of the non-canonical NF-κB pathway. Therefore, our data showing that over-expression of RelB protects cells from AUR + BSO-induced toxicity in the MM cell line, U266, is interesting, especially considering the results of a very recent study suggesting that the NF-κB inducing kinase (NIK), an important regulator of the non-canonical NF-κB pathway, is a potential therapeutic target for MCL treatment [53].
In summary, we show that the combination of AUR and BSO elicits significant cell death of MCL, DLCL and MM cells through the potent inhibition of both TrxR and NF-κB. Indeed, the synergistic effect of BSO on AUR induced cell death may be due, in part, to our novel findings that BSO augments AUR induced inhibition of TrxR and NF-κB activity. Importantly, BSO does not seem to increase the cytotoxicity of AUR in normal peripheral blood B-cells. Such selective tumor toxicity speaks to the potential safety of this therapeutic strategy. AUR has an established history as a well-tolerated oral agent and BSO has been studied in phase I trials where it was found to be safe and well tolerated. This was demonstrated at doses that resulted in serum levels of 50–100 mM and which inhibited cellular glutathione levels to < 10% of pre-treatment levels [54, 55]. Notably, we observed synergy with AUR in vitro at BSO concentrations of 5–15 μM. Taken together, combining AUR and BSO may prove to be a safe, effective treatment strategy against B-cell malignancies and warrants further investigation in prospective clinical trials.
Highlights.
B-cell malignancies are a common type of cancer.
We investigated the effect of auranofin + BSO on malignant B-cell survival.
AUR interacted synergistically with BSO to trigger death in malignant B-cell types.
There was less toxicity toward normal B-cells.
Toxicity is mediated by dual inhibition of TrxR and NF-κB.
Acknowledgments
We thank Dr. James Kobie for his assistance with human B-cell isolation.
Funding
Supported by grants from the National Institutes of Health, USA (R01 NS054578 and R01 NS066801 to SBM, R03 DA035086 to MK, R01 HL071158 to PSB), also by a Leukemia and Lymphoma Society Translational Grant (6344–11), a U.S. Public Health Service Grant (RO1-CA122645), and a SPORE in Lymphoma Grant (P50-CA130805) to SHB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
Authors have no competing interests.
References
- 1.Sharma SV, Settleman J. Exploiting the balance between life and death: targeted cancer therapy and “oncogenic shock”. Biochem Pharmacol. 2010;80:666–673. doi: 10.1016/j.bcp.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Champion GD, Cairns DR, Bieri D, et al. Dose response studies and longterm evaluation of auranofin in rheumatoid arthritis. J Rheumatol. 1988;15:28–34. [PubMed] [Google Scholar]
- 4.Williams HJ, Ward JR, Egger MJ, et al. Auranofin, gold sodium thiomalate, and placebo in the treatment of rheumatoid arthritis. Cooperative systematic studies of rheumatic diseases. Clin Rheumatol. 1984;3(Suppl 1):39–50. doi: 10.1007/BF03342621. [DOI] [PubMed] [Google Scholar]
- 5.Furst DE. Mechanism of action, pharmacology, clinical efficacy and side effects of auranofin. An orally administered organic gold compound for the treatment of rheumatoid arthritis. Pharmacotherapy. 1983;3:284–298. doi: 10.1002/j.1875-9114.1983.tb03277.x. [DOI] [PubMed] [Google Scholar]
- 6.Brewer EJ, Jr, Giannini EH, Person DA. Early experiences with auranofin in juvenile rheumatoid arthritis. Am J Med. 1983;75:152–156. doi: 10.1016/0002-9343(83)90490-4. [DOI] [PubMed] [Google Scholar]
- 7.Hull RG, Morgan SH, Parke AL, Childs L, Goldman M, Hughes GR. A double-blind study comparing sodium aurothiomalate and auranofin in patients with rheumatoid arthritis previously stabilized on sodium aurothiomalate. Int J Clin Pharmacol Res. 1984;4:395–401. [PubMed] [Google Scholar]
- 8.Jessop JD, O’Sullivan MM, Lewis PA, et al. A long-term five-year randomized controlled trial of hydroxychloroquine, sodium aurothiomalate, auranofin and penicillamine in the treatment of patients with rheumatoid arthritis. Br J Rheumatol. 1998;37:992–1002. doi: 10.1093/rheumatology/37.9.992. [DOI] [PubMed] [Google Scholar]
- 9.Mustacich D, Powis G. Thioredoxin reductase. Biochem J. 2000;346(Pt 1):1–8. [PMC free article] [PubMed] [Google Scholar]
- 10.Kim NH, Park HJ, Oh MK, Kim IS. Antiproliferative effect of gold(I) compound auranofin through inhibition of STAT3 and telomerase activity in MDA–MB 231 human breast cancer cells. BMB Rep. 2013;46:59–64. doi: 10.5483/BMBRep.2013.46.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello MP. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic Biol Med. 2007;42:872–881. doi: 10.1016/j.freeradbiomed.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 12.Nakaya A, Sagawa M, Muto A, Uchida H, Ikeda Y, Kizaki M. The gold compound auranofin induces apoptosis of human multiple myeloma cells through both down-regulation of STAT3 and inhibition of NF-kappaB activity. Leuk Res. 2011;35:243–249. doi: 10.1016/j.leukres.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 13.Zalewski PD, Forbes IJ, Valente L, Hurst NP. Auranofin increases the affinity of phorbol dibutyrate receptors in chronic lymphocytic leukemia cells (B cells) J Immunol. 1987;138:3005–3009. [PubMed] [Google Scholar]
- 14.Mandal PK, Schneider M, Kolle P, et al. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res. 2010;70:9505–9514. doi: 10.1158/0008-5472.CAN-10-1509. [DOI] [PubMed] [Google Scholar]
- 15.Griffith OW. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J Biol Chem. 1982;257:13704–13712. [PubMed] [Google Scholar]
- 16.Sobhakumari A, Love-Homan L, Fletcher EV, et al. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS One. 2012;7:e48175. doi: 10.1371/journal.pone.0048175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fath MA, Ahmad IM, Smith CJ, Spence J, Spitz DR. Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Clin Cancer Res. 2011;17:6206–6217. doi: 10.1158/1078-0432.CCR-11-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antelmann H, Helmann JD. Thiol-based redox switches and gene regulation. Antioxid Redox Signal. 2011;14:1049–1063. doi: 10.1089/ars.2010.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeon KI, Jeong JY, Jue DM. Thiol-reactive metal compounds inhibit NF-kappa B activation by blocking I kappa B kinase. J Immunol. 2000;164:5981–5989. doi: 10.4049/jimmunol.164.11.5981. [DOI] [PubMed] [Google Scholar]
- 20.Jeon KI, Byun MS, Jue DM. Gold compound auranofin inhibits IkappaB kinase (IKK) by modifying Cys-179 of IKKbeta subunit. Exp Mol Med. 2003;35:61–66. doi: 10.1038/emm.2003.9. [DOI] [PubMed] [Google Scholar]
- 21.Namba M, Ohtsuki T, Mori M, et al. Establishment of five human myeloma cell lines. In Vitro Cell Dev Biol. 1989;25:723–729. doi: 10.1007/BF02623725. [DOI] [PubMed] [Google Scholar]
- 22.Sniderhan LF, Garcia-Bates TM, Burgart M, Bernstein SH, Phipps RP, Maggirwar SB. Neurotrophin signaling through tropomyosin receptor kinases contributes to survival and proliferation of non-Hodgkin lymphoma. Exp Hematol. 2009;37:1295–1309. doi: 10.1016/j.exphem.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skalska J, Brookes PS, Nadtochiy SM, et al. Modulation of cell surface protein free thiols: a potential novel mechanism of action of the sesquiterpene lactone parthenolide. PLoS One. 2009;4:e8115. doi: 10.1371/journal.pone.0008115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai Y, Landowski TH, Rosen ST, Dent P, Grant S. Combined treatment with the checkpoint abrogator UCN-01 and MEK1/2 inhibitors potently induces apoptosis in drug-sensitive and -resistant myeloma cells through an IL-6-independent mechanism. Blood. 2002;100:3333–3343. doi: 10.1182/blood-2002-03-0940. [DOI] [PubMed] [Google Scholar]
- 25.Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramirez SH, Sanchez JF, Dimitri CA, Gelbard HA, Dewhurst S, Maggirwar SB. Neurotrophins prevent HIV Tat-induced neuronal apoptosis via a nuclear factor-kappaB (NF-kappaB)-dependent mechanism. J Neurochem. 2001;78:874–889. doi: 10.1046/j.1471-4159.2001.00467.x. [DOI] [PubMed] [Google Scholar]
- 27.Maggirwar SB, Ramirez S, Tong N, Gelbard HA, Dewhurst S. Functional interplay between nuclear factor-kappaB and c-Jun integrated by coactivator p300 determines the survival of nerve growth factor-dependent PC12 cells. J Neurochem. 2000;74:527–539. doi: 10.1046/j.1471-4159.2000.740527.x. [DOI] [PubMed] [Google Scholar]
- 28.Kiebala M, Polesskaya O, Yao Z, Perry SW, Maggirwar SB. Nuclear factor-kappa B family member RelB inhibits human immunodeficiency virus-1 Tat-induced tumor necrosis factor-alpha production. PLoS One. 2010;5:e11875. doi: 10.1371/journal.pone.0011875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laska EM, Meisner M, Siegel C. Simple designs and model-free tests for synergy. Biometrics. 1994;50:834–841. [PubMed] [Google Scholar]
- 31.Dasmahapatra G, Lembersky D, Son MP, et al. Obatoclax interacts synergistically with the irreversible proteasome inhibitor carfilzomib in GC- and ABC-DLBCL cells in vitro and in vivo. Mol Cancer Ther. 2012;11:1122–1132. doi: 10.1158/1535-7163.MCT-12-0021. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Lu G, Xiao H, You H, et al. Synergistic inhibition of lung tumorigenesis by a combination of green tea polyphenols and atorvastatin. Clin Cancer Res. 2008;14:4981–4988. doi: 10.1158/1078-0432.CCR-07-1860. [DOI] [PubMed] [Google Scholar]
- 33.Liu T, Yacoub R, Taliaferro-Smith LD, et al. Combinatorial effects of lapatinib and rapamycin in triple-negative breast cancer cells. Mol Cancer Ther. 2011;10:1460–1469. doi: 10.1158/1535-7163.MCT-10-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cox AG, Brown KK, Arner ES, Hampton MB. The thioredoxin reductase inhibitor auranofin triggers apoptosis through a Bax/Bak-dependent process that involves peroxiredoxin 3 oxidation. Biochem Pharmacol. 2008;76:1097–1109. doi: 10.1016/j.bcp.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 35.Rigobello MP, Bindoli A. Mitochondrial thioredoxin reductase purification, inhibitor studies, and role in cell signaling. Methods Enzymol. 2010;474:109–122. doi: 10.1016/S0076-6879(10)74007-6. [DOI] [PubMed] [Google Scholar]
- 36.Rigobello MP, Callegaro MT, Barzon E, Benetti M, Bindoli A. Purification of mitochondrial thioredoxin reductase and its involvement in the redox regulation of membrane permeability. Free Radic Biol Med. 1998;24:370–376. doi: 10.1016/s0891-5849(97)00216-5. [DOI] [PubMed] [Google Scholar]
- 37.Rigobello MP, Folda A, Baldoin MC, Scutari G, Bindoli A. Effect of auranofin on the mitochondrial generation of hydrogen peroxide. Role of thioredoxin reductase. Free Radic Res. 2005;39:687–695. doi: 10.1080/10715760500135391. [DOI] [PubMed] [Google Scholar]
- 38.Liu JJ, Liu Q, Wei HL, Yi J, Zhao HS, Gao LP. Inhibition of thioredoxin reductase by auranofin induces apoptosis in adriamycin-resistant human K562 chronic myeloid leukemia cells. Pharmazie. 2011;66:440–444. [PubMed] [Google Scholar]
- 39.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 40.Chen C, Edelstein LC, Gelinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L) Mol Cell Biol. 2000;20:2687–2695. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harhaj EW, Sun SC. Regulation of RelA subcellular localization by a putative nuclear export signal and p50. Mol Cell Biol. 1999;19:7088–7095. doi: 10.1128/mcb.19.10.7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weir SJ, DeGennaro LJ, Austin CP. Repurposing approved and abandoned drugs for the treatment and prevention of cancer through public-private partnership. Cancer Res. 2012;72:1055–1058. doi: 10.1158/0008-5472.CAN-11-3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spyrou G, Holmgren A. Deoxyribonucleoside triphosphate pools and growth of glutathione-depleted 3T6 mouse fibroblasts. Biochem Biophys Res Commun. 1996;220:42–46. doi: 10.1006/bbrc.1996.0353. [DOI] [PubMed] [Google Scholar]
- 44.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5:417–421. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 46.O’Connor OA, Wright J, Moskowitz C, et al. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol. 2005;23:676–684. doi: 10.1200/JCO.2005.02.050. [DOI] [PubMed] [Google Scholar]
- 47.Richardson PG, Xie W, Mitsiades C, et al. Single-agent bortezomib in previously untreated multiple myeloma: efficacy, characterization of peripheral neuropathy, and molecular correlations with response and neuropathy. J Clin Oncol. 2009;27:3518–3525. doi: 10.1200/JCO.2008.18.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fabre C, Mimura N, Bobb K, et al. Dual inhibition of canonical and noncanonical NF-kappaB pathways demonstrates significant antitumor activities in multiple myeloma. Clin Cancer Res. 2012;18:4669–4681. doi: 10.1158/1078-0432.CCR-12-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gloire G, Piette J. Redox regulation of nuclear post-translational modifications during NF-kappaB activation. Antioxid Redox Signal. 2009;11:2209–2222. doi: 10.1089/ars.2009.2463. [DOI] [PubMed] [Google Scholar]
- 50.Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005;7:395–403. doi: 10.1089/ars.2005.7.395. [DOI] [PubMed] [Google Scholar]
- 51.Sun SC. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shih VF, Tsui R, Caldwell A, Hoffmann A. A single NFkappaB system for both canonical and non-canonical signaling. Cell Res. 2011;21:86–102. doi: 10.1038/cr.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rahal R, Frick M, Romero R, et al. Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nat Med. 2014;20:87–92. doi: 10.1038/nm.3435. [DOI] [PubMed] [Google Scholar]
- 54.Bailey HH, Ripple G, Tutsch KD, et al. Phase I study of continuous-infusion L-S,R-buthionine sulfoximine with intravenous melphalan. J Natl Cancer Inst. 1997;89:1789–1796. doi: 10.1093/jnci/89.23.1789. [DOI] [PubMed] [Google Scholar]
- 55.O’Dwyer PJ, Hamilton TC, LaCreta FP, et al. Phase I trial of buthionine sulfoximine in combination with melphalan in patients with cancer. J Clin Oncol. 1996;14:249–256. doi: 10.1200/JCO.1996.14.1.249. [DOI] [PubMed] [Google Scholar]