

Abstract
The physiological control of cortisol synthesis in the adrenal cortex involves stimulation of adrenocorticotrophic hormone (ACTH) by hypothalamic corticotrophin-releasing hormone (CRH) and then stimulation of the adrenal by ACTH. The control loop of the hypothalamic–pituitary–adrenal (HPA) axis is closed by negative feedback of cortisol on the hypothalamus and pituitary. Understanding this system is required to master the diagnosis, differential diagnosis and treatment of endogenous hypercortisolism – Cushing's syndrome. Endogenous Cushing's syndrome is caused either by excess ACTH secretion or by autonomous cortisol release from the adrenal cortex. Diagnosis of cortisol excess exploits three physiological principles: failure to achieve the normal nadir in the cortisol diurnal rhythm, loss of sensitivity of ACTH-secreting tumours to cortisol negative feedback, and increased excretion of free cortisol in the urine. Differentiating a pituitary source of excess ACTH (Cushing's disease) from an ectopic source is accomplished by imaging the pituitary and sampling for ACTH in the venous drainage of the pituitary. With surgical removal of ACTH or cortisol-secreting tumours, secondary adrenal insufficiency ensues because of the prior suppression of the HPA axis by glucocorticoid negative feedback. Medical therapy is targeted to the anatomical location of the dysregulated component of the HPA axis. Future research will focus on new diagnostics and treatments of Cushing's syndrome. These are elegant examples of translational research: understanding basic physiology informs the development of new approaches to diagnosis and treatment. Appreciating pathophysiology generates new areas for inquiry of basic physiological and biochemical mechanisms.
Introduction
The Nobel Prize in Medicine or Physiology makes it abundantly clear that an understanding of physiological principles is paramount to excellence in clinical medicine (de Herder, 2014). One of the best examples of this translational interplay is the physiology of the hypothalamic–pituitary–adrenal (HPA) axis and the control of its individual components, as well as the pathophysiology of the axis (Raff, 1993; Raff & Findling, 2003; Raff et al. 2014). The Nobel Prize in Physiology or Medicine in 1977 was divided, one half jointly, to Roger Guillemin and Andrew V. Schally ‘for their discoveries concerning the peptide hormone production of the brain’, and the other half to Rosalyn Yalow ‘for the development of radioimmunoassays of peptide hormones’ (de Herder, 2014). These two towering achievements in Medicine and Physiology have revolutionized the diagnosis, differential diagnosis, and treatment of endogenous hypercortisolism – Cushing's syndrome. These advances led to our current understanding of the control of adrenocorticotrophic hormone (ACTH) synthesis from proopiomelanocortin (POMC), and hence cortisol release, by the hypothalamic peptide corticotrophin-releasing hormone (CRH) as well as to the ability to measure ACTH.
This review briefly summarizes the physiological control systems necessary to understand the pathophysiology, diagnosis, differential diagnosis, and treatment of Cushing's syndrome. Only through a thorough understanding of the physiology of the system can one properly diagnose and care for patients with Cushing's syndrome. Conversely, understanding the pathophysiology of Cushing's syndrome illuminates the normal control of the HPA axis.
Physiology of the hypothalamic–pituitary–adrenal axis
The HPA axis exists primarily to generate a basal cortisol rhythm and a cortisol response to a wide variety of stimuli collectively, but somewhat imprecisely, called ‘stress’ (McEwen & Sapolsky, 1995; Jacobson, 2005). The axis encompasses physiological feedback and central integrative control, and the control of the synthesis of CRH in the hypothalamus, ACTH (also known as corticotrophin) in the anterior pituitary, and cortisol in the adrenal cortex.
Hypothalamic–pituitary–adrenal axis (Fig.1)
Figure 1. The hypothalamic-pituitary-adrenal axis.
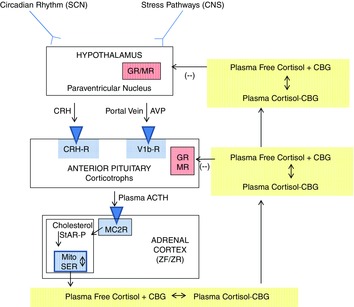
The suprachiasmatic nucleus (SCN) has circadian rhythm inputs and neural stress pathways in the central nervous system (CNS) have inputs into the corticotrophin-releasing hormone (CRH) and vasopressin (AVP) neuronal cell bodies in the paraventricular nucleus. CRH and AVP are released into capillaries in the median eminence that merge to form the portal veins; these drain into the anterior pituitary where another capillary network is formed. CRH and AVP stimulate the corticotrophs in the anterior pituitary to release adrenocorticotropic hormone (ACTH). ACTH stimulates the melanocortin 2 receptors (MC2R; also known as the ACTH receptors) on the steroidogenic cells of the zona fasciculata (ZF) and zona reticularis (ZR). Binding of ACTH to the MC2R stimulates the production of cAMP, which activates StAR-protein to mediate cholesterol transport into the mitochondria (the rate-limiting step of steroidogenesis). Cholesterol is the substrate for the initial step of steroidogenesis (see Fig. 2). Cortisol is released into the plasma where it binds reversibly to corticosteroid-binding globulin (CBG; also known as cortisol-binding globulin). CBG-bound plasma cortisol circulates throughout the body. In the capillaries in target tissue, it dissociates from CBG and diffuses into the target cell. Cortisol negative feedback inhibition is exerted in the anterior pituitary and the hypothalamus via the binding of cortisol to glucocorticoid (GR) and mineralocorticoid (MR) receptors. Reproduced with permission from Raff et al. (2014).
Circadian and stress inputs alter the activity of CRH (parvocellular) neurons in the paraventricular nucleus of hypothalamus. Many hypothalamic neurons that synthesize CRH also synthesize arginine vasopressin (Whitnall et al. 1987; Mouri et al. 1993); it is thought that increased vasopressin (AVP) release from these hypophysiotropic nerves, along with CRH, is necessary for the generation of a full ACTH response to a variety of stimuli (Whitnall et al. 1987; Mouri et al. 1993; Raff, 1993; Jacobson, 2005). CRH and AVP in the portal blood vessels enters the anterior pituitary and stimulates the activity of the corticotrophs – cells in the anterior pituitary that synthesize the large precursor molecule proopiomelanocortin (POMC). Furthermore, magnocellular AVP neurons with axons that terminate in the posterior pituitary also participated in the control of ACTH via short portal veins from the posterior to the anterior pituitary, and by axons that terminate in the median eminence (Raff, 1987, 1993).
The pituitary corticotrophs preferentially produce ACTH by post-translational processing of POMC (Raff et al. 2014), and ACTH is released into the pituitary capillaries from which it drains into the systemic circulation. ACTH stimulates the adrenal cortex to produce cortisol through processes described below. Cortisol is released from the adrenal cortex in the free form most of which binds to carrier proteins in the plasma compartment. Corticosteroid-binding globulin (CBG) is the high specificity–low capacity binding protein produced by the liver (Lin et al. 2010). Cortisol also binds to albumin in the plasma. At physiological cortisol secretion rates, most (∼95%) of the circulating cortisol is bound to CBG in the plasma (Lin et al. 2010). Cortisol must dissociate from CBG in the capillaries of target tissue in order to diffuse into the target tissue and bind to the glucocorticoid and, in some cases, the mineralocorticoid receptor.
An absolutely critical component of the HPA axis that must be understood from a physiological and clinical point of view is cortisol (glucocorticoid) negative feedback, the ultimate goal of which is to prevent ACTH over-secretion and to terminate the response to stressful stimuli (Keller-Wood & Dallman, 1984; Raff, 1987). This feedback is exerted at both the hypothalamic and pituitary levels, and perhaps within other areas of the central nervous system. The dynamics of cortisol negative feedback are quite complex. For the purposes of this review, we will focus on classic ‘delayed’ negative feedback that requires the genomic actions of cortisol and is expressed over hours (Keller-Wood & Dallman, 1984). It is this negative feedback that can be exploited in clinical testing of Cushing's syndrome.
Adrenal steroidogenesis (Fig.2)
Figure 2. Simplified schematic diagram of cortisol synthesis in the zona fasciculata cell.
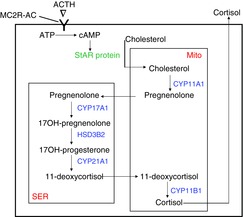
As shown in Fig. 1, ACTH binds to the melanocortin-2 receptor (MC2R), activates adenylate cyclase (AC), and increases production of cAMP. This activates a phosphorylation cascade leading to the activation of the steroidogenic acute regulatory (StAR) protein, which mediates cholesterol transport from the cytoplasm to the inner mitochondrial (Mito) membrane. There, cholesterol is converted to pregnenolone by CYP11A1 (P450scc; side chain cleavage). Pregnenolone diffuses out of the mitochondria through the cytosol into the smooth endoplasmic reticulum (SER) where enzymatic conversion occurs sequentially to 17OH-pregnenolone by CYP17A1 (P450c17; 17-hydroxylase) to 17OH-progesterone by HSD3B2 (3-beta-hydroxylase) to 11-deoxycortisol by CYP21A1 (P450c21; 21-hydroxylase). 11-deoxycortisol then diffuses into the mitochondria for the last step in which cortisol is produced by CYP11B1 (P450c11; 11-beta-hydroxylase). Cortisol is not stored in the cell; rather, it diffuses into the extracellular fluid and then to the capillaries (see Fig. 1).
There are three anatomical and functional zones of the adrenal cortex. The zona glomerulosa synthesizes the mineralocorticoid aldosterone, and the zona fasciculata and reticularis synthesize cortisol and the adrenal androgens. These steroidogenic pathways have been extensively studied and reviewed (Gallo-Payet & Battista, 2014), and only the pathway for the synthesis of cortisol (‘glucocorticoid pathway’) in the zona fasciculata cell will be briefly summarized here.
ACTH binds to the melanocortin 2 receptor (MC2R – a G-protein coupled receptor) and activates the production of cAMP by adenylate cyclase (Gallo-Payet & Battista, 2014). There are probably other second messengers that can influence the activation of the pathway, but cAMP is considered the most important (Gallo-Payet & Battista, 2014). Increases in cytosolic cAMP lead to a phosphorylation cascade that activates the steroidogenic acute regulatory (StAR) protein (Gallo-Payet & Battista, 2014). StAR protein facilitates the transport of lipophilic cholesterol (substrate for the initial step of steroidogenesis) from the cytosol to the inner mitochondrial membrane where it is converted to pregnenolone by CYP1A1 (P450scc; ‘side chain cleavage’). From there on, steroidogenesis occurs obligatorily by the shuttling of steroid products out of, and then back into the mitochondria where cortisol is produced in the final step by CYP11B1 (P450c11; ‘11β-hydroxylase’). Because cortisol is a steroid, it is not stored within the cell; it diffuses into the extracellular fluid and then into the capillaries of the adrenal cortex. Knowledge of the steroidogenic pathway will become important when we discuss drugs that can be used to lower cortisol synthesis in patients with Cushing's syndrome.
Basal activity – circadian rhythm
Like most endocrine systems, the HPA axis exhibits a basal (unstressed) circadian rhythm with cortisol peaking soon after awakening (usually 06.00–08.00 h in people with a diurnal lifestyle), and at its nadir at or a little after midnight (Spiga et al. 2014). It is important to note, however, that like other endocrine rhythms, there is pulsatile, more ultradian-like rhythm superimposed on this circadian rhythm (Lightman & Conway-Campbell, 2010). Therefore, whenever studying the HPA axis whether experimentally or clinically, careful attention has to be paid to the time of day and the conditions under which the studies are done (Raff et al. 2014). In fact, as described below, evaluation of alterations in the circadian rhythm of the HPA axis can be exploited in the diagnosis of Cushing's syndrome (Raff et al. 2014). It is important to emphasize this is a basal, unstressed property of the HPA axis.
Stimuli – ‘stress and stressors’
The concept of a stress-response has been used for decades to characterize the HPA axis (McEwen & Sapolsky, 1995). Many have tried to define ‘stress’ in terms of the physiological response. It is important to point out that none of the definitions of stress are all encompassing. We propose that the use of the term ‘stimuli’ may be more precise than ‘stressor’, because there is obviously great individuality of what a person finds stressful. Regardless, there is a long list of stimuli that activate the HPA axis including neurogenic/psychological factors (such as pain, anxiety, public speaking) and physiological (such as hypotension, hypoxia, infection, hypoglycaemia), and then some that are a mixture of neurogenic and physiological stimuli (e.g. trauma, burn) (Jacobson, 2005). Some of these can be classified as real or perceived threats to homeostasis. In fact, an operational definition of ‘stress’ could be anything that increases the activity of the HPA axis. Ultimately, the increase in cortisol improves the ability to respond to the stressor.
Effects of cortisol: glucocorticoid vs. mineralocorticoid receptors
The classic response elicited by cortisol is mediated by binding to intracellular receptors – the glucocorticoid and mineralocorticoid receptors, which are part of the nuclear receptor family (Vandevyver et al. 2014). At first, one might think that all of the effects of cortisol should be mediated by the glucocorticoid receptor (GR). However, cortisol also can bind to the mineralocorticoid receptor (MR) in the kidney and brain. What, then, prevents cortisol from being a major physiological mineralocorticoid considering that free cortisol circulates in concentrations far greater than free aldosterone? It turns out that the renal MR is protected from the effects of cortisol by virtue of the conversion of cortisol to the inactive molecule cortisone by the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11BHSD2; Tomlinson & Stewart, 2001). This explains why, when cortisol is pathologically increased (as in Cushing's syndrome), sodium retention, potassium wasting, and hypertension can ensue – cortisol has increased to levels that overwhelm the ability of 11BHSD2 to render it inactive, and cortisol exerts aldosterone-like effects in the kidney. It also helps to explain why hypokalaemia is more common in ectopic ACTH in which cortisol secretion is usually more prodigious than in Cushing's disease (Aron et al. 1997).
Cushing's syndrome
This eponym honours the seminal description of endogenous hypercortisolism by Dr Harvey Cushing, one of the giants of modern neurosurgery (Newell-Price et al. 2006). This review will primarily focus on endogenous (i.e. spontaneous) Cushing's syndrome, although exogenous (iatrogenic) Cushing's syndrome due to pharmacological doses of synthetic glucocorticoids is a far more common problem.
There are several subtypes of Cushing's syndrome that can be partitioned by whether the cause of increased cortisol secretion is ACTH dependent or ACTH independent (Fig.3).
Figure 3. Subtypes of endogenous Cushing's syndrome.
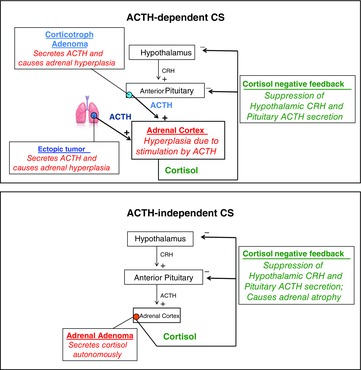
ACTH-dependent Cushing's syndrome is usually caused by a benign ACTH-secreting corticotroph (pituitary) adenoma. More rarely, a non-pituitary (ectopic) tumour can synthesize and release ACTH. Either way, the adrenal cortex becomes hyperplastic and the increased cortisol negative feedback suppresses hypothalamic CRH release and the otherwise normal corticotrophs in the anterior pituitary. In ACTH-independent Cushing's syndrome, the cause is usually a benign adenoma that autonomously secretes cortisol. This suppresses CRH and ACTH release through negative feedback and leads to atrophy of the otherwise normal cortisol-synthesizing adrenocortical cells.
ACTH-dependent Cushing's syndrome
In this form, the adrenal cortex has received excessive stimulation of the MC2R, leading to inappropriate and pathological adrenal cortisol secretion (Fig.3). The two sources of this excessive ACTH are (a) an ACTH-secreting pituitary adenoma and (b) ectopic (non-pituitary) ACTH secretion, usually due to a neuroendocrine tumour, (small cell lung carcinoma or carcinoid tumours of the lungs being most common; Isidori & Lenzi, 2007). Pituitary adenomas are by far the most common cause of ACTH-dependent Cushing's syndrome and are referred to as Cushing's disease (Raff et al. 2014).
A variety of cellular mechanisms have been proposed for the development of ACTH-secreting tumours. It is generally acknowledged that pituitary corticotroph adenomas are clonal in nature (Zhou et al. 2014). In this regard, genetic and epigenetic mutations of tumour suppressor genes are of great interest as potential mechanisms of tumorogenesis (Zhou et al. 2014). Among the genes reported to be altered in corticotroph tumours are cyclin 1, aryl hydrocarbon receptor interacting protein (AIP), bone morphogenic protein-4 (BMP-4), cadherin 1 (CDH1), and suppressor of cytokine signalling 1 (SOCS1) (Zhou et al. 2014).
Another area of interest relates to the relative resistance of corticotroph tumours to glucocorticoid negative feedback inhibition. Brg 1 is a protein involved in the feedback of glucocorticoid-induced POMC expression and is inappropriately expressed in corticotroph tumours (Bilodeau et al. 2006). An interesting possible mechanism relates to the role of oestrogens in the development of Cushing's disease. There is a high female:male ratio in Cushing's disease and a majority of male cases are prepubertal. This suggests that oestrogens may have a role in the pathogenesis of Cushing's disease (Storr et al. 2004). Finally, polymorphisms in growth factor receptors such as FGFR4 may also be involved in the alterations in growth and sensitivity to glucocorticoid negative feedback in corticotroph adenomas (Nakano-Tateno et al. 2014).
ACTH-independent Cushing's syndrome
There are two general types of ACTH-independent Cushing's syndrome. In both types, circulating glucocorticoid activity is increased without ACTH stimulation. In endogenous ACTH-independent Cushing's syndrome, the adrenal gland(s) produce cortisol autonomously without ACTH stimulation. This is most commonly due to a benign adrenocortical adenoma, but can be from adrenal cortical carcinoma, or other more rare forms of bilateral adrenal disease (Carroll & Findling, 2009).
In the second type of ACTH-independent Cushing's syndrome, excessive glucocorticoid activity is produced by administration of exogenous glucocorticoids. The most common cause is oral glucocorticoid therapy (e.g. prednisone) in supraphysiological doses for the treatment of a wide variety of inflammatory diseases (Stahn & Buttgereit, 2008). The use of oral glucocorticoid therapy is common and, as a consequence, a common cause of serious side-effects including hypertension, obesity, osteoporosis, and glaucoma. Other routes of glucocorticoid therapy have also been shown to cause exogenous or iatrogenic Cushing's syndrome including intraarticular, intrathecal, inhaled, epidural, and even topical or ocular glucocorticoids (Hopkins & Leinung, 2005; Chiang et al. 2006; Abraham, 2007; Christensson et al. 2008). The effects of exogenous glucocorticoids can be exacerbated by drugs that decrease the metabolism of glucocorticoids, such as antiretroviral therapy (Valin et al. 2009).
As shown in Fig.3, long-term suppression of the HPA axis due to an adrenal adenoma suppresses ACTH secretion (negative feedback) and leads to atrophy of the remaining normal adrenal cortices. Therefore, removal of an adrenal adenoma can lead to a precipitous and, if untreated, life-threatening decrease in cortisol secretion. It may take months for the HPA axis to recover, so physiological glucocorticoid replacement is necessary in this scenario. Following the same logic, one of the more relevant clinical challenges from a physiological point of view is the approach to wean patients from long-term glucocorticoid therapy (Hopkins & Leinung, 2005). Just as with an autonomous cortisol secreting adenoma, the HPA axis may be suppressed by supraphysiological doses of glucocorticoids and the adrenal cortices can atrophy. This is why weaning patients from long-term glucocorticoid therapy can be quite challenging.
Genetic adrenal alterations causing cortisol excess (Fig.4)
Figure 4. Summary of different pathways leading to increased proliferation and function of cortisol-synthesizing cells in the adrenal cortex.
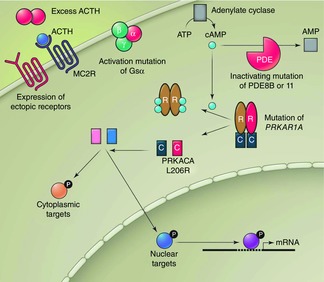
Factors associated with increased activity are shown in red and include excess ACTH stimulating the MC2R (see Fig. 3). Other ACTH-independent causes include expression of receptors to other secretagogues, activating mutation of the G-protein subunit Gs-alpha, inactivating mutations of phosphodiesterases PDE8B or 11, a mutation of PRKAR1A (Carney complex), and mutations in protein kinase A catalytic subunit PRKACA. Reproduced with permission from Kirschner (2014).
Several genetic alterations in the adrenal glands have been identified as causing adrenal enlargement or excess cortisol secretion from cortisol-secreting adrenal adenomas (Yaneva et al. 2010). For example, in McCune-Albright syndrome, an activating mutation of the G-protein subunit Gsα in the adrenal gland leads to the development of Cushing's syndrome early in life. In the Carney complex, a mutation of PRKAR1A causes altered regulation of protein kinase A (PKA) and subsequent adrenal nodules and cortisol excess.
More recently, the work of four different groups from around the world has identified the same mutation in the PKA signalling pathway for cortisol production (Beuschlein et al. 2014; Cao et al. 2014; Goh et al. 2014; Sato et al. 2014). PKA is a cAMP-dependent kinase that is critical in many pathways including adrenal signalling and regulation. PKA is a heterotetrameric complex consisting of two catalytic and two regulatory subunits. In the normal situation, ACTH binds to MC2R and increases cytosolic cAMP levels (see Fig.2). This intracellular cAMP increase causes binding of the PKA regulatory subunit and release of the catalytic subunit of PKA (PRKACA), which then phosphorylates targets in the cytoplasm and nucleus. The recent work of four groups independently identified the same L206R mutation in PRKACA in cortisol-secreting adrenal adenomas (Giordano, 2014; Kirschner, 2014). This mutation was found in 35 to 69% of the cortisol-secreting adenomas that were studied. The discovery of this mutation may hold far-reaching implications for identifying the genetic cause of other forms of Cushing's syndrome.
Plasma ACTH concentrations in the various forms of Cushing's syndrome
Assessing plasma ACTH levels in patients with the various forms of Cushing's syndrome, shown in Fig.3, is an excellent physiological–clinical correlation. At first blush, it would seem logical that patients with ACTH-dependent Cushing's syndrome should have circulating plasma ACTH concentrations well above the reference range. Otherwise, how can one explain the cortisol hypersecretion and adrenocortical hyperplasia that occurs? This is one of the most important phenomena in endocrine physiology and medicine, and its appreciation requires an understanding of the subtleties of glucocorticoid negative feedback.
As shown in Fig.5, plasma ACTH concentrations are often within the reference range in patients with Cushing's disease. The reference range is often mistakenly referred to in general parlance as a ‘normal’ range, which is not the case. It is common for pituitary adenomas to express the glucocorticoid receptor and still be sensitive to cortisol negative feedback (Yanovski et al. 1998). However, their sensitivity to cortisol inhibition is attenuated compared to normal corticotrophs. This restrains the tumour from secreting even more ACTH and from more rapid growth. However, over time, ACTH secretion creeps up and induces adrenal hypertrophy. Probably the best way to understand this is to compare the plasma ACTH levels in patients with Cushing's disease (pituitary ACTH-dependent Cushing's syndrome) to those achieved in patients with ACTH-independent Cushing's (i.e. adrenal tumours) in whom ACTH is clearly suppressed (Fig.5). In patients with Cushing's disease, the plasma ACTH is considered ‘inappropriately not-suppressed’ (Raff & Findling, 2003; Raff et al. 2014). If the pituitary corticotrophs were all completely normal, ACTH should be suppressed in the presence of cortisol hypersecretion. The restraining effect of cortisol negative feedback on pituitary adenoma growth is further highlighted by the enlargement of these tumours in Nelson's syndrome (also known as corticotroph tumour progression; Karl et al. 1996). Nelson's syndrome occurs in patients with Cushing's disease in whom bilateral adrenalectomy has been performed to treat the hypercortisolism. Even with physiological glucocorticoid replacement, pituitary tumours can grow considerably and become problematic. A possible mechanism of this is a somatic frame-shift mutation in the glucocorticoid receptor gene in corticotroph tumours that evolve into ACTH-secreting macroadenomas causing Nelson's syndrome (Karl et al. 1996).
Figure 5. Plasma ACTH in Cushing's syndrome subtypes.
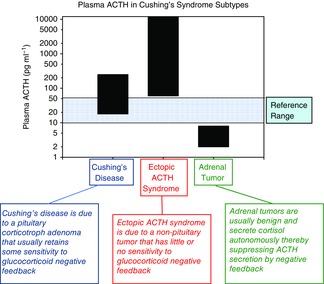
Plasma ACTH concentrations (pg ml−1) in patients with ACTH-dependent Cushing's syndrome (Cushing's disease (pituitary corticotroph adenomas) and ectopic ACTH) and ACTH-independent Cushing's syndrome (adrenal tumour). Note that plasma ACTH is within the reference range (blue shading) in many patients with Cushing's disease and that, on average, patients with ectopic ACTH have very high plasma ACTH. Furthermore, patients with adrenal autonomy usually have suppressed plasma ACTH due to increased cortisol negative feedback suppression of the hypothalamus and pituitary. To convert to pmol l−1, multiply pg ml−1 by 0.2202. From Raff et al. (2014) with permission.
Ectopic ACTH-secreting tumours tend to be more dysregulated and much less sensitive to cortisol negative feedback, and so plasma ACTH is usually frankly increased (Fig.5) and the hypercortisolism more prodigious (Aron et al. 1997; Isidori & Lenzi, 2007).
A physiological approach to diagnosis
The features of Cushing's syndrome, such as obesity, hypertension, hirsutism, depression, and dyslipidaemia, are ubiquitous in the general population (Findling & Raff, 2005). Because endogenous Cushing's syndrome is relatively uncommon, it is critical that the diagnostic approach is inexpensive and efficient so that large number of patients can be screened, but also has an excellent diagnostic performance. This topic has been recently reviewed in great detail (Raff, 2009; Raff et al. 2014), so this section will briefly describe the physiological concepts behind each test.
Late-night salivary cortisol
One of the earliest disruptions of the HPA axis in Cushing's syndrome of any aetiology is the failure to achieve the full decrease in cortisol secretion at its normal diurnal nadir (between bedtime and 02.00 h). It was convincingly demonstrated that measuring an increase in unstressed serum cortisol level at midnight has high sensitivity for identification of patients with Cushing's syndrome (Newell-Price et al. 1995; Papanicolaou et al. 1998). However, obtaining a late-night unstressed blood sample in routine clinical practice is not feasible. For that reason, home sampling of salivary cortisol at bedtime has become the test of choice for the initial screening of patients with suspected Cushing's syndrome (Raff et al. 1998; Raff, 2012). This test, with a sensitivity and specificity both >90–95%, evolved directly from basic physiological principles (i.e. the sometimes subtle disruption of the circadian rhythm) and has revolutionized the approach to the patient suspected of having endogenous Cushing's syndrome (Carroll et al. 2008, 2009).
Low-dose dexamethasone suppression test
This test also arose out of basic physiological principles – that abnormal ACTH-secreting tumours (whether pituitary or ectopic) lose sensitivity to glucocorticoid negative feedback. This is intuitively obvious since, if these ACTH-secreting cells had normal sensitivity to cortisol negative feedback, the patients would not have clinical Cushing's syndrome! This is exploited with the use of a low dose of the pure glucocorticoid receptor agonist dexamethasone. In the most common approach, a small dose of dexamethasone (1 mg) is given at 23.00 h, and serum cortisol is measured at 08.00 h the next morning (Findling et al. 2004b). If the patient does not have endogenous Cushing's syndrome, the morning cortisol concentration should be suppressed. This test is particularly useful if it is not convenient to assess salivary cortisol, if the patient does not have a regular diurnal lifestyle, or if one suspects very mild Cushing's syndrome because of incidentally discovered adrenal neoplasms (adrenal incidentalomas; Reimondo et al. 2011).
Urine free cortisol
The concept of clearance is well known to physiologists. Although most cortisol is cleared in the kidney after glucuronidation in the liver, the free cortisol in the plasma does appear in the urine, theoretically in proportion to the secretion rate from the adrenal cortex (Finken et al. 1999). Although 24 h urine collections for the analysis of free cortisol excretion are widely used, they are cumbersome for the patient and perform least well as a screening test, particularly if the Cushing's syndrome is mild (Alexandraki & Grossman, 2011; Petersenn et al. 2013; Elias et al. 2014). However, they can be useful as a confirming test before differential diagnosis is entertained.
Differential diagnosis
As described above and shown in Fig.4, the measurement of a frankly suppressed plasma ACTH is usually sufficient to identify patients with an autonomous cortisol-secreting adrenal tumour. After ACTH-independent Cushing's syndrome is confirmed, a CT scan should be performed to lateralize the source of adrenal cortisol hypersecretion.
Pituitary (Cushing's disease) vs. ectopic ACTH
It is commonly assumed that imaging techniques (e.g. MRI or CT) will discriminate between pituitary and ectopic ACTH-secreting tumours. When the diagnosis of ACTH-dependent Cushing's syndrome is made, it is typical to first perform a very high quality, pituitary directed magnetic resonance imaging (MRI) study. If a pituitary neoplasm is clear and unequivocal, it is common to refer the patient directly to the neurosurgeon (Raff & Findling, 2003). Unfortunately, pituitary tumours are often too small to visualize with even the most sophisticated imaging modalities (Hall et al. 1994); ectopic tumours are also often not identified on imaging and remain occult in many patients (Zemskova et al. 2010).
If imaging is equivocal or negative, the best approach to differential diagnosis of ACTH-dependent Cushing's syndrome has, of course, a physiological solution – selective venous sampling of the pituitary venous effluent to determine if it is the source of ACTH hypersecretion. The logic of this approach is intuitive and relies on the classic Fick equation that relates (a) the concentration gradient between inflow (arterial blood) and outflow (venous blood) of an organ secreting a hormone, (b) the secretion rate of the hormone, and (c) the total blood flow through the organ. If one holds secretion rate constant, the concentration gradient will be inversely proportional to the blood flow. Therefore, by sampling the venous effluent of organs with a higher secretion rate relative to a lower blood flow, one may be able to locate the source of the secretory product of the organ.
This physiological concept has been exploited in a clinical test called inferior petrosal sinus (IPS) sampling for ACTH (Findling et al. 1991; Oldfield et al. 1991; Fig.6). Briefly, catheters are placed in both inferior petrosal sinuses that are the direct recipient of the venous drainage from the anterior pituitary. A reference catheter is placed in the inferior vena cava (IVC) to assess the ACTH concentration in the systemic circulation. Typically, exogenous CRH is injected to maximally stimulate the abnormal (tumoral) pituitary corticotrophs, and blood samples for the measurement of plasma ACTH concentration are obtained from both petrosal sinuses and the IVC. If there is a significant ACTH gradient from the IPS to the IVC, then the pituitary is the source of ACTH hypersecretion. If there is not a significant gradient, then the excess ACTH is coming from a non-pituitary (ectopic) source, assuming the IPS catheters were placed correctly and there is not anomalous venous drainage from the pituitary (Doppman et al. 1999; Findling et al. 2004a).
Figure 6. Inferior petrosal sinus sampling to differentiate Cushing's disease from ectopic ACTH syndrome.
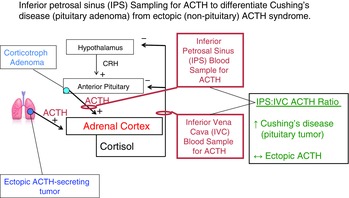
The use of inferior petrosal sinus (IPS) sampling for ACTH (after administration of corticotrophin-releasing factor (CRH)) in the differentiation of pituitary vs. ectopic ACTH-dependent Cushing's syndrome. A pituitary tumour will secrete ACTH into the normal venous drainage of the anterior pituitary that drains into the inferior petrosal sinuses bilaterally. Catheters are placed in the IPS and a peripheral vein (usually the inferior vena cava (IVC)) as a reference. If the pituitary is the source of excess ACTH, there will be an increased gradient of ACTH concentration in the IPS relative to the IVC reference sample. If the ACTH is coming from a non-pituitary source, the IPS ACTH concentration will not be significantly increased because the cortisol excess has suppressed CRH secretion as well as ACTH secretion from the otherwise non-diseased anterior pituitary.
IPS sampling for ACTH indicates whether the pituitary is the source of ACTH hypersecretion. If it is not, then one must search for the occult ectopic ACTH-secreting tumour (Doppman et al. 1989; Ilias et al. 2005; Isidori et al. 2006). It would seem logical that extension of this application of the Fick principle to venous sampling of every likely organ to localize a source of ectopic ACTH secretion would be useful. However, from a physiological point of view, venous catheterization of the likely organs is usually not helpful, primarily because their blood flows are too high to generate a consistent diagnostic arterial–venous ACTH gradient (Doppman et al. 1989).
Treatment
Once the tumoral cause of cortisol excess is identified, the typical first step is surgical removal. In the case of a cortisol secreting adrenal adenoma, the best approach is unilateral adrenalectomy, usually performed laparoscopically with minimal morbidity and mortality (Young & Thompson, 2005). One should remember the physiological concept of cortisol negative feedback post-operatively as prior, chronic cortisol excess leads to the suppression of ACTH and undoubtedly has led to atrophy of the contralateral adrenal gland. This requires post-operative glucocorticoid replacement to prevent sudden, potentially lethal secondary adrenal insufficiency. As described above for exogenous glucocorticoid therapy, weaning from glucocorticoid replacement may take months to allow sufficient time for the hypothalamus and pituitary to ‘wake up’ from long-term suppression thus increasing ACTH secretion and inducing growth and recovery of the remaining adrenal gland.
For Cushing's disease, the surgical treatment of choice is transphenoidal pituitary surgery with the removal of the pituitary corticotroph adenoma without disrupting other pituitary function. Ectopic ACTH production is best treated with appropriate surgical removal of the causative tumour. Complete removal of the chronically ACTH-secreting tumour also results in secondary adrenal insufficiency (Isidori et al. 2006; Isidori & Lenzi, 2007). In this case, the adrenal cortices are hypertrophied, not atrophied. However, the cortisol excess has suppressed CRH secretion from the hypothalamus as well as directly suppressed ACTH secretion from the non-tumoral, previously normal corticotrophs in the anterior pituitary. Therefore, glucocorticoid therapy must be given post-operatively to these patients as well.
The surgical failure rate for Cushing's disease is significant as is the recurrence rate many years after successful pituitary surgery (Atkinson et al. 2005; Abdelmannan et al. 2013; Alexandraki et al. 2013; Barbot et al. 2013; Hameed et al. 2013). For that reason, patients should be followed for many, many years to screen for recurrence (Raff, 2012, 2013). Reoperation of the causative tumour is often not feasible so other therapeutic measures must be employed. Fortunately, our understanding of the physiology of the HPA axis and cortisol production has made a number of pharmacological agents available to treat cortisol excess.
At present, three targets of medical therapy for Cushing's syndrome exist (Fig.7): (a) pituitary (ACTH production), (b) adrenal (cortisol production,) and (c) peripheral tissues (glucocorticoid effects). Of the agents available in these three groups, only two medications are currently approved for use by the US Food and Drug Administration (FDA). The first of these is mifepristone, a glucocorticoid receptor antagonist. It is a competitive inhibitor of cortisol at the GR and is effective because its affinity for the GR is higher than cortisol. When administered, ACTH and cortisol levels increase due to blockade of the GR on the hypothalamic CRH cell bodies and the pituitary corticotrophs (reduction in negative feedback agonism). However, at sufficient doses of mifepristone, the effects of even this increased cortisol are blocked (Fleseriu & Petersenn, 2012; Nieman, 2013). The increase in cortisol secretion due to a reduction in negative feedback with GR receptor blockade obviates the monitoring of serum cortisol levels to determine the effectiveness of treatment. Because of this, one has to rely on more subjective clinical assessments to determine if cortisol excess is being over- or under-treated.
Figure 7. Mechanism of action and targets for therapy in Cushing disease.
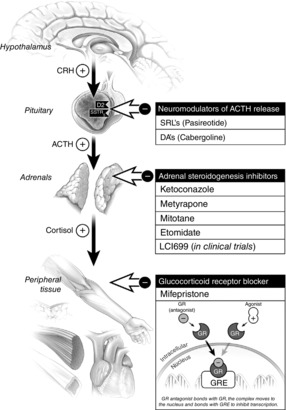
The medical therapies shown in the figure can be exerted at the pituitary, adrenal gland, or by blocking the binding of cortisol to the glucocorticoid receptor in the target tissue. ACTH, adrenocorticotropic hormone; DA, dopamine agonist; GR, glucocorticoid receptor; GRE, glucocorticoid response elements; SRL, somatostatin receptor ligand. Reproduced with permission from Fleseriu (2012).
The second FDA approved medication for Cushing's syndrome is pasireotide, a somatostatin receptor analogue that targets corticotroph adenoma ACTH production. Corticotroph adenomas express somatostatin receptors, specifically type 5, in higher density. Pasireotide binds preferentially to the somatostatin type 5 receptor and decreases ACTH secretion from corticotroph adenomas (Colao et al. 2012). Pasireotide has not been extensively studied in ectopic ACTH secretion but is probably not effective due to receptor expression differences in ectopic ACTH-secreting tumours.
The oldest medications used to treat Cushing's syndrome are directed at the adrenal gland. These medications decrease cortisol production by interfering with steroidogenesis, and hence are called steroidogenesis inhibitors. Metyrapone (11β-hydroxylase inhibitor) either alone, or in combination with other inhibitors of steroidogenesis, has been used for many decades to decrease adrenal steroidogenesis (Fleseriu & Petersenn, 2012; Fleseriu, 2012). The obvious challenge of using these drugs also illustrates how important understanding cortisol negative feedback is in developing medical therapy. As mentioned previously, corticotroph adenomas are restrained somewhat by glucocorticoid negative feedback, so decreasing cortisol with drugs can result in increases in ACTH secretion which can ‘break through’ the pharmacological enzyme block. Also, metyrapone is not commercially available in the United States (Fleseriu & Petersenn, 2012).
Over the years, the most frequently used steroidogenesis inhibitor is the antifungal agent ketoconazole. Ketoconazole exerts its antifungal effects by inhibiting ergosterol which is vital to the fungal cell membrane (Newell-Price, 2014). Ketoconazole also inhibits several enzymes involved in the steroid biosynthesis pathway (C17–20 lyase, 17α-hydroxylase, and 11β-hydroxylase). The inhibition of 11β-hydroxylase is key in its use for Cushing's syndrome as 11β-hydroxylase is the enzyme responsible for catalysing the final step in cortisol synthesis. Other adrenocortical directed inhibitors are listed in Fig.7, including a new agent (LC1699) under investigation that shows promise (Fleseriu, 2012).
Commentary on areas of future research
The diagnosis and treatment of Cushing's syndrome continues to be among the most challenging in medicine. There are several areas of research that we hope will receive attention and improve in the next decade. The first is continued improvement in imaging modalities. Even with the newest techniques, small pituitary adenomas are difficult to visualize and impossible to differentiate from benign, non-functional pituitary neoplasms that occur with significant frequency in the general population (Hall et al. 1994). Localization of otherwise occult ectopic ACTH-secreting tumours is even more challenging. The development of more functional nuclear imaging techniques targeting POMC production would be of great benefit in this regard.
Many ectopic tumours produce large amounts of POMC intermediates, and it has been suggested that adoption of assays for POMC and its fragments may improve the diagnosis of ectopic ACTH and reduce the need for invasive selected venous sampling (Page-Wilson et al. 2014).
One area that can certainly improve is treatment. Recurrence or failure to obtain a cure during pituitary neurosurgery continues to be a serious problem even in the best of neurosurgical hands (Atkinson et al. 2005; Abdelmannan et al. 2013; Alexandraki et al. 2013; Barbot et al. 2013; Hameed et al. 2013). It is hoped that improved advances in endoscopic techniques may improve immediate cure and perhaps even reduce the rate of recurrence (Tabaee et al. 2009). It is also possible that pre- and post-operative radiotherapy may also improve the treatment of pituitary tumours (Jagannathan et al. 2007). Finally, after many years of little progress, the development of new and improved medical therapies should continue. New compounds that inhibit ACTH-secreting tumours are being identified and may be of possible use in treating patients (Bangaru et al. 2010).
Conclusion and summary statement
This review has stressed how important it is to have a thorough knowledge of all aspects of the physiology of the HPA axis in order to understand the diagnosis, differential diagnosis, and treatment of Cushing's syndrome. Understanding that the normal diurnal nadir in cortisol is not achieved and that abnormal ACTH-secreting cells are not fully sensitive to cortisol negative feedback is the basis of the modern approach to diagnosis. It is often difficult to localize a radiologically unapparent source of ACTH, so knowledge of the Fick principle is useful to understand how sampling the venous drainage of the pituitary for ACTH is critical. An appreciation of the long-term effects of cortisol negative feedback in suppressing HPA function is paramount to knowing how to treat patients post-operatively with glucocorticoid replacement, as well as how to properly wean patients from this replacement. Finally, targets for pharmacological medical therapy of Cushing's syndrome are based on the pathophysiology of the syndrome.
In summary, Cushing's syndrome is an important example of bidirectional, translational research where knowledge of the physiology of the HPA axis informs the development of new diagnostic and treatment approaches, whereas a thorough understanding of the pathophysiology of Cushing's syndrome is a great stimulus to developing new concepts of the control of pituitary and adrenal function.
Glossary
Abbreviations
- AC
adenylate cyclase
- ACTH
adrenocorticotrophic hormone
- AVP
vasopressin
- CBG
corticosteroid-binding globulin
- CRH
corticotrophin-releasing hormone
- DA
dopamine agonist
- GR
glucocorticoid receptor
- GRE
glucocorticoid response elements
- HPA
hypothalamic–pituitary–adrenal
- IPS
inferior petrosal sinus
- IVC
inferior vena cava
- MC2R
melanocortin-2 receptor
- MR
mineralocorticoid receptor
- POMC
proopiomelanocortin
- SCN
suprachiasmatic nucleus
- SER
smooth endoplasmic reticulum
- SRL
somatostatin receptor ligand
- ZF
zona fasciculata
- ZR
zona reticularis
Biography
Hershel Raff received his PhD in Environmental Physiology from the Johns Hopkins University in the laboratory of Robert S. Fitzgerald and was an NRSA Post-Doctoral Fellow at University of California San Francisco in the laboratory of Mary F. Dallman. He is currently Professor of Medicine, Surgery, and Physiology at the Medical College of Wisconsin and Scientific Director and Clinical Supervisor of the Endocrine Research Laboratory at Aurora St Luke’s Medical Center. His basic research focuses on neonatal endocrinology and his clinical research focuses on the laboratory diagnosis of disorders of the pituitary–adrenal system. Ty Carroll received his MD, and did his Residency in InternalMedicine and a Fellowship in Endocrinology at the Medical College of Wisconsin. He is currently Assistant Professor of Medicine at the Medical College ofWisconsin. His research focuses on the diagnosis and treatment of Cushing’s syndrome.
 |
Additional information
Competing interests
Hershel Raff has no competing interests. Ty Carroll is a consultant for Corcept Therapeutics.
Author contributions
H.R. created the initial outline. H.R. and T.C. wrote sections of the initial draft, edited multiple drafts, and approved the submitted version. H.R. created the figures and T.C. approved the submitted version. H.R. and T.C. both edited and approved the revised version.
References
- Abdelmannan D, Chaiban J, Selman WR. Arafah BM. Recurrences of ACTH-secreting adenomas after pituitary adenomectomy can be accurately predicted by perioperative measurements of plasma ACTH levels. J Clin Endocrinol Metab. 2013;98:1458–1465. doi: 10.1210/jc.2012-3910. [DOI] [PubMed] [Google Scholar]
- Abraham G. Exogenous Cushing's syndrome induced by surreptitious topical glucocorticosteroid overdose in infants with diaper dermatitis. J Pediatr Endocrinol Metab. 2007;20:1169–1171. doi: 10.1515/jpem.2007.20.11.1169. [DOI] [PubMed] [Google Scholar]
- Alexandraki KI. Grossman AB. Is urinary free cortisol of value in the diagnosis of Cushing's syndrome? Curr Opin Endocrinol Diabetes Obes. 2011;18:259–263. doi: 10.1097/MED.0b013e3283487193. [DOI] [PubMed] [Google Scholar]
- Alexandraki KI, Kaltsas GA, Isidori AM, Storr HL, Afshar F, Sabin I, Akker SA, Chew SL, Drake WM, Monson JP, et al. Long-term remission and recurrence rates in Cushing's disease: predictive factors in a single-centre study. Eur J Endocrinol. 2013;168:639–648. doi: 10.1530/EJE-12-0921. [DOI] [PubMed] [Google Scholar]
- Aron DC, Raff H. Findling JW. Effectiveness versus efficacy: the limited value in clinical practice of high dose dexamethasone suppression testing in the differential diagnosis of adrenocorticotropin-dependent Cushing's syndrome. J Clin Endocrinol Metab. 1997;82:1780–1785. doi: 10.1210/jcem.82.6.3991. [DOI] [PubMed] [Google Scholar]
- Atkinson AB, Kennedy A, Wiggam MI, McCance DR. Sheridan B. Long-term remission rates after pituitary surgery for Cushing's disease: the need for long-term surveillance. Clin Endocrinol (Oxf) 2005;63:549–559. doi: 10.1111/j.1365-2265.2005.02380.x. [DOI] [PubMed] [Google Scholar]
- Bangaru ML, Woodliff J, Raff H. Kansra S. Growth suppression of mouse pituitary corticotroph tumor AtT20 cells by curcumin: a model for treating Cushing's disease. PLoS One. 2010;5:e9893. doi: 10.1371/journal.pone.0009893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbot M, Albiger N, Koutroumpi S, Ceccato F, Frigo AC, Manara R, Fassina A, Gardiman MP, Scanarini M, Mantero F. Scaroni C. Predicting late recurrence in surgically treated patients with Cushing's disease. Clin Endocrinol (Oxf) 2013;79:394–401. doi: 10.1111/cen.12133. [DOI] [PubMed] [Google Scholar]
- Beuschlein F, Fassnacht M, Assie G, Calebiro D, Stratakis CA, Osswald A, Ronchi CL, Wieland T, Sbiera S, Faucz FR, et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing's syndrome. N Engl J Med. 2014;370:1019–1028. doi: 10.1056/NEJMoa1310359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilodeau S, Vallette-Kasic S, Gauthier Y, Figarella-Branger D, Brue T, Berthelet F, Lacroix A, Batista D, Stratakis C, Hanson J, et al. Role of Brg1 and HDAC2 in GR trans-repression of the pituitary POMC gene and misexpression in Cushing disease. Genes Dev. 2006;20:2871–2886. doi: 10.1101/gad.1444606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, He M, Gao Z, Peng Y, Li Y, Li L, Zhou W, Li X, Zhong X, Lei Y, et al. Activating hotspot L205R mutation in PRKACA and adrenal Cushing's syndrome. Science. 2014;344:913–917. doi: 10.1126/science.1249480. [DOI] [PubMed] [Google Scholar]
- Carroll T, Raff H. Findling JW. Late-night salivary cortisol measurement in the diagnosis of Cushing's syndrome. Nat Clin Pract Endocrinol Metab. 2008;4:344–350. doi: 10.1038/ncpendmet0837. [DOI] [PubMed] [Google Scholar]
- Carroll T, Raff H. Findling JW. Late-night salivary cortisol for the diagnosis of Cushing syndrome: a meta-analysis. Endocr Pract. 2009;15:335–342. doi: 10.4158/EP09023OR. [DOI] [PubMed] [Google Scholar]
- Carroll TB. Findling JW. Cushing's syndrome of nonpituitary causes. Curr Opin Endocrinol Diabetes Obes. 2009;16:308–315. doi: 10.1097/MED.0b013e32832d8950. [DOI] [PubMed] [Google Scholar]
- Chiang MY, Sarkar M, Koppens JM, Milles J. Shah P. Exogenous Cushing's syndrome and topical ocular steroids. Eye (Lond) 2006;20:725–727. doi: 10.1038/sj.eye.6701956. [DOI] [PubMed] [Google Scholar]
- Christensson C, Thoren A. Lindberg B. Safety of inhaled budesonide: clinical manifestations of systemic corticosteroid-related adverse effects. Drug Saf. 2008;31:965–988. doi: 10.2165/00002018-200831110-00002. [DOI] [PubMed] [Google Scholar]
- Colao A, Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, Schoenherr U, Mills D, Salgado LR. Biller BM. A 12-month phase 3 study of pasireotide in Cushing's disease. N Engl J Med. 2012;366:914–924. doi: 10.1056/NEJMoa1105743. [DOI] [PubMed] [Google Scholar]
- de Herder WW. Heroes in endocrinology: Nobel Prizes. Endocr Connect. 2014;3:R94–R104. doi: 10.1530/EC-14-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doppman JL, Chang R, Oldfield EH, Chrousos G, Stratakis CA. Nieman LK. The hypoplastic inferior petrosal sinus: a potential source of false-negative results in petrosal sampling for Cushing's disease. J Clin Endocrinol Metab. 1999;84:533–540. doi: 10.1210/jcem.84.2.5475. [DOI] [PubMed] [Google Scholar]
- Doppman JL, Nieman L, Miller DL, Pass HI, Chang R, Cutler GB, Jr, Schaaf M, Chrousos GP, Norton JA. Ziessman HA. Ectopic adrenocorticotropic hormone syndrome: localization studies in 28 patients. Radiology. 1989;172:115–124. doi: 10.1148/radiology.172.1.2544919. [DOI] [PubMed] [Google Scholar]
- Elias PC, Martinez EZ, Barone BF, Mermejo LM, Castro M. Moreira AC. Late-night salivary cortisol has a better performance than urinary free cortisol in the diagnosis of Cushing's syndrome. J Clin Endocrinol Metab. 2014;99:2045–2051. doi: 10.1210/jc.2013-4262. [DOI] [PubMed] [Google Scholar]
- Findling JW, Kehoe ME. Raff H. Identification of patients with Cushing's disease with negative pituitary adrenocorticotropin gradients during inferior petrosal sinus sampling: prolactin as an index of pituitary venous effluent. J Clin Endocrinol Metab. 2004a;89:6005–6009. doi: 10.1210/jc.2004-1378. [DOI] [PubMed] [Google Scholar]
- Findling JW, Kehoe ME, Shaker JL. Raff H. Routine inferior petrosal sinus sampling in the differential diagnosis of adrenocorticotropin (ACTH)-dependent Cushing's syndrome: early recognition of the occult ectopic ACTH syndrome. J Clin Endocrinol Metab. 1991;73:408–413. doi: 10.1210/jcem-73-2-408. [DOI] [PubMed] [Google Scholar]
- Findling JW. Raff H. Screening and diagnosis of Cushing's syndrome. Endocrinol Metab Clin North Am. 2005;34:385–402. doi: 10.1016/j.ecl.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Findling JW, Raff H. Aron DC. The low-dose dexamethasone suppression test: a reevaluation in patients with Cushing's syndrome. J Clin Endocrinol Metab. 2004b;89:1222–1226. doi: 10.1210/jc.2003-030207. [DOI] [PubMed] [Google Scholar]
- Finken MJ, Andrews RC, Andrew R. Walker BR. Cortisol metabolism in healthy young adults: sexual dimorphism in activities of A-ring reductases, but not 11β-hydroxysteroid dehydrogenases. J Clin Endocrinol Metab. 1999;84:3316–3321. doi: 10.1210/jcem.84.9.6009. [DOI] [PubMed] [Google Scholar]
- Fleseriu M. Medical management of persistent and recurrent Cushing disease. Neurosurg Clin N Am. 2012;23:653–668. doi: 10.1016/j.nec.2012.06.012. [DOI] [PubMed] [Google Scholar]
- Fleseriu M. Petersenn S. Medical management of Cushing's disease: what is the future. Pituitary. 2012;15:330–341. doi: 10.1007/s11102-012-0397-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo-Payet N. Battista MC. Steroidogenesis – adrenal cell signal transduction. Compr Physiol. 2014;4:889–964. doi: 10.1002/cphy.c130050. [DOI] [PubMed] [Google Scholar]
- Giordano TJ. Genetics: Pinpointing a hotspot in adrenal Cushing syndrome. Nat Rev Endocrinol. 2014;10:447–448. doi: 10.1038/nrendo.2014.89. [DOI] [PubMed] [Google Scholar]
- Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, Kuntsman JW, Korah R, Suttorp AC, Dietrich D, et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet. 2014;46:613–617. doi: 10.1038/ng.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall WA, Luciano MG, Doppman JL, Patronas NJ. Oldfield EH. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med. 1994;120:817–820. doi: 10.7326/0003-4819-120-10-199405150-00001. [DOI] [PubMed] [Google Scholar]
- Hameed N, Yedinak CG, Brzana J, Gultekin SH, Coppa ND, Dogan A, Delashaw JB. Fleseriu M. Remission rate after transsphenoidal surgery in patients with pathologically confirmed Cushing's disease, the role of cortisol, ACTH assessment and immediate reoperation: a large single center experience. Pituitary. 2013;16:452–458. doi: 10.1007/s11102-012-0455-z. [DOI] [PubMed] [Google Scholar]
- Hopkins RL. Leinung MC. Exogenous Cushing's syndrome and glucocorticoid withdrawal. Endocrinol Metab Clin North Am. 2005;34:371–384. doi: 10.1016/j.ecl.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA. Nieman LK. Cushing's syndrome due to ectopic corticotropin secretion: twenty years' experience at the National Institutes of Health. J Clin Endocrinol Metab. 2005;90:4955–4962. doi: 10.1210/jc.2004-2527. [DOI] [PubMed] [Google Scholar]
- Isidori AM, Kaltsas GA, Pozza C, Frajese V, Newell-Price J, Reznek RH, Jenkins PJ, Monson JP, Grossman AB. Besser GM. The ectopic adrenocorticotropin syndrome: clinical features, diagnosis, management, and long-term follow-up. J Clin Endocrinol Metab. 2006;91:371–377. doi: 10.1210/jc.2005-1542. [DOI] [PubMed] [Google Scholar]
- Isidori AM. Lenzi A. Ectopic ACTH syndrome. Arq Bras Endocrinol Metabol. 2007;51:1217–1225. doi: 10.1590/s0004-27302007000800007. [DOI] [PubMed] [Google Scholar]
- Jacobson L. Hypothalamic–pituitary–adrenocortical axis regulation. Endocrinol Metab Clin North Am. 2005;34:271–292. doi: 10.1016/j.ecl.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Jagannathan J, Sheehan JP, Pouratian N, Laws ER, Steiner L. Vance ML. Gamma knife surgery for Cushing's disease. J Neurosurg. 2007;106:980–987. doi: 10.3171/jns.2007.106.6.980. [DOI] [PubMed] [Google Scholar]
- Karl M, Von WG, Kempter E, Katz DA, Reincke M, Monig H, Ali IU, Stratakis CA, Oldfield EH, Chrousos GP. Schulte HM. Nelson's syndrome associated with a somatic frame shift mutation in the glucocorticoid receptor gene. J Clin Endocrinol Metab. 1996;81:124–129. doi: 10.1210/jcem.81.1.8550738. [DOI] [PubMed] [Google Scholar]
- Keller-Wood ME. Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocr Rev. 1984;5:1–24. doi: 10.1210/edrv-5-1-1. [DOI] [PubMed] [Google Scholar]
- Kirschner LS. Medicine. A unified cause for adrenal Cushing's syndrome. Science. 2014;344:804–805. doi: 10.1126/science.1254901. [DOI] [PubMed] [Google Scholar]
- Lightman SL. Conway-Campbell BL. The crucial role of pulsatile activity of the HPA axis for continuous dynamic equilibration. Nat Rev Neurosci. 2010;11:710–718. doi: 10.1038/nrn2914. [DOI] [PubMed] [Google Scholar]
- Lin HY, Muller YA. Hammond GL. Molecular and structural basis of steroid hormone binding and release from corticosteroid-binding globulin. Mol Cell Endocrinol. 2010;316:3–12. doi: 10.1016/j.mce.2009.06.015. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Sapolsky RM. Stress and cognitive function. Curr Opin Neurobiol. 1995;5:205–216. doi: 10.1016/0959-4388(95)80028-x. [DOI] [PubMed] [Google Scholar]
- Mouri T, Itoi K, Takahashi K, Suda T, Murakami O, Yoshinaga K, Andoh N, Ohtani H, Masuda T. Sasano N. Colocalization of corticotropin-releasing factor and vasopressin in the paraventricular nucleus of the human hypothalamus. Neuroendocrinology. 1993;57:34–39. doi: 10.1159/000126339. [DOI] [PubMed] [Google Scholar]
- Nakano-Tateno T, Tateno T, Hlaing MM, Zheng L, Yoshimoto K, Yamada S, Asa SL. Ezzat S. FGFR4 polymorphic variants modulate phenotypic features of Cushing disease. Mol Endocrinol. 2014;28:525–533. doi: 10.1210/me.2013-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell-Price J. Ketoconazole as an adrenal steroidogenesis inhibitor: effectiveness and risks in the treatment of Cushing's disease. J Clin Endocrinol Metab. 2014;99:1586–1588. doi: 10.1210/jc.2014-1622. [DOI] [PubMed] [Google Scholar]
- Newell-Price J, Bertagna X, Grossman AB. Nieman LK. Cushing's syndrome. Lancet. 2006;367:1605–1617. doi: 10.1016/S0140-6736(06)68699-6. [DOI] [PubMed] [Google Scholar]
- Newell-Price J, Trainer P, Perry L, Wass J, Grossman A. Besser M. A single sleeping midnight cortisol has 100% sensitivity for the diagnosis of Cushing's syndrome. Clin Endocrinol (Oxf) 1995;43:545–550. doi: 10.1111/j.1365-2265.1995.tb02918.x. [DOI] [PubMed] [Google Scholar]
- Nieman LK. Update in the medical therapy of Cushing's disease. Curr Opin Endocrinol Diabetes Obes. 2013;20:330–334. doi: 10.1097/MED.0b013e3283631809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield EH, Doppman JL, Nieman LK, Chrousos GP, Miller DL, Katz DA, Cutler GB., Jr Loriaux DL. Petrosal sinus sampling with and without corticotropin-releasing hormone for the differential diagnosis of Cushing's syndrome. N Engl J Med. 1991;325:897–905. doi: 10.1056/NEJM199109263251301. [DOI] [PubMed] [Google Scholar]
- Page-Wilson G, Freda PU, Jacobs TP, Khandji AG, Bruce JN, Foo ST, Meece K, White A. Wardlaw SL. Clinical utility of plasma POMC and AgRP measurements in the differential diagnosis of ACTH-dependent Cushing's syndrome. J Clin Endocrinol Metab. 2014;99:E1838–E1845. doi: 10.1210/jc.2014-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanicolaou DA, Yanovski JA, Cutler GB, Jr, Chrousos GP. Nieman LK. A single midnight serum cortisol measurement distinguishes Cushing's syndrome from pseudo-Cushing states. J Clin Endocrinol Metab. 1998;83:1163–1167. doi: 10.1210/jcem.83.4.4733. [DOI] [PubMed] [Google Scholar]
- Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, Sen K, Salgado LR, Colao A. Biller BM. High variability in baseline urinary free cortisol values in patients with Cushing's disease. Clin Endocrinol (Oxf) 2013;80:261–269. doi: 10.1111/cen.12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff H. Glucocorticoid inhibition of neurohypophysial vasopressin secretion. Am J Physiol Regul Integr Comp Physiol. 1987;252:R635–R644. doi: 10.1152/ajpregu.1987.252.4.R635. [DOI] [PubMed] [Google Scholar]
- Raff H. Interactions between neurohypophysial hormones and the ACTH–adrenocortical axis. Ann NY Acad Sci. 1993;689:411–425. doi: 10.1111/j.1749-6632.1993.tb55564.x. [DOI] [PubMed] [Google Scholar]
- Raff H. Utility of salivary cortisol measurements in Cushing's syndrome and adrenal insufficiency. J Clin Endocrinol Metab. 2009;94:3647–3655. doi: 10.1210/jc.2009-1166. [DOI] [PubMed] [Google Scholar]
- Raff H. Cushing's syndrome: diagnosis and surveillance using salivary cortisol. Pituitary. 2012;15:64–70. doi: 10.1007/s11102-011-0333-0. [DOI] [PubMed] [Google Scholar]
- Raff H. Update on late-night salivary cortisol for the diagnosis of Cushing's syndrome: methodological considerations. Endocrine. 2013;44:346–349. doi: 10.1007/s12020-013-0013-0. [DOI] [PubMed] [Google Scholar]
- Raff H. Findling JW. A physiologic approach to diagnosis of the Cushing syndrome. Ann Intern Med. 2003;138:980–991. doi: 10.7326/0003-4819-138-12-200306170-00010. [DOI] [PubMed] [Google Scholar]
- Raff H, Raff JL. Findling JW. Late-night salivary cortisol as a screening test for Cushing's syndrome. J Clin Endocrinol Metab. 1998;83:2681–2686. doi: 10.1210/jcem.83.8.4936. [DOI] [PubMed] [Google Scholar]
- Raff H, Sharma ST. Nieman LK. Physiological basis for the etiology, diagnosis, and treatment of adrenal disorders: Cushing's syndrome, adrenal insufficiency, and congenital adrenal hyperplasia. Compr Physiol. 2014;4:739–769. doi: 10.1002/cphy.c130035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimondo G, Allasino B, Bovio S, Saba L, Ardito A, Angeli A. Terzolo M. Pros and cons of dexamethasone suppression test for screening of subclinical Cushing's syndrome in patients with adrenal incidentalomas. J Endocrinol Invest. 2011;34:e1–e5. doi: 10.1007/BF03346701. [DOI] [PubMed] [Google Scholar]
- Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y, Yoshida K, Nagata Y, Sato-Otsubo A, Yoshizato T, et al. Recurrent somatic mutations underlie corticotropin-independent Cushing's syndrome. Science. 2014;344:917–920. doi: 10.1126/science.1252328. [DOI] [PubMed] [Google Scholar]
- Spiga F, Walker JJ, Terry JR. Lightman SL. HPA axis-rhythms. Compr Physiol. 2014;4:1273–1298. doi: 10.1002/cphy.c140003. [DOI] [PubMed] [Google Scholar]
- Stahn C. Buttgereit F. Genomic and nongenomic effects of glucocorticoids. Nat Clin Pract Rheumatol. 2008;4:525–533. doi: 10.1038/ncprheum0898. [DOI] [PubMed] [Google Scholar]
- Storr HL, Isidori AM, Monson JP, Besser GM, Grossman AB. Savage MO. Prepubertal Cushing's disease is more common in males, but there is no increase in severity at diagnosis. J Clin Endocrinol Metab. 2004;89:3818–3820. doi: 10.1210/jc.2003-031531. [DOI] [PubMed] [Google Scholar]
- Tabaee A, Anand VK, Barron Y, Hiltzik DH, Brown SM, Kacker A, Mazumdar M. Schwartz TH. Endoscopic pituitary surgery: a systematic review and meta-analysis. J Neurosurg. 2009;111:545–554. doi: 10.3171/2007.12.17635. [DOI] [PubMed] [Google Scholar]
- Tomlinson JW. Stewart PM. Cortisol metabolism and the role of 11β-hydroxysteroid dehydrogenase. Best Pract Res Clin Endocrinol Metab. 2001;15:61–78. doi: 10.1053/beem.2000.0119. [DOI] [PubMed] [Google Scholar]
- Valin N, De CN, Garrait V, Bergeron A, Bouche C. Molina JM. Iatrogenic Cushing's syndrome in HIV-infected patients receiving ritonavir and inhaled fluticasone: description of 4 new cases and review of the literature. J Int Assoc Physicians AIDS Care (Chic) 2009;8:113–121. doi: 10.1177/1545109709332019. [DOI] [PubMed] [Google Scholar]
- Vandevyver S, Dejager L. Libert C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr Rev. 2014;35:671–693. doi: 10.1210/er.2014-1010. [DOI] [PubMed] [Google Scholar]
- Whitnall MH, Smyth D. Gainer H. Vasopressin coexists in half of the corticotropin-releasing factor axons present in the external zone of the median eminence in normal rats. Neuroendocrinology. 1987;45:420–424. doi: 10.1159/000124768. [DOI] [PubMed] [Google Scholar]
- Yaneva M, Vandeva S, Zacharieva S, Daly AF. Beckers A. Genetics of Cushing's syndrome. Neuroendocrinology. 2010;92(Suppl. 1):6–10. doi: 10.1159/000314215. [DOI] [PubMed] [Google Scholar]
- Yanovski JA, Cutler GB, Jr, Chrousos GP. Nieman LK. The dexamethasone-suppressed corticotropin-releasing hormone stimulation test differentiates mild Cushing's disease from normal physiology. J Clin Endocrinol Metab. 1998;83:348–352. doi: 10.1210/jcem.83.2.4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young WF., Jr Thompson GB. Laparoscopic adrenalectomy for patients who have Cushing's syndrome. Endocrinol Metab Clin North Am. 2005;34:489–499. doi: 10.1016/j.ecl.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Zemskova MS, Gundabolu B, Sinaii N, Chen CC, Carrasquillo JA, Whatley M, Chowdhury I, Gharib AM. Nieman LK. Utility of various functional and anatomic imaging modalities for detection of ectopic adrenocorticotropin-secreting tumors. J Clin Endocrinol Metab. 2010;95:1207–1219. doi: 10.1210/jc.2009-2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhang X. Klibanski A. Genetic and epigenetic mutations of tumor suppressive genes in sporadic pituitary adenoma. Mol Cell Endocrinol. 2014;386:16–33. doi: 10.1016/j.mce.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]