

INTRODUCTION
The initial description of Fuchs’ endothelial corneal dystrophy (FECD) in 19101 was made without a slit-lamp biomicroscope and, thus, did not include ‘drop-like’ excrescences beneath the endothelium observed by Vogt in 1921 who is credited with coining the term ‘guttae’ (Latin for drops).2 The origin of gutta has remained obscure. Possible explanations could be that they arise as cellular secretions3 or as extrusions from weak areas in Descemet membrane (DM).4
Limited reports describe morphologies of guttae in FECD. Laing et al described five stages distinguished by size, abnormalities of cells, coalescence of multiple guttae, and contour. They observed several stages in the same cornea.3 Gottsch et al suggest distinct guttae morphologies arise from specific genetic causes of FECD.4
Our studies of a transgenic mouse model of early onset FECD5 suggest novel insights into the origin of guttae.
METHODS
Transgenic mice harbouring the Q455 K mutation in the α 2 collagen VIII gene, confocal microscopy, periodic acid Schiff staining, and transmission electron microscopy (TEM) were described previously.5
RESULTS
Confocal microscopy of a homozygous Q455 K mouse shows endothelial polymegathism and pleomorphism (figure 1A). Laing et al’s stage 1 and stage 2 guttae are present. Also seen is a distinct, sharply raised gutta occurring at a cell border, a morphology ascribed by Gottsch et al to late onset FECD. Histologic sections show differently sized guttae (figure 1B) as seen in figure 1A.
Figure 1.
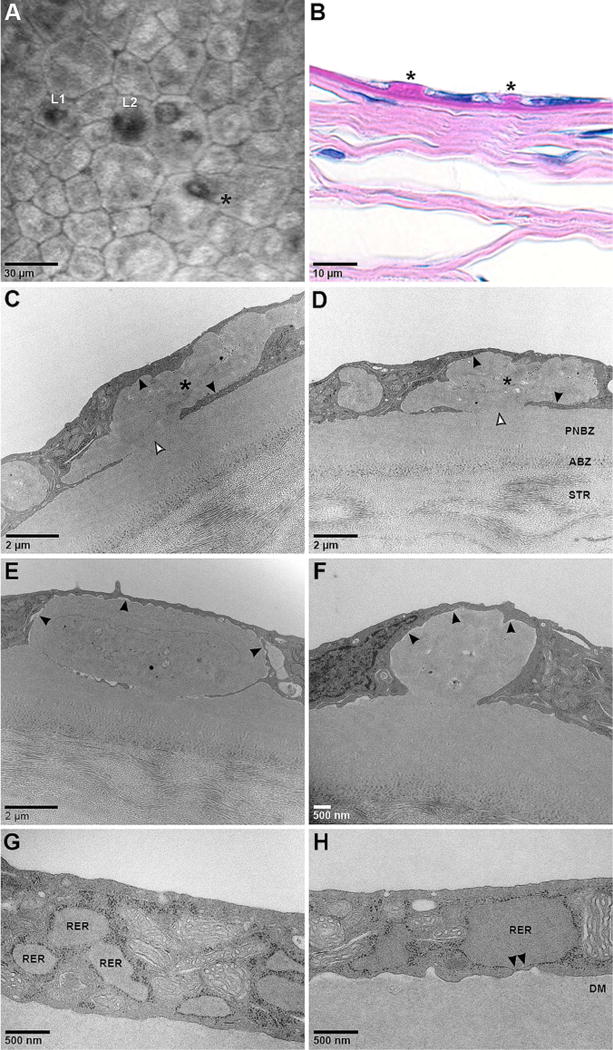
Varying appearances of guttae in homozygous Q455 K mutant mouse corneas. (A) Corneal confocal microscopy shows endothelial cell polymegathism and pleomorphism. Multiple forms of guttae are present in close proximity including Laing et al’s stages 1 (L1) and 2 (L2), and a distinct, sharply raised gutta suggested by Gottsch et al to be a characteristic of late onset FECD (*). (B) Periodic acid Schiff staining shows differently sized guttae (*). (C,D) Transmission electron microscopy shows large, irregularly shaped masses of material (*) with similar electron density and quality as the posterior non-banded zone (PNBZ) of Descemet membrane (DM) which appear to be intracellular as there is no obvious separation with the adjacent cytoplasm (closed arrowheads). Also shown is the anterior banded zone (ABZ) of DM adjacent to the stroma (STR). Internal features suggestive of cellular structures are present as are areas of focal attachment to DM (open arrowheads). (E,F) The other major form of this material shows more regular borders, more homogeneous internal features, and areas of clear separation from the cytoplasm (closed arrowheads). (G) Endothelial cells show areas of markedly dilated rough endoplasmic reticulum (RER). (H) In some areas the dilated RER becomes closely approximated to the basal cell membrane with loss of ribosomes (arrowheads) suggesting potential areas of membrane fusion which would enable contact and attachment between DM and RER contents. These areas of attachment could become the stalks of guttae.
TEM of homozygous Q455 K mouse corneas show large, irregular masses with similar characteristics as the posterior non-banded zone of DM (figure 1C,D). These masses appear intracellular without obvious separation between the adjacent cytoplasm, although they could be extracellular, invaginating the cell membrane in the plane of the section. Focal attachments to DM are present. The irregular border of these masses, internal features consistent with cellular structures, and lack of clear separation with adjacent cytoplasm (figure 1C,D) suggest an intracellular location.
The other major form of this material shows a smoother border with more homogeneous internal features, focal attachment to DM, and areas of separation with the cytoplasm (figure 1E,F), suggesting an extracellular location either by extrusion or death of the cell containing it.
DISCUSSION
A variety of guttae morphologies are seen in human FECD patients and our mouse models. The presence of multiple guttae forms in the same cornea of mice carrying a defined genetic defect suggests variation results from factors beyond the primary gene mutation.
A question arises regarding the origin of these presumably early stage, guttae-like structures in our mouse model. A previously reported feature of FECD endothelium is expanded rough endoplasmic reticulum (RER) (figure 1G).5, 6 In some areas, the dilated RER becomes closely approximated to the basal cell membrane with loss of ribosomes (figure 1H), giving an effaced appearance that suggests the potential for fusion of the cell and RER membranes which could enable attachment between DM and the RER contents. These areas would become the stalk of the gutta (figure 1C–F).
Thus, one mechanism (figure 2A) involves collections of membrane-bound intracellular material which coalesce and possibly fuse with the basal cell membrane (figure 2A). Ultimately, the gutta assumes an extracellular location either by extrusion or death of the cell containing it. An alternative involves localised, cellular secretion of material onto DM (figure 2B).
Figure 2.
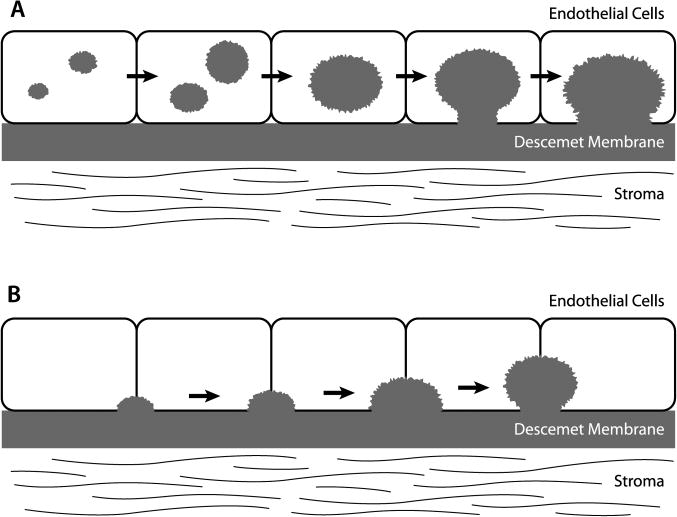
Schematic models of potential mechanisms of gutta formation. (A) One model involves collections of membrane-bound, intracellular, proteinaceous material which form, grow and potentially fuse with other similar structures giving rise to the bulk of a gutta. The collections come into close contact with the basal cell membrane with fusion of apposing membranes creating a ‘pore’ through which the proteinaceous material can attach to Descemet membrane, leading to the ‘stalk’ of a gutta. Over time, the appearance of the gutta can change with remodelling or addition of new material. (B) An alternate model involves secretion of proteinaceous material by the endothelial cells directly onto Descemet membrane, such that the bulk of the gutta is formed outside the cell with ongoing deposition resulting in growth of the gutta.
The similar appearances of guttae in our defined genetic model (figure 1A) with those described in human late onset FECD suggest that significant aspects of the disease are shared across species and genetic aetiologies. If so, elucidation of the origins of guttae in mice and men may provide insights into pathophysiology which could enhance development of novel FECD treatments.
Acknowledgments
Funding Grants from the National Institutes of Health (EY019874), J Willard and Alice S Marriott Foundation, Edward Colburn, Lorraine Collins, Richard Dianich, Mary Finegan, Barbara and Peter Freeman, Stanley Friedler, MD, Herbert Kasoff, Diane Kemker, Jean Mattison, Lee Silverman, Norman Tunkel, PhD (all to ASJ), and Research to Prevent Blindness (to Wilmer Eye Institute).
Footnotes
Correction notice This article has been corrected since it was published Online First. The word ‘gutta’ has been amended to ‘guttae’ in the article title.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data are available upon request from the corresponding author.
References
- 1.Fuchs E. Dystrophia epitheliali corneae. Graefes Arch Clin Exp Ophthalmol. 1910;76:478–508. [Google Scholar]
- 2.Friedenwald H, Friedenwald JS. Epithelial dystrophy of the cornea. Br J Ophthalmol. 1925;9:14–20. doi: 10.1136/bjo.9.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laing RA, Leibowitz HM, Oak SS, et al. Endothelial mosaic in Fuchs’ dystrophy. A qualitative evaluation with the specular microscope. Arch Ophthalmol. 1981;99:80–3. doi: 10.1001/archopht.1981.03930010082007. [DOI] [PubMed] [Google Scholar]
- 4.Gottsch JD, Sundin OH, Liu SH, et al. Inheritance of a novel COL8A2 mutation defines a distinct subtype of Fuchs corneal dystrophy. Investig Ophthalmol Vis Sci. 2005;46:1934–9. doi: 10.1167/iovs.04-0937. [DOI] [PubMed] [Google Scholar]
- 5.Jun AS, Meng H, Ramanan N, et al. An alpha 2 collagen VIII transgenic knock-in mouse model of Fuchs endothelial corneal dystrophy shows early endothelial cell unfolded protein response and apoptosis. Hum Mol Genet. 2012;21:384–93. doi: 10.1093/hmg/ddr473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engler C, Kelliher C, Spitze AR, et al. Unfolded Protein Response in Fuchs Endothelial Corneal Dystrophy: a Unifying Pathogenic Pathway? Am J Ophthalmol. 2010;149:194–202. doi: 10.1016/j.ajo.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]