

Abstract
In Cystic Fibrosis (CF) patients, hyper-inflammation is a key factor in lung destruction and disease morbidity. We have previously demonstrated that macrophages drive the lung hyper-inflammatory response to LPS in CF mice, due to reduced levels of the scaffold protein CAV1 with subsequent uncontrolled TLR4 signaling.
Here we show that reduced CAV1 and, consequently, increased TLR4 signaling, in human and murine CF macrophages and murine CF lungs, is caused by high microRNA-199a-5p levels, which are PI3K/AKT-dependent. Down-regulation of microRNA-199a-5p or increased AKT signaling restores CAV1 expression and reduces hyper-inflammation in CF macrophages. Importantly, the FDA approved drug celecoxib reestablishes the AKT/miR-199a-5p/CAV1 axis in CF macrophages, and ameliorates lung hyper-inflammation in Cftr-deficient mice.
Thus, we identify the AKT/miR-199a-5p/CAV1 pathway as a regulator of innate immunity, which is dysfunctional in CF macrophages contributing to lung hyper-inflammation. Importantly, this pathway is targeted by celecoxib.
Introduction
Lung hyper-inflammation, characterized by increased production of IL-8, IL-6, and other pro-inflammatory cytokines, decreased anti-inflammatory cytokines and a greater number of immune cells 1, is recognized as a key factor in Cystic Fibrosis (CF) lung destruction. Therefore, several anti-inflammatory drugs have been tested in CF patients. Among these, high-dose treatment with the non-steroidal anti-inflammatory drug (NSAID) ibuprofen is, to date, the only drug proven to be effective in slowing the progression of lung disease 2, and increasing life expectancy in CF patients 3. Unfortunately, the high doses needed for achieving a beneficial outcome are associated with several side effects, limiting its use among CF patients.
In the last few years, tremendous progress has been made in identifying small molecules (potentiators) that effectively increase the activity of the defective cystic fibrosis transmembrane conductance regulator (CFTR) channel, the protein affected in CF, which has resulted in therapy for a small number of CF patients. In addition, other small molecules effective in improving the plasma membrane localization (correctors) of the most common CFTR mutant (F508del) are currently in clinical trials. Nevertheless, it is not yet clear whether CFTR corrector and potentiator therapies will suppress inflammation in CF 4. Thus, there is a need to identify new therapeutic interventions for controlling hyper-inflammation in CF.
Although it is thought that hyper-inflammation in CF is a consequence of chronic infection, growing evidence suggests that CFTR, in addition to its very well established role in ion transport, also regulates signaling in receptors involved in the immune response 5–7. CFTR also modulates signaling in MΦs 8, which are central mediators of the inflammatory response, contributing both to initiation and resolution of inflammation. Indeed, we and others have demonstrated that CF MΦs contribute directly to the increased production of pro-inflammatory cytokines in mouse lungs, and that expression of wild-type (WT) CFTR in MΦs is sufficient to ameliorate the exaggerated lung inflammation in CF mice 9, 10. We were the first group to report that CFTR modulates the trafficking and signaling of the innate immune receptor Toll like receptor 4 (TLR4) in MΦs. We found that in the absence of CFTR, TLR4 signaling is increased leading to an enhanced pro-inflammatory response 7. More recently, these findings were corroborated in CF airway epithelial cells, suggesting that CFTR controls TLR4 signaling in several cell types 11. Subsequently, we determined that murine and human CF MΦs, mucosal MΦs from human CF nasal polyp biopsies, and lung tissues from knockout (KO) and F508del/F508del mice fail to induce expression of the scaffold protein caveolin 1 (CAV1) in response to lipopolysaccharides (LPS) 12. Due to reduced CAV1, heme-oxygenase 1 (HO-1), a key stress response protein involved in balancing cellular redox status and inflammation, does not translocate to the plasma membrane, where it normally acts to downregulate TLR4 signaling 12, 13. Thus, the CAV1/HO-1 protective pathway is altered in CF cells and, by preventing down-regulation of TLR4 signaling, this defect contributes to the perpetuation of the vicious cycle of hyper-inflammation and oxidative stress in CF 12. The mechanism/s by which dysfunctional CFTR alters CAV1 induction in response to inflammatory triggers remains to be elucidated.
MicroRNAs (miRNAs) are small noncoding RNAs that exert post-transcriptional gene regulation activity by targeting messenger RNAs. MiRNAs are involved in various fundamental biological processes and deregulation of miRNAs results in pathological conditions. Importantly, miRNAs play a central role in the regulation of innate host defense and the inflammatory response 14 and their disregulation contributes to chronic inflammatory lung diseases, including asthma, chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis, cystic fibrosis, and bronchiectasis, which are all characterized by an abnormal innate host defense response to environmental stimuli and infection 15. Several miRNAs are abnormally expressed in CF-affected primary airway epithelial cells. These miRNAs fall into two main categories: miRNAs that modulate levels of functional CFTR directly (e.g. miRs 145, 223, 494, 509, 101) or indirectly (e.g. miR138), and several others that affect the innate immune response by altering downstream signaling (e.g. miRs 126, 93, 155) of TLRs or by weakening humoral defense (miR-31) (summarized in references 16–19).
We hypothesized that reduced CAV1 induction in CF MΦs, which affects TLR4 signaling, could be due to altered miRNA-dependent post-transcriptional regulation. Among miRNAs that are predicted to target the 3′-UTR of CAV1 and that are conserved among species, microRNA 199a-5p (miR-199a-5p) has been validated for its ability to target CAV1 20. Based on this evidence, we studied miR-199a-5p expression in CF. Here, we demonstrate, using a combination of pharmacological and genetic tools that lack of CFTR in MΦs is associated with high levels of miR-199a-5p due to blunted phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling in response to TLR4 activation. Increased levels of miR-199a-5p, targeting the 3′-UTR of CAV1, interfere with induction of CAV1 in response to inflammatory triggers, ultimately leading to ineffective negative feedback of TLR4 signaling and hyper-inflammation.
Thus, we have identified a novel dysregulated signaling pathway in CF that contributes to lung hyper-inflammation. Importantly, we show that celecoxib (Celebrex), an FDA-approved NSAID used in the treatment of osteoarthritis and rheumatoid arthritis and not yet tested for efficacy in CF patients or in other chronic inflammatory lung diseases, restores the AKT/miR-199a-5p/CAV1 pathway in CF MΦs and ameliorates lung hyper-inflammation in Cftr-deficient mice.
Given the leading role of TLR4 signaling in MΦs to control lung innate host defense, dysfunctions in the AKT/miR-199a-5p/CAV1 pathway in MΦs may play a role in the pathobiology of several chronic inflammatory lung diseases.
Results
Increased levels of miR-199a-5p in CF MΦs and lung tissues
We have previously shown that CF MΦs treated with LPS have increased TLR4 signaling due to reduced CAV1 expression 12. The mechanism/s by which lack of functional CFTR in MΦs interferes with CAV1 induction in response to inflammatory triggers remained to be investigated. In lung fibroblasts 20 and myoblasts 21, microRNA-199a-5p was recently identified as a potent regulator of CAV1 expression by specifically binding its 3′-untranslated region (UTR). Thus, we tested whether increased levels of miR-199a-5p are responsible for reduced CAV1 in bone marrow derived (BMD) MΦs isolated from Cftr-deficient mice (referred to hereafter as CF MΦs), and whether miR-199a-5p plays a role in regulating TLR4 signaling. We observed that LPS exposure decreased miR-199a-5p by 50% in WT murine MΦs after 2 hours (h), and this effect persisted for 4 h (Fig. 1A). In contrast, in CF MΦs the LPS-induced down-regulation of miR-199a-5p was completely abrogated, and CF MΦs had a modest, but statistically significant, increase in miR-199a-5p (Fig. 1A). In contrast, miR-802, another miRNA with a predicted recognition site for CAV1, was not affected by LPS stimulation and no differences were observed between WT and CF MΦs (Supplementary Fig. 1A). Importantly, expression of the hypoxia-inducible factor 1alpha (Hif-1α), which is a known target of miR-199a-5p in lung tissues 22 and myocytes 23, was decreased 2-fold in CF MΦs exposed to LPS compared to WT cells (Supplementary Fig. 1B), indicating that miR-199a-5p is differentially upregulated.
Figure 1. miR-199a-5p, an upstream regulator of CAV1 and TLR4 signaling, is increased in CF.

(A) MiR-199a-5p relative expression (qPCR) in murine WT (white bars) and CF (black bars) BM-derived MΦs, untreated or treated with LPS; (B) Cartoon representation of the in vivo treatment and qPCR for miR-199a-5p in lung lysates from WT and CF-KO mice untreated or treated with LPS; (C) Flow cytometry dot-plots of WT murine MΦs infected with RV-miR-GFP (miR-CTR) vector control (top) or RV-miR-199a-5p-GFP (bottom), showing FACS sorting scheme; (D) qPCR for miR-199a-5p (top), CAV1 (middle) and IL-6 (bottom) in RVs infected and sorted WT MΦs, untreated or treated with LPS; data are compared to uninfected WT MΦs; (E) WB and densitometric analysis for HO-1 in uninfected or RVs infected WT MΦs, untreated or treated with LPS; (F) Flow cytometry dot-plots of CF murine MΦs infected with RV-sponge (SPG)-control, RV-199a-3p-SPG or RV-199a-5p-SPG, showing puromycin-selected-GFP positive population expressing the MΦs marker MAC-1 (top panel) and MΦs cytospun cells stained with Giemsa, demonstrating absence of morphological abnormalities among different MΦ populations (bottom panel); qPCR for miR-199a-5p and for CAV1 (G) and IL-6 (H) in RV-SPG infected and puromycin-selected CF MΦs, untreated or treated with LPS; (I) WB and densitometric analysis for COX-2 in RV-SPG infected and puromycin-selected CF MΦs, untreated or treated with LPS. For qPCR, miR-199a levels are normalized to RNU6B and CAV1/IL-6 expression to S18. For WB, protein fold increase is normalized to B-actin. Unless indicated differently, for each experiment the data are the result of three experimental biological repeats or are representative of three experimental biological repeats. Statistical analyses were conducted using one-sided two-sample t-tests. Error bars indicate standard deviation. Symbols * and # indicate a statistically significant difference among the experimental group and control group with a P value <0.05.
MiR-199a-5p/CAV1 levels were also modulated by the TLR5 ligand flagellin, which together with LPS plays a major role in driving inflammation in CF. As observed for LPS, CF MΦs exposed to flagellin have reduced CAV1 expression and increased levels of miR-199a-5p, suggesting that the miR-199a-5p/CAV1 pathway is downstream of the MyD88-dependent innate immune response (Supplementary Fig. 1C).
MiR-199a-5p has recently been linked to α1-antitrypsin deficiency and to the unfolded protein response (UPR) 24. At the experimental conditions used and at the time points assessed, we did not observe statistically significant differences between WT and CF MΦs in the induction of the UPR regulator gene Grp78 or of the signal transducer ATF6 (Supplementary Fig. 1D). This does not exclude that miR-199a-5p may affect the levels of UPR-associated transcriptome after prolonged exposure to LPS or in the presence of the F508del protein.
The miR-199a-5p and miR-199a-3p mature miRNA sequences arise from a common stem loop structure that is highly conserved across vertebrate species. Although to a lesser extent than miR-199a-5p, miR199a-3p, which arises from the 3′ arm of the miR199a hairpin and does not have the predicted consensus sequences for the CAV1 3′-UTR 21, was also modulated by LPS and increased levels were observed in CF MΦs compared to controls (Supplementary Fig. 1E, left panel). MiR-199b-5p, another miRNA with similar sequence homology to miR-199a-5p but transcribed from a different genetic locus 21, was expressed at very low levels compared to miR-199a-5p in MΦs. LPS induced a modest and transient increase in miR-199b-5p but no differences were observed between WT and CF cells (Supplementary Fig. 1E, right panel).
We then assessed the transcriptional levels of the stem-loop miR-199a precursors. Expression of both the a-1 and a-2 precursors was modulated by LPS. MiR-199a-2 stem loop precursors had a pattern of expression comparable to that observed for miR-199a-5p, with decreased levels in response to LPS in WT MΦs. Furthermore, miR-199a-2 down-regulation was absent in CF MΦs (Supplementary Fig. 1F). Thus, the variation in mature miR-199a-5p levels between WT and CF MΦs in response to LPS may be predominantly due to differential expression of the miR-199a-2 stem loop precursor sequence.
We next tested the expression of miR-199a-5p in the lungs of CF mice in response to inhaled LPS, which we have already reported have reduced CAV-1 expression 9, 12 (see also Fig. 3H). Consistent with the in vitro results, we found that LPS challenge caused miR-199a-5p down-regulation, which was not observed in CF lung tissues (Fig. 1B). A similar pattern of expression was observed for miR-199a-3p, while miR-802 levels, although increased by LPS, were not different between WT and CF mice (Supplementary Fig. 1G). Taken together, these results suggest that miR-199a is dysregulated in CF MΦs and in CF murine lungs, and that elevated levels of miR-199a may play an important role in CF-related lung hyper-inflammation.
Figure 3. Celecoxib rescues the miR-199a-5p/CAV1 pathways by stimulating PI3K-AKT signaling in CF MΦs, and decreases the lung hyper-inflammatory response to LPS in CF-affected mice.

(A) qPCR for IL-6 (left), miR-199a-5p (middle) and CAV1(right) and (B) WB and densitometric analysis for HO-1 in CF murine MΦs untreated or treated with LPS, in absence or presence of celecoxib; (C) qPCR for miR-199a-5p (left) and CAV1 (right) in WT and CF murine MΦs treated with LPS, in presence or absence of celecoxib and of PI3K-AKT inhibitor LY94002; (D) WB and densitometric analysis for AKT, pAKT and B-actin in WT and CF murine MΦs treated with LPS for 4h in presence or absence of celecoxib; (E) In vivo experiment schematic representation; total and differential BAL fluid cell number (F), hematoxylin/eosin staining in paraffin embedded lung tissues (G), qPCR for IL-6, miR-199a-5p and CAV1 (H) and body weigh loss (I) from WT and CF mice treated chronically (10 days) with celecoxib (Celebrex) and then challenged with LPS for three days. Mice were sacrificed 24h after last nebulization. For qPCR, miR-199a levels are normalized to RNU6B and CAV1 and IL-6 expression to S18. For WB, protein fold increase is normalized to B-actin or total AKT (pAKT). For the in vitro experiments, the data are the result of three experimental biological repeats; for the in vivo study, three mice were used for each group, and the experiment was repeated independently twice (total: 6 mice per group). Statistical analyses were conducted using one-sided two-sample t-tests (q-PCR) or two-sample unequal variance t-tests (BAL fluid cell numbers). Error bars indicate standard deviation. The symbol * indicates a statistically significant difference among groups with a P values <0.05. Images were acquired using an Olympus BX51 microscope with a 4× objective; scale bar is 100μm.
MiR-199a-5p regulates CAV1 levels and TLR4 signaling in MΦs
To prove that miR-199a-5p regulates CAV1 expression in LPS-stimulated MΦs, and modulates the inflammatory response, we over-expressed miR-199a-5p in WT cells. Murine WT bone marrow-derived (BMD)-progenitor cells were infected with the retrovirus vector (RV) pMSCV-miR-199a5p-PGK-EGFP (RV-miR-199a-5p) or pMSCV-PGK-EGFP control (RV-miR-CTR) and differentiated in MΦ-colony stimulating factor. Over-expression of miR-199a-5p had no effect on MΦ differentiation, as demonstrated by the unchanged MAC-1 expression between infected (GFP-pos) and uninfected (GFP-neg) cells (Supplementary Fig. 1H). GFP-positive cells were sorted (Fig. 1C), plated and treated with LPS for 2h. RV-miR-199a-5p infected cells highly up-regulated miR-199a-5p in both untreated and LPS treated conditions with a very slight increase in miR-199a-5p levels in RV-miR-CTR infected cells (Fig. 1D, upper panel). While LPS treatment upregulated CAV1 expression 15-fold, overexpression of miR-199a-5p resulted in a dramatic decrease in CAV1 expression in response to LPS. A small decrease of CAV1 expression was also observed in RV-miR-CTR infected cells (Fig. 1D, middle panel). Consistent with our previous findings, decreased CAV1 expression was associated with a hyper-inflammatory response to LPS, as shown by increased IL-6 (Fig. 1D, lower panel) and decreased amounts of the downstream target HO-1 (Fig. 1E).
Next, we tested whether knocking down miR-199a-5p would abrogate the hyper-inflammatory response in CF MΦs. We used RV vectors that expressed RNA containing complementary binding sites for miR-199a-5p (microRNA sponge). The miR-199a-5p sponge (RV-miR-199a-5p-SPG) should specifically bind and competitively inhibit miR-199a-5p binding to mRNA targets, thus providing stable and specific miR-199a-5p inhibition 25. As a control, we used a RV- control- sponge (RV-CTR-SPG), which does not target miRNAs, or a RV-miR199a-3p-sponge (RV-miR-199a-3p-SPG), which specifically inhibits miR-199a-3p but not miR-199a-5p. These vectors also encode EGFP and puromycin-resistance genes, allowing for selection of infected MΦs. Up to 89% of the cells were positive for GFP and for the MΦ markers MAC1 (Fig. 1F, upper panel) or F4/80 (Supplementary Fig. 1I), and no alterations of MΦ morphology were observed after infection (Fig. 1F, lower panel). The sequestration of microRNAs by sponges can trigger their degradation 25. Accordingly, the miR-199a-5p sponge caused a 30% decrease of miR-199a-5p expression in primary transduced CF MΦs in the presence or absence of LPS, but had no effect on miR-199a-3p (Fig. 1G, upper panel). Similarly, miR-199-3p sponge decreased miR-199a-3p levels, but had no effect on miR-199a-5p (Supplementary Fig. 1J). CF MΦ transduction with miR-199a-5p sponge (but not with miR-199a-3p sponge) led to a 2.2-fold increase in CAV1 expression in response to LPS compared to control vectors (Fig. 1G, lower panel). Consistent with induction of CAV1 expression, sequestration of miR-199a-5p in CF-MΦs reduced TLR4 signaling, as demonstrated by 2.5-fold decreased IL-6 expression (Fig. 1H) and 2.5-fold decreased COX-2 protein 3 (Fig. 1I). Interestingly, down-regulation of miR-199a-3p has some anti-inflammatory effects, which are CAV1-independent (Fig. 1G–I).
In summary, elevated miR-199a-5p level in WT MΦs is sufficient to abrogate CAV1 expression in response to LPS and to promote the hyper-inflammatory response, while down-regulation of miR-199a-5p in CF MΦs reestablishes CAV1 expression and reduces the pro-inflammatory response.
AKT signaling is required for miR-199a-5p/CAV1 modulation
Next we investigated the signaling pathway required for miR-199a-5p down-regulation in response to LPS. In cardiac myocytes, down-regulation of miR-199a-5p in response to oxygen-related stressors is dependent on phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) (PI3K-AKT) pathway activation 26. In addition, increased levels of miR-199a-5p and decreased pAKT were observed in lung tissue samples of patients with COPD 22. In MΦs, the PI3K-AKT pathway is induced by LPS and its activation regulates different immune functions 27. Among these, PI3K-AKT negatively regulates TLR4 signaling 28, 29. Thus, we investigated whether the down-regulation of miR-199a-5p in WT MΦs in response to LPS was dependent on PI3K-AKT pathway activation and whether this pathway was altered in CF MΦs.
WT and CF MΦs treated with the PI3K inhibitor (LY94002) had increased miR-199a-5p (Fig. 2A, upper panel), and reduced CAV1 (Fig. 2A; lower panel), both at steady-state and in response to LPS. HO-1 induction in response to LPS was affected by LY94002 treatment as well, while its very low expression at steady-state was not altered (Fig. 2B and Supplementary Fig. 2A). LY94002 also increased miR-199a-3p levels in response to LPS (Supplementary Fig. 2B), suggesting that the PI3K/AKT regulates miR-199a at the expression level.
Figure 2. Induction of the PI3K-AKT signaling in CF MΦs rescues the miR-199a-5p/CAV1 axis and reduces hyper-inflammation.

(A) qPCR for miR-199a-5p (top panel) and CAV1 (bottom panel) or (B) WB and densitometric analysis for HO-1 in WT and CF-KO murine MΦs untreated or treated with LPS, in absence or presence of the PI3K-AKT inhibitor LY94002; (C) qPCR for miR-199a-5p (top panel) and CAV1 (bottom panel) or (D) WB and densitometric analysis for HO-1 and COX-2 in WT and AKT1-KO murine MΦs untreated or treated with LPS; (E) WB and densitometric analysis for phospho-AKT and total AKT in WT and CF-KO murine MΦs untreated or treated with LPS; (F) qPCR for miR-199a-5p (top panel), CAV1(middle panel) and IL-6 (bottom panel) or WB and densitometric analysis for HO-1 (G) in WT, WT+CFTR inhibitor CFTRinh172, CF-KO, PTEN-KO and PTEN-KO+ CFTRinh172 MΦs untreated or treated with LPS. Symbols * indicates a statistically significant difference among WT+CFTRinh172 and PTEN-KO+ CFTRinh172 MΦ groups.
For qPCR, miR199a levels are normalized to RNU6B and CAV1 and IL-6 expression to S18. For WB, protein fold increase is normalized to B-actin or AKT (pAKT). Unless indicated differently, for each experiment the data are the result of three experimental biological repeats or are representative of three experimental biological repeats. Statistical analyses were conducted using one-sided two-sample t-tests. Error bars indicate standard deviation. Symbols * and # indicate a statistically significant difference among the experimental group and control group with a P values <0.05.
In mammals, the AKT family consists of three isoforms, AKT1/PKBα, AKT2/PKBβ and AKT3/PKBγ. We chose to test the AKT1 isoform since this isoform is involved in regulation of LPS tolerance in MΦs via microRNAs 30. We show that MΦs differentiated from the BM of homozygous AKT1-KO mice have increased levels of miR-199a-5p and decreased CAV1 expression in response to LPS compared to WT cells (Fig. 2C). Consistent with defective TLR4 negative feedback, AKT1-KO MΦs have increased IL-6 (Supplementary Fig. 2C) and COX-2 expression (Fig. 2D) in response to LPS. HO-1 protein level was elevated in untreated AKT1-KO MΦs, but its induction in response to LPS was blunted compared to WT cells (Fig. 2D). Together, these data suggest that negative regulation of TLR4 signaling via miR199-5p/CAV1 is, at least in part, PI3K/AKT1-dependent.
These results led us to assess AKT phosphorylation in CF MΦs challenged with LPS. We found that CF MΦs, although they expressed abundant total AKT, had a modest but statistically significant reduction of AKT phosphorylation at serine 473 compared to WT MΦs after 1h and 2h. In addition, this difference persisted at 4h of LPS challenge (Fig. 3D). Thus, blunted PI3K-AKT signaling in response to LPS observed in CF MΦs may contribute to defective regulation of the miR199-5p/CAV1 axis and hyper-inflammation. To test the direct effect of AKT phosphorylation on miR-199a levels, CF MΦs were treated with increasing doses of the AKT agonist SC79 for 4 hours. SC79 decreased miR-199a-5p levels in a dose-dependent manner, which correlated with augmented AKT signaling. High doses of SC79 also had effects on miR-199-3p, but not on miR-802 (Supplementary Fig. 2D).
Next, we tested whether rescuing PI3K-AKT signaling in CF environment would restore a controlled inflammatory response. To this end, we used BMD-MΦs from phosphatase and tensin homolog (PTEN) KO mice, which have constitutive up-regulation of phosphoAKT and are hypo-responsive to signals associated with MΦ activation 31. PTEN KO MΦs (Fig. 2F, blue bars) have slightly lower miR-199a-5p levels and slightly higher CAV1 expression than WT cells in response to LPS (Fig. 2F, white bars). Both WT cells treated with the CFTR inhibitor CFTRinh172 32 (Fig. 2F, grey bars) and CF cells (Fig. 2F, black bars) had high miR-199a-5p and reduced CAV1 expression compared to WT controls. Therefore, chemical inhibition of CFTR in WT cells recapitulates the CF phenotype, as we have also demonstrated previously 7. Importantly, CFTR inhibition in a PTEN-KO background (Fig. 2F, violet bars) caused reduction of miR-199a-5p and increased in CAV1 expression compared to WT MΦs treated with CFTRinh172 (Fig. 2F, gray bars). In addition, CFTR inhibition in PTEN-KO MΦs also decreased LPS-induced inflammation, as shown by lowered IL-6 expression (Fig. 2F, bottom panel), and increased HO-1 protein production (Fig. 2G), in a PI3K/AKT-dependent manner (Supplementary Fig. 2E). These data suggest that stimulation of the PI3K-AKT signaling pathway in CF MΦs rescues the hyper-inflammatory response to LPS.
Celecoxib targets the AKT/miR-199a-5p/CAV1 pathway
Celecoxib (Celebrex), an FDA-approved selective COX-2 inhibitor used in the treatment of osteoarthritis, rheumatoid arthritis and pain management, has been reported to activate PI3K/AKT and mitochondrial redox signaling to enhance HO-1-mediated anti-inflammatory activity in vascular endothelium 33. Celecoxib also induces CAV1 expression 34, potentially in a COX-2 inhibition-independent manner 35. Thus, we tested the hypothesis that celecoxib could decrease TLR4 signaling in CF MΦs via modulation of the miR-199-5p/CAV1 axis. CF MΦs were pretreated overnight with celecoxib prior to LPS treatment. In addition to the expected down-regulation of the pro-inflammatory response, as shown by reduced IL-6 expression (Fig. 3A, left panel, dark red bars), celecoxib treated CF MΦs had a statistically significant decrease in miR-199a-5p expression (Fig. 3A, middle panel, blue bars), as well as increased CAV1 mRNA (Fig. 3A, right panel, orange bars) and HO-1 protein production (Fig. 3B) compared to vehicle-treated (DMSO) CF cells. The highest induction of the HO-1 pathway without cell toxicity was observed at a dose of 25μM (Supplementary Fig. 2F),..
The ability of celecoxib to downregulate miR-199a-5p and increase CAV1 was prevented by pre-treatment of CF MΦs with the PI3K/AKT inhibitor LY94002 (Fig. 3C), and activation of the PI3K/AKT pathway was increased in celecoxib pre-treated CF MΦs challenged with LPS (Fig. 3D). Therefore, celecoxib modulates the miR-199a-5p/CAV1 signaling pathway in CF MΦs challenged with LPS in a PI3K/AKT dependent manner.
Unlike celecoxib, at 25μM, the classic NSAID ibuprofen fails to efficiently rescue the miR-199a/CAV1 pathway in CF MΦs treated with LPS. Modulation of miR-199a/CAV1 was observed at a higher ibuprofen dosage (100μM), which, however, had more broad and nonspecific effects, as it also affected miR-802 expression (Supplementary Fig. 2G). Ibuprofen was also less efficient at inducing HO-1 compared to celecoxib (Supplementary Fig. 2H).
Because celecoxib abrogated the hyper-inflammatory response of CF MΦs to LPS, we next tested its effect on LPS-induced hyper-inflammation in CF murine lungs 7, 9. Mice were treated orally with the FDA approved drug celecoxib (Celebrex) for 10 days (25mg/kg/day) or with vehicle (apple juice) alone (Supplementary Table 1). At this dosage, Celebrex has anti-inflammatory effects in rat lungs with smoke-induced emphysema 36. Mice received nebulized LPS, as previously described 9, from day 8 to day 10 and sacrificed 24h after the last nebulization (day 11) (Fig. 3E). CF mice treated with celecoxib had reduced inflammatory responses to LPS compared to vehicle-treated CF mice, as demonstrated by reduced total bronchoalveolar lavage (BAL) fluid cell number (TOT), macrophages (MAC), and neutrophils (PMN) (Fig. 3F). In addition, histological analysis of lung tissues stained with hematoxylin and eosin (H&E) confirmed decreased inflammation in mice treated with celecoxib (Fig. 3G). Decreased inflammatory BAL cells were associated with reduced IL-6 expression in celecoxib-treated CF lung tissues (Fig. 3H, left panel). Importantly, celecoxib treatment in LPS-challenged CF mice reduced miR-199a-5p (Fig. 3H, middle panel), and increased CAV1 expression (Fig. 3H, right panel) in lung tissues compared to vehicle-treated CF tissues. Pre-treatment with celecoxib also reduced weight loss associated with LPS challenge in CF mice (Fig. 3I). Celecoxib had minimal effects on WT mice (Fig. 3G–I).
In contrast to celecoxib, ibuprofen treatment (50mg/Kg for 10 days), despite some effects on the inflammatory response to LPS compared to vehicle-treated CF animals, was not effective in modulating the miR-199a-5p/CAV1 pathway or in preventing LPS-induced weight loss in CF mice (Supplementary Fig. 3C–D and supplementary Table 1).
Finally, to test whether celecoxib’s protective action to LPS is due, at least in part, to the AKT-dependent down-regulation of the miR-199a-5p in vivo, AKT1-KO mice (supplementary Table 1) were treated with celecoxib or vehicle alone and then challenged with LPS, as described in Fig. 4E. As previously reported 37, AKT1-KO mice had robust lung neutrophilic inflammation in response to LPS (Supplementary Fig. 3B). Up-regulation of miR-199a-5p and down-regulation of CAV1 were even more pronounced than what was observed in CF mice, as was LPS-induced weight loss. Celecoxib treatment in AKT1-KO mice had a modest effect in reducing BAL cell number and IL-6 expression, but no restoration of the miR-199a-5p/CAV1 pathway and no protection against weight loss were observed (Supplementary Fig. 3A–D). Thus, celecoxib-dependent protection against weight loss and lung hyper-inflammation in response to LPS requires activation of the AKT/miR-199a-5p/CAV1 pathway.
Figure 4. miR-199a-5p/CAV1 axis is defective in human CF MΦs and can be modulated by celecoxib pre-treatment.

(A) qPCR for miR-199a-5p (left) and CAV1(right) in PB-derived MΦs isolated from four healthy donors (HD) (each donor sample is indicated with a white dot) and eight CF patients (each patient sample is indicated with a black triangle), untreated or treated with LPS for 2 and 4 hours (h). Relative expression is calculated over untreated controls and the average relative expression is indicated by a black (HD) or a red (CF) bar; (B) qPCR for miR-199a-5p and CAV1 in human CF MΦs (CF hMΦs) treated with LPS without (left panels) or with celecoxib (25μM) pretreatment (right panels; indicated as CF+C); each color represents a distinct CF patient; (C) qPCR for miR-199a-5p (left) and CAV1 (right) in human MΦs differentiated from non-CF CD34-positive mobilized PB cells and conditioned with the CFTR inhibitor CFTRinh172 (HD+ CFTRinh) or with vehicle alone (HD+DMSO); (D) qPCR for miR-199a-5p (left) and CAV1(right) in HD+DMSO and HD+ CFTRinh pretreated with celecoxib (25μM) (HD+ CFTRinh+C). The miR-199a-5p levels are normalized to RNU6B and CAV1 expression to S18. For the experiments with CD34-positive mobilized PB cells the data are the result of three experimental biological repeats. Statistical analyses were conducted using one-sided two-sample t-tests. Error bars indicate standard deviation. The symbol * indicates a statistically significant difference among groups with a P values <0.05.
Celecoxib restores the miR-199a-5p/CAV1 axis in human CF MΦs
We then tested whether miR-199a-5p levels are also elevated in human MΦs isolated from CF patients. We isolated PB-derived MΦs from eight CF donors (supplementary Table 2) and from four healthy donor controls (HD). Cells were treated with LPS for 2h and 4h. The fold change in expression of miR-199a-5p and CAV1 in response to LPS for each individual’s MΦs was compared to its own untreated control. Despite variability in response to LPS across individuals, we found that miR-199a-5p levels were elevated, and CAV1 expression was decreased in MΦs from CF individuals when compared to HD MΦs, as we have observed in murine cells (Fig. 4A). Pre-treatment of CF MΦs with celecoxib (color lines indicate three different donors) decreased miR-199a-5p levels, and increased CAV1 expression (Fig. 4B).
To investigate whether the inability of CF MΦs to up-regulate CAV1 in response to LPS is a consequence of nonfunctional CFTR, we treated human MΦs differentiated from CD34-positive mobilized PB cells with CFTRinh172 or with vehicle (DMSO) alone. Non-CF human MΦs treated with CFTRinh172 phenocopied CF MΦs, with increased miR-199a-5p, decreased CAV1 induction (Fig. 4C) and increased IL-6 expression (Supplementary Fig. 3E) in response to LPS. Thus, in human MΦs the hyper-inflammatory dysfunction is directly linked to the presence of functional CFTR. Furthermore, overnight pre-treatment with celecoxib decreased miR-199a-5p, and increased CAV1 expression in non-CF human MΦs treated with CFTRinh172 (Fig. 4D). In summary, we have validated that the miR-199a-5p/CAV1 axis is defective in primary human diseased CF MΦs and that celecoxib treatment can rescue this defect.
Discussion
Hyper-inflammation is recognized as a critical factor in lung tissue deterioration, and lung function decline in CF patients, which contributes to morbidity and mortality 1. Although CF mice do not fully recapitulate human CF lung disease, they are a very good model for studying CF-related GI manifestations, ion transport defects and systemic inflammation 38. In fact, CF mice manifest hyper-inflammation in the lungs 9, 39, pancreas 40, and intestine 41 at baseline, or after exposure to various inflammatory triggers. Thus, CF mouse models recapitulate aspects found in human CF hyper-inflammation.
The CFTR gene is expressed in secretory epithelia of the body and, at lower levels, in other cell. CFTR protein is detected in murine 9, 42 and human 43, 44 MΦs and CFTR-like Cl− conductance has been recorded in monocytes/MΦs 7, 42, 43, suggesting that in these cells CFTR functions as a cAMP-dependent chloride channel. Several studies have shown that CF MΦs contribute directly to lung hyper-inflammation 9, 10. CFTR MΦs also display defective bacterial killing 42, 44, 45 due to dysfunctional autophagy 46. Thus, CF MΦs exhibit several cell-autonomous immune dysfunctions that contribute to CF lung disease, which make MΦs potential targets for effective CF treatments.
We have shown that hyper-secretion of pro-inflammatory cytokines in CF MΦs is due to reduced levels of the scaffold protein caveolin 1 (CAV1) that causes TLR4 signaling upregulation 7, 12. CAV1 is a multi-functional structural protein involved in the formation of specialized plasma membrane lipid rafts 47, in mediating clathrin-independent endocytosis and cholesterol trafficking, and in mediating bacterial and viral immunity 48, 49. CAV1 also binds to and controls the location, and activity of several signaling molecules, including TNFR1, ERK1/2, Src, nitric oxide synthase, and glucocorticoid receptors 50. Importantly, CAV1 binds to, and negatively regulates TLR4 in MΦs 13, and this protective mechanism is defective in CF MΦ 12. CAV1 also down-regulates TGF-β signaling 51, a key pathway with relevance to lung fibrosis. Consistent with the observed interdependency between decreased CAV1 and TGF-β-driven lung fibrosis, increased TGF-β signaling has recently been found in the lungs of CF patients 52.
Here we show that miR-199a-5p, an upstream direct regulator of CAV1, is increased in CF MΦ and CF murine lungs exposed to inflammatory triggers. MiR-199a-5p modulates several signaling pathways such TGF-β 20 and WNT 21 and elevated levels of miR-199a-5p have been linked to alteration of various cellular processes, such as suppressed autophagy 53, increased apoptosis 54, and defective hypoxic response,23 most of which are also dysregulated in CF 55, 56. Here, for the first time, we link miR-199a-5p levels with TLR4 signal regulation in MΦs showing that elevated levels of miR-199a-5p, by impairing CAV1-dependent TLR4 signaling suppression, contribute to CF-related lung hyper-inflammation (Fig. 1). Furthermore, using genetic and biochemical techniques, we demonstrated that the miR-199a-5p/CAV1 axis in MΦs and mouse lung tissue is modulated by PI3K-AKT signaling and that in CF MΦs, the blunted AKT phosphorylation in response to LPS contributes to cellular accumulation of miR-199a-5p and decreased CAV1. Thus, the miR-199a-5p/CAV1 axis participates in the hyper-inflammatory response to LPS (Fig. 2, Supplementary Fig. 2 and 3). This is an intriguing observation, since TLR4 activation in MΦs induces the binding between MyD88 and the PI3K regulatory subunit, which ultimately activates AKT signaling. The PI3K-AKT signaling pathway plays a critical role in feedback inhibition of TLR4 signaling 28, 30, and dysfunctional PI3K-AKT activation in response to LPS is sufficient to enhance the MAPK pathway and NF-kB nuclear translocation 28, 29, which we previously reported is enhanced in LPS-treated CF MΦs 7. Thus, although uncontrolled AKT signaling can lead to a variety of cancers, induction of this pathway in response of inflammatory stressors is fundamental for balancing immune cell functions. Importantly, genetic restoration of the AKT signaling in a CF background is sufficient for decreasing the hyper-inflammatory response to LPS (Fig. 2). Treatment of alveolar MΦs isolated from CF patients with the insulin-like growth factor 1 (IGF-1), a potent AKT activator, enhances their capability to kill bacteria 57. Thus, blunted AKT signaling may also play a role in the defective bactericidal activity of CF MΦs. This pathway may also be defective in CF airway epithelial cells since a recent study demonstrated that pharmacological or genetic inhibition of CFTR in airway epithelial cells prevents PI3K plasma membrane localization 58, which is necessary for AKT phosphorylation. Our data in genetic mouse models suggests that, among the different AKT isoforms, AKT1 signaling is involved in regulating miR-199-5p/CAV1 and TLR4 signaling, which is consistent with its leading role in orchestrating neutrophil recruitment in mice37 and in modulating the abundance of several miRNAs that regulate TLR4 signaling (e.g. miR-155, let7e and miR-125b) 30.
Our data provide evidence that the FDA approved nonsteroidal anti-inflammatory drug celecoxib (Celebrex), which is currently not considered for treatment in CF or other chronic lung inflammatory diseases, stimulates the AKT/miR-199a-5p/CAV1 signaling pathway, decreasing lung hyper-inflammation and preventing the excessive weight loss associated with LPS exposure in Cftr-deficient mice (Fig. 3). The fact that this drug has favorable effects in Cftr-null mice suggests that celecoxib’s mechanism/s of action circumvents the need for functional CFTR. Thus, this drug may have a broad therapeutic application, which may include benefits for CF patients with nonsense mutations. Importantly, at the doses used, celecoxib had minimal effects in modulating lung inflammation in WT mice (Fig. 3), suggesting a CF environment-dependent mode of action.
Our data show that celecoxib’s beneficial effects are, at least in part, due to modulation of the AKT/miR-199a-5p/CAV1 pathway, in addition to COX-2 inhibition. Also, genetic down-regulation of miR-199a-5p in CF MΦs decreased COX-2 protein levels (Fig. 1), suggesting that celecoxib regulates not only COX-2 enzymatic function, but also its expression. Highlighting the relevance of this observation, Cftr-null mice have inherently higher levels of COX-2 59. Celecoxib is an intriguing small molecule also because it appears to bind to the hydrophobic acyl core of phospholipid membranes, changing physical properties of membranes and potentially modifying the activity of membrane bound proteins,60, 61 which may explain in part celecoxib’s effect on TLR4 signaling. However, further studies are necessary to better understand the COX-2-independent mechanisms of celecoxib. Celecoxib is more efficient than ibuprofen in modulating the miR199-5p/CAV1 pathway, in reducing lung hyper-inflammation and in preventing LPS-induced weight loss in CF mice (Supplementary Fig. 3C–D). In CF patients, ibuprofen has been proven to be efficient in preserving lung function and weight loss, but this is achieved with high doses that maintain the drug plasma concentration at 50–100μg/ml 2. We do not know whether in our mouse study the ibuprofen plasma concentration was maintained substantially overtime.
Our in vivo studies support the hypothesis that AKT1 is required for miR-199a-5p down-regulation in response to LPS and that the celecoxib-dependent protection of weight loss and, partially, of hyper-inflammation in response to LPS, requires activation of this pathway. In CF patients, maintenance of body weight is critical for lung function preservation 62, and impaired AKT signaling may contribute to the low body mass index in patients with CF, which may not only be caused by the loss of exocrine pancreatic function or by gut malabsorption 63. In addition, AKT is a key metabolic modulator and defective AKT signaling is associated with insulin resistance 64. Thus, this finding may be also relevant in the context of CF-related diabetes.
In conclusion, our data provide mechanism-based evidence that celecoxib is a potential candidate for controlling lung hyper-inflammation in CF. Although Celebrex has a warning of increased risk of myocardial infarction, these adverse cardiovascular events appear similar among users of celecoxib and other classical NSAIDs 65. In a large randomized clinical trial centered at the Cleveland Clinic, celecoxib is currently being compared to naproxen and ibuprofen for its overall benefit vs. risk profile for treating osteoarthritis and rheumatoid arthritis (NCT00346216).
In the context of chronic inflammatory lung diseases, elevated levels of miR-199a-5p or reduced expression of CAV1 has been reported in lung fibroblasts and monocytes of idiopathic pulmonary fibrosis 20, scleroderma 66 and asthma 67 patients, and in lung endothelial cells of COPD patients 22. Based on the evidence that components of the AKT/miR-199a-5p/CAV1 pathway are defective in several chronic inflammatory lung diseases, and given the leading role of MΦs’ TLR4 signaling to innate host defense response to environmental stimuli and infection, this study provides novel insights into the development and treatment of these inflammatory-based lung diseases. This study may be of particular relevance for COPD. In fact, cigarette smoke, the major cause of COPD, leads to systemic CFTR dysfunction 68.
In conclusion, we have identified the AKT/miR-199a-5p/CAV1 axis as an important regulatory pathway in MΦs that serves as a cellular protective mechanism and ensures immune-regulation in response to inflammatory triggers. Importantly, we demonstrate that this pathway is defective in CF, contributing to the hyper-inflammatory lung disease in Cftr-deficient mice. These dysfunctions in CF murine and human MΦs and in CF murine lungs are reversed by celecoxib treatment (summarized in Fig. 5). Our pre-clinical studies provide data suggesting that celecoxib would be beneficial for controlling lung disease in CF patients, thus supporting further testing in the clinical setting.
Figure 5. TLR4 signaling negative feedback is regulated by the miR-199a-5p/CAV1 axis in an AKT-dependent manner and this pathway is altered in CF MΦs.
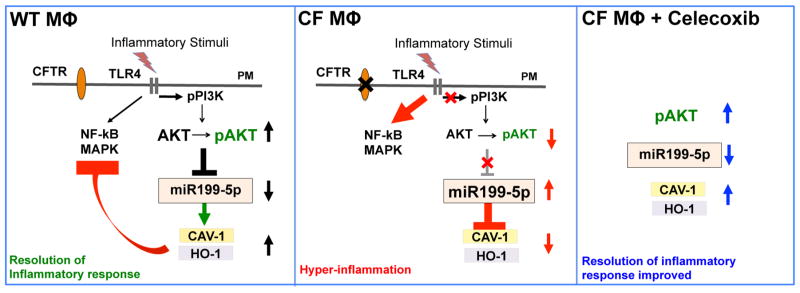
TLR4 activation induces the PI3K-AKT pathway in MΦs, which down-regulates microRNA miR-199a-5p, allowing expression of CAV-1 and subsequent negative feedback on the TLR4 pro-inflammatory cascade (NF-kB/MAPK signaling). In absence of functional CFTR, MΦs have a blunted PI3K/pAKT signaling in response to TLR4 activation, which leads to accumulation of miR-199a-5p. Increased levels of miR-199a-5p, targeting the 3′-UTR of CAV1, interfere with induction of CAV1 in response to inflammatory triggers, ultimately leading to ineffective negative feedback of TLR4 signaling and hyper-inflammation. Celecoxib stimulates the AKT/miR-199a-5p/CAV1 pathway in CF murine and human MΦs, and decreases lung hyper-inflammation in Cftr-deficient mice.
Methods
Constructs
The miR199a expression construct for miR-199a was cloned by PCR-amplification of genomic DNA with the following primers: CAGAACTTTCTCCAGATGCG and ATGGCAGACTGATAGGGCC PCR product was cloned into the pMIRWAY-puro and/or pMIRWAY-GFP vectors 69(with MSCV LTR driving miRNA or sponge, and with PGK promoter driving puromycin resistance gene or GFP), through GateWay cloning (Invitrogen).
The miRNA-inhibiting sponge sequences were obtained by oligonucleotide synthesis of 5 copies of either miR-199a-5p sites or miR-199a-3p sites, and cloned to generate sponges with 10 copies of bulged binding sites. Specifically, the sequences were digested with either SalI or XhoI and then ligated. Ten-copy sponge sequences were placed downstream of EGFP 25 and cloned into pMIRWAY-puro vector. Five-copy sponge oligo sequences are listed below.
miR-199a-5p:
ATACAGATCTCCGTCGACGAACAGGTAGAAATAACACTGGGAAGAACAGATAGCAACAACACTGGGTTGAACAGGTAGGACTAACACTGGGACGAACAGATAGATACAACACTGGGCAGAACAGGTAGGAATAACACTGGGCTCGAGTAGGTGACACTATAGTTTCTCCAGATGCGAGCCGGGCGATCGCAGCACCATG
miR-199a-3p:
ATACAGATCTCCGTCGACTAACCAATGAAATGACTACTGTAAAAAACCAATCCTACCACTACTGCATTTAACCAATGGAGTGACTACTGTGACAAACCAATCATAGCACTACTGCCACTAACCAATGGTATGACTACTGTCTCGAGTAGGTGACACTATAGGCAGCAAATGTGCCACGTCAGCGATCGCAGCACCATG
Control vectors for miRNA are pMIRWAY-puro or pMIRWAY-GFP vectors without miRNA.
Retroviral Preparation and Cell Transduction
Packing cells 293T (5×106) were transfected with plasmid mixture containing 10ug of retroviral construct, 10ug of gag/pol and 10ug of VSVG constructs using FuGENE HD transfection reagent (Promega) following the instructions. Retrovirus was harvested in viral collection medium (DMEM with 10% heat-inactivated FBS+1%PSG+20mM Hepes) for 3 days after transfection, twice a day. The viral collection medium was spun down at 1500rpm for 5 mins to remove 293T cells and concentrated with Amicon Ultra centrifugal filter-10K (Millipore), at 4000rpm for 2hrs at 4 °C. Then, viral vectors were aliquoted and stored at −80 °C until use.
Titer assessment: Retrovirus titer was assessed using 293T cells. 1X106 cells were plated in 1.25ml medium (DMEM supplemented with 10% heat-inactivated FBS; antibiotics and L-glutamine) together with 1μl polybrene (10mg/ml, American Bioanalytical) and with differing amounts of viral solution (none, 5μl, 15μl and 30μl). After spininfection (1800rpm for 90 min), cells were cultured for 36–48hrs before flow cytometry analysis for GFP positive cells. The virus titer is calculated as follows: (GFP positive/100 X cell number)/Volume of virus (ml).
For murine bone marrow cell transduction we use a multiplicity of infection (MOI) virus:cells of 20:1. Briefly, 5 ×105 cells were cultured in 6-well plates and exposed to each virus in 1 ml of serum-free medium. After spin infection (as above), cells were washed and incubated in serum-containing medium and in M-CSF for 7 days. The GFP positive cells were sorted (or selected with puromycin) and GFP-positive cells were then plated and subjected to treatments as indicated.
Chemicals and Reagents
The following antibodies were used: rabbit anti-HO-1 (1:2000 for western blot, Abcam); rabbit anti-COX-2 (1:1000, Cell Signaling Technology); rabbit polyclonal anti-AKT and anti-pAKT (1:1000, Cell Signaling Technology); rabbit anti-actin (1:5000, Santa Cruz); anti-rabbit-HRP (1:2000, Santa Cruz). Pseudomonas aeruginosa (PA) LPS (Sigma-Aldrich) was prepared in PBS at 100X stock solution and used at a concentration of 100 ng/mL. Flagellin (Imgenex, San Diego CA) was used at a concentration of 100 ng/ml). CFTR inhibitor CFTRinh172, a kind gift from Dr. Alan Verkman)32, was freshly prepared in DMSO and used at concentration of 20μM. The PI3K/AKT inhibitor LY94002 (Cell Signaling Technology) was prepared in DMSO and used at a concentration of 20μM. The AKT Activator II, SC79 (Merck Millipore) was dissolved in DMSO and used at the final concentrations indicated. Celecoxib (Sigma-Aldrich, St Louis MO) was dissolved in DMSO (stock solution 20 mM) and used at a final concentration of 25μM. For the in vivo study we used the FDA approved branded celecoxib (Celebrex) at concentration of 25mg/kg/mouse/day. Ibuprofen (Sigma-Aldrich, St Louis MO) was dissolved in DMSO (stock solution 50 mM) and used at a final concentration of 25μM or 100μM. For the in vivo study we used Walgreens Children’s Ibuprofen Suspension (20mg/ml) at a final concentration of 50 mg/kg/mouse/day.
Isolation and culture of macrophages
BM-derived murine macrophages
BM collection was performed as described in ref.9. Briefly, BM cells were flushed with DMEM medium from the medullary cavities of the tibias and femurs using 25G needle in sterile conditions. The BM suspension was filtered through a 70 micron cell strainer, and then mononuclear cells were enriched by the Ficoll-Paque method (Histopaque 1077 Sigma H8889). After overnight culture, the non-adherent cells were differentiated for 7 days in 20ng/ml recombinant M-CSF (ConnStem Inc., CT, USA). After 7 days, cells were detached and characterized by flow cytometry (F4-80+/MAC-1+ population). The day before experiments, cells were plated according to experimental design.
Human macrophages
Blood was obtained from healthy donors (HD) or from patients (supplementary Table 2) with CF during their annual check-up with informed consent in accordance with the Yale University Medical School Human Investigation Committee. MΦs were cultured as described in ref. 7. Briefly, human mononuclear cells were isolated from whole blood by Ficoll-Paque method (Histopaque 1077 Sigma H8889), and seeded at 2×106 cells/well in 12 well plates in RPMI supplemented with 10% FBS and 40ng/ml recombinant human M-CSF (ConnStem Inc., CT, USA). Cells were fed every other day and split 1:1 every 4 days. After 1–2 weeks, cells were characterized by flow cytometry (CD14+/CD45+) and morphology analyzed on cytospin. Before LPS treatment, cells were washed extensively with PBS, detached with Accutase (Innovative Cell Technology), and seeded at 0.5×106 cells per well across 12 well plates before LPS treatment.
Human CD34+ cells were isolated by immunomagnetic selection (Miltenyi CliniMacs) from granulocyte colony-stimulating factor mobilized peripheral blood collected via apheresis with donor consent and differentiated into MΦs by 7 days of culture in human M-CSF (ConnStem Inc., CT, USA). In the experiments in which we use CFTRinh172 (20μM) or celecoxib (25μM), cells were pre-treated overnight with drugs or vehicle alone (DMSO) before adding LPS (100 ng/ml). For inhibition of the PI3K/AKT pathway, cells were pre-treated with LY94002 (20μM) or DMSO control for 1h before LPS challenge.
Mouse Models
All procedures were performed in compliance with relevant laws and institutional guidelines, and were approved by the Yale University Institutional Animal Care and Use Committee. Age and gender of all experimental mice are listed in Table S1.Transgenic CFTR−/− (B6.129P2-KOCftrtm1UNC) mice were purchased from the Jackson Laboratory and bred in the Yale University Animal Facility. Mice are fully back-crossed to the C57BL/6 background. Littermate CFTR−/− mice were fed a liquid diet (Peptamen, Nestle, Deerfield, Illinois) as previously described 9. Non-CF littermate control mice used in the experiments were maintained on an identical diet to the CFTR−/− mice to eliminate nutritional status as a potential confounder (supplementary Table 1). The homozygous AKT1-KO mice are on a C57B/6 background; they were generated as previously described 70, and provided by Dr. Patty J. Lee. Mice (AKT1-KO and WT) used for the in vivo study are listed in supplementary Table 1. The PTEN-KO mice (B6.129S4-Ptentm1Hwu) For obtaining deletion of PTEN in blood cells, mice were crossed with VAV-Cre mice (B6.Cg-Tg-Vav1-cre-A2Kio/J) (early stage of hematopoiesis). Both transgenic mice were originally purchased from the Jackson Laboratory and provided by Dr. Diane S. Krause. BM cells were isolated at 6–8 weeks of mouse age.
In vivo experiment and BAL fluid collection and analysis
For the in vivo study, WT, CF or AKT1-KO mice were treated orally with Celebrex, ibuprofen or with vehicle alone (supplementary Table 1). One capsule of Celebrex (100 mg) was dissolved in 14ml of apple juice, and diluted in liquid food (Peptamen) at a dose of 25mg/kg per mouse; ibuprofen suspension (20mg/ml) was diluted in Peptamen at a final concentration of 50 mg/kg/mouse/day. Food was changed daily for the time indicated (Fig. 3E). On day 8, WT, CF and AKT1-KO mice received LPS (Sigma L8643) over 3 days (a dose a day). LPS (12.5 mg) was administered with a nebulizer (Pulmo-Aide Compressor, Natallergy). Five ml of solution were nebulized over 15 minutes 9. During the 3 days of LPS nebulization, Celebrex was administered (25mg/kg) via oral gavage 4h after each nebulization. At the times indicated, mice were sacrificed. Bronchoalveolar lavage (BAL) fluid was collected using standard methods. The total and differential cell count in the BAL of LPS-treated mice was assessed by counting, by morphological analysis on BAL cell cytospin preps and by flow cytometry analysis. After cardiac perfusion with PBS supplemented with heparin, the left lung lobes were inflated, harvested and paraffin embedded; the right lobes were snap frozen in liquid nitrogen and used for RNA/protein isolation.
RT-PCR and expression analysis
Cells were lysed in Triazol and total RNA was isolated from 1×106 cells using QiagenRNAMini Kits™ (Qiagen), following the instructions. Lung tissues were homogenized before RNA isolation. For cDNA analysis (Roche Molecular Biochemical), 2 μg total RNA was reverse-transcribed using iScript™ cDNA Synthesis Kit (Bio-Rad) following the manufacturer’s specifications. Real-time PCR analysis was performed with a Bio-Rad iCycler using TaqMan technology. Copy number was normalized by 18S and the relative expression to 0 (untreated cells) was calculated by ΔΔCt method. All TaqMan primers and probes were purchased from Applied Biosystem (Life Technology).
For miRNA analysis, RNA was isolated from cells or tissues using the miRNeasy Mini Kit (Qiagen), and the reverse transcribed using the miSCript II RT kit (Qiagen). All miRNAs primers were purchased from Qiagen and qPCR was performed using miScript SYBR green PCR kit (Qiagen). Copy number was normalized by RNU6B and the relative expression to untreated cells was calculated by ΔΔCt method.
Unless indicated, the data are the result of at list three experimental biological repeats or are representative of three experimental biological repeats.
Protein isolation and Western Blot
Cold RIPA lysis buffer containing phosphatase inhibitors was added to cells. After 30min of incubation in ice, the lysates were centrifuged and the supernatants recovered. An equal amount of protein was separated by electrophoresis on 10% Mini PROTEAN Gels (BIO-RAD), transferred to nitrocellulose membrane (Bio-Rad Laboratories, CA) and hybridized using standard procedures. Horseradish Peroxidase-conjugated to IgG secondary antibodies (1:5000, Santa Cruz) and Biorad ECL was used for detection. The chemiluminescence imaging system ChemiDoc (Biorad) and the Image lab software (Biorad) were used for image acquisition and for signal quantification. Protein relative expression is normalized to beta-actin or AKT (for pAKT). Images have been cropped for presentation. Full size images are presented in supplementary Fig. 4.
Statistical Analysis
Statistical analyses were conducted using one-sided two-sample t-tests or two-sample unequal variance t-tests (in vivo studies). All experiments were performed in triplicate, unless indicated differently. Data are expressed as mean ± standard deviation. A P value <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Stephanie Donaldson for helping with the mouse husbandry and Christina Barone for technical advice. Peiying Shan and Dr. Maor Sauler for helping with the AKT1-KO mouse study. Drs. Chad Sanada, Alexandra Teixeira, Shangin Guo, Brain D. Adams, Seyedtaghi Takyar, Lauren Cohn and Clemente J. Britto for valuable suggestions. This work was supported by National Institutes of Health Grants RO1 HL093004 to M.E.E. and E.M.B. The Italian Cystic Fibrosis Research Foundation, grant FFC #15/2012 to E.M.B (collaborator); The American Cystic Fibrosis Foundation grant CF BRUSCI14G0 to E.M.B.
Footnotes
Author contributions
P.Z. and J.C. performed experiments; J.L. provided expertise and valuable material for experiments using miRNA vectors; S.Z. helped with viral vectors experiments; A.D.S. helped to design experiments; J.L.K. provided human blood samples from the Adult CF Center at Yale and reviewed the manuscript; P.J.L., D.S.K. and M.E.E. provided important reagents, provided advice and reviewed the manuscript; E.M.B. initiated and designed the study, developed assays, performed experiments, analyzed data and wrote the manuscript.
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Chmiel JF, Davis PB. State of the art: why do the lungs of patients with cystic fibrosis become infected and why can’t they clear the infection? Respir Res. 2003;4:8. doi: 10.1186/1465-9921-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Konstan MW, Schluchter MD, Xue W, Davis PB. Clinical use of Ibuprofen is associated with slower FEV1 decline in children with cystic fibrosis. Am J Respir Crit Care Med. 2007;176:1084–1089. doi: 10.1164/rccm.200702-181OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.VanDevanter D, et al. High dose ibuprofen significantly improves long-term CF survival. PEDIATRIC PULMONOLOGY. 2012;47:354–354. [Google Scholar]
- 4.Rowe SM, et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med. 2014;190:175–184. doi: 10.1164/rccm.201404-0703OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dudez T, et al. CFTR in a lipid raft-TNFR1 complex modulates gap junctional intercellular communication and IL-8 secretion. Biochim Biophys Acta. 2008;1783:779–788. doi: 10.1016/j.bbamcr.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vij N, Mazur S, Zeitlin PL. CFTR is a negative regulator of NFkappaB mediated innate immune response. PLoS ONE. 2009;4:e4664. doi: 10.1371/journal.pone.0004664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruscia EM, et al. Abnormal trafficking and degradation of TLR4 underlie the elevated inflammatory response in cystic fibrosis. J Immunol. 2011;186:6990–6998. doi: 10.4049/jimmunol.1100396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartl D, et al. Innate immunity in cystic fibrosis lung disease. J Cyst Fibros. 2012 doi: 10.1016/j.jcf.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Bruscia EM, et al. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator−/− mice. Am J Respir Cell Mol Biol. 2009;40:295–304. doi: 10.1165/rcmb.2008-0170OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonfield TL, Hodges CA, Cotton CU, Drumm ML. Absence of the cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infection. J Leukoc Biol. 2012 doi: 10.1189/jlb.0412188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelly C, et al. Toll-like receptor 4 is not targeted to the lysosome in cystic fibrosis airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2013;304:L371–382. doi: 10.1152/ajplung.00372.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang PX, et al. Reduced Caveolin-1 Promotes Hyperinflammation due to Abnormal Heme Oxygenase-1 Localization in Lipopolysaccharide-Challenged Macrophages with Dysfunctional Cystic Fibrosis Transmembrane Conductance Regulator. J Immunol. 2013;190:5196–5206. doi: 10.4049/jimmunol.1201607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang XM, Kim HP, Nakahira K, Ryter SW, Choi AM. The heme oxygenase-1/carbon monoxide pathway suppresses TLR4 signaling by regulating the interaction of TLR4 with caveolin-1. J Immunol. 2009;182:3809–3818. doi: 10.4049/jimmunol.0712437. [DOI] [PubMed] [Google Scholar]
- 14.Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839–845. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 15.Foster PS, et al. The emerging role of microRNAs in regulating immune and inflammatory responses in the lung. Immunol Rev. 2013;253:198–215. doi: 10.1111/imr.12058. [DOI] [PubMed] [Google Scholar]
- 16.McKiernan PJ, Cunningham O, Greene CM, Cryan SA. Targeting miRNA-based medicines to cystic fibrosis airway epithelial cells using nanotechnology. Int J Nanomedicine. 2013;8:3907–3915. doi: 10.2147/IJN.S47551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramachandran S, et al. Post-transcriptional regulation of cystic fibrosis transmembrane conductance regulator expression and function by microRNAs. Am J Respir Cell Mol Biol. 2013;49:544–551. doi: 10.1165/rcmb.2012-0430OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fabbri E, et al. Expression of microRNA-93 and Interleukin-8 during Pseudomonas aeruginosa-mediated induction of proinflammatory responses. Am J Respir Cell Mol Biol. 2014;50:1144–1155. doi: 10.1165/rcmb.2013-0160OC. [DOI] [PubMed] [Google Scholar]
- 19.Weldon S, et al. miR-31 dysregulation in cystic fibrosis airways contributes to increased pulmonary cathepsin S production. Am J Respir Crit Care Med. 2014;190:165–174. doi: 10.1164/rccm.201311-1986OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lino Cardenas CL, et al. miR-199a-5p Is upregulated during fibrogenic response to tissue injury and mediates TGFbeta-induced lung fibroblast activation by targeting caveolin-1. PLoS genetics. 2013;9:e1003291. doi: 10.1371/journal.pgen.1003291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexander MS, et al. MicroRNA-199a is induced in dystrophic muscle and affects WNT signaling, cell proliferation, and myogenic differentiation. Cell Death Differ. 2013;20:1194–1208. doi: 10.1038/cdd.2013.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizuno S, et al. MicroRNA-199a-5p is associated with hypoxia-inducible factor-1alpha expression in lungs from patients with COPD. Chest. 2012;142:663–672. doi: 10.1378/chest.11-2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rane S, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. 2009;104:879–886. doi: 10.1161/CIRCRESAHA.108.193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hassan T, et al. miR-199a-5p silencing regulates the unfolded protein response in chronic obstructive pulmonary disease and alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2014;189:263–273. doi: 10.1164/rccm.201306-1151OC. [DOI] [PubMed] [Google Scholar]
- 25.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rane S, et al. An antagonism between the AKT and beta-adrenergic signaling pathways mediated through their reciprocal effects on miR-199a-5p. Cellular signalling. 2010;22:1054–1062. doi: 10.1016/j.cellsig.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, et al. Kinase AKT controls innate immune cell development and function. Immunology. 2013 doi: 10.1111/imm.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laird MH, et al. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J Leukoc Biol. 2009;85:966–977. doi: 10.1189/jlb.1208763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32132. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- 30.Androulidaki A, et al. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity. 2009;31:220–231. doi: 10.1016/j.immuni.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luyendyk JP, et al. Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J Immunol. 2008;180:4218–4226. doi: 10.4049/jimmunol.180.6.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sonawane ND, Verkman AS. Thiazolidinone CFTR inhibitors with improved water solubility identified by structure-activity analysis. Bioorg Med Chem. 2008;16:8187–8195. doi: 10.1016/j.bmc.2008.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamdulay SS, et al. Celecoxib activates PI-3K/Akt and mitochondrial redox signaling to enhance heme oxygenase-1-mediated anti-inflammatory activity in vascular endothelium. Free Radic Biol Med. 2010;48:1013–1023. doi: 10.1016/j.freeradbiomed.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 34.Kim SR, et al. Selective COX-2 inhibitors modulate cellular senescence in human dermal fibroblasts in a catalytic activity-independent manner. Mech Ageing Dev. 2008;129:706–713. doi: 10.1016/j.mad.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Han S, Roman J. COX-2 inhibitors suppress lung cancer cell growth by inducing p21 via COX-2 independent signals. Lung Cancer. 2006;51:283–296. doi: 10.1016/j.lungcan.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 36.Roh GS, et al. Anti-inflammatory effects of celecoxib in rat lungs with smoke-induced emphysema. Am J Physiol Lung Cell Mol Physiol. 2010;299:L184–191. doi: 10.1152/ajplung.00303.2009. [DOI] [PubMed] [Google Scholar]
- 37.Liu G, et al. Kinase AKT1 negatively controls neutrophil recruitment and function in mice. J Immunol. 2013;191:2680–2690. doi: 10.4049/jimmunol.1300736. [DOI] [PubMed] [Google Scholar]
- 38.Egan M. How useful are cystic fibrosis mouse models? Drug Discovery Today: Disease Models. 2009;6:35–41. doi: 10.1016/j.ddmod.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Heeckeren A, et al. Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas aeruginosa. J Clin Invest. 1997;100:2810–2815. doi: 10.1172/JCI119828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dimagno MJ, et al. A proinflammatory, antiapoptotic phenotype underlies the susceptibility to acute pancreatitis in cystic fibrosis transmembrane regulator (−/−) mice. Gastroenterology. 2005;129:665–681. doi: 10.1016/j.gastro.2005.05.059. [DOI] [PubMed] [Google Scholar]
- 41.Becker KA, Tummler B, Gulbins E, Grassme H. Accumulation of ceramide in the trachea and intestine of cystic fibrosis mice causes inflammation and cell death. Biochem Biophys Res Commun. 2010;403:368–374. doi: 10.1016/j.bbrc.2010.11.038. [DOI] [PubMed] [Google Scholar]
- 42.Di A, et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol. 2006;8:933–944. doi: 10.1038/ncb1456. [DOI] [PubMed] [Google Scholar]
- 43.Sorio C, et al. Defective CFTR Expression and Function Are Detectable in Blood Monocytes: Development of a New Blood Test for Cystic Fibrosis. PLoS One. 2011;6:e22212. doi: 10.1371/journal.pone.0022212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van de Weert-van Leeuwen PB, et al. Optimal Complement-Mediated Phagocytosis of Pseudomonas aeruginosa by Monocytes is CFTR-Dependent. Am J Respir Cell Mol Biol. 2013 doi: 10.1165/rcmb.2012-0502OC. [DOI] [PubMed] [Google Scholar]
- 45.Del Porto P, et al. Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas aeruginosa. PLoS One. 2011;6:e19970. doi: 10.1371/journal.pone.0019970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdulrahman BA, et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy. 2011;7:1359–1370. doi: 10.4161/auto.7.11.17660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harris J, Werling D, Hope JC, Taylor G, Howard CJ. Caveolae and caveolin in immune cells: distribution and functions. Trends Immunol. 2002;23:158–164. doi: 10.1016/s1471-4906(01)02161-5. [DOI] [PubMed] [Google Scholar]
- 48.Gadjeva M, Paradis-Bleau C, Priebe GP, Fichorova R, Pier GB. Caveolin-1 modifies the immunity to Pseudomonas aeruginosa. J Immunol. 2010;184:296–302. doi: 10.4049/jimmunol.0900604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan K, et al. Elevated inflammatory response in caveolin-1-deficient mice with Pseudomonas aeruginosa infection is mediated by STAT3 protein and nuclear factor kappaB (NF-kappaB) J Biol Chem. 2011;286:21814–21825. doi: 10.1074/jbc.M111.237628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peffer ME, et al. Caveolin-1 Regulates Genomic Action of the Glucocorticoid Receptor in Neural Stem Cells. Molecular and cellular biology. 2014 doi: 10.1128/MCB.01121-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Le Saux CJ, et al. Down-regulation of caveolin-1, an inhibitor of transforming growth factor-beta signaling, in acute allergen-induced airway remodeling. J Biol Chem. 2008;283:5760–5768. doi: 10.1074/jbc.M701572200. [DOI] [PubMed] [Google Scholar]
- 52.Harris WT, et al. Myofibroblast differentiation and enhanced TGF-B signaling in cystic fibrosis lung disease. PLoS One. 2013;8:e70196. doi: 10.1371/journal.pone.0070196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yi H, et al. Differential roles of miR-199a-5p in radiation-induced autophagy in breast cancer cells. FEBS Lett. 2013;587:436–443. doi: 10.1016/j.febslet.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 54.Eto K, Goto S, Nakashima W, Ura Y, Abe SI. Loss of programmed cell death 4 induces apoptosis by promoting the translation of procaspase-3 mRNA. Cell Death Differ. 2012;19:573–581. doi: 10.1038/cdd.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luciani A, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol. 2010;12:863–875. doi: 10.1038/ncb2090. [DOI] [PubMed] [Google Scholar]
- 56.Legendre C, Mooij MJ, Adams C, O’Gara F. Impaired expression of hypoxia-inducible factor-1alpha in cystic fibrosis airway epithelial cells - a role for HIF-1 in the pathophysiology of CF? J Cyst Fibros. 2011;10:286–290. doi: 10.1016/j.jcf.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 57.Bessich JL, Nymon AB, Moulton LA, Dorman D, Ashare A. Low levels of insulin-like growth factor-1 contribute to alveolar macrophage dysfunction in cystic fibrosis. J Immunol. 2013;191:378–385. doi: 10.4049/jimmunol.1300221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Villella VR, et al. Disease-relevant proteostasis regulation of cystic fibrosis transmembrane conductance regulator. Cell Death Differ. 2013 doi: 10.1038/cdd.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen J, et al. CFTR negatively regulates cyclooxygenase-2-PGE(2) positive feedback loop in inflammation. J Cell Physiol. 2012;227:2759–2766. doi: 10.1002/jcp.23020. [DOI] [PubMed] [Google Scholar]
- 60.Maier TJ, et al. Cellular membranes function as a storage compartment for celecoxib. J Mol Med (Berl) 2009;87:981–993. doi: 10.1007/s00109-009-0506-8. [DOI] [PubMed] [Google Scholar]
- 61.Sade A, Banerjee S, Severcan F. Effects of the non-steroidal anti-inflammatory drug celecoxib on cholesterol containing distearoyl phosphatidylcholine membranes. Spectroscopy. 2011;25:177–185. [Google Scholar]
- 62.Zemel BS, Jawad AF, FitzSimmons S, Stallings VA. Longitudinal relationship among growth, nutritional status, and pulmonary function in children with cystic fibrosis: analysis of the Cystic Fibrosis Foundation National CF Patient Registry. J Pediatr. 2000;137:374–380. doi: 10.1067/mpd.2000.107891. [DOI] [PubMed] [Google Scholar]
- 63.Rosenberg LA, Schluchter MD, Parlow AF, Drumm ML. Mouse as a model of growth retardation in cystic fibrosis. Pediatr Res. 2006;59:191–195. doi: 10.1203/01.pdr.0000196720.25938.be. [DOI] [PubMed] [Google Scholar]
- 64.Guo S. Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J Endocrinol. 2014;220:T1–T23. doi: 10.1530/JOE-13-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McGettigan P, Henry D. Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA. 2006;296:1633–1644. doi: 10.1001/jama.296.13.jrv60011. [DOI] [PubMed] [Google Scholar]
- 66.Tourkina E, et al. Caveolin-1 regulates leucocyte behaviour in fibrotic lung disease. Ann Rheum Dis. 2010;69:1220–1226. doi: 10.1136/ard.2009.117580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bains SN, et al. Loss of caveolin-1 from bronchial epithelial cells and monocytes in human subjects with asthma. Allergy. 2012;67:1601–1604. doi: 10.1111/all.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rab A, et al. Cigarette smoke and CFTR: implications in the pathogenesis of COPD. Am J Physiol Lung Cell Mol Physiol. 2013;305:L530–541. doi: 10.1152/ajplung.00039.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheng J, et al. An Extensive Network of TET2-Targeting MicroRNAs Regulates Malignant Hematopoiesis. Cell reports. 2013;5:471–481. doi: 10.1016/j.celrep.2013.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.