

Abstract
We previously characterized a H+ transport pathway in medullary thick ascending limb nephron segments that when activated stimulated the production of superoxide by NAD(P)H oxidase. Importantly, the activity of this pathway was greater in Dahl salt-sensitive rats than salt-resistant (SS.13BN) rats, and superoxide production was enhanced in low Na+ media. The goal of this study was to determine the molecular identity of this pathway and its relationship to Na+. We hypothesized that the voltage-gated proton channel, HV1, was the source of superoxide-stimulating H+ currents. In order to test this hypothesis, we developed HV1−/− null mutant rats on the Dahl salt-sensitive rat genetic background using zinc-finger nuclease gene targeting. HV1 could be detected in medullary thick limb from wild-type rats. Intracellular acidification using an NH4Cl prepulse in 0 sodium/BaCl2 containing media resulted in superoxide production in thick limb from wild-type but not HV1−/− rats (P<0.05), and more rapid recovery of intracellular pH in wild-type rats (ΔpHi 0.005U/sec vs. 0.002U/sec, p=0.046 respectively). Superoxide production was enhanced by low intracellular sodium (<10mM) in both thick limb and peritoneal macrophages only when HV1 was present. When fed a high salt diet, blood pressure, outer-medullary renal injury (tubular casts) and oxidative stress (4-Hydroxynonenal staining) were significantly reduced in HV1−/− rats compared to wild-type Dahl salt-sensitive rats. We conclude that HV1 is expressed in medullary thick ascending limb and promotes superoxide production in this segment when intracellular Na+ is low. HV1 contributes to the development of hypertension and renal disease in Dahl salt-sensitive rats.
Keywords: FREE RADICALS, HYPERTENSION, HVCN1, OXIDATIVE STRESS, PROTON CHANNEL
Introduction
The Dahl salt-sensitive (SS) rat is a naturally occurring model of salt-sensitive hypertension that closely mimics human forms of disease observed in the African American population 1–6. A number of previous studies indicate that oxidative stress, specifically within the renal outer medullary region, promotes the development of hypertension and renal injury in the SS rat model 6–8. Within this region, renal medullary thick ascending limb tubular segments (mTAL) are thought to be the greatest source of reactive oxygen species and much of the superoxide produced by this segment is derived from NAD(P)H oxidase 9. As we have shown superoxide production is enhanced in mTAL of SS rats compared to salt-resistant animals 10–12, it is likely that superoxide production by this segment contributes to the development of disease in these animals. The cellular mechanisms responsible for augmented superoxide production in mTAL of salt-sensitive rats when fed a high salt diet however, remain unclear.
Superoxide production in mTAL in response to cellular H+ efflux was initially reported by Li et al 13. We have since characterized a novel H+ transport pathway in mTAL, that when activated stimulated the production of superoxide by NAD(P)H oxidase 10. Importantly this pathway was enhanced in SS rats compared to salt-resistant control animals (SS.13BN)10. A number of indicators suggested that HV1, a voltage-gated H+ channel expressed highly in immune cells 14, could be the unidentified H+ transport pathway we characterized in mTAL. First, as charged protons cannot readily cross the plasma membrane 15 we suspected an ion channel. Second, superoxide production was only observed in the presence of a large outward pH gradient, not following an inward pH gradient 10. Third, proton efflux was linked to cellular depolarization and activation of the NAD(P)H oxidase 10. HV1 fulfilled all these criteria. HV1 is specific to protons 15. The opening of HV1 is pH dependent with the channel only mediating H+ efflux, not influx, under physiological conditions 15, 16. HV1 opening is voltage-dependent, opening in response to cellular depolarization, and HV1 has been associated with NADPH oxidase 15, 17, 18.
Given the number of characteristics shared between HV1 and the unidentified H+ transport pathway we characterized in mTAL, we hypothesized that ‘HV1 is present in mTAL and contributes to superoxide producing H+ currents following cellular acidification. Further, as high salt feeding is likely to promote both depolarization as well as acidification of mTAL secondary to enhanced apical Na reabsorption 19, 20 and increased acid delivery to this segment 21, 22, (factors we predict would promote opening of HV1 in vivo) we hypothesized that ‘activation of Hv1 contributes to the development of outer-medullary oxidative stress, hypertension and renal injury in vivo following high salt feeding in Dahl salt-sensitive rats’. As, specific pharmacological inhibitors of HV1 are not yet available23, we tested this hypothesis by developing an HV1−/− null mutant rat on the Dahl salt-sensitive rat (Medical College of Wisconsin) genetic background.
As our previous work indicated reactive oxygen species (ROS) production in response to H+ efflux was greatly enhanced by low extracellular Na+ 10, we also investigated the relationship between Na concentration, HV1 and NADPH oxidase activity in mTAL as alterations in Na handling could potentially be an important regulatory feedback system for ROS production in this segment24–27.
Methods
Studies used adult SS rats (Medical College of Wisconsin) maintained ad libitum on water and a standard pellet diet containing 0.4% NaCl since weaning. All studies were conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. All of the protocols were approved in advance by the institutional animal care committee at Georgia Regents University or the Medical college of Wisconsin.
Bulk mTAL isolation
mTALs were isolated using a bulk dissection method as described previously 28 except two sieves rather than one were used. Briefly, the kidneys were flushed with 10ml of cold saline followed by 10ml of HEPES buffered Hanks Balanced salt solution (HBSS) containing 200 U/ml of type II collagenase (Worthington Biochemical). The kidneys were collected and cut into thin slices transversely along the cortical-medullary axis. The inner stripe of the outer medulla was isolated and incubated with collagenase solution at 37°C for 30minutes with intermittent pipetting. Every 5 mins, the digested tissue was pipetted out and passed through a 100µm and 70 µm sieve. mTAL segments were collected on the 70µm sieve, and digestion stopped by 1% BSA in pH 7.4 HBSS. We have previously demonstrated the collected tissue contains ~95% mTAL29.
Respiratory Burst Assay
Peritoneal macrophages (MØ) cells were collected as previously described 8. In brief, rats were anesthetized with isoflurane (2–5%) and 50mL of HBSS was injected in to the abdominal cavity followed by a small midline incision. The excess fluid was collected by syringe. The collected fluids were centrifuged at 400 g for 10 min. Collected cell pellets which contain predominantly MØ 8 were resuspended and aliquoted onto a clear-bottom 96-well plate (Bioexpress) at ~1*106 cells per well. 1 mM L-012 (Wako Pharmaceuticals) was used to determine superoxide production using a FLUOstar Omega plate reader (BMG Labtech). Cells were maintained at 37°C and luminescence measured for 30 minutes at 2 minute intervals. Addition of 100µM of the PKC activator phorbol 12-myristate 13-acetate (PMA) was used to stimulate the respiratory burst and maximal luminescence (arbitrary units) recorded.
In order to set intracellular Na+ concentration, cells were pre-incubated for 15min in solutions containing; 70mM N-methyl-D-glucamine, 5.5 mM D-glucose, 1.3mM CaCl2, 1mM MgSO4, 5M HEPES at pH 7.4. NaCl levels were altered to maintain intracellular [Na+] ([Na+]I at either 5, 10, 15, 20, or 25mM and balanced to a total of 70mM with the addition of KCl. The ionophore nystatin (135 U/mL) was prepared daily to permeablise cells to Na+ 30. In order to determine whether effects were due to differences in activity of Na/H exchangers (NHE), 100µM of the NHE inhibitor cariporide was added. ZnCl2 (100µM) was added to some wells to acutely inhibit HV1 activity. Maximal superoxide production rate was normalized to solutions containing 25mM NaCl within each plate.
Transfection of Peritoneal Macrophages
Cells were obtained as above. After 2 h incubation, the cells were washed once with serum-free medium to eliminating non-adherent lymphocytes and polymorphonuclear leukocytes from the adherent macrophages, then cultured in 200 ul of 10% FBS DMEM supplemented with 500 U/ml rat Granulocyte Macrophage Colony Stimulating Factor (rGM-CSF) (GenScript, Cat# Z02992-10). The primary macrophages were cultured in the presence of rGM-CSF for 7 to 10 days prior to transfection.
hHvcn1 transfection: When the cells were reached 50–60% confluence, the medium was removed and 100 ul of fresh no pen/strep and no FBS DMEM added. For each well of 96 well plates, we diluted 150 ng of hHvcn1 (human Hvcn1 cDNA was cloned into pQBI25-fC3) into 7.5 ul of Opti-MEM (Gibco, Cat# 31985-062), with 0.3 ul of PromoFectin-Macrophage Transfection solution (PromoCell GmbH, Cat# PK-CT-2000-MAC-10). We then added PromoFectin-Macrophage solution to the DNA solution, vortex-mixed the solution and spun down briefly before incubating for 30 minutes at room temperature. We then added 15 ul of this mixture drop-wise into each well. After 4 hours incubation, we add 85 ul of no pen/strep 10% FBS DMEM to each well and put in a 37C incubator. After 48 hours of hHvcn1 transfection the respiratory burst assay was performed as detailed above.
Micro dissection and imaging of mTAL
Rats were anesthetized with isoflurane (2–5%) and isolation of mTAL tissue strips performed as described previously31. Thin tissue strips containing mTALs were placed on a glass coverslip coated with the tissue adhesive Cell-Tak (BD Biosciences) for fluorescence imaging. Tissue strips containing mTALs were loaded with either dihydroethidium DHE (50 mmol/L) or 2',7'-Bis-(2-Carboxyethyl) -5-(And-6) - carboxyfluorescein (BCECF; 6 µmol/L) in HBSS for 1 hour at room temperature. Coverslips were placed on a heated imaging chamber maintained at 37°C (Warner Instruments) that allowed the rapid exchange of superfusion buffer and mounted on the stage of an inverted microscope (IX81 Olympus). mTAL were visualized with a ×40 water immersion objective lens. The signal was detected using a high-resolution digital camera (Photometrics Evolve, Roper Scientific). Excitation was provided by a Sutter DG-4 175W xenon arc lamp (Sutter Instruments) that allowed high-speed excitation wavelength switching. 3 to 10 mTAL epithelial cells were selected within each tissue strip to quantify changes in fluorescent intensity of dyes using Metafluor imaging software (Universal Imaging). Intracellular [Na+] was set at either 5 or 30mM using the same solutions as that used to set [Na+]I in MØ.
Whole cell voltage-clamp recordings
Proton currents from HV1 were recorded as previously described 32. Traces shown in Figure 1 were not corrected for liquid junction potential, and leak currents were not subtracted.
Figure 1. Confirmation of loss of HV1 expression in mutant rats.
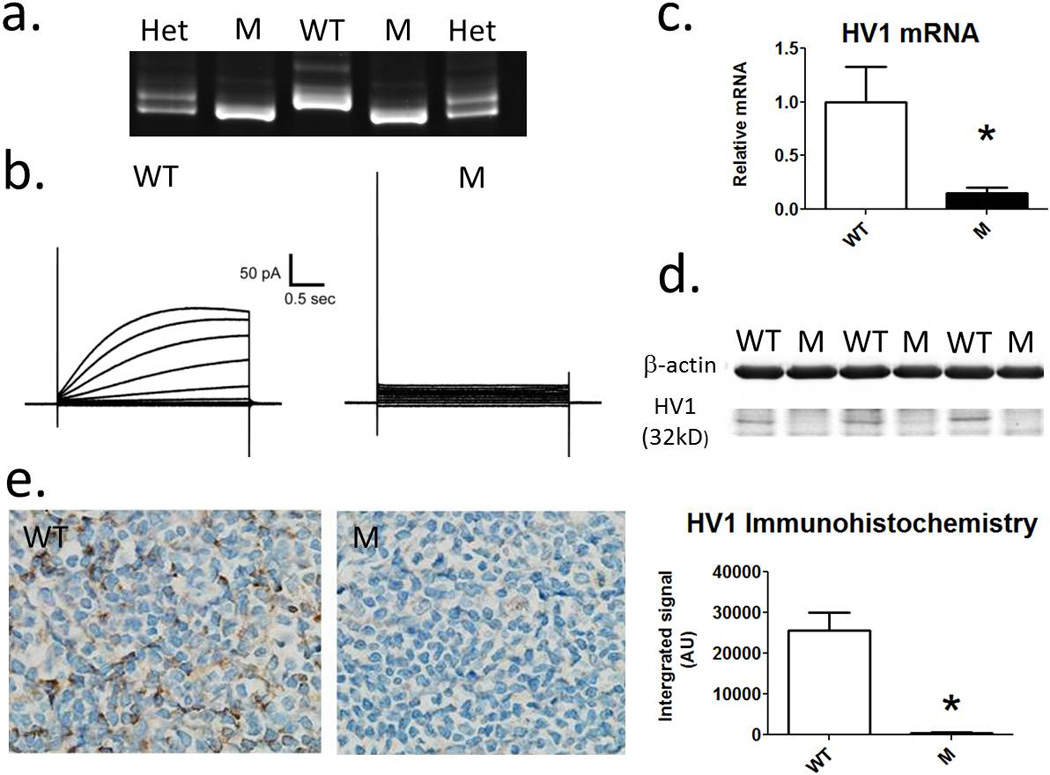
a) Representative gel for genotyping rats demonstrating 8bp deletion in gene encoding HV1. Lanes 1 and 5, heterozygous HV1−/+ mutants (Het); lanes 2 and 3, homozygous HV1−/− Mutants (M); Lane 3, Wild-type (WT). b) A representative trace demonstrating proton currents in response to step changes in membrane potential in single peritoneal macrophages from wild-type (right) and mutant (left) rats. c) relative HV1 mRNA expression from spleen homogenates from WT (open columns) and M (closed columns) rats compared to GAPDH mRNA (n=4). d) Western blot for HV1 in membrane fraction of WT and M rats. A band representing HV1 is shown at ~32kd. e) Immunohistochemical staining of spleen using anti-HV1 in WT and M rats (n=4). Left, representative image of anti-HV1 stained tissue from WT and M rats at 40X. Right, quantification of anti-HV1 staining (integrated signal (arbitrary units (AU)). * = p<0.05 using unpaired t-test. Data are mean±SE.
In vivo studies
Male 9 week male old WT SS and HV1−/− SS rats were anesthetized (2–5% isoflurane) and blood pressure telemeters (Data Sciences International, St Paul, MN) surgically implanted. Following 1 week of recovery, animals were again anesthetized and the left femoral vein catheterized for infusion of fluids. Surgically implanted catheters were then tunneled under the skin and exposed between the shoulder blades. Catheters were protected by a light wire spring attached to a swivel (Instech Laboratories Inc, Plymouth Meeting, PA), which allows free 360-degree movement of the animal. Rats were individually housed and allowed to recover for 1 week before beginning experimental measurements. Blood pressure was recorded 24 hr daily. Rats were initially be maintained on a low Na+ (0.4% NaCl diet) before being switched to a 8% NaCl diet for 2 weeks. On day 3 of high NaCl feeding, rats were either maintained on vehicle infusions (1% DMSO in saline i.v. at a rate of 6.9 µL/min) of KR32568 (Sigma) added to the infusion media (2mg/Kg/day) to stimulate HV1. On day 15, rats were anesthetized and the kidneys excised for histological analysis. After processing, histological samples were blinded to the investigator and images quantified using Metamorph analysis software.
Results
Confirmation of HV1 deletion in mutant rats
HV1 mutant Dahl salt-sensitive rats were generated with zinc-finger nuclease technology (13, 16). Genomic DNA of homozygous HV1 mutant (HV1−/−) rats was sequenced to reveal an 8 bp deletion, resulting in a frame-shift mutation and predicted loss of full length HVCN1 protein (Figure 1.a). Loss of HVCN1 was validated functionally using whole cell patch (Figure 1.b). Step changes in applied voltage between −60 + 100mV induced H+ currents in 7 of 7 MØ cells from three wild-type rats while no current was observed in any of 9 cells studied from four HV1−/− mutant rats. These data are consistent with a single gene for HV1 and confirmed complete functional ablation in HV1−/− mutant rats. As HV1 is highly expressed in immune cells of the spleen and its localization known, we first confirmed the specificity of our mRNA primer and anti-HV1 antibody using whole spleen homogenates of Wild-type and HV1−/− mutant rats as in situ positive and negative controls, respectively. mRNA encoding HV1 was greatly reduced in spleen of HV1−/− mutant rats compared to wild-type rats (Figure 1.c). Western blot detected a band representing HV1 at the predicted molecular weight (32Kd) in the membrane fraction of spleen from wild-type but not HV1−/− mutant rats (Figure 1.d). Immunohistochemical staining of the spleen demonstrated numerous darkly stained cells within the spleen of wild-type rats, consistent with known distribution of HV1 in splenic tissue (Figure 1.e). HV1 protein staining was also nearly undetectable in whole cell homogenates of spleen from HV1−/− mutant rats compared to wild-type.
HV1 expression in mTAL
In order to determine whether HV1 was expressed in mTAL of SS rats, we quantified mRNA and protein expression specifically in mTAL using the same PCR primers and antibodies used in spleen. mTAL from mutant rats served as a negative control. HV1 mRNA was 6 fold higher than presumed background in bulk isolated mTAL of wild-type compared to HV1−/− mutant rats (please see http://hyper.ahajournals.org). We could not detect HV1 by Western in membrane fractions of bulk isolated mTAL in either genotype (data not shown); however, HV1 staining was identified specifically within mTAL in outer-medullary sections (Figure 2) and this was significantly greater than background staining in mTAL from mutant rats (4382±920AU vs 1458±709AU p=0.036, n=5). Anti-Hv1 immuno-gold staining and transmission electron microscopy localized Hv1 staining largely to the apical membrane of thick ascending limb (Figure 2).
Figure 2. HV1 expression in mTAL.

Legend does not match figure Left, representative immunohistochemical staining of renal outer medulla using anti-HV1 in WT and M rats at 100X magnification; Right, Electron micrograph demonstrating Hv1 localization to the apical membrane of renal medullary thick ascending limb cells using anti-HV1 immune-gold staining. Arrows indicate clumps of gold particles indicating HV1 expression.
H+ efflux mediated superoxide production in mTAL
In order to test our hypothesis that ‘HV1 contributes to superoxide-enhancing H+ currents in mTAL’ we determined superoxide production (ΔEth/ΔDHE) in response to cellular acidification using an NH4Cl prepulse (20mM) under the conditions in which we previously used to characterize superoxide-stimulating H+ currents 10. Consistent with our previous work, superoxide production was stimulated following acidification in 0 Na+, BaCl2 (100µM) media in micro dissected mTAL from wild-type SS rats (Figure 3). In contrast, under the same conditions, cellular acidification with 20mM NH4Cl did not promote detectable changes in the rate of superoxide production in mTAL micro-dissected from HV1−/− mutant Dahl rats (Figure 3; PSTRAIN*TIME<0.001). Consistent with our hypothesis that H+ efflux through HV1 promotes superoxide production in mTAL, intracellular pH ([pHI]) recovery rate was significantly greater (p=0.046) and plateaued at higher [pHI] in mTAL from wild-type rats compared to HV1−/− mutant animals (slope = 0.005U/sec & 0.002U/sec; plateau= pH 7.35±0.29 vs. 7.08±0.04, respectively).
Figure 3. Superoxide production and intracellular pH recovery in mTAL following NH4Cl prepulse.
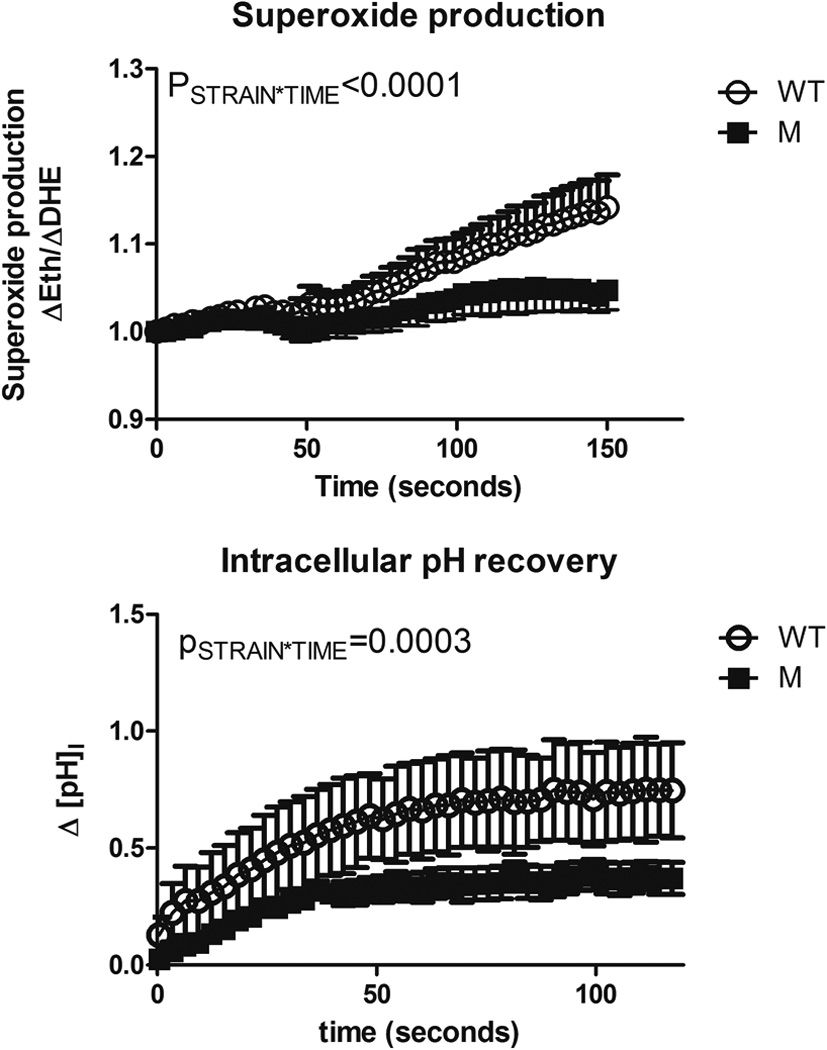
(Top panel); superoxide production in isolated medullary thick ascending limb from wild-type (WT – open circles; n=6) and HV1−/− mutant (M – closed squares; n=7) rats loaded with the superoxide sensitive dye, dihydrethidium. × axis, time in seconds. NH4Cl (20mM) was removed from the bath at t=30 seconds. Y axis, superoxide production (ΔEth/ΔDHE) normalized to 1 at t=0. (Bottom panel); Intracellular pH recovery following removal of NH4Cl from the media in isolated medullary thick ascending limb WT (n=8) and M (n=6) rats. × axis, time in seconds following maximal acidification. y axis, change in intracellular pH (ΔpHI) from maximal acidification. PSTRAIN*TIME, result of a two-way repeated measures ANOVA comparing response to WT and M rats. Media contained 0 Na+, 100mM BaCl2 (see methods). Data are mean±SE.
HV1 and the Na+ dependence of NAD(P)H oxidase in MØ
We next investigated the relationship between HV1, Na+ concentration and NAD(P)H oxidase activity. In order to investigate this relationship in detail, we first utilized rat peritoneal MØ. These cells can be easily collected from the peritoneal cavity in large numbers, and express high levels of both HV1 and NAD(P)H oxidase. To determine whether changes in intracellular Na+ concentration may have a direct effect on MØ superoxide production, intracellular Na+ was set between (5–25mM) using the cation ionophore nystatin, and the NAD(P)H oxidase dependent respiratory burst was stimulated in MØ with PMA. Responses are shown normalized to the maximal response at 25mM [Na]I (Figure 4). We found that low Na+ (5–10mM) greatly enhanced superoxide production in response to PMA (Figure 4; PGROUP*SODIUM=0.02). The effect was due to intracellular Na+, as no significant relationship with Na+ was observed in the absence of the ionophore nystatin (Figure 4). Further, the effect was independent of Na/H exchange, as the relationship was maintained in the presence of the potent Na/H exchange inhibitor cariporide (Figure 4).
Figure 4. Effect of intracellular Na+ concentration on the respiratory burst in macrophages.
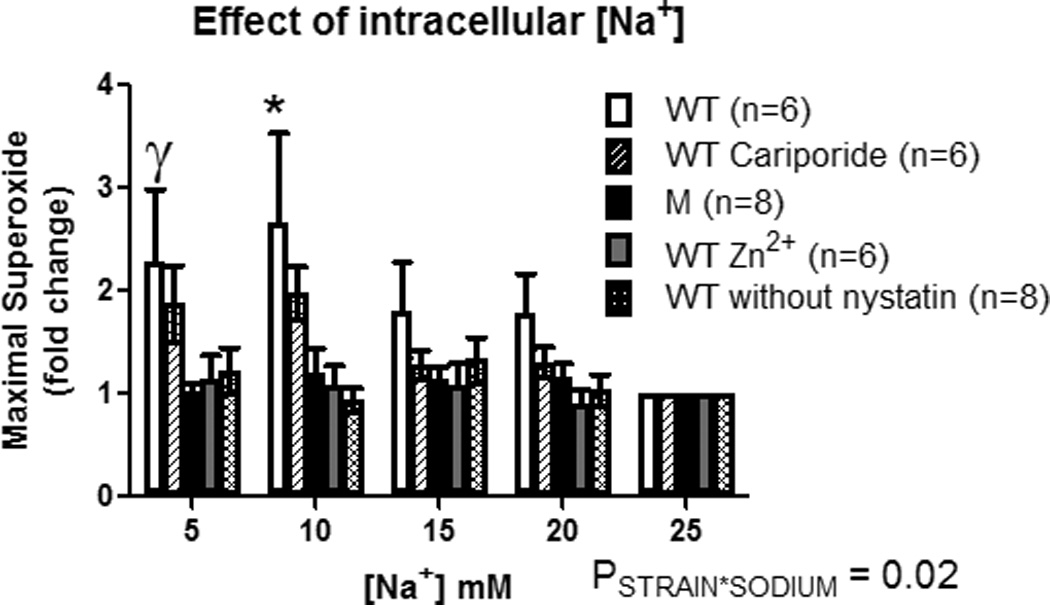
X-axis, intracellular Na+ concentration ([Na+]I). Y-axis, maximal superoxide production in freshly isolated peritoneal rat macrophages following addition of PMA (100mM) determined by L-012 luminescence (data normalized to the response at 25mM [Na+]I within each animal). Wild-type (open columns; n=6); Wild-type + 100µM cariporide (hatched columns; n=6); HV1 mutant (closed columns; n=8); Wild-type + 100µM ZnCl2 (grey filled columns; n=6); Wild-type without addition of Nystatin (double hatched columns; n=8). Data were compared using 2-way ANOVA with Bonferroni post-hoc test comparing Wild-type to all other groups. * = p<0.05 comparing Wild-type to HV1 mutant, Wild-type + ZnCl2 and Wild-type without Nystatin. ϒ =p<0.05 comparing Wild-type to HV1 mutant only. Data are mean±SE.
We measured superoxide production in MØ from HV1−/− mutant rats, and in MØ from WT rats in the presence of 100µM Zn2+ (which acutely inhibits HV1 activity), to determine whether enhancement of NAD(P)H oxidase activity at low intracellular Na+ is mediated by HV1. In cells from the same WT animal at 25mM [Na+]I, maximal raw signal was reduced from 4605±2006 arbitrary units (AU) in the absence of Zn2+ to 1868±837AU in the presence of Zn2+. In addition, no relationship between [Na+] and superoxide production was observed in Zn2+ treated MØ (Figure 4). Consistent with these data, no relationship between [Na+]I and maximal superoxide production was observed during the respiratory burst in MØ harvested from HV1−/− mutant rats (Figure 4; raw signal at 25mM 5091±1551AU).
To determine whether the effect of Na on NAD(P)H oxidase depended on HV1 expression or was secondary to altered expression of other proteins during development, we transfected MØ harvested from HV1−/− mutant rats with human HV1. A significant relationship was observed between intracellular [Na] and PMA stimulated superoxide production in primary cultures of peritoneal MØ harvested from wild-type rats (Figure 5). Consistent with studies performed in freshly isolated MØ, this relationship was absent in cells from HV1−/− mutant rats (Figure 5). Transfection of MØ from HV1−/− mutant rats with human HV1 increased human HV1 expression ~2400 fold and restored the relationship between intracellular [Na] and superoxide production observed in wilt-type MØ (Figure 5). Apocyanin treatment (100µM) of WT cells diminished overall superoxide production (1513AU vs 126AU, for vehicle and apocynin treated primary cultured MØ at 25mM [Na]I, respectively) and abolished the relationship between intracellular [Na] and superoxide production in wild-type cells (Figure 5), confirming superoxide production was mediated by NAD(P)H oxidase.
Figure 5. Contribution of Hv1 and NAD(P)H oxidase toward Na sensing in primary cultured macrophages.

X-axis, intracellular Na+ concentration ([Na+]I). Y-axis, maximal superoxide production in freshly isolated peritoneal rat macrophages following addition of PMA (100mM) determined by L-012 luminescence (data normalized to the response at 25mM [Na+]I within each animal). HV1−/− control (closed columns; n=5); Wild-type (WT) control (open columns; n=5); Wild-type + apocynin 100µM (striped columns; n=5); HV1−/− transfected with human Hv1 (checked columns; n=5). Data were compared using 2-way ANOVA with Bonferroni post-hoc test comparing HV1−/− control to all other groups. * = p<0.05. Data are mean±SE.
Intracellular Na+, HV1 and superoxide production in mTAL
To determine whether HV1 promotes superoxide in mTAL when intracellular Na+ levels are low, we determined superoxide production (ΔEth/ΔDHE) in freshly micro-dissected live mTAL from wild-type and HV1−/− mutant rats at both 5mM and 30mM intracellular [Na+]. In response to acidification using an NH4Cl (20mM) prepulse, the rate of superoxide production was enhanced in mTAL from wild-type rats at 5mM but not 30mM [Na+]I (Figure 6; PSODIUM*GROUP<0.0001). Importantly, in mTAL from HV1−/− mutant rats no significant difference was observed in superoxide production following acidification at either 5 or 30mM Na+ (Figure 6).
Figure 6. Superoxide production in response to H+ efflux at low and high intracellular Na+ in mTAL.
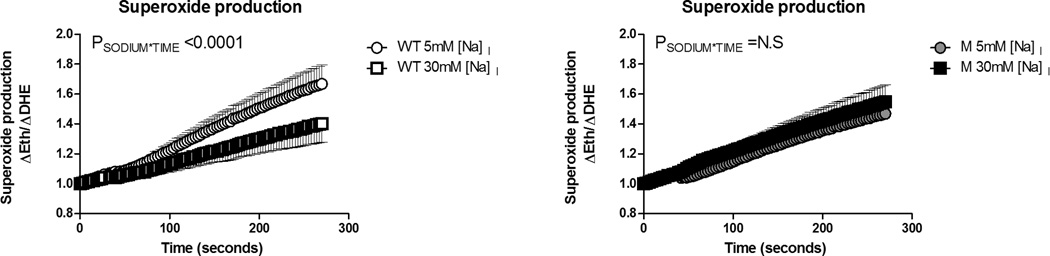
Superoxide production in isolated medullary thick ascending limb from wild-type (WT; left panel; n=6) and HV1−/− mutant (M; right panel; n=7) rats loaded with the superoxide sensitive dye, dihydrethidium. Open circles, wild type at 5mM intracellular [Na+]; open squares, wild type at 30mM intracellular [Na+]; closed circles, mutant at 5mM intracellular [Na+]; grey filled squares, mutant at 30mM intracellular [Na+]. × axis, time in seconds. NH4Cl (20mM) was removed from the bath at t=30 seconds. Y axis, superoxide production (ΔEth/ΔDHE) normalized to 1 at t=0. PSODIUM*TIME, result of a two-way repeated measures ANOVA comparing response within strain at 5 and 30mM intracellular [Na+]. N.S = not significant. Data are mean±SE.
24 hour mean arterial pressure measured by radio-telemetry was significantly lower in HV1−/− rats compared to wild-type animals following high salt feeding (8% NaCl) for 2 weeks (PSTRAIN=0.046). Outer-medullary tubular casts were markedly lower in HV1−/− rats compared to wild-type SS rats (Figure 7). Outer-medullary 4-HNE staining (oxidized lipids) was also markedly reduced in HV1−/− rats when compared to wild-type animals indicating reduced outer-medullary oxidative stress. Administration of KR32568 to activate HV1 in vivo did not alter blood pressure or outer-medullary injury markers. 4HNE staining tended to be elevated in KR32568 treated wild-type animals compared to vehicle, but this did not reach statistical significance (P=0.11).
Figure 7. Blood pressure, renal outer-medullary oxidative stress and tubulointerstitial injury in wild-type and HV1 mutant Dahl salt-sensitive rats.
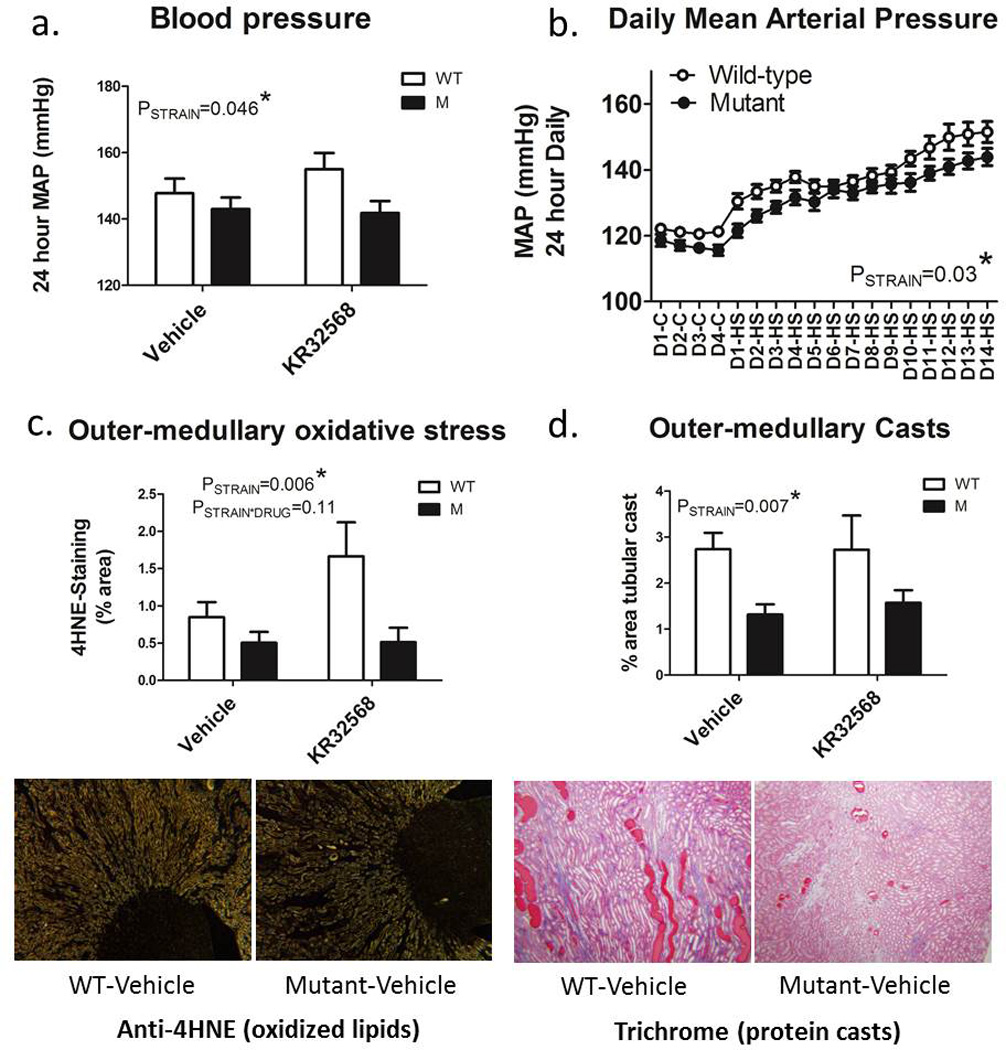
a) average 24 hour mean arterial pressure as measured by radio-telemetry over the final 3 days of high (8%) NaCl feeding for two weeks in wild-type (WT; open columns) and HV1−/− mutant (M; closed columns) maintained on either vehicle (right) or given the NHE-1 inhibitor KR32568 (2mg/kg/day) from day 4 of high salt feeding (left). b) 24 hour daily mean arterial blood pressures (mmHg) as measured by radio-telemetry during 0.4% NaCl control (C) and 8% high salt (HS) feeding for 14 days (D). Open circles, wild type (n=12); closed circles, HV1−/− mutant (n=12). All animals received only vehicle infusions until day 3 of high salt feeding (D3-HS). Drug or vehicle infusions were maintained until day 14 of high salt feeding (D14-HS). c) outer-medullary oxidative stress as determined by anti-4-HNE staining of the outer-medullary region. Shown are representative images of anti-4HNE staining in the outer-medulla and papilla of vehicle treated wild-type (left) and HV1−/− mutant rats (right) demonstrating more intense membrane staining of mTAL in wild-type animals. d) outer-medullary protein casts (% outer-medullary area). Shown are representative trichrome stained images of the outer-medulla vehicle treated wild-type (left) and HV1−/− mutant rats (right) demonstrating greater % area containing of brightly colored protein casts in wild-type animals. All histological measurements were assessed at the end of the study after 14 days of high salt feeding. Data are mean±SE. All data were compared with 2-way ANOVA.
Discussion
HV1 is a voltage-gated proton channel highly expressed in immune cells where it acts to maintain NAD(P)H oxidase activity during the respiratory burst 14. HV1 has been found in a number of cell types outside the immune system 23 including lung epithelial cells 33, 34 and spermatozoa 35. However, no studies have identified the presence of HV1 in the mammalian kidney or have demonstrated a physiological link to ROS production in renal epithelial cells. This study demonstrates that HV1 is present in mTAL and that it contributes to H+ efflux- dependent superoxide production. Genetic deletion of HV1 in the SS rat was confirmed by sequence analysis revealing an 8-bp deletion in a key coding region of HV1. Complete functional knockout of the gene was confirmed by whole-cell voltage-clamp technique. Because murine gene knockout models have revealed lack of specificity of many commonly used antibodies 36, we first verified the specificity of a commercial antibody to HV1 in situ in spleen, a tissue in which HV1 expression is high and its localization known. Anti-HV1 from abcam (Cat# ab117520) clearly demonstrated expression of HV1 in spleen from WT but not from HV1−/− rats (Figure 1). When we used this antibody for Western blots, we found that detection depended on use of membrane fractions which are presumably enriched in HV1. Even in these enriched samples, the band for HV1 was weak, suggesting a limited sensitivity of this antibody for detection via Western blot. Immunohistochemical analysis of kidney sections using this antibody also detected HV1 (primarily around the apical membrane of thick ascending limb) in wild-type but not HV1−/− Dahl SS. The localization of HV1 to the apical membrane was also confirmed by transmission electron microscopy using immuno-gold stained sections. Although we were unable to detect HV1 protein expression in mTAL by western blot, the presence of HV1 in mTAL is supported by PCR, immunohistochemistry and functional analysis (pHI).
Functional analysis of both superoxide production and pHI recovery following NH4Cl prepulse under the same conditions used to characterize superoxide-stimulating H+ currents in our previous study (0 Na+ media, BaCl2)10 demonstrate that HV1 is required for superoxide production following cellular acidification in mTAL 10. Li et al first reported that stimulating H+ efflux, either by rapidly increasing the pH of the extracellular media or by intracellular acidification using an NH4Cl prepulse promoted the production of superoxide by NAD(P)H oxidase in mTAL13. While Li et al concluded that any H+ efflux promoted NAD(P)H oxidase activity including via Na/H exchangers, our own subsequent data did not support a direct link between Na/H exchanger activity and NAD(P)H oxidase activation. Instead, our data indicated that, in response to intracellular acidification, activation of a second unidentified H+ transport pathway mediated superoxide production by the oxidase in this segment 10. In both studies NADPH oxidase was confirmed as the source of H+ efflux mediated ROS production using either apocynin or diphenylene iodonium 10, 13. Our current data confirm our previous conclusions and identify this transport pathway as the voltage-gated proton channel HV1.
Another major finding of the current study is that HV1 modulates NAD(P)H oxidase activity in response to differing levels of intracellular [Na+]. In our previous study we found that superoxide production following cellular acidification by NH4Cl was greatly enhanced in 0 Na+ media 10. In order to elucidate the relationship between Na+, HV1 and NAD(P)H oxidase we first performed experiments in isolated peritoneal MØ from SS rats. MØ provided an excellent platform to investigate these relationships as they express high levels of HV1, can easily be isolated in large numbers, ROS production from NAD(P)H oxidase can be induced by addition of PMA and phagocytes are a well-established model to study the function of both HV1 and NAD(P)H oxidase 8, 37–39. We found that low intracellular Na+, between the ranges of 5–10mM, greatly enhanced ROS production during the PMA stimulated respiratory burst in MØ relative to 25mM [Na+]I. The effect was due to intracellular Na+ as in the absence of the ionophore nystatin, an increase in ROS production at low Na+ levels was not observed. Importantly, the effect of low intracellular Na+ to enhance maximal superoxide production in mTAL depended on the activity of HV1. In MØ from wild-type SS rats, addition of 100mM Zn2+, which inhibits HV1, completely abolished the relationship between Na+ and superoxide production. Further, in MØ isolated from HV1−/− rats no relationship between intracellular Na+ and maximal superoxide production was observed unless these cells were transfected with human HV1 which restored the ‘Na sensing’ phenotype observed in wild-type cells. We conclude that HV1 must act to ‘sense’ intracellular Na+ either directly or act as a signal to modulate the activity of NAD(P)H oxidase secondary to changes in intracellular Na+. We confirmed these relationships in isolated mTAL from wild-type and HV1−/− rats using a second indicator of ROS production, dihydroethidium.
We and others have previously identified NADPH oxidase as the source of ROS following H+ efflux in mTAL and we confirmed NADPH oxidase as the source of Na-sensitive superoxide production in MØ in the current study. Loss of ‘Na sensing’ was not due to loss of NAD(P)H oxidase activity as significant superoxide production was still detected in the absence of HV1. Further, expression of NADPH oxidase subunits was not altered between mTAL from wild-type and HV1−/− mutant rats (please see http://hyper.ahajournals.org) indicating systemic loss of HV1 did not alter NADPH oxidase expression in TAL during development. Our novel finding that HV1 acts to sense intracellular Na+ and modulate NAD(P)H oxidase activity has broad implications to the study of the function of NAD(P)H oxidase activity in cells that express HV1.
Our finding that HV1 senses intracellular Na+ and modulates NAD(P)H oxidase activity may be of particular relevance to mTAL in which changes in intracellular Na+ are known to occur in response to alterations in Na+ transport activity. Basal levels of intracellular Na+ in mTAL have been reported between ~10–20mM, increase with the inhibition of Na/K/ATPase and decrease with inhibition of apical NKCC2 transporter activity 30, 40. Importantly, based on our current data, we would predict that physiologically relevant changes in intracellular Na+ of ± 5 to10mM from rest have the potential to markedly alter superoxide production via HV1 signaling. Given the important role of superoxide in stimulating the activity of Na+ transporters in mTAL41, Na+ sensing by HV1 and subsequent alteration in local NAD(P)H oxidase activity may play an important role in the feedback regulation of Na+ reabsorption by this segment.
While our study is the first to identify HV1 in mTAL, proton currents attributed to HV1 have previously been identified in proximal tubular cells of Frog (Rana pippens)42. In regard to their activation however, the authors of this study concluded that ‘the role of depolarization activated GH (HV1) in proximal tubule is not entirely clear, since proximal tubule cells do not normally experience drastic changes in either pH or membrane potential’42. This brings into question the physiological relevance of HV1 in mTAL. While we observed functional effects attributed to HV1 in our studies, most of our experiments were performed under somewhat contrived conditions directed toward maximal activation and identification of the channel, including 0 Na+, bicarbonate free media and rapid cellular acidification with NH4Cl. What evidence is there then that HV1 would be activated in mTAL under physiological conditions? Resting membrane potential in mTAL has been reported between −80mV to −40mV depending on transport conditions 19. The trans-membrane voltage threshold for opening of HV1 can be described by Vthreshold = 20mV – 40ΔpH 33. This voltage dependence is shifted −30 to −40mV during enhanced gating conditions associated with activation of NAD(P)H oxidase 15, 18. Therefore, even under ‘enhanced gating’ conditions, in the absence of an outward pH gradient, HV1 within mTAL would remain in the closed state. Importantly, in regards to activation of HV1 in mTAL, a large outward pH gradient exists in this segment under normal physiological conditions. mTAL actively reabsorb much of the NH4+ secreted by the proximal tubule 43–45. Due to the more rapid uptake of NH4+ across the apical membrane, and relatively slower loss of NH4+ relative to NH3 across the basolateral membrane, under normal physiological conditions where the tubular perfusate contains ~4mM NH4+, an outward pH gradient of 0.8–0.9 units is present 43. Such pH gradient would be predicted to result in a shift ~ 34mV toward more negative membrane potentials, within the range observed in mTAL. Therefore, due to the unique physiology of mTAL which results in a large outward pH gradient, we speculate that, even under normal physiological conditions, HV1 would be poised at or near its activated state. Differences in oxidative stress within the outer-medulla observed in vivo between wild-type and HV1−/− mutant rats further supports the concept HV1 in mTAL is active under physiological conditions (Figure 7).
Our in vivo data support a role of HV1 in the development of hypertension and kidney injury in Dahl SS rats. Blood pressure, was significantly lower in HV1−/− rats fed a high salt diet compared to wild-type SS rats, with HV1 accounting for about ~25% of the rise in arterial pressure. While this reduction in blood pressure is relatively modest, it is possible that it reflects a significant change in mTAL function. In regard to the final control of urinary Na excretion and blood pressure regulation, Na transport in mTAL is not thought to be as important as transport in more distal nephron segments. As evidence of this, heterozygous deletion of ROMK, which is critical for mTAL Na reabsorption, produces a relatively mild reduction in blood pressure in Dahl SS rats 46 similar to that observed in the current study following Hv1 deletion. In contrast, inhibition of epithelial Na channels in more distal nephron segments has a much greater effect 47. A significant alteration in mTAL function is also consistent with our findings of marked reductions in outer-medullary oxidative stress and tubular injury, which were all more than 50% lower in HV1−/− animals and suggest an important role of HV1 in the development of tubulo-interstitial injury. Further studies are required to determine whether lower blood pressure in HV1−/− mutant rats is a consequence of altered mTAL function or related to other potential mediators of blood pressure such as immune cells which highly express Hv1 and have been demonstrated to be important in the development of hypertension in Dahl salt-sensitive rats48.
Apical Na reabsorption is normally tightly coupled to basolateral NaKATPase activity, therefore despite enhancing Na transport, it is unlikely that a high salt diet would greatly alter intracellular [Na] in mTAL in the absence of other factors. Rather, as high NaCl delivery to the TAL promotes cellular depolarization secondary to increased NKCC2 activity 19, 20, 49, we speculate that HV1 may be activated by depolarization of the apical membrane. High salt diet has also been shown to increase acid excretion in Dahl SS rats21 which has been shown to promote both intracellular acidification 43 and cellular depolarization in mTAL secondary to low pHI inhibition of ROMK 22. Therefore increased acid delivery to mTAL during high salt feeding may also promote opening of Hv1 in vivo.
As inhibition of Na/H exchangers has been demonstrated to activate HV1, we examined the effect of Na/H exchanger 1 (NHE) inhibition using KR32568 on in vivo responses to high NaCl diet in both wild-type and HV1−/− rats. In vivo administration of KR32568 during high NaCl feeding did not alter blood pressure or renal injury in either wild-type or HV1−/− rats and although oxidative stress (4HNE-staining) tended to be increased in wild-type animals, this did not reach significance. These data may suggest that either HV1 is already activated in vivo, or that greater activation of HV1 does not alter blood pressure/renal injury further. While our data confirm a role of HV1 in the development of hypertension and renal injury in Dahl SS rats, further studies are required to confirm whether this is mediated by mTAL HV1 expression or other sources such as circulating immune cells.
Current dogma holds that H+ transport by HV1 allows NADPH to operate at a high rate by limiting cellular acidosis and depolarization which feedback upon activation of the oxidase to inhibit the enzyme 14. A number of indicators suggest it is unlikely that superoxide production in mTAL would require H+ efflux in order to prevent feedback inhibition of oxidase activity. Firstly, unlike phagocytic NADPH oxidase, which generates extracellular superoxide and requires H+ efflux to balance the electron current, there is evidence epithelial NADPH oxidase generates largely intracellular superoxide and therefore would not require H+ efflux across the cell membrane to balance charge 50, 51. Secondly, superoxide production by NADPH oxidase in mTAL is much lower than that produced during the respiratory burst in phagocytes and significant cellular depolarization/acidification is not known to occur upon activation of the oxidase in these cells. An alternative explanation for our data is that, rather than solely preventing feedback inhibition of the oxidase, HV1 activation secondary to cellular depolarization in the presence of an outward pH gradient directly promotes the formation of superoxide by stimulating NADPH oxidase activity. Further studies will be required to resolve these key issues.
Summary and Perspectives
In summary, we conclude that 1) HV1 is present in mTAL and contributes to superoxide production in response to H+ efflux; 2) that HV1 modulates NADPH oxidase activity in response to the level of intracellular [Na+]. 3) That HV1 contributes to the development of outer-medullary oxidative stress, hypertension and renal disease in high salt fed Dahl SS rats. This is the first functional evidence for a role of HV1 in the kidney and identifies HV1 as a potential target to limit renal oxidative stress and the development of salt-sensitive hypertension and renal injury. Our finding that HV1 regulates NAD(P)H oxidase activity in response to intracellular Na+ levels may have important implications in regard to tubular Na+ reabsorption and kidney function. This finding is also likely to have broad implications for other organ systems where HV1 is expressed. The development and characterization of a HV1−/− mutant rat model is likely to provide an important tool for the study of HV1 including its potential importance to cardiovascular disease.
Supplementary Material
Novelty and Significance: 1) What Is New 2) What Is Relevant? Summary.
What is new? This study demonstrates that the voltage-gated proton channel HV1 is expressed in medullary thick ascending limb, that HV1 contributes to superoxide production in this segment, that HV1 acts to modulate superoxide production in response to changes in intracellular [Na+] and that HV1 contributes to blood pressure regulation and renal injury in the Dahl salt-sensitive rat model.
What is Relevant? Superoxide production within the renal outer medulla by NAD(P)H oxidase is important in long-term blood pressure control and the development of renal injury via effects on Na+ transport and renal hemodynamics and is augmented in Dahl salt-sensitive rats. We now identify HV1 as a critical component contributing to enhanced superoxide production in mTAL of Dahl salt-sensitive rats and a novel molecular target for the treatment of renal oxidative stress. Demonstration of HV1 as a Na+ sensor, contributes to our understanding of the physiological stimuli that promote activation of NAD(P)H oxidase within the kidney and in other tissues expressing HV1.
In summary, we determined HV1 as the molecular identity of a source of superoxide in mTAL which is augmented in Dahl-salt-sensitive rats. Further, we demonstrate that HV1 acts to modulate the activity of NAD(P)H oxidase in both mTAL and MØ in response to physiologically relevant changes in intracellular Na+ and that HV1 contributes to blood pressure regulation and renal injury in the Dahl salt-sensitive rat model. These data indicate that HV1 is likely to play an important physiological function as a Na+ sensor and may provide a novel target for the treatment of renal oxidative stress, hypertension and kidney injury.
Acknowledgments
The authors would like to thank the PhysGen KO program (http://rgd.mcw.edu/wg/physgenknockouts) for development of HV1−/− mutant rats. The authors would like to thank Kelly Hyndman for her careful review of the manuscript and the Electron Microscopy Core Services at Georgia Regents University.
Source(s) of Funding:
This work was supported by an American Heart Association Scientist Development Grant to Paul O’Connor (10SDG4150061), NIH GO grant RC2 HL101681 (Howard Jacob) and NIH GM078319 (Nevin Lambert).
Footnotes
Conflict(s) of Interest/Disclosure(s):
None
References
- 1.Sullivan JM. Salt sensitivity Definition, conception, methodology, and long-term issues. Hypertension. 1991;17:I61–I68. doi: 10.1161/01.hyp.17.1_suppl.i61. [DOI] [PubMed] [Google Scholar]
- 2.Kotchen TA, Zhang HY, Covelli M, Blehschmidt N. Insulin resistance and blood pressure in dahl rats and in one-kidney, one-clip hypertensive rats. Am J Physiol. 1991;261:E692–E697. doi: 10.1152/ajpendo.1991.261.6.E692. [DOI] [PubMed] [Google Scholar]
- 3.Kidambi S, Kotchen JM, Krishnaswami S, Grim CE, Kotchen TA. Cardiovascular correlates of insulin resistance in normotensive and hypertensive african americans. Metabolism. 2010;60:835–842. doi: 10.1016/j.metabol.2010.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bloch MJ, Basile J. African american patients with hypertensive chronic kidney disease receive no benefit on kidney disease progression from the currently recommended blood pressure goal of <130/80 mm hg unless there is significant proteinuria at baseline: Long-term follow-up of the aask study. J Clin Hypertens (Greenwich) 2011;13:214–216. doi: 10.1111/j.1751-7176.2010.00409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campese VM. Salt sensitivity in hypertension Renal and cardiovascular implications. Hypertension. 1994;23:531–550. doi: 10.1161/01.hyp.23.4.531. [DOI] [PubMed] [Google Scholar]
- 6.Cowley AW., Jr Renal medullary oxidative stress, pressure-natriuresis, and hypertension. Hypertension. 2008;52:777–786. doi: 10.1161/HYPERTENSIONAHA.107.092858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor NE, Glocka P, Liang M, Cowley AW., Jr Nadph oxidase in the renal medulla causes oxidative stress and contributes to salt-sensitive hypertension in dahl s rats. Hypertension. 2006;47:692–698. doi: 10.1161/01.HYP.0000203161.02046.8d. [DOI] [PubMed] [Google Scholar]
- 8.Feng D, Yang C, Geurts AM, Kurth T, Liang M, Lazar J, Mattson DL, O'Connor PM, Cowley AW., Jr Increased expression of nad(p)h oxidase subunit p67(phox) in the renal medulla contributes to excess oxidative stress and salt-sensitive hypertension. Cell Metab. 2012;15:201–208. doi: 10.1016/j.cmet.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li N, Yi FX, Spurrier JL, Bobrowitz CA, Zou AP. Production of superoxide through nadh oxidase in thick ascending limb of henle's loop in rat kidney. Am J Physiol Renal Physiol. 2002;282:F1111–F1119. doi: 10.1152/ajprenal.00218.2001. [DOI] [PubMed] [Google Scholar]
- 10.O'Connor PM, Lu L, Liang M, Cowley AW., Jr A novel amiloride-sensitive h+ transport pathway mediates enhanced superoxide production in thick ascending limb of salt-sensitive rats, not na+/h+ exchange. Hypertension. 2009;54:248–254. doi: 10.1161/HYPERTENSIONAHA.109.134692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Connor PM, Lu L, Schreck C, Cowley AW., Jr Enhanced amiloride-sensitive superoxide production in renal medullary thick ascending limb of dahl salt-sensitive rats. Am J Physiol Renal Physiol. 2008;295:F726–F733. doi: 10.1152/ajprenal.00137.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mori T, O'Connor PM, Abe M, Cowley AW., Jr Enhanced superoxide production in renal outer medulla of dahl salt-sensitive rats reduces nitric oxide tubular-vascular cross-talk. Hypertension. 2007;49:1336–1341. doi: 10.1161/HYPERTENSIONAHA.106.085811. [DOI] [PubMed] [Google Scholar]
- 13.Li N, Zhang G, Yi FX, Zou AP, Li PL. Activation of nad(p)h oxidase by outward movements of h+ ions in renal medullary thick ascending limb of henle. Am J Physiol Renal Physiol. 2005;289:F1048–F1056. doi: 10.1152/ajprenal.00416.2004. [DOI] [PubMed] [Google Scholar]
- 14.DeCoursey TE. Voltage-gated proton channels find their dream job managing the respiratory burst in phagocytes. Physiology (Bethesda) 2010;25:27–40. doi: 10.1152/physiol.00039.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeCoursey TE. Voltage-gated proton channels: Molecular biology, physiology, and pathophysiology of the h(v) family. Physiol Rev. 2013;93:599–652. doi: 10.1152/physrev.00011.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musset B, Cherny VV, Morgan D, Okamura Y, Ramsey IS, Clapham DE, DeCoursey TE. Detailed comparison of expressed and native voltage-gated proton channel currents. J Physiol. 2008;586:2477–2486. doi: 10.1113/jphysiol.2007.149427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Musset B, Clark RA, DeCoursey TE, Petheo GL, Geiszt M, Chen Y, Cornell JE, Eddy CA, Brzyski RG, El Jamali A. Nox5 in human spermatozoa: Expression, function, and regulation. J Biol Chem. 2012;287:9376–9388. doi: 10.1074/jbc.M111.314955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeCoursey TE, Cherny VV, Zhou W, Thomas LL. Simultaneous activation of nadph oxidase-related proton and electron currents in human neutrophils. Proc Natl Acad Sci U S A. 2000;97:6885–6889. doi: 10.1073/pnas.100047297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schlatter E, Greger R. Camp increases the basolateral cl- -conductance in the isolated perfused medullary thick ascending limb of henle's loop of the mouse. Pflugers Arch. 1985;405:367–376. doi: 10.1007/BF00595690. [DOI] [PubMed] [Google Scholar]
- 20.Liu R, Garvin JL, Ren Y, Pagano PJ, Carretero OA. Depolarization of the macula densa induces superoxide production via nad(p)h oxidase. Am J Physiol Renal Physiol. 2007;292:F1867–F1872. doi: 10.1152/ajprenal.00515.2006. [DOI] [PubMed] [Google Scholar]
- 21.Batlle DC, Sharma AM, Alsheikha MW, Sobrero M, Saleh A, Gutterman C. Renal acid excretion and intracellular ph in salt-sensitive genetic hypertension. J Clin Invest. 1993;91:2178–2184. doi: 10.1172/JCI116444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bleich M, Kottgen M, Schlatter E, Greger R. Effect of nh4+/nh3 on cytosolic ph and the k+ channels of freshly isolated cells from the thick ascending limb of henle's loop. Pflugers Arch. 1995;429:345–354. doi: 10.1007/BF00374149. [DOI] [PubMed] [Google Scholar]
- 23.Capasso M, DeCoursey TE, Dyer MJ. Ph regulation and beyond: Unanticipated functions for the voltage-gated proton channel, hvcn1. Trends Cell Biol. 2010;21:20–28. doi: 10.1016/j.tcb.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ortiz PA, Garvin JL. Superoxide stimulates nacl absorption by the thick ascending limb. Am J Physiol Renal Physiol. 2002;283:F957–F962. doi: 10.1152/ajprenal.00102.2002. [DOI] [PubMed] [Google Scholar]
- 25.Garvin JL, Ortiz PA. The role of reactive oxygen species in the regulation of tubular function. Acta Physiol Scand. 2003;179:225–232. doi: 10.1046/j.0001-6772.2003.01203.x. [DOI] [PubMed] [Google Scholar]
- 26.Juncos R, Garvin JL. Superoxide enhances na-k-2cl cotransporter activity in the thick ascending limb. Am J Physiol Renal Physiol. 2005;288:F982–F987. doi: 10.1152/ajprenal.00348.2004. [DOI] [PubMed] [Google Scholar]
- 27.Juncos R, Hong NJ, Garvin JL. Differential effects of superoxide on luminal and basolateral na+/h+ exchange in the thick ascending limb. Am J Physiol Regul Integr Comp Physiol. 2006;290:R79–R83. doi: 10.1152/ajpregu.00447.2005. [DOI] [PubMed] [Google Scholar]
- 28.Trinh-Trang-Tan MM, Bouby N, Coutaud C, Bankir L. Quick isolation of rat medullary thick ascending limbs Enzymatic and metabolic characterization. Pflugers Arch. 1986;407:228–234. doi: 10.1007/BF00580681. [DOI] [PubMed] [Google Scholar]
- 29.Yang C, Stingo FC, Ahn KW, Liu P, Vannucci M, Laud PW, Skelton M, O'Connor P, Kurth T, Ryan RP, Moreno C, Tsaih SW, Patone G, Hummel O, Jacob HJ, Liang M, Cowley AW., Jr Increased proliferative cells in the medullary thick ascending limb of the loop of henle in the dahl salt-sensitive rat. Hypertension. 2012;61:208–215. doi: 10.1161/HYPERTENSIONAHA.112.199380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ortiz PA, Hong NJ, Garvin JL. No decreases thick ascending limb chloride absorption by reducing na(+)-k(+)-2cl(−) cotransporter activity. Am J Physiol Renal Physiol. 2001;281:F819–F825. doi: 10.1152/ajprenal.2001.281.5.F819. [DOI] [PubMed] [Google Scholar]
- 31.Dickhout JG, Mori T, Cowley AW., Jr Tubulovascular nitric oxide crosstalk: Buffering of angiotensin ii-induced medullary vasoconstriction. Circ Res. 2002;91:487–493. doi: 10.1161/01.res.0000035243.66189.92. [DOI] [PubMed] [Google Scholar]
- 32.Ramsey IS, Ruchti E, Kaczmarek JS, Clapham DE. Hv1 proton channels are required for high-level nadph oxidase-dependent superoxide production during the phagocyte respiratory burst. Proc Natl Acad Sci U S A. 2009;106:7642–7647. doi: 10.1073/pnas.0902761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cherny VV, Markin VS, DeCoursey TE. The voltage-activated hydrogen ion conductance in rat alveolar epithelial cells is determined by the ph gradient. J Gen Physiol. 1995;105:861–896. doi: 10.1085/jgp.105.6.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iovannisci D, Illek B, Fischer H. Function of the hvcn1 proton channel in airway epithelia and a naturally occurring mutation, m91t. J Gen Physiol. 2010;136:35–46. doi: 10.1085/jgp.200910379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lishko PV, Kirichok Y. The role of hv1 and catsper channels in sperm activation. J Physiol. 2010;588:4667–4672. doi: 10.1113/jphysiol.2010.194142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herrera M, Sparks MA, Alfonso-Pecchio AR, Harrison-Bernard LM, Coffman TM. Lack of specificity of commercial antibodies leads to misidentification of angiotensin type 1 receptor protein. Hypertension. 2012;61:253–258. doi: 10.1161/HYPERTENSIONAHA.112.203679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morgan D, Capasso M, Musset B, Cherny VV, Rios E, Dyer MJ, DeCoursey TE. Voltage-gated proton channels maintain ph in human neutrophils during phagocytosis. Proc Natl Acad Sci U S A. 2009;106:18022–18027. doi: 10.1073/pnas.0905565106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El Chemaly A, Okochi Y, Sasaki M, Arnaudeau S, Okamura Y, Demaurex N. Vsop/hv1 proton channels sustain calcium entry, neutrophil migration, and superoxide production by limiting cell depolarization and acidification. J Exp Med. 2009;207:129–139. doi: 10.1084/jem.20091837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kapus A, Romanek R, Qu AY, Rotstein OD, Grinstein S. A ph-sensitive and voltage-dependent proton conductance in the plasma membrane of macrophages. J Gen Physiol. 1993;102:729–760. doi: 10.1085/jgp.102.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu M, Lopez B, Dos Santos EA, Falck JR, Roman RJ. Effects of 20-hete on na+ transport and na+ -k+ -atpase activity in the thick ascending loop of henle. Am J Physiol Regul Integr Comp Physiol. 2007;292:R2400–R2405. doi: 10.1152/ajpregu.00791.2006. [DOI] [PubMed] [Google Scholar]
- 41.Schreck C, O'Connor PM. Nad(p)h oxidase and renal epithelial ion transport. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1023–R1029. doi: 10.1152/ajpregu.00618.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu X, Sackin H. Effect of ph on potassium and proton conductance in renal proximal tubule. Am J Physiol. 1995;269:F289–F308. doi: 10.1152/ajprenal.1995.269.3.F289. [DOI] [PubMed] [Google Scholar]
- 43.Watts BA, 3rd, Good DW. Effects of ammonium on intracellular ph in rat medullary thick ascending limb: Mechanisms of apical membrane nh4+ transport. J Gen Physiol. 1994;103:917–936. doi: 10.1085/jgp.103.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Attmane-Elakeb A, Karim Z, Bichara M. [role of the na(+)-k+(nh4+)-2cl cotransporter of the medullary ascending limb in the regulation of renal acid-base equilibrium] Nephrologie. 2002;23:209–211. [PubMed] [Google Scholar]
- 45.Karim Z, Attmane-Elakeb A, Bichara M. Renal handling of nh4+ in relation to the control of acid-base balance by the kidney. J Nephrol. 2002;(15 Suppl 5):S128–S134. [PubMed] [Google Scholar]
- 46.Zhou X, Zhang Z, Shin MK, Horwitz SB, Levorse JM, Zhu L, Sharif-Rodriguez W, Streltsov DY, Dajee M, Hernandez M, Pan Y, Urosevic-Price O, Wang L, Forrest G, Szeto D, Zhu Y, Cui Y, Michael B, Balogh LA, Welling PA, Wade JB, Roy S, Sullivan KA. Heterozygous disruption of renal outer medullary potassium channel in rats is associated with reduced blood pressure. Hypertension. 2013;62:288–294. doi: 10.1161/HYPERTENSIONAHA.111.01051. [DOI] [PubMed] [Google Scholar]
- 47.Pavlov TS, Levchenko V, O'Connor PM, Ilatovskaya DV, Palygin O, Mori T, Mattson DL, Sorokin A, Lombard JH, Cowley AW, Jr, Staruschenko A. Deficiency of renal cortical egf increases enac activity and contributes to salt-sensitive hypertension. J Am Soc Nephrol. 2013;24:1053–1062. doi: 10.1681/ASN.2012080839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the dahl salt-sensitive rat. Hypertension. 2006;48:149–156. doi: 10.1161/01.HYP.0000228320.23697.29. [DOI] [PubMed] [Google Scholar]
- 49.Haque MZ, Ares GR, Caceres PS, Ortiz PA. High salt differentially regulates surface nkcc2 expression in thick ascending limbs of dahl salt-sensitive and salt-resistant rats. Am J Physiol Renal Physiol. 2011;300:F1096–F1104. doi: 10.1152/ajprenal.00600.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of nadph oxidases in the cardiovascular system. Circ Res. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li JM, Shah AM. Intracellular localization and preassembly of the nadph oxidase complex in cultured endothelial cells. J Biol Chem. 2002;277:19952–19960. doi: 10.1074/jbc.M110073200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.