

Summary
Bacterial RNA polymerase is the first point of gene expression and a validated target for antibiotics. Studied for several decades, the Escherichia coli transcriptional apparatus is by far the best characterized, with numerous RNA polymerase mutants and auxiliary factors isolated and analyzed in great detail. Since the E. coli enzyme was refractory to crystallization, structural studies have been focused on Thermus RNA polymerase s, revealing atomic details of the catalytic center and RNA polymerase interactions with nucleic acids, antibiotics, and regulatory proteins. However, numerous differences between these enzymes, including resistance of Thermus RNA polymerases to some antibiotics, underscored the importance of the E. coli enzyme structures. Three groups published the se long awaited structures in 2013, enabling functional and structural studies of the same model system. This progress was made possible, in large part, by the use of multicistronic vectors for expression of the E. coli enzyme in large quantities and in a highly active form. Here we describe the commonly used vectors and procedures for purification of the E. coli RNA polymerase.
Keywords: Transcription, RNA polymerase, Expression, Purification
1. Introduction
In bacteria, a single multisubunit RNA polymerase (RNAP) transcribes all genes. The holoenzyme is composed of a (most commonly) α2ββ′ω core, which carries out all types of catalysis but cannot specifically bind to or melt the DNA, and one of σ factors, which recognize distinct promoter elements and facilitate DNA strand separation near the transcription start site. Understanding the regulation of gene expression requires studies of the basic molecular mechanism of RNAP and the ways accessory factors interact with the enzyme during all stages of transcription. Bacterial RNAP is also a validated target for antibiotic therapies, but only two RNAP inhibitors are currently used in clinical practice.
Among many methods that have been applied to study transcription, structural analysis is indispensable in providing the atomic level of detail required for understanding of catalysis and design of antibiotics targeting the transcription apparatus. After the first structure of T. aquaticus RNAP was solved by the Darst lab in 1999 (Zhang et al., 1999), Thermus enzymes became the structural model of choice. Many key features have been characterized using these thermophilic RNAPs, including the active site geometry and conformational transitions during the nucleotide addition cycle (Vassylyev et al., 2007b), RNAP interactions with the nucleic acids in initiation (Zhang et al., 2012) and elongation complexes (Vassylyev et al., 2007a), and binding sites for σ (Vassylyev et al., 2002), elongation factors (Tagami et al., 2010), and antibiotics (Belogurov et al., 2009; Campbell et al., 2001; Vassylyev et al., 2005; Vassylyev et al., 2007b).
However, the gap between the structural analysis and the functional studies of bacterial transcription, carried out predominantly with mesophilic enzymes, was expanding. Thermus enzymes are an excellent model for the dissection of the catalytic mechanism, which is highly conserved among multisubunit RNAPs. In contrast, RNAP regions distant from the active site are quite divergent, and many species-specific insertions are located in the β and β′ subunits (Lane and Darst, 2010), complicating sequence alignments. Regulatory strategies and auxiliary factors, which in many cases bind to divergent sites, appear to be quite distinct, with Thermus species lacking many of the accessory proteins characterized in E. coli. Thermus RNAPs are resistant to several inhibitors of the E. coli enzyme, including fidaxomycin, a recently approved by the FDA antibiotic for treatment of Clostridium difficile infections. Finally, even when a ligand binds to both RNAPs, the binding sites could be far apart; for example, the regulator of stringent response ppGpp binds to different sites, and has different effects, on the E. coli and T. thermophilus enzymes (Artsimovitch et al., 2004; Ross et al., 2013; Zuo et al., 2013).
Ideally, functional and structural studies should be carried out on the same model system. By obtaining the structure of the E. coli RNAP, the Murakami (Murakami, 2013), Steitz (Zuo et al., 2013) and Darst (Bae et al., 2013)labs have made this possible. This breakthrough will facilitate mechanistic analysis of transcriptional machinery, aid in improvement of existing RNAP inhibitors, and may lead to development of novel antibiotics which are urgently needed to treat all infections, and those caused by Gram-negative pathogens in particular.
The success in obtaining these structures came from purification of a recombinant E. coli RNAP core assembled in vivo from co-expressed subunits. The first multicistronic vector (Fig. 1) contained rpoA, rpoB and rpoC genes (encoding the α, β and β′ subunits, respectively) expressed from the T7 gene 10 promoter (Artsimovitch et al., 2003). The wild-type spacer between the rpoB and rpoC genes, which are likely assembled co-translationally in vivo, was maintained and a purification tag was placed at the C-terminus of β′ to purify the complete core, which is assembled in the order of α2→α2β→α2ββ′. Using this vector, it was possible to purify active RNAP, yet no well-diffracting crystals could be obtained. Surprisingly, a nonessential 91-residue ω subunit proved to be the key missing factor. After it became clear that ω is essential for RNAP regulation by ppGpp (Vrentas et al., 2005), we added rpoZ to the rpoABC cassette to yield pVS10 (Belogurov et al., 2007), a version that was used by the Steitz lab (Zuo et al., 2013), a similar construct was used by Darst a co-workers (Bae et al., 2013; Twist et al., 2011) co-expressing ω from a compatible plasmid in the Murakami lab (Murakami, 2013) worked equally well.
Fig. 1. Polycistronic vectors for expression of E. coli core RNAP.

Representative examples of plasmids used most often are shown, other variants may be more suitable for a particular application.
A. pIA423 contains rpoA, Band C genes. An intein-chitin binding protein tag (IMPACT-Nitride;, NEB) allows for easy, one-step purification of the core RNAP on chitin matrix.
B. pVS10 (rpoA, B, C and Z) encodes a C-terminally His-tagged β′.
C. pIA900 (rpoA, B, C and Z) encodes a C-terminally His-tagged β′ which can be removed using TEV protease.
D. pIA1000 (rpoA, B, C and Z) encodes an N-terminally His-tagged β.
E. pIA1198 (rpoA, B, C and Z) encodes a β′ with C-terminal HA and His tags.
F. pIA1202 (rpoA, B, C and Z) encodes a β′ with a C-terminal minimal biotin tag (AVI tag) followed by a His tag ; the latter can be removed using TEV protease, leaving the AVI tag intact.
These structures will provide a framework for the analysis of the basic mechanism of catalysis and its regulation. In the course of this analysis, construction of many variant RNAPs with substitutions at chosen locations will be indispensable to test molecular models, design enzymes for specific applications, and perhaps to improve the resolution of the structures. Here we describe the commonly used vectors and procedures for purification of the E. coli RNAP that will facilitate these studies.
2. Vectors for RNAP expression and mutagenesis
Many vectors are available for mutagenesis, subcloning, and expression of the E. coli RNAP subunits. Here, we provide examples of the vectors we are used routinely in several laboratories. Some of these have been published, and all are available upon request.
2.1. Polycistronic vectors
These vectors (see Table 1) allow for efficient expression of the RNAP core enzyme, α2ββ′ω, either from a single vector or from two compatible vectors, one of which carries the rpoZ gene encoding the ω subunit.
Table 1.
Polycistronic E. coli RNAP expression vectors.
| Plasmid | Tag and AbR allele | Features | Origin & Resistance |
|---|---|---|---|
| Vectors without the rpoZ gene | |||
| pIA581 | His6:β | T7P—α—His6:β—β′; the His6 tag at the N-terminus of rpoB | pMB1 Ampicillin |
| pIA299 | β′:His6 | T7P—α—β—β′:His6; the His6 tag at the C-terminus of rpoC | pMB1 Ampicillin |
| pIA423 | β′:CBP | T7P—α—β—β′:CBP; a chitin-binding protein/intein tag (CBP; Impact-Nitride;, NEB) fused to the C-terminus of rpoC | pMB1 Ampicillin |
| pIA468 | β′:BCCP | T7P—α—β—β′:BCCP; residues 71–156 of the biotin carboxyl carrier protein (BCCP) fused to the C-terminus of rpoC | pMB1 Ampicillin |
| pIA439 | His6:β RifR β′:CBP |
T7P—α—His6:βS531F—β′:CBP; His6 tag at the N-terminus of rpoB; RifR S531F allele; CBP tag at the C-terminus of rpoC | pMB1 Ampicillin |
| pIA358 | α:His6 β′:CBP |
T7P—α:His6—β—β′:CBP; the His6 tag at the C-terminus of rpoA; CBP tag at the C-terminus of rpoC | pMB1 Ampicillin |
| pIA477 | His6:β RifR β′:BCCP |
T7P—α—His6:βH526Y—β′:BCCP: His6 tag at the N-terminus of rpoB; RifR H526Y allele; BCCP tag at the C-terminus of rpoC | pMB1 Ampicillin |
| pIA570 | His6:β RifR |
T7P—α—His6:βS531F—β′; the His6 tag at the N-terminus of rpoB; RifR S531F allele | pMB1 Ampicillin |
| pIA639 | β′:His6 StlR |
T7P—α—β—β′S793F:His6; the His6 tag at the C-terminus of rpoC; StlR S793F allele | pMB1 Ampicillin |
| Vectors with the rpoZ gene | |||
| pIA1158 | α:His6 | T7P—α:His6—β—β′—ω; the His6 tag at the C-terminus of rpoA | pMB1 Kanamycin |
| pIA1000 | His6:β | T7P—α—His6:β—β′—ω; the His6 tag at the rpoB N-terminus; unique silent AvrII and MfeI sites at β residues 477 and 555 | pMB1 Kanamycin |
| pIA1070 | His6:β RifR |
T7P—α—His6:βD516V—β′—ω; the His6 tag at the N-terminus of rpoB; RifR D516V allele | pMB1 Ampicillin |
| pVS10 | β′:His6 | T7P—α—β—β′:His6—ω; the His6 tag at the C-terminus of rpoC | pMB1 Ampicillin |
| pIA787 | β′:His6 β′:PKA |
T7P—α—β—β′:RRASV:His6—ω; the His6 tag and a protein kinase A (PKA) site at the C-terminus of rpoC | pMB1 Ampicillin |
| pIA821 | His6:β PKA:β RifR |
T7P—α—His6:RRASV:βS531F —β′—ω; His6 tag and PKA site at the N-terminus of rpoB; RifR S531F allele eliminates an internal kinase site | pMB1 Kanamycin |
| pIA900 | β′:TEV:His10 | T7P—α—β—β′:TEV:His10—ω; a removable via TEV protease cleavage His10 tag at the C-terminus of rpoC | pMB1 Ampicillin |
| pIA999 | β′:biotin | T7P—α—β—β′:GLNDIFEAQKIEWH—ω; a minimal biotin (AVI) substrate at the C-terminus of rpoC | pMB1 Ampicillin |
| pIA1198 | β′:HA:His7 | T7P—α—β—β′:YPYDVPDYA:His7—ω; the HA and His7 tags at the C-terminus of rpoC | pMB1 Ampicillin |
| pIA1202 | β′:biotin: TEV:His7 | T7P—α—β—β′:GLNDIFEAQKIEWH:TEV:His7—ω; an AVI tag followed by a removable via TEV protease cleavage His7 tag at the C-terminus of rpoC at the C-terminus of rpoC | pMB1 Ampicillin |
All vectors have the rpoA, rpoB and rpoC genes expressed under the control of the phage T7 gene 10 promoter. A subset of vectors also have the rpoZ gene; for the rest, the ω subunit can be co-expressed from a compatible pIA839 plasmid.
All rpo genes are expressed from T7 gene 10 promoter in a “standard” pET vector background, with T7 terminator downstream from rpoZ. The levels of expression follow the order ω>α2>β>β′, thus only the fully-assembled, ω-saturated core is purified when a purification tag is located at the C-terminus of the β′ subunit. Other advantages of the co-overexpression system are (i) a streamlined purification protocol; (ii) relatively high yields; (iii) ability to purify the core enzyme that contains toxic and assembly-defective mutations, such as the deletions of “dispensable regions” (Artsimovitch et al., 2003); and (iv) a “ crystallizable” protein (Murakami, 2013; Zuo et al., 2013).
The purified core enzyme is catalytically active on nucleic acid scaffolds but not in promoter-dependent initiation, which requires an initiation factor σ (typically σ70) added in trans; see chapter 11 for an example of (a variant) E. coli σ70 purification protocol.
2.2. Single-subunit vectors
However, these vectors are large (15+ kb) and have very few remaining restriction sites that can be used for subcloning of the altered fragments. Therefore, we carry out mutagenesis in smaller vectors, encoding single subunits or their fragments (see Table 2 and Fig. 2).
Table 2.
Vectors for site-directed mutagenesis or expression of single E. coli RNAP subunits
| Plasmid | Subunit & AbR | Features | Origin & Resistance |
|---|---|---|---|
| pIA281 | αWT — |
T7P—rpoA; α subunit under control of the bacteriophage T7 gene 10 promoter; HindIII site in rpoA is removed | pMB1 Ampicillin |
| pIA545 | βWT — |
Ptrc—His6-rpoB WT; N-terminally tagged β under control of IPTG-inducible Ptrc promoter; a silent MfeI @ 555 | pMB1 Ampicillin |
| pIA178 | βS531F RifR |
Ptrc—His6-rpoB S531F; a rifampicin-resistant (RifR) S531F allele in the Ptrc—rpoB construct | pMB1 Ampicillin |
| pIA223 | βD516V RifR |
Ptrc—His6-rpoB D516V; a RifR D516V (marked with a silent BsgI site) allele in the Ptrc—rpoB construct | pMB1 Ampicillin |
| pIA661 | β′ WT — |
Ptrc—rpoC WT-His6; C-terminally tagged β′ under control of an IPTG-inducible Ptrc promoter; a silent EagI @ 675 | pMB1 Ampicillin |
| pIA635 | β′ S793F StlR |
Ptrc—rpoC S793F-His6; a streptolydigin-resistant S793F (marked with a silent EcoRV site) allele in the Ptrc—rpoC construct | pMB1 Ampicillin |
| pIA388 | β′I774S CBRR |
Ptrc—rpoC I774S-His6; a CBR-resistant I774S (marked with a silent EarI site) allele in the Ptrc—rpoC construct | pMB1 Ampicillin |
| pIA839 | ωWT — |
PBAD—rpoZ; ω subunit under control of arabinose-inducible PBAD promoter | P15A Chloramphenicol |
| pIA586 | σWT — |
T7P—His6-rpoD; N-terminally tagged σ70 subunit under control of the T7 gene 10 promoter | pMB1 Kanamycin |
| pIA1127 | σWT — |
T7P—His6-rpoD; N-terminally tagged σ70 under control of the T7 gene 10 promoter; removable His6 tag | pMB1 Kanamycin |
| pIA458 | βC & β′N — |
A fragment encompassing the C-terminal part of rpoB, the linker with an SpeI site, and the N-terminal part of rpoC | pUC Ampicillin |
Fig. 2. Plasmids for rpoB and rpoC mutagenesis.

mutant versions of shorter rpoA and rpoZ can be obtained synthetically from commercial sources. We introduce a desired substitution by site-directed mutagenesis (for example, QuikChange), adding a silent restriction site (if possible) for easy screening. We sequence the mutant fragment between two closest convenient restriction sites and then clone this fragment back into the original plasmid (to make sure there are no mutations elsewhere). Finally, we reclone a fragment flanked by two sites that are unique in pVS10 (or another similar vector).
A. Mutagenesis of the β subunit. We use three vectors that encode the N-terminally His6-tagged β subunit under control of Ptrc promoter most frequently. The wild-type plasmid pIA545 (shown)encodes the wild-type rpoB except a silent MfeI site at β residue 555 ; it complements a temperature sensitive defect in the rpoB gene. Two other plasmids have the same structure but lack the MfeI site; instead, they encode rifampicin-resistant rpoB alleles: βD516V in pIA223 and βS531F in pIA178. The activity of altered RNAPs can be monitored in the presence of rifampicin, to eliminate concerns of contamination with the wild-type core. Following mutagenesis, we reclone the NcoI-SbfI fragment, which encompasses almost entire rpoB)into a desired vector (e.g., pIA821).
B. Mutagenesis of the C-terminus of β and the N-terminal half of β′. pIA458 is a Litmus-based plasmid that does not encode any complete RNAP subunit. Mutagenized fragments can be transferred into a vector of choice (e.g., pIA900 for β′ mutants) on the SbfI-BsmI fragment. pIA458 has as an SpeI site in the intergenic region separating the rpoB and rpoC genes that can be used for screening .
C. Mutagenesis of the C-terminal half of the β subunit. We use pIA661, which encodes the wild-type rpoC except for a silent EagI site at β′ residue 675, most frequently. Two variants carrying antibiotic-resistant alleles, pIA388 and pIA635 (see Table 2) can be used instead. These plasmids encode the C-terminally His6 tagged β′ subunit under control of Ptrc promoter. The wild-type plasmid complements a temperature-sensitive defect in the rpoC gene. Following mutagenesis, we reclone the BsmI-HindIII fragment into a desired vector (e.g., pVS10).
In these vectors, an rpo gene is expressed under control of T7 gene 10 promoter (rpoA and rpoD), IPTG-inducible Ptrc promoter (rpoB and rpoC), or arabinose-inducible PBAD promoter (rpoZ).
The single-subunit vectors can be used to express altered subunits alone, most commonly to test their ability to complement defects conferred by temperature-sensitive chromosomal rpo alleles (Weilbaecher et al., 1994)or to confer resistance to an antibiotic (Yuzenkova et al., 2002).
3. Materials
All solutions are prepared using ultrapure water (resistivity of 18 MΩ, e.g. MilliQ) and analytical/molecular biology grade reagents and filtered through bottle-top 0.2 μm PES filter to remove particulate contaminations (unless indicated otherwise). Bacterial growth media are made using purified (by deionization or reverse osmosis to resistivity of >5 MΩ, e.g. Elix) water and biotechnology grade media components and sterilized by autoclaving. All sterilized media can be stored at room temperature (unless indicated otherwise), all protein purification and chromatographic solutions are stored at 4 °C (unless indicated otherwise). We list the manufacturers for supplies and reagents that we currently use; alternative sources can be tried but we cannot guarantee that they will work identically.
3.1. Bacterial growth media and supplements
LB growth medium for liquid culture: weigh 125 g of Difco LB Miller (Luria-Bertani) dry pre-mix, dissolve in 4.5 L of water, add water to 5 L total volume, and mix. Dispense 600 mL of prepared LB per 2 L Erlenmeyer flask (total of 8), cover with heavy duty aluminum foil, secure the foil to the flask with the autoclave tape. Dispense the remaining LB (~200 mL) into an autoclavable media bottle. Autoclave media and store at room temperature.
100 mg/mL stock carbenicillin solution: weigh 0.5 g carbenicillin, dissolve in 4 mL of water, add water to 5 mL total, filter through 0.2 μm PES syringe filter, aliquot 1 mL per 1.5 ml microcentrifuge tube, and store at −20°C (see Note 1).
LB+carbenicillin agar plates: weigh 40 of Difco LB Agar, place into 1 L bottle, add magnetic stir bar and water to 1 L total, cover with foil, secure the foil to the flask with the autoclave tape. Bring the mix to boiling on the hot plate with heavy duty aluminum stirring, and sterilize by autoclaving. Cool autoclaved medium to 50–60°C while stirring, add 1 mL of 100 mg/mL carbenicillin stock solution, continue stirring for another 5 min, and pour the plates. Store plates at 4°C.
1 M stock solution of IPTG: weigh 1.19 g of IPTG, dissolve in 4 mL of water, add water to 5 mL total, filter through 0.2 μm PES syringe filter, make immediately before use or aliquot 1 mL per 1.5 ml microcentrifuge tube, and store at −20°C.
3.2. Protein purification and chromatographic solutions
1.1 M stock solution of Tris-HCl (pH 6.9): weigh 121.14 g of Tris base (e.g. Trizma base), dissolve in 900 ml of water, adjust pH to 6.9 with concentrated HCl (~80 ml), add water to 1 L total.
Cell lysis buffer (50 mM Tris-HCl (pH 6.9), 500 mM NaCl, 5% glycerol): weigh 29.22 g of NaCl, dissolve in 500 mL of water, add 50 mL of 1 M Tris-HCl (pH 6.9), add 50 mL of glycerol, add water to 1 L total.
4 M stock solution of imidazole: weigh 68.08 g of imidazole, dissolve in 200 mL of water, adjust pH to 7.4 with HCl, add water to 250 mL total.
0.5 M stock solution of EDTA (pH 8.0): weigh 186.12 g of Na2EDTA·2H2O, dissolve in 800 mL of water, adjust pH to 8.0 by adding NaOH (~20 g of pellets), add water to 1 L total.
1 M stock solution of DTT: weigh 1.54 g of DL-DTT, dissolve in 8 mL of water, add water to 10 mL total, filter through 0.2 um PES syringe filter, aliquot 1 mL per 1.5 ml microcentrifuge tube, and store at −20°C.
50 mg/mL stock solution of lysozyme: weigh 0.5 g of lysozyme (e.g. human recombinant lysozyme, Sigma-Aldrich), dissolve in 4 ml of water, add water to 5 mL total. Make immediately before use, do not store.
Buffer A (50 mM Tris-HCl (pH 6.9), 5% glycerol, 0.5 mM EDTA, 1 mM DTT): combine 500 mL water, 50 mL of 1 M Tris-HCl (pH 6.9), 50 mL of glycerol, 1 mL of 0.5 M EDTA (pH 8.0), 1 mL of 1 M DTT, 1 tablet of complete EDTA-free protease inhibitors cocktail (Roche Applied Science). Add water to 1 L total.
Buffer B (50 mM Tris-HCl (pH 6.9), 1.5 M NaCl, 5% glycerol, 0.5 mM EDTA, 1 mM DTT): weigh 87.66 g of NaCl, dissolve in 500 mL water, add 50 mL of 1 M Tris-HCl (pH 6.9), 50 mL of glycerol, 1 mL of 0.5 M EDTA (pH 8.0), 1 mL of 1 M DTT, 1 tablet of complete EDTA-free protease inhibitors cocktail (Roche Applied Science). Add water to 1 L total.
1 M stock solution of Tris-HCl (pH 7.5): weigh 121.14 g of Tris base (e.g. Trizma base), dissolve in 900 ml of water, adjust pH to 7.5 with concentrated HCl (~65 ml), add water to 1 L total.
Ni binding buffer (cell lysis buffer + 20 mM imidazole): combine 0.25 mL of 4 M imidazole solution and 49.75 mL of cell lysis buffer in a 50 mL conical tube (e.g. BD Falcon), mix by vortexing and keep on ice. Make immediately prior to use, do not store.
Ni elution buffer (cell lysis buffer + 250 mM imidazole): combine 0.625 mL of 4 M imidazole solution and 9.375 mL of cell lysis buffer, mix by vortexing and keep on ice. Make immediately prior to use, do not store.
Dialysis buffer AB5 (50 mM Tris-HCl (pH 6.9), 75 mM NaCl, 5% glycerol, 0.5 mM EDTA, 1 mM DTT): weigh 4.38 g of NaCl, dissolve in 500 mL water, add 50 mL of 1 M Tris-HCl (pH 6.9), 50 mL of glycerol, 1 mL of 0.5 M EDTA (pH 8.0), 1 mL of 1 M DTT. Add water to 1 L total.
RNAP storage buffer (10 mM Tris-HCl (pH 7.5), 50% glycerol, 100 mM NaCl, 0.1 mM EDTA, 0.1 mM DTT): weigh 5.85 g of NaCl, dissolve in 300 mL of water, add 10 mL 1 M Tris-HCl (pH 7.5), 0.2 mL 500 mM EDTA (pH 8.0), 0.1 mL 1 M DTT, 500 mL glycerol. Add water to 1 L total.
Commercial ready-made materials for running and staining gels. We use NuPAGE MES SDS Running Buffer, NuPAGE LDS Loading Buffer, NuPAGE 4–12% Bis-Tris Gel (all – Life Technologies), Precision Plus Protein Dual Color Standards (Bio-Rad), and GelCode Blue Stain Reagent (Thermo).
4. Methods
All protein purification procedures are performed with refrigeration (on ice, in cold room or in a chromatographic refrigerator, as indicated). All time estimates are given only as a general guidance.
4.1. Over-expression of E. coli RNAP core
Transform T7-based over-expression strain of E. coli (e.g., BL21 (λDE3))with pVS10 plasmid by electroporation or using chemically competent cells, plate on LB+carbenicillin agar, incubate overnight at 37°C. Remove plate to 4°C to suppress growth of satellite colonies.
Inoculate single colony of pVS10-transformed over-expression strain into 100 mL of LB supplemented with 0.1 mL 100 mg/mL carbenicillin solution (0.1 mg/L final concentration) in 250 mL Erlenmeyer flask. Incubate with agitation (250 rpm) overnight at 37°C.
Inoculate each of the 2 L Erlenmeyer flasks (600 ml LB) with overnight culture (1:100, v:v), supplement with 0.6 mL carbenicilin stock solution, and incubate with agitation (250 rpm) at 37°C. Monitor culture growth periodically by measuring OD600.
Induce RNAP core expression by adding 0.6 mL of 1 M IPTG (to final concentration of 1 mM) once OD600 reaches 0.75 (~4 hours after inoculation). Continue incubation for another 3 hours (see Note 2).
Harvest cells by centrifugation (6000 g, 10 min at 4 °C, e.g. using 1 L bottles in F10S-4X1000 LEX rotor and Sorvall RC6+ centrifuge). Decant and discard the supernatant. Proceed to RNAP core purification or store cell pellets at −80°C (thaw pellets on ice before proceeding with protein purification).
4.2. Cell lysis and clearing of the cell lysate
Place centrifuge bottles with cell pellets on ice.
Dispense 100 mL of cell lysis buffer into 2 50 mL conical tubes, add 1 tablet of complete EDTA-free protease inhibitors cocktail (Roche Applied Science) into each tube, dissolve by vortexing. Incubate tubes on ice for 15 min.
Resuspend cell pellets in cell lysis buffer on ice, using wide tip 10 mL pipette. Estimate final volume (e.g., using graduations on the tubes) and supplement cell suspension with freshly made 50 mg/mL solution of lysozyme to final concentration of 1 mg/mL. Incubate suspension on ice for 30 min (see Note 3).
Disrupt cells by ultrasonication on ice; e.g., using Tapped Bio Horn and Branson Sonifier Digital 450 at 60% power output, 8×30 sec continuous applications with 2 min cooling intermissions (see Note 4).
Remove cell debris by centrifugation (29000 g, 30 min at 40°C, e.g. using 50 mL Oak Ridge Tubes (Nalgene) in F21S-8X50y BioSEAL rotor and Sorvall RC6+ centrifuge). Transfer supernatants into clean tubes (e.g., Oak Ridge) by aspiration, discard the pellets, and repeat centrifugation (29000 g, 45 min at 4°C; see Note 5). Discard the pelleted debris, transfer supernatants into 50 mL conical tubes, keep on ice.
4.3. Ni-affinity chromatography of His6-tagged RNAP core
All steps take place in the cold room or in a chromatographic refrigerator.
Estimate volume of cleared cell lysate using volumetric graduation of conical tubes. Add 4 M imidazole stock solution to each tube to final concentration of 20 mM.
Set a His GraviTrap column (GE Healthcare Life Science) into a vertical rack (e.g., Bio-Rad Poly Column Rack) with flow-through collection reservoir for each 50 ml of lysate (see Note 6). Attach Labmate (GE Healthcare Life Science) buffer extension reservoir to each column. The rest of the instructions apply to each column.
Wash column by gravity flow with 10 mL of ultrapure water.
Wash column with 10 mL of Ni binding buffer.
Apply 50 mL of cleared extract (supplemented with 20 mM imidazole) to the column, drain completely.
Wash column with 40 mL of Ni binding buffer.
Replace collection reservoir with microcentrifuge tube rack. Label 8 1.5 mL microcentrifuge tubes (e.g., Eppendorf Protein LoBind) 1 through 8 for fraction collection, and place them under the column.
Apply 0.5 mL of Ni Elution buffer to the column. Collect flow-through into tube 1.
Sequentially apply 7 1 mL aliquots of Ni Elution buffer to the column. Collect flow-through into tubes 2–8. Place tubes on ice.
Confirm the presence of RNAP core in fractions 2–8 by SDS-PAGE; use 10–20 μL of each fraction.
Combine all RNAP core-containing fractions and dialyze them (e.g., using 3.500 MWCO Slide-A-Lyzer Dialysis Cassette, Thermo) overnight against 100 volumes of dialysis buffer AB5.
4.4. Heparin-affinity chromatography of RNAP core
All the following steps can be implemented in instrument-specific fashion on any medium-pressure liquid chromatography system, the method as written is realized on AKTA FPLC (GE Healthcare Life Science); see Note 7.
Place pump inlet tubing A and B into buffers A and B, respectively. Run the pump (4 mL/min) without at 100% B until conductivity stabilizes at ~95 mS/cm (~10–15 min). Switch the pump to 0% B and continue running until conductivity stabilizes at ~4 mS/cm (~10–15 min). Lower the rate to 1 mL/min and connect HiPrep Heparin FF 16/10 column (GE Healthcare Life Science); see Note 8.
To equilibrate the column set the pump at 2 mL/min and 5% B, run until conductivity stabilizes at ~9.5 mS/cm.
Load dialyzed sample onto the column at 1 mL/min, wash the column by 20 mL of 5% B at the same rate.
Apply gradient 5% B to 100% B over 200 mL at 1 mL/min, monitor protein elution from the column by UV absorbance at 280 or 215 nm. Begin collecting 1.5 mL fractions when conductivity reaches 20 mS/cm. RNAP core usually elutes from heparin column at conductivity between 30 and 45 mS/cm. Stop collecting fractions once conductivity reaches 60 mS/cm. At this time switch the pump to 100% B and 2 mL/min, continue running for 20 min, switch the pump to 5% B, run for another 20 min and end the run.
Based on UV absorbance and conductivity profiles identify RNAP core peak between 30 and 45 mS/cm. Select fractions with UV absorbance within 20% of the maximum value, confirm presence of RNAP core there by SDS-PAGE (use 30–45 μL of each fraction).
Combine selected fractions and dialyze them overnight against 100 volumes of dialysis buffer AB5.
4.5. Ion-exchange chromatography of RNAP core
Start the pump at 1 mL/min and 5% B, replace heparin column with MonoQ 10/100 GL (GE Healthcare Life Science); see Note 9. Continue running till conductivity stabilizes at ~9.5 mS/cm (~15 min).
Load dialyzed sample onto the column at 1 mL/min, wash the column by 20 mL of 5% B at the same rate.
Apply gradient 5% B to 100% B over 200 mL at 1 mL/min, monitor protein elution from the column by UV absorbance at 280 or 215 nm. Begin collecting 0.8 mL fractions when conductivity reaches 15 mS/cm. RNAP core usually elutes from MonoQ column at conductivity between 20 and 30 mS/cm. Stop collecting fractions once conductivity reaches 50 mS/cm. At this time switch the pump to 100% B, continue running for 20 min, switch the pump to 5% B, run for another 20 min and end the run.
Based on UV absorbance and conductivity profiles identify RNAP core peak between 20 and 30 mS/cm. Select fractions with UV absorbance within 20% of the maximum value, confirm presence of RNAP core there in and their purity by SDS-PAGE (use 20 μL of each fraction); see Figure 3 for an example.
Combine the fractions with highest concentration and purity of RNAP core, change the buffer as per downstream application (see Note 10). For storage, dialyze overnight against 500 volumes of RNAP storage buffer, aliquot as needed into screw-top gasketed microcentrifuge tubes, flash-freeze aliquots in liquid nitrogen, and store at −80°C.
Fig. 3. Sample purification of the E. coli RNAP.
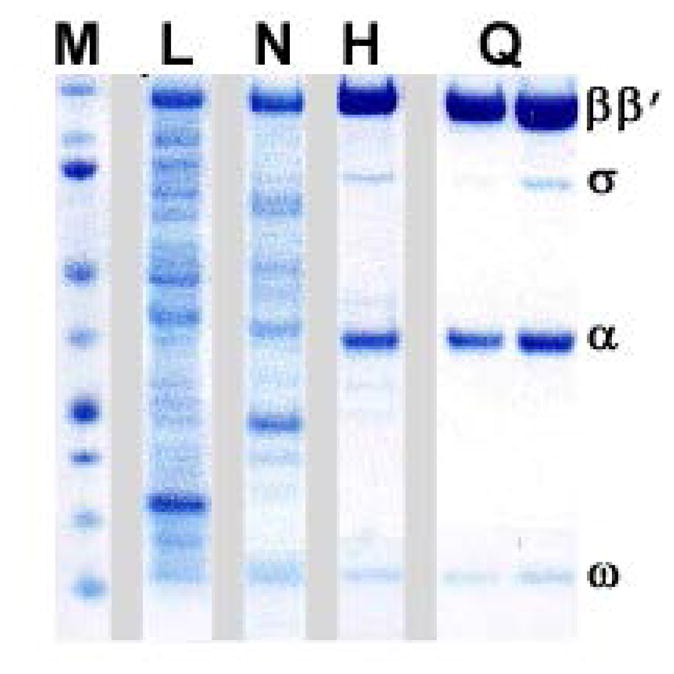
A variant with a deletion in the β′ jaw (β′ residues 1149–1190) was used. Samples of the cell lysate (L), Ni-NTA eluate (N; see section 4.3), heparin peak fractions (H; see section 4.4), and MonoQ peak fractions (H; see section 4.5) were analyzed by SDS-PAGE using a 4–12% NuPAGE® Bis-Tris pre-cast polyacrylamide gel (Life Technologies). Chromatography through the MonoQ column separates the core enzyme (left) from the holoenzyme (right). The lane marked M was loaded with the molecular weight markers; the positions of the RNAP subunits are indicated on the right.
Acknowledgments
This work was supported by the National Institutes of Health GM67153 grant.
Footnotes
Carbenicillin can be substituted by ampicillin, but the former performs better due to its higher stability.
Wild-type E. coli RNAP core expresses well when induced from pVS10 plasmid by IPTG at 37 °C, whereas some “mutant” forms (such as large indels) express better at lower temperatures (e.g., 30 °C). Use longer induction times if the cultures are grown at temperatures below 37 °C (e.g., induce for 5 hours at 30 °C as opposed to 3 hours at 37 °C). As an alternative to improve yield and solubility of expressed proteins, use auto-induction protocol, which utilizes expression induction in the stationary phase (Studier, 2005). We have used it extensively with pVS10 and derivative plasmids and a variety of expression strains with great success.
Lysozyme activity varies greatly with source and preparation. We find human recombinant lysozyme presently to be the best value for large scale preparations, based on price and specific activity.
As alternative for cell disruption use French press. We would not recommend any of the commercially available cell lysis formulations as they didn’t perform on par with sonication or the French press. Cell disruptors such as EmulsiFlex (Avestin) used in lieu of sonication tend to produce less active preparations of RNAP core.
Clearing extract by high-speed centrifugation is essential for efficient gravity flow chromatography, as cell debris when not removed clogs the columns filters and bed, interfering with the flow rate and uniformity, even if when column is advertised as capable of handling “uncleared” cell extracts. If you do not have access to centrifuge capable of delivering 29000 g acceleration, perform batch-binding of His6-tagged RNAP core to Ni Sepharose Fast Flow and pack it into PD-10 gravity column (both GE Healthcare Life Science) per manufacturer’s instructions.
His Gravitrap column is packed with Ni Sepharose Fast Flow beads (GE Healthcare Life Science), a medium density (15 μM of Ni++ per ml of beads) affinity resin for purification of His6-tagged proteins. As alternative for purification of His6-tagged RNAP core a number of Ni++ or Co++ resins can be used without significant changes in protocol or drastic drop in efficiency; note that Co++ affinity media usually have lower protein-binding capacity than Ni++ ones. Of interest is high density Ni Agarose from Gold Biotechnology, which features 20–40 μM of Ni++ per ml of beads.
Heparin-affinity chromatography removes most of the nucleic acid contaminations present in RNAP preparation after the Ni-affinity step. Performing it prior to ion-exchange chromatography is thus very important since nucleic acids have higher affinity to the MonoQ column than does RNAP and if not removed they will interfere with core binding.
Pre-packed HiTrap Heparin FF 16/10 is the preferred column for preparative scale heparin-affinity chromatography, both in terms of capacity and reproducibility. It can be substituted with similar size column packed in-house with Heparin Sepharose 6 Fast Flow (GE Healthcare Life Science); another pre-packed heparin-affinity column from GE, 5 mL HiTrap Heparin HP has much lower capacity and reduced resolution. Connecting several HiTrap columns head-to-tail may approach 16/10 in protein-binding capacity but compromises resolution even further.
MonoQ columns provide the best resolution for RNAP core purification. Q Sepharose is not a recommended substitute due to its significantly reduced resolution. With some success, SOURCE 15Q 4.6/100 PE (GE Healthcare Life Science) can replace MonoQ 10/100 GL, albeit with lower resolution which may require additional polishing steps.
E. coli RNAP core purified according to the preceding streamlined protocol is as a rule 95+% pure and 80+% active in in vitro transcription applications. For additional polishing steps, repeat ion-exchange chromatographic step or perform size-exclusion chromatography (e.g., using Superdex 200 10/300 GL (GE Healthcare Life Science) in 50% buffer B).
References
- Artsimovitch I, Patlan V, Sekine S, Vassylyeva MN, Hosaka T, Ochi K, Yokoyama S, Vassylyev DG. Structural basis for transcription regulation by alarmone ppGpp. Cell. 2004;117:299–310. doi: 10.1016/s0092-8674(04)00401-5. [DOI] [PubMed] [Google Scholar]
- Artsimovitch I, Svetlov V, Murakami KS, Landick R. Co-overexpression of Escherichia coli RNA polymerase subunits allows isolation and analysis of mutant enzymes lacking lineage-specific sequence insertions. J Biol Chem. 2003;278:12344–12355. doi: 10.1074/jbc.M211214200. [DOI] [PubMed] [Google Scholar]
- Bae B, Davis E, Brown D, Campbell EA, Wigneshweraraj S, Darst SA. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of sigma70 domain 1.1. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:19772–19777. doi: 10.1073/pnas.1314576110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belogurov GA, Vassylyeva MN, Sevostyanova A, Appleman JR, Xiang AX, Lira R, Webber SE, Klyuyev S, Nudler E, Artsimovitch I, et al. Transcription inactivation through local refolding of the RNA polymerase structure. Nature. 2009;457:332–335. doi: 10.1038/nature07510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belogurov GA, Vassylyeva MN, Svetlov V, Klyuyev S, Grishin NV, Vassylyev DG, Artsimovitch I. Structural basis for converting a general transcription factor into an operon-specific virulence regulator. Mol Cell. 2007;26:117–129. doi: 10.1016/j.molcel.2007.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell. 2001;104:901–912. doi: 10.1016/s0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- Lane WJ, Darst SA. Molecular evolution of multisubunit RNA polymerases: structural analysis. J Mol Biol. 2010;395:686–704. doi: 10.1016/j.jmb.2009.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami KS. X-ray crystal structure of Escherichia coli RNA polymerase sigma70 holoenzyme. J Biol Chem. 2013;288:9126–9134. doi: 10.1074/jbc.M112.430900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W, Vrentas CE, Sanchez-Vazquez P, Gaal T, Gourse RL. The magic spot: a ppGpp binding site on E. coli RNA polymerase responsible for regulation of transcription initiation. Mol Cell. 2013;50:420–429. doi: 10.1016/j.molcel.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Tagami S, Sekine S, Kumarevel T, Hino N, Murayama Y, Kamegamori S, Yamamoto M, Sakamoto K, Yokoyama S. Crystal structure of bacterial RNA polymerase bound with a transcription inhibitor protein. Nature. 2010;468:978–982. doi: 10.1038/nature09573. [DOI] [PubMed] [Google Scholar]
- Twist KA, Husnain SI, Franke JD, Jain D, Campbell EA, Nickels BE, Thomas MS, Darst SA, Westblade LF. A novel method for the production of in vivo-assembled, recombinant Escherichia coli RNA polymerase lacking the alpha C-terminal domain. Protein science : a publication of the Protein Society. 2011;20:986–995. doi: 10.1002/pro.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassylyev DG, Sekine S, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 A resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- Vassylyev DG, Svetlov V, Vassylyeva MN, Perederina A, Igarashi N, Matsugaki N, Wakatsuki S, Artsimovitch I. Structural basis for transcription inhibition by tagetitoxin. Nat Struct Mol Biol. 2005;12:1086–1093. doi: 10.1038/nsmb1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassylyev DG, Vassylyeva MN, Perederina A, Tahirov TH, Artsimovitch I. Structural basis for transcription elongation by bacterial RNA polymerase. Nature. 2007a;448:157–162. doi: 10.1038/nature05932. [DOI] [PubMed] [Google Scholar]
- Vassylyev DG, Vassylyeva MN, Zhang J, Palangat M, Artsimovitch I, Landick R. Structural basis for substrate loading in bacterial RNA polymerase. Nature. 2007b;448:163–168. doi: 10.1038/nature05931. [DOI] [PubMed] [Google Scholar]
- Vrentas CE, Gaal T, Ross W, Ebright RH, Gourse RL. Response of RNA polymerase to ppGpp: requirement for the omega subunit and relief of this requirement by DksA. Genes Dev. 2005;19:2378–2387. doi: 10.1101/gad.1340305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilbaecher R, Hebron C, Feng G, Landick R. Termination-altering amino acid substitutions in the beta′ subunit of Escherichia coli RNA polymerase identify regions involved in RNA chain elongation. Genes Dev. 1994;8:2913–2927. doi: 10.1101/gad.8.23.2913. [DOI] [PubMed] [Google Scholar]
- Yuzenkova J, Delgado M, Nechaev S, Savalia D, Epshtein V, Artsimovitch I, Mooney RA, Landick R, Farias RN, Salomon R, et al. Mutations of bacterial RNA polymerase leading to resistance to microcin j25. J Biol Chem. 2002;277:50867–50875. doi: 10.1074/jbc.M209425200. [DOI] [PubMed] [Google Scholar]
- Zhang G, Campbell EA, Minakhin L, Richter C, Severinov K, Darst SA. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 A resolution. Cell. 1999;98:811–824. doi: 10.1016/s0092-8674(00)81515-9. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Feng Y, Chatterjee S, Tuske S, Ho MX, Arnold E, Ebright RH. Structural basis of transcription initiation. Science. 2012;338:1076–1080. doi: 10.1126/science.1227786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Wang Y, Steitz TA. The mechanism of E. coli RNA polymerase regulation by ppGpp is suggested by the structure of their complex. Mol Cell. 2013;50:430–436. doi: 10.1016/j.molcel.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]