

Abstract
The low-conductance, highly calcium-selective channels encoded by the Orai family of proteins represent a major pathway for the agonist-induced entry of calcium associated with the generation and modulation of the key intracellular calcium signals that initiate and control a wide variety of physiologically important processes in cells. There are two distinct members of this channel family that co-exist endogenously in many cell types: the store-operated Ca2+ release-activated CRAC channels and the store-independent arachidonic acid-regulated ARC channels. Although the activities of both channels are regulated by the stromal-interacting molecule-1 (STIM1) protein, two distinct pools of this protein are responsible, with the major pool of STIM1 in the endoplasmic reticulum membrane regulating CRAC channel activity, whilst the minor pool of plasma membrane STIM1 regulates ARC channel activity. We now show that a critical feature in determining this selective activation of the two channels is the phosphorylation status of a single threonine residue (T389) within the extensive (∼450 residue) cytosolic domain of STIM1. Specifically, protein kinase A (PKA)-mediated phosphorylation of T389 of STIM1 is necessary for effective activation of the ARC channels, whilst phosphorylation of the same residue actually inhibits the ability of STIM1 to activate the CRAC channels. We further demonstrate that the PKA-mediated phosphorylation of T389 occurs at the plasma membrane via the involvement of the anchoring protein AKAP79, which is constitutively associated with the pool of STIM1 in the plasma membrane. The novel mechanism we have described provides a means for the cell to precisely regulate the relative activities of these two channels to independently modulate the resulting intracellular calcium signals in a physiologically relevant manner.
Key points
Although both the calcium store-dependent CRAC channels and the store-independent ARC channels are regulated by the protein STIM1, CRAC channels are regulated by STIM1 in the endoplasmic reticulum, whilst ARC channels are regulated by the STIM1 constitutively resident in the plasma membrane.
We now demonstrate that activation of the ARC channels, but not CRAC channels, is uniquely dependent on phosphorylation of a single residue (T389) in the extensive cytosolic domain of STIM1 by protein kinase A.
We further demonstrate that the phosphorylation of the T389 residue by protein kinase A is mediated by the association of plasma membrane STIM1 with the scaffolding protein AKAP79.
Together, these findings indicate that the phosphorylation status of this single residue in STIM1 represents a key molecular determinant of the relative activities of these two co-existing Ca2+ entry channels that are known to play critical, but distinct, roles in modulating a variety of physiologically relevant activities.
Introduction
The low conductance, highly calcium-selective ion cha-nnels formed by the Orai family of proteins (Orai1–3) represent the major pathway of the agonist-induced entry of extracellular calcium that is an essential component in the generation of the calcium signals that specifically regulate a multitude of essential cellular responses in electrically non-excitable cells. To date, two such Orai channels have been described as being endogenously present in a variety of these cell types – the store-operated CRAC channels, and the store-independent arachidonic acid-activated ARC channels. In addition to their distinct modes of activation, these two Orai channels differ in their molecular composition, with CRAC channels being formed exclusively from Orai1 subunits, whilst ARC channels are a heteromeric assembly of Orai1/Orai3 subunits. Consequently, both CRAC channels and the ARC channels share a requirement for Orai1 in their molecular composition. However, they also share a requirement for stromal interacting molecule 1 (STIM1) for their activation. Because of this, definitive identification and isolation of the specific functional roles of these co-existing channels requires the clear identification and characterization of their unique features and modes of regulation, as well as the development of relevant approaches and molecular tools for the analysis of the resulting physiological responses. The importance of this is only emphasized by the existing focus on the CRAC channels which has resulted in claims, frequently seen in the current Orai channel literature, that the demonstration of an essential requirement for Orai1 and/or STIM1 in any particular cellular response specifically implies the involvement of the CRAC channels. Indeed, this assumption has even extended to efforts to develop compounds or reagents that target these molecules (particularly Orai1) as potential drugs that might impact clinically relevant activities of the CRAC channels largely for their potential use in CRAC-dependent autoimmune diseases and allergic responses (Yoshino et al. 2007; Di Sabatino et al. 2009; Chen et al. 2013; Cox et al. 2013; Lin et al. 2013; Rice et al. 2013; see also Derler et al. 2008). However, because the Orai1 protein is not exclusive to CRAC channels, such agents would not target the CRAC channels alone. Consequently, the definitive evaluation of the distinct physiologically relevant roles of these two co-existing, and molecularly related, conductances can only be revealed by examination of a range of appropriate unique features in their activation and regulation.
In this context, we have previously shown that, whilst the store-operated CRAC channels are activated by STIM1 located in the membrane of the endoplasmic reticulum, activation of the store-independent ARC channels depends on the discrete pool of STIM1 that constitutively resides in the plasma membrane (PM) (Mignen et al. 2007; Thompson & Shuttleworth, 2012). Moreover, the initial step in the activation of CRAC channels is the loss of Ca2+ from the N-terminal EF-hand, located in the endoplasmic reticulum (ER)-luminal domain of STIM1, as these stores become depleted of Ca2+ (Liou et al. 2005, 2007; Zhang et al. 2005; Wu et al. 2006; Luik et al. 2008). However, the equivalent loss of Ca2+ from the EF-hand of STIM1 in plasma membrane fails to result in activation of either the ARC channels or CRAC channels (Thompson & Shuttleworth, 2013). Consequently, the evidence indicates that the two pools of cellular STIM1 are functionally entirely distinct, thereby allowing the two endogenous Orai channels to operate under discrete specific conditions of stimulation and to serve unique roles in the regulation of agonist-activated Ca2+ signals (Mignen et al. 2001; Thompson & Shuttleworth, 2011). However, it is unclear how this ‘discrimination’ between the activities of the two pools of cellular STIM1, and their effects on the different endogenous Orai channels, is achieved.
Of potential relevance in this context was an earlier finding that the ARC channels are uniquely modulated by the balance between a stimulatory PKA-mediated phosphorylation and an inhibitory calcineurin-mediated dephosphorylation (Mignen et al. 2003), and that these effects were apparently co-ordinated via an A-kinase anchoring protein (AKAP), possibly AKAP79 (Mignen et al. 2005a). In contrast, store-operated CRAC channel activity was entirely unaffected by such AKAP-mediated phosphorylation/dephosphorylation events. Together, these findings form the basis for the following investigation of the molecular processes responsible for this unique, subtype-specific regulation of the activities of these two co-existing, functionally important, endogenous Orai channels.
Methods
Cells lines, constructs and transfections
Culture conditions for the Flp-In-293 cells (Invitrogen, Grand Island, NY, USA), a cell line derived from HEK-293 cells, and procedures for the depletion of endogenous STIM1 using small interfering RNA (siRNA) duplexes were as previously described (Thompson & Shuttleworth, 2012). Details of the specific constructs used in this report can be found in the online Supporting information. Constructs were prepared using a variety of molecular approaches including sub-cloning, QuikChange II (Stratagene, Agilent Technologies, Santa Clara, CA, USA) site-directed mutagenesis and PCR. All PCR-modified constructs were confirmed by sequencing. Cells were transfected using an Amaxa Nucleofector II (Lonza, Allendale, NJ, USA) following the manufacturer's guidelines.
Electrophysiology
Procedures for patch clamp experiments were essentially as previously described (Thompson & Shuttleworth, 2011, 2013), with whole-cell current recordings performed on cells at room temperature (20–22ºC), using 250 ms voltage pulses to −80 mV delivered every 2 s from a holding potential of 0 mV. Current–voltage relationships were obtained by applying 10 ms pulses to potentials between −100 mV and +80 mV at 20 mV intervals. The standard internal (pipette) solution contained 140 mm CsC2H3O2, 3.5 mm CaCl2, 3.72 mm MgCl2, 10 mm EGTA, and 10 mm Hepes (pH 7.2). The calculated free Ca2+ concentration of this solution was 100 nm. For measurement of CRAC channel currents, the above internal solution was changed by removing all CaCl2 and increasing MgCl2 to 6.77 mm. The standard extracellular solution contained 140 mm NaCl, 5 mm CsCl, 1.2 mm MgCl2, 10 mm CaCl2, 30 mm glucose and 10 mm Hepes (pH 7.4). As before, activation of the ARC channels was induced by exogenous addition of arachidonic acid (AA, 8 μm) to the bath solution, and CRAC channel activation was induced by addition of the InsP3 receptor agonist adenophostin-A (2 μm) via the patch pipette (Thompson & Shuttleworth, 2011). For ARC channel currents, the initial currents obtained before activation of the channel were routinely used for leak subtraction. For CRAC channel currents, leak subtraction was obtained from measurements at the end of each experiment in an external solution containing La3+ (100 μm).
Cell imaging
All images were obtained on an Olympus Fluoview 1000 multiphoton/confocal laser scanning microscope system (FV1000 MPE, Center Valley, PA, USA), with an Olympus BX61WI upright microscope coupled to a suite of diode and gas lasers (405, 440, 515, 565 and 633 nm) operated and controlled by the Olympus FV10-ASW software. Imaging and data acquisition for fluorescence resonance energy transfer (FRET) analysis was performed using a ×60 water-immersion objective (UPLSAPO, numeric aperture 1.2).
FRET assays
Image analysis and determination of FRET signals were carried out using the Fluoview software (v. 2.0a), as previously described (Thompson & Shuttleworth, 2013), using the sensitized emission approach for FRET analysis to obtain a value of FRET efficiency (E%) (Elangovan et al. 2003; Sekar & Periasamy, 2003). Analysis of the FRET efficiency data for the plasma membrane-associated proteins were performed in ImageJ (NIH, Bethesda, MD, USA) as previously described (Thompson & Shuttleworth, 2013). The mean data presented were recorded for the number of cells indicated (n), obtained from a minimum of three separate transfections per experimental treatment.
In vitro phosphorylation and Phos-tag analysis
Western blot experiments designed to identify in vitro PKA-mediated hyperphosphorylation of STIM1 were performed using the Phos-tag acrylamide SDS-PAGE technique (Wako, Richmond, VA, USA) following the manufacturer's instructions. Essentially, this involves inclusion of MnCl2 in the SDS-PAGE acrylamide solution which results in a decreased migration of a phosphorylated protein as a result of the binding of Mn2+ to the phosphate group via the associated Phos-tag complex. For these experiments, the Flp-In 293 cells were nucleofected (Amaxa Nucleofector) following the manufacturer's instructions with 0.75 μg of either a flag-tagged STIM1-Δ448 or the same construct bearing the T389A mutation. After 48 h of incubation, the cells were washed with Ca2+/Mg2+-free PBS, followed by trituration in fresh Ca2+/Mg2+-free PBS, and then centrifuged (100 g for 5 min). After removal of the supernatant, 1 ml ice-cold lysis buffer (25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% NP-40, 1% sodium deoxycholate) was added along with protease inhibitors and phosphatase inhibitors (cOmplete mini EDTA-free and PhosSTOP, respectively, Roche, Indianapolis, IN, USA). The samples were homogenized in ground glass homogenizers on ice, and left on ice for 15 min before centrifuging (16,000 g at 4°C for 15 min). The supernatant was transferred to fresh 1.5 ml centrifuge tubes and 5 μl of a Flag-mAb (Origene, Rockville, MD, USA 1:200) added and rotated overnight at 4°C. Immobilized protein A/G beads (100 μl, Pierce, Rockford, IL, USA) were transferred to Pierce Spin Columns and washed three times with 500 μl lysis buffer, followed by 500 μl of the same buffer containing protease and phosphatase inhibitors. The samples (500 μl) were added and rotated at 4°C in the cold room for 2.5 h. The samples were then centrifuged (1000 g for 20 s), washed three times with lysis buffer, and then twice with 50 mm Tris pH 7.4, followed by 50 mm Tris pH 7.4 with 10 mm MgCl2 containing the protease and phosphatase inhibitors. A PKA reaction mix (300 μl) comprising a cAMP-dependent protein kinase catalytic subunit reaction buffer and kinase (6 μl in 700 μl reaction buffer, NEB Ipswich, MA, USA), 500 μm ATP, along with protease and phosphatase inhibitors was prepared. A 300 μl volume of this kinase/ATP buffer was added to the samples, and incubated under rotation for 60 min at 30°C. Parallel control incubations used the same reaction buffer, without ATP and the kinase catalytic subunit. After washing twice (600 μl 50 mm Tris pH 7.4), 150 μl sample buffer with DTT was added, followed by heating to 90°C for 5 min. After addition of bromophenol blue, the samples were loaded onto a 7% phosphate affinity SDS–PAGE gel (Phos-Tag, Wako) prepared according to the manufacturer's instructions, and run at constant current (30 mA) for 2 h. The gels were then transferred onto a polyvinylidene difluoride (PDVF) membrane and probed with 1:1000 Flag-pAb for 3 h, followed by 1:10,000 anti-rabbit IRDye secondary Ab (Li-Cor, 800CW, Lincoln, NE, USA) for 60 min. Blots were analysed on an Odyssey Scanner (Li-Cor).
Western blotting and co-immunoprecipitation
Endogenous AKAP79 was immunoprecipitated from cell lysates obtained from approximately 107 cells stably expressing STIM1. To minimize non-specific associations, lysates were prepared in an immunoprecipitation buffer containing 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm sodium orthovanadate, 1 μg ml–1 leupeptin and protease inhibitors (cOmplete mini EDTA free, Roche). Unsolubilized membrane fragments were removed by centrifugation (100,000 g for 40 min). A volume of 400 μl (680 μg protein) lysate/sample was used. Ten microlitres (2.5 μg) of an AKAP79 mAb (Transduction Labs, Lexington, KY, USA) or STIM1 mAb (BD Biosciences, San Jose, CA, USA) were added and samples incubated overnight at 4°C. Fifty microlitres of prewashed A/G agarose beads were added and incubated at 4°C for 2.5 h. The beads were then washed six times with lysis buffer and the protein eluted with 50 μl of 2X sample loading buffer plus β-mercaptoethanol, and heated to 70°C for 10 min. The immunoprecipitation (IP) samples (50 μl) and lysate (10 μl) were run on a 7% SDS–PAGE gel, transferred to nitrocellulose membrane and blotted with 1:300 of a STIM1 mAb (BD Bioscience 610954) followed by 1:2000 of a goat anti-mouse horseradish peroxidase-linked secondary Ab, and detected using Pierce enhanced chemiluminescence.
Statistical analyses
In all cases, P values were obtained by paired two-tailed Student's t test in GraphPad Prism 5. All error bars represent mean ± SEM based on the number of independent experiments, indicated by the n value.
Results
As noted above, our previous studies have demonstrated that ARC channel activity is uniquely impacted by a balance between PKA-mediated phosphorylation and calcineurin-mediated dephosphorylation, with the ‘phosphorylated state’ apparently predominating under resting conditions (Mignen et al. 2001, 2003). However, the actual specific target(s) of this phosphorylation are unknown. We therefore began by examining potential PKA phosphorylation sites in the proteins known to be essential for functional ARC channel activity. Because Orai3 is a unique component of these channels, it would seem likely that this protein might represent the potential target for the PKA-dependent phosphorylation. Indeed, in silico examination of potential PKA-dependent sites in the Orai3 sequence revealed five such sites. However, four of these are conserved in Orai1, and CRAC channel activity is not modulated by the above PKA-dependent phosphorylation (Parekh & Penner, 1995; Bödding, 2001), and the fifth is an extracellular site located within the large loop between transmembrane domains 3 and 4. Based on this, we concluded that Orai3 is unlikely to be the relevant target of PKA-dependent regulation of ARC channel activity.
In contrast, the original identification of STIM1 as a PM-resident protein showed that it was constitutively phosphorylated, predominantly on serine and/or threonine residues (Manji et al. 2000), raising the possibility that the target for PKA phosphorylation might be the STIM1. Correspondingly, in silico analyses of the STIM1 sequence indicated multiple potential PKA-dependent phosphorylation sites within its extensive cytosolic domain. However, most of these are clustered within the more C-terminal regions beyond residue 448 (Fig.1A), and we have previously shown that expression of a STIM1 construct that is terminated at this residue has no significant effect on normal ARC activity (Thompson & Shuttleworth, 2013). Examination of the remaining region of the STIM1 cytosolic domain indicated residue T389 as having a high probability of being a potential PKA phosphorylation site (Fig.1A). To confirm this possibility, we performed an in vitro PKA-mediated hyperphosphorylation analysis using a STIM1 construct terminated at residue 448 (STIM1-Δ448) bearing a C-terminal FLAG tag. The results obtained demonstrated a clear phosphorylation-dependent mobility shift that was lost on mutation of the T389 residue to alanine (Fig.1B), confirming that the T389 residue of STIM1 was indeed a potential PKA-dependent phosphorylation site.
Figure 1. Phosphorylation sites in the cytosolic domain of STIM1.

A, amino acid sequence of the cytosolic region of STIM1 indicating potential PKA-mediated serine and threonine phosphorylation sites, as determined by in silico prediction. The shaded area indicates those residues that would be absent in the STIM1-Δ448 construct that is terminated at residue His448. B, results of a representative in vitro PKA-mediated hyperphosphorylation assay. Shown are the effects of PKA activation on wild-type STIM1-Δ448 (WT versus WT + PKA), and the corresponding absence of any PKA effect in cells expressing STIM1-Δ448 bearing the T389A mutation (T389A + PKA).
Effects of the phosphorylation of STIM1 residue T389 on Orai channel activities
To examine the possible effects of phosphorylation of the T389 residue on ARC channel activity, we began by expressing an siRNA-resistant STIM1-Δ448 construct bearing the T389A mutation in cells treated with an siRNA to endogenous STIM1. The results obtained showed that such expression reduced inward AA-activated ARC channel currents measured at −80 mV by some 85% when compared to the corresponding cells expressing an siRNA-resistant wild-type STIM1-Δ448 (0.09 ± 0.03 pA pF–1 versus 0.61 ± 0.04 pA pF–1, n = 6). In marked contrast, the corresponding store-operated CRAC channel currents were entirely unaffected by expression of the STIM1-Δ448(T389A) mutant construct (0.60 ± 0.05 pA pF–1, versus 0.64 ± 0.08 pA pF–1, n = 7 in the unmutated STIM1-Δ448, P = 0.7) (Fig.2A and B). Importantly, the above effect of the T389A mutation on ARC channel currents was not an artefact resulting from the absence of any additional C-terminal phosphorylation sites in the STIM1-Δ448 construct, as incorporation of the same T389A mutation in full-length STIM1 produced a similar specific loss of ARC channel currents measured at −80 mV from a value of 0.57 ± 0.03 pA pF–1 (n = 7) in cells expressing the wild-type STIM1, to only 0.19 ± 0.03 pA pF–1 (n = 6) in cells expressing the STIM1(T389A) mutant construct (Fig.2C). Correspondingly, CRAC channel currents measured at −80 mV in cells expressing a full-length version of the STIM1(T389A) mutant (0.55 ± 0.03 pA pF–1, n = 6) were not significantly different from those obtained on expression of full-length wild-type STIM1 (0.51 ± 0.03 pA pF–1, n = 7, P = 0.3) (Fig.2D).
Figure 2. Effect of the T389A mutation in STIM1 on CRAC and ARC channel currents.
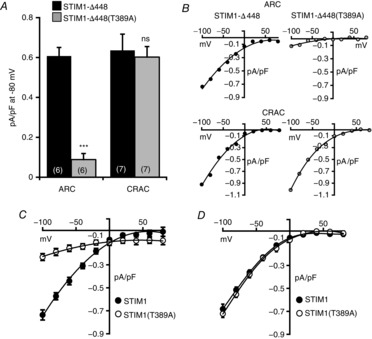
A, inward ARC channel and CRAC channel currents measured at −80 mV in cells expressing a STIM1 construct terminated at residue 448 (STIM1-Δ448, black columns) or the same construct bearing the T389A mutation (grey columns). Values are mean ± SEM with values of n (number of cells) shown in parentheses. ***P < 0.0001, and ‘ns’ (not significant), as indicated by Student's unpaired two-tailed t test. B, representative current–voltage relationships for the corresponding ARC channel currents (top row), and CRAC channel currents (bottom row). C, mean current–voltage relation for ARC channels in cells expressing full-length STIM1 (filled circles, n = 7), and a full-length STIM1 construct bearing the T389A mutation (open circles, n = 6). D, mean (±SEM) current–voltage relation for CRAC channels in cells expressing full-length STIM1 (filled circles, n = 7), and a full-length STIM1 construct bearing the T389A mutation (open circles, n = 6).
To further characterize the effect of the phosphorylation status of the T389 residue of STIM1 on ARC channel activity, we next examined the effect of mutation of the T389 residue to a glutamate (T389E) in STIM1-Δ448, thereby mimicking the phosphorylation of this residue. Expression of an siRNA-resistant STIM1-Δ448(T389E) construct in cells depleted of endogenous STIM1 resulted in an AA-induced ARC channel activity at −80 mV of 0.47 ± 0.01 pA pF–1 (n = 6) (Fig.3A and B), a value that was only slightly smaller than that seen with the wild-type STIM1-Δ448 above. To confirm that the currents measured with this mutant STIM1 reflected genuine ARC channel activity, we examined the effect of siRNA-induced knockdown of Orai3 (Mignen et al. 2008). Expression of an siRNA-resistant phosphomimetic STIM1(T389E) construct in cells depleted of endogenous STIM1 and Orai3 by treatment with appropriate siRNAs resulted in AA-activated currents of only 0.20 ± 0.02 pA pF–1 at −80 mV (n = 6) (Fig.3C).
Figure 3. Effect of the T389E mutation in STIM1 on CRAC and ARC channel currents.
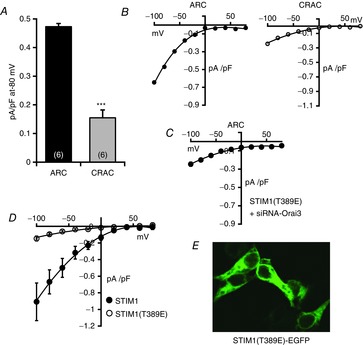
A, mean (±SEM) inward ARC channel and CRAC channel currents measured at −80 mV in cells expressing the STIM1-Δ448 construct bearing the T389E mutation. Values are mean ± SEM with values of n (number of cells) shown in parentheses. ***P < 0.0001 as indicated by Student's unpaired two-tailed t test. B, representative current–voltage relationships for the corresponding ARC channel currents (left), and CRAC channel currents (right). C, mean current–voltage relation for ARC channel currents in cells expressing the phosphomimetic STIM1(T389E) after knockdown of endogenous STIM1 and Orai3. Values are mean (±SEM), n = 6. D, mean (±SEM) current–voltage relation for CRAC channel in cells expressing full-length STIM1 (filled circles, n = 4), and a full-length STIM1 construct bearing the T389E mutation (open circles, n = 4). E, representative image showing the cytosolic distribution of an expressed fluorescently tagged STIM1 bearing the T389E mutation.
In marked contrast to the ARC channel data above, examination of the store-operated CRAC channel currents in cells expressing the STIM1-Δ448(T389E) mutant resulted in a clear inhibition of these currents to a value of only 0.15 ± 0.03 pA pF–1 (n = 6) (Fig.3A and B), equivalent to a 77% inhibition when compared to CRAC channel currents in the wild-type STIM1-Δ448 (see above). Again this effect was shown to be unrelated to the STIM1-Δ448 deletion, as expression of the same mutation in full-length STIM also resulted in a similar reduction in store-operated CRAC channel currents from 0.67 ± 0.14 to 0.09 ± 0.02 pA pF–1 at −80 mV (n = 4) (Fig.3D). Together, the implication from these data is that the phosphorylation status of this single residue in STIM1, by uniquely modulating activation of the ARC channels, essentially determines the relative activities of the co-existing CRAC and ARC channels. Given our previous demonstration that the regulation of these two channels involves distinct pools of cellular STIM1, specifically the ER versus PM pools, respectively (Mignen et al. 2007; Thompson & Shuttleworth, 2012), the above data clearly raise the question of whether the phosphorylation status of STIM1, specifically on the T389 residue, directly determines the relative activity of these channels, or whether it simply determines the physical distribution of STIM1 between these two pools. However, arguing against any such dynamic redistribution of STIM1 between the ER and PM pools are data previously obtained where a biotinylation assay failed to indicate any significant change in the levels of PM STIM1 under resting conditions compared to that seen following maximal activation of the ARC channels (Mignen et al. 2007). Consistent with this, expression of a fluorescently-tagged STIM1 construct bearing the T389E phosphomimetic mutation that selectively enabled activation of the ARC channels whilst inhibiting CRAC channel activity (Fig.3A and B) indicated an essentially uniform distribution throughout the cytosol, rather than being primarily localized to the plasma membrane (Fig.3E).
We next turned to the Lck-STIM1-C construct that consists of just the cytosolic portion of STIM1 that is constitutively anchored, in the appropriate orientation, to the inner face of the plasma membrane by the N-terminal Lck-sequence (Thompson & Shuttleworth, 2012). As we have previously shown, this construct allows the exclusive activation of the ARC channels whether by exogenous AA, or by addition of an appropriate agonist that would normally activate both ARC and CRAC channels (Thompson & Shuttleworth, 2012, 2013). Using this approach, we considered whether effectively ‘forcing’ the STIM1-T389A mutant to the PM by expression of a Lck-STIM1-Δ448 construct incorporating the T389A mutation, would be sufficient to restore normal ARC channel activity. However, expression of the relevant siRNA-resistant Lck-STIM1-Δ448(T389A) construct in cells depleted of endogenous STIM1 by appropriate siRNA resulted in AA-activated ARC channel currents of only 0.17 ± 0.04 pA pF–1 at −80 mV (n = 7). This compares to a value of 0.48 ± 0.05 pA pF–1 at −80 mV (n = 9) for the similarly activated ARC channel currents recorded in corresponding cells expressing an unmutated Lck-STIM1-Δ448 (Fig.4A and B). Confirmation that the above observed inhibition of AA-activated ARC channel currents was not due to a failure of the Lck-attached (T389A) construct to be successfully delivered to the PM was demonstrated by confocal imaging of a fluorescently-tagged version of this phospho-mutant construct (Fig.4C). Together, the above data indicate that, whilst the phosphorylation of the T389 residue of STIM1 is the primary determinant of effective ARC channel activation, the key role of such phosphorylation is not the specific delivery of STIM1 to the PM, nor the maintenance of its stability at the membrane.
Figure 4. Effect of the T389A mutation in a Lck-STIM1-Δ448 construct on ARC channel currents.
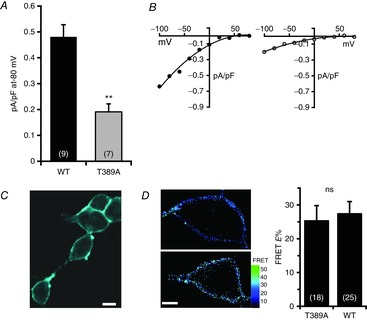
A, mean (±SEM) inward ARC channel currents measured at −80 mV in cells expressing the Lck-STIM1-Δ448 construct, or the same construct bearing the T389A mutation. Values are mean ± SEM with values of n (number of cells) shown in parentheses. **P = 0.0005 as indicated by Student's unpaired two-tailed t test. B, representative current–voltage relationships for the corresponding ARC channel currents in cells expressing the Lck-STIM1-Δ448 construct (left), or the same construct bearing the T389A mutation (right). C, representative image of cells confirming the plasma membrane expression of a Cerulean-tagged Lck-STIM1-Δ448 construct bearing the T389A mutation. Scale bar = 10 μm. D, representative images of the cellular FRET signal obtained on co-expression of an N-terminal Venus-tagged Orai3 construct with either a C-terminal Cerulean-tagged Lck-STIM1-Δ448(T389A) construct (top), or the wild-type Lck-STIM1-Δ448 construct (bottom). Scale bar = 5 μm. Also shown are the mean (±SEM) of the corresponding FRET values.
Finally, we have previously shown that STIM1 in the plasma membrane appears to be constitutively associated with the Orai3 within the ARC channels (Thompson & Shuttleworth, 2013). Given this, we considered whether phosphorylation of the T389 residue in STIM1 might be required for this interaction, with the specific inhibitory effect of expression of the T389A mutation in STIM1 on ARC channel activity reflecting a disruption of this association. To examine this, we measured FRET signals between co-expressed Lck-STIM1-Δ448(T389A) and Orai3 constructs bearing C-terminal Cerulean and N-terminal Venus fluorescent tags, respectively. Such expression resulted in a FRET signal (E%) of 25.3 ± 4.5 (n = 18) (Fig.4D, top), a value that is not significantly different from that seen in the corresponding control experiment where the T389A mutant Lck-STIM1-Δ448 was replaced with a wild-type Lck-STIM1-Δ448 (E% = 27.4 ± 3.6, n = 25, P = 0.7) (Fig.4D, bottom). Clearly, these data indicate that the inhibition of ARC channel currents induced by the T389A mutation in STIM1 does not appear to result from any disruption of the constitutive association of plasma membrane STIM1 and Orai3.
Together, the above experiments have demonstrated that STIM1 can be phosphorylated (in vitro) by PKA, that this phosphorylation is absent in a STIM1 construct in which the T389 residue is mutated to an alanine, and that such phosphorylation has profound, yet opposite, effects on the activities of the CRAC and ARC channels.
The role of AKAP79 in mediating the phosphorylation status of residue T389
Of potential relevance in the context of these new findings are data from our previous studies demonstrating that the PKA (phosphorylation) and calcineurin (dephosphorylation) effects on ARC channel activity are ‘integrated’ by an AKAP, most likely AKAP79 (Mignen et al. 2005a). Given the above demonstration that activation of the ARC channels depends on the presence of STIM1 in the plasma membrane that is phosphorylated on residue T389, we examined whether this phosphorylation involved AKAP79. To explore this possible functional association of STIM1 with AKAP79, and its impact of the phosphorylation status of STIM1, we began by examining the effect of the expression of an AKAP79 construct that terminates at residue 361 (Oliveria et al. 2003), thereby removing the PKA-binding site (Carr et al. 1992; Glantz et al. 1993). Because HEK293 cells are known to endogenously express AKAP79 (Hoshi et al. 2005; Mignen et al. 2005a), we expressed an siRNA-resistant version of the AKAP79-ΔPKA construct (bearing a C-terminal fluorescent Venus tag) along with the STIM1-Δ448 construct in cells in which both endogenous AKAP79 and STIM1 had been depleted by appropriate siRNAs. Such expression resulted in AA-activated ARC channels currents of only 0.19 ± 0.03 pA pF–1 at −80 mV (n = 7) (Fig.5A and B), demonstrating that expression of the mutant AKAP79 markedly reduced the normal activation of these currents. In contrast, however, co-expression of the phosphomimetic STIM1-Δ448(T389E) construct with the same PKA mutant AKAP79 now resulted in ARC channel currents of 0.54 ± 0.02 pA pF–1 at −80 mV (n = 7), a value that is not significantly different from that reported above for control cells (P = 0.15) (see Fig.2A).
Figure 5. Effect of expression of an AKAP79 construct lacking the PKA-binding region on ARC channel currents.
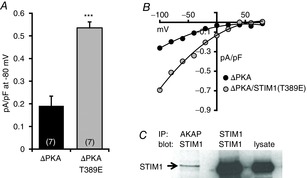
A, mean (±SEM) inward ARC channel currents measured at −80 mV in cells expressing the Lck-STIM1-Δ448 construct with an AKAP79 construct lacking the PKA-binding domain (ΔPKA), or the same construct with a STIM1-Δ448 construct bearing the T389E mutation (ΔPKA/T389E). Values are mean ± SEM with values of n (number of cells) shown in parentheses. ***P < 0.0001 as indicated by Student's unpaired two-tailed t test. B, representative current–voltage relationships for the corresponding ARC channel currents in cells expressing the AKAP79-ΔPKA construct along with Lck-STIM1-Δ448 (black symbols), or the same construct along with a Lck-STIM1-Δ448 construct bearing the T389E mutation (grey symbols). C, representative Western blot demonstrating the co-immunoprecipitation of endogenous STIM1 with AKAP79.
Consistent with the above findings, co-immunoprecipitation assays indicated that STIM1 can interact with AKAP79 (Fig.5C). Although the extent of such co-IP is clearly relatively modest, it should be noted that, if the AKAP79–STIM1 interaction is unique to the pool of STIM1 that is resident in the PM, as we would predict, then the fact that this pool represents only ∼10–25% of total cellular STIM1 could explain the modest co-IP response observed. Therefore, to further confirm the critical association between STIM1 and AKAP79, we examined FRET signals between co-expressed AKAP79 and STIM1-Δ448 constructs bearing C-terminal Venus and Cerulean fluorescent tags, respectively. It should be noted that these experiments required that we use the fluorescently tagged Lck-STIM1 construct, because detection of the small pool of PM-located STIM1 in the presence of a fluorescently-tagged wild-type STIM1 is not feasible given that ∼80–90% of the cellular STIM1 is located in the ER membrane. Using this approach, experiments revealed that co-expression of the AKAP79-Ven and Lck-STIM1-Δ448-Cer constructs resulted in clear FRET signals essentially confined to the PM (Fig.6A, top row) with a mean E% value of 39.0 ± 3.8 (n = 14). As a negative control for these data we co-expressed the AKAP79-Ven with a Lck-STIM1-Δ448-Cer construct bearing the so-called C2A mutations (W430A, I433A and L436A) that prevent the constitutive dimerization of STIM1 molecules rendering it into an inactive state (Yang et al. 2012), as previously used by us (Thompson & Shuttleworth, 2013). Analysis of FRET signals between the AKAP79-Ven and this C2A-mutant Lck-STIM1-Δ448-Cer construct resulted in a significantly reduced mean E% value of only 15.9 ± 2.9 (n = 18), equivalent to a 60% reduction in the FRET signal (P < 0.0001). Importantly, imaging of the individual fluorescence signals from the two co-expressed constructs revealed that this result was not due to any failure of these constructs to express in the appropriate location within the cell (Fig.6A, bottom row). Taken together, the above data indicate that an AKAP79-associated PKA mediates the phosphorylation of the T389 residue in STIM1 that is essential for the activation of the ARC channels by AA.
Figure 6. FRET between AKAP79 and Lck-STIM1-Δ448, and between AKAP79 and Orai3.
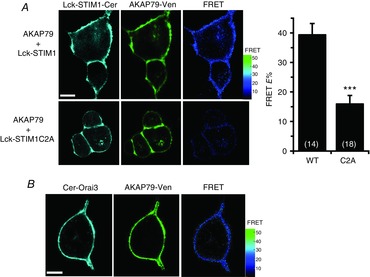
A, top row, representative images of cells co-expressing Lck-STIM1-Δ448 and AKAP79 constructs bearing C-terminal Cerulean and Venus tags, respectively, together with the resulting FRET signal (right); bottom row, corresponding representative images for the same experiment except that the co-expressed Lck-STIM1-Δ448 construct carried the C2A mutations shown previously to prevent STIM1 oligomerization. Scale bars are 10 μm. Also shown are the mean (±SEM) of the corresponding FRET values. ***P < 0.0001 as indicated by Student's unpaired two-tailed t test. B, representative images of a cell co-expressing N-terminally Cerulean-tagged Orai3 and N-terminally Venus-tagged AKAP79, together with the resulting FRET image. Scale bar is 10 μm.
As already noted, we have previously demonstrated that the plasma membrane STIM1 and Orai3 exist in a constitutive association even under resting conditions (Thompson & Shuttleworth, 2013). Given this, we examined the possibility that the AKAP79 might also be associated with Orai3, thereby potentially forming a trimeric assembly. Co-expression of the AKAP79-Ven construct with an N-terminally Cerulean-tagged Orai3 construct (Cer-Orai3) resulted in the generation of a plasma membrane-located FRET signal with a mean E% value of 25.8 ± 2.0 (n = 15) (Fig.6B). Although this is significantly less than the FRET seen between AKAP79 and Lck-STIM1 above, these data suggest that plasma membrane STIM1, AKAP79 and the Orai3 subunits, presumably within the ARC channels, might form a constitutive interactive trimeric complex under resting conditions.
Finally, although the experiments described so far have shown that the T389 residue in STIM1 represents the relevant ‘target’ of the AKAP79-dependent PKA-mediated phosphorylation, it remains unclear whether this residue is also the relevant target of the previously identified calcineurin-mediated dephosphorylation (Mignen et al. 2003). To examine this, we applied the approach used in our earlier study in which we demonstrated that simply raising cytosolic Ca2+ levels to 400 nm via the applied patch pipette was sufficient to induce the activation of endogenous calcineurin resulting in the subsequent inhibition of ARC channel currents measured at −80 mV by some 65% (Mignen et al. 2003). In marked contrast, examination of store-operated CRAC channel currents induced by ER Ca2+ store depletion are entirely unaffected by this increase in cytosolic Ca2+, indicating that such a ‘global’ activation of calcineurin has no significant effect on CRAC channel activity (Mignen et al. 2003).
We therefore began by confirming that the STIM1-Δ448 also showed a similar calcineurin-induced, ARC channel-specific inhibitory effect to that seen in the full-length STIM1. Examination of the AA-activated ARC channel currents in cells expressing the STIM1-Δ448 construct showed that raising cytosolic Ca2+ from 100 nm to 400 nm resulted in the inhibition of the currents from a value of 0.61 ± 0.04 pA pF–1 at −80 mV (n = 6) to 0.16 ± 0.03 pA pF–1, n = 6 (Fig.7A and B), equivalent to a 74% reduction, similar to that seen previously with the full-length STIM1 (Mignen et al. 2003). In marked contrast, in cells expressing the equivalent phosphomimetic STIM1-Δ448(T389E) construct, raising cytosolic Ca2+ to 400 nm failed to have any significant effect on ARC channel currents (mean inward currents at −80 mV with 100 nm cytosolic Ca2+ were 0.47 ± 0.01 pA pF–1, versus 0.48 ± 0.02 pA pF–1 with 400 nm cytosolic Ca2+, n = 6) (Fig.7C). Together, these data indicate that the same T389 residue in STIM1 is the likely target for both the PKA-mediated activation and the calcineurin-mediated inhibition of ARC channel activity, consistent with the idea that these opposing effects are the result of a functional association between AKAP79 and STIM1 in the plasma membrane.
Figure 7. Effect of intracellular calcineurin activation on ARC channel currents in cells expressing the T389E mutation of STIM1.
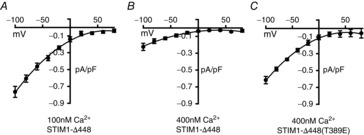
Shown are mean (±SEM) ARC channel current–voltage relationships in cells expressing the STIM1-Δ448 construct at a ‘normal’ cytosolic calcium concentration of 100 nm (A), the same following calcineurin activation by an elevated calcium concentration of 400 nm (B), and in cells expressing the STIM1-Δ448 construct bearing the T389E mutation at the same elevated (400 nm) calcium concentration (C).
Discussion
Together, we have demonstrated that the PKA-dependent phosphorylation status of the T389 residue in STIM1, as mediated via the scaffolding protein AKAP79, represents an essential feature in determining the activity of the ARC channels. As already noted, the evidence suggests that the effect of the phosphorylation status of the T389 residue is unlikely to be related to an influence on the distribution of cellular STIM1 between the ER and plasma membranes. Instead, the data indicate that the phosphorylation status of this residue appears to have its effects directly on the ability of STIM1 to regulate the activities of the Orai channels. Thus, it would seem that endogenous STIM1 is spontaneously distributed between the ER and plasma membrane domains, perhaps in proportions that depend on glycosylation. As a consequence of this distribution, the STIM1 in the plasma membrane is phosphorylated by PKA on the T389 residue presumably because, in this location, the STIM1 is able interact with the co-localized AKAP79. Critically, the data obtained with the T389E STIM1 mutant show that whilst phosphorylation is required for ARC channel activity, it is not in itself sufficient to induce activity as this still requires AA. However, precisely how the phosphorylation status of this residue induces these effects on channel activation remains unclear.
Examination of the location of the T389 residue in the overall sequence of the cytosolic portion of STIM1 indicates that it lies within the series of three coiled-coil domains of which the last two form the so-called CAD (or SOAR) domain that is known to be the minimal region for successful interaction of STIM1 with both the CRAC channels (Frischauf et al. 2009; Park et al. 2009) and the ARC channels (Thompson & Shuttleworth, 2013). The location of the T389 residue at the C-terminal end of the second coiled-coil in this structure (Fig.8A) raises the possibility that the phosphorylation status of this residue might impact the coiled-coil structure of this region of STIM1. However, examination of the sequence that includes this second coiled-coil domain in STIM1 (residues 352–394) using the ‘MARCOILS’ program (Delorenzi & Speed, 2002) revealed that incorporation of the T389A mutation had a negligible effect on the predicted coiled-coil probability of this region of STIM1 (61.4% for wild-type sequence versus 64.7% for the T389A mutant). Moreover, incorporation of the T389E mutation moderately increased the coiled-coil probability to 79.4%. Based on this analysis, it would seem that changes in the predicted structure of the second coiled-coil domain of STIM1 induced by the phosphorylation status of the T389 residue are unlikely to explain the observed profound, and reciprocal, effects on the respective Orai channel activities.
Figure 8. Diagrams indicating the location of the critical threonine 389 residue in STIM1.
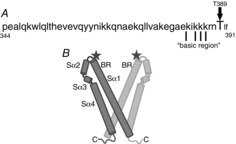
A, amino acid sequence of the region of STIM1 that forms the ‘second coiled-coil’ domain that is thought to be involved in the activation of Orai channels, and the position of the T389 residue in this sequence. Indicated are the series of critical lysine residues within the so-called ‘basic domain’, mutation of which blocks activation of both CRAC and ARC channels (see text for details). B, diagram indicating the location of the T389 residue (‘star’) at the apex of the proposed V-shape cleft formed by the predicted crystal structure of the CAD/SOAR region of a STIM1 dimer (BR, basic region; Sα1-4, sequential predicted α-helical domains). Based on the structures reported by Yang et al. (2012).
An additional ‘structural’ issue of potential relevance is the fact that the T389 residue lies immediately adjacent to a series of four lysine residues within the so-called ‘basic region’ in STIM1 (residues 382–387) (Calloway et al. 2010; Korzeniowski et al. 2010) (Fig.8A). In the activated STIM1 dimer, these residues are thought to line the inner surface of a V-shaped cleft structure formed by the second and third coiled-coil regions of the CAD/SOAR domain and which represents the site for interaction between STIM1 and the C-terminus of Orai1 (Yang et al. 2012; Stathopulos et al. 2013). Specifically, the T389 residue lies at the extreme apex of each branch of this V-shaped dimer structure (Fig.8B). Given this, it might seem that phosphorylation of the T389 residue located in this position so close to the critical lysines could impact the overall basic nature of this region. However, just as previous studies have demonstrated that mutation of these lysine residues to alanines results in the inhibition of CRAC channel activity (Calloway et al. 2010; Korzeniowski et al. 2010; Yang et al. 2012), we have shown that the same mutations incorporated into the Lck-STIM1-C construct results in an essentially identical inhibition of ARC channel activity (Thompson & Shuttleworth, 2013). Such findings are therefore difficult to reconcile with the uniquely selective, and entirely distinct, effects of the phosphorylation status of the T389 residue on ARC versus CRAC channel activity.
Several previous reports have demonstrated the phosphorylation of STIM1, particularly at serine residues, which result in important consequences for STIM1 functions. However, in these studies, the relevant sites are all located in the more C-terminal regions of the protein, beyond residue 448, and involve the action of a variety of kinases (CDK1, ERK1/2) distinct from PKA (Smyth et al. 2009; Yu et al. 2009; Pozo-Guisado et al. 2010; Lopez et al. 2012; Sheridan et al. 2013). Potentially more relevant are studies in a variety of cell types that have indicated that phosphatase inhibition results in reduced store-operated Ca2+ entry (Berlin & Preston, 1993; Koike et al. 1994; Montero et al. 1994; Tojyo et al. 1995; Murphy et al. 1996; Sakai & Ambudkar, 1996). Although the bases for these effects were not resolved, they would be consistent with our finding that phosphorylation of the T389 residue results in an inhibition of CRAC channel activity. Interestingly, a recent report has demonstrated an additional, but very different, example of an interaction between AKAP79 and Orai channels that results in the highly localized activation of the nuclear factor of activated T cells (NFAT) (Kar et al. 2014). Here, AKAP79 is recruited specifically to the CRAC channels, but only after store depletion, where it is exposed to a pool of calmodulin associated with the channel. Activation of this calmodulin within a microdomain generated by the entering Ca2+ results in the activation of AKAP79-associated calcineurin, dephosphorylation of NFAT and its translocation to the nucleus.
Finally, our studies have revealed a molecular basis for our previous demonstration that the activation of CRAC versus ARC channels depends on two distinct pools of cellular STIM1 (Mignen et al. 2007), as well as the demonstrated ‘reciprocal switch’ in the mode of Ca2+ entry as agonist concentration is increased (Mignen et al. 2001, 2003). Moreover, whilst we have previously demonstrated the exclusive activation of the ARC channels by an expressed Lck-STIM1 construct, this construct lacks both the transmembrane domain of STIM1 and its entire N-terminal region, including the Ca2+-binding EF-hand that is essential in inducing the activation of the CRAC channels on store depletion. In contrast, the current study demonstrates that exclusive activation of either the ARC channels or the CRAC channels can be demonstrated with a normal full-length STIM1 construct bearing appropriate single phosphomimetic or phospho-mutant mutations of the T389 residue in the cytosolic domain of STIM1. Critically, such a ‘phosphoSTIM1-dependent’ effect is likely to have significant functional implications, in that it provides a functional basis for the modulation of the relative activities of these two co-existing Ca2+ entry pathways in a manner appropriate to induce their demonstrated distinct roles in the generation and regulation of agonist-activated Ca2+ signals in cells (Shuttleworth, 1996; Mignen et al. 2005b; Thompson & Shuttleworth, 2011).
Glossary
Abbreviations
- AA
arachidonic acid
- AKAP
A-kinase anchoring protein
- ARC channel
arachidonate-regulated Ca2+ channel
- CRAC channel
Ca2+ release-activated Ca2+ channel
- E%
fluorescence resonance energy transfer efficiency
- ER
endoplasmic reticulum
- FRET
fluorescence resonance energy transfer
- PKA
protein kinase A
- PM
plasma membrane
- STIM1
stromal interacting molecule 1
Additional information
Competing interests
None declared.
Author contributions
J.L.T. and T.J.S. both contributed to the conception, design, analysis and interpretation of the data. T.J.S. drafted the manuscript, with assistance from J.L.T. Both authors have approved the final version.
Funding
This work was supported by National Institutes of Health grant GM040457 to T.J.S.
References
- Berlin RD. Preston SF. Okadaic acid uncouples calcium entry from depletion of intracellular stores. Cell Calcium. 1993;14:379–386. doi: 10.1016/0143-4160(93)90042-5. [DOI] [PubMed] [Google Scholar]
- Bödding M( Activation of store-operated Ca2+ entry in RBL cells without the contribution of protein kinases. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:633–638. doi: 10.1007/s002100100420. [DOI] [PubMed] [Google Scholar]
- Calloway N, Holowka D. Baird B. A basic sequence in STIM1 promotes Ca2+ influx by interacting with the C-terminal acidic coiled coil of Orai1. Biochemistry. 2010;49:1067–1071. doi: 10.1021/bi901936q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DW, Stofko-Hahn RE, Fraser ID, Cone RD. Scott JD. Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins. Characterization of AKAP 79. J Biol Chem. 1992;267:16816–16823. [PubMed] [Google Scholar]
- Chen G, Panicker S, Lau KY, Apparsundaram S, Patel VA, Chen SL, Soto R, Jung JK, Ravindran P, Okuhara D, Bohnert G, Che Q, Rao PE, Allard JD, Badi L, Bitter HM, Nunn PA, Narula SK. DeMartino JA. Characterization of a novel CRAC inhibitor that potently blocks human T cell activation and effector functions. Mol Immunol. 2013;54:355–367. doi: 10.1016/j.molimm.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Cox JH, Hussell S, Søndergaard H, Roepstorff K, Bui JV, Deer JR, Zhang J, Li ZG, Lamberth K, Kvist PH, Padkjaer S, Haase C, Zahn S. Odegard VH. Antibody-mediated targeting of the Orai1 calcium channel inhibits T cell function. PLoS One. 2013;8:e82944. doi: 10.1371/journal.pone.0082944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorenzi M. Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics. 2002;18:617–625. doi: 10.1093/bioinformatics/18.4.617. [DOI] [PubMed] [Google Scholar]
- Derler I, Fritsch R, Schindl R. Romanin C. CRAC inhibitors: identification and potential. Expert Opin Drug Discov. 2008;3:787–800. doi: 10.1517/17460441.3.7.787. [DOI] [PubMed] [Google Scholar]
- Di Sabatino A, Rovedatti L, Kaur R, Spencer JP, Brown JT, Morisset VD, Biancheri P, Leakey NA, Wilde JI, Scott L, Corazza GR, Lee K, Sengupta N, Knowles CH, Gunthorpe MJ, McLean PG, MacDonald TT. Kruidenier L. Targeting gut T cell Ca2+ release-activated Ca2+ channels inhibits T cell cytokine production and T-box transcription factor T-bet in inflammatory bowel disease. J Immunol. 2009;183:3454–3462. doi: 10.4049/jimmunol.0802887. [DOI] [PubMed] [Google Scholar]
- Elangovan M, Wallrabe H, Chen Y, Day RN, Barroso M. Periasamy A. Characterization of one- and two-photon excitation fluorescence resonance energy transfer microscopy. Methods. 2003;29:58–73. doi: 10.1016/s1046-2023(02)00283-9. [DOI] [PubMed] [Google Scholar]
- Frischauf I, Muik M, Derler I, Bergsmann J, Fahrner M, Schindl R, Groschner K. Romanin C. Molecular determinants of the coupling between STIM1 and Orai channels: differential activation of Orai1–3 channels by a STIM1 coiled-coil mutant. J Biol Chem. 2009;284:21696–21706. doi: 10.1074/jbc.M109.018408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz SB, Li Y. Rubin CS. Characterization of distinct tethering and intracellular targeting domains in AKAP75, a protein that links cAMP-dependent protein kinase IIβto the cytoskeleton. J Biol Chem. 1993;268:12796–12804. [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK. Scott JD. Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat Cell Biol. 2005;7:1066–1073. doi: 10.1038/ncb1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar P, Samanta K, Kramer H, Morris O, Bakowski D. Parekh AB. Dynamic assembly of a membrane signaling complex enables selective activation of NFAT by Orai1. Curr Biol. 2014;24:1361–1368. doi: 10.1016/j.cub.2014.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike Y, Ozaki Y, Qi R, Satoh K, Kurota K, Yatomi Y. Kume S. Phosphatase inhibitors suppress Ca2+ influx induced by receptor-mediated intracellular Ca2+ store depletion in human platelets. Cell Calcium. 1994;15:381–390. doi: 10.1016/0143-4160(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Korzeniowski MK, Manjarres IM, Varnai P. Balla T. Activation of STIM1-Orai1 involves an intramolecular switching mechanism. Sci Signal. 2010;3:ra82. doi: 10.1126/scisignal.2001122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FF, Elliott R, Colombero A, Gaida K, Kelley L, Moksa A, Ho SY, Bykova E, Wong M, Rathanaswami P, Hu S, Sullivan JK, Nguyen HQ. McBride HJ. Generation and characterization of fully human monoclonal antibodies against human Orai1 for autoimmune disease. J Pharmacol Exp Ther. 2013;345:225–238. doi: 10.1124/jpet.112.202788. [DOI] [PubMed] [Google Scholar]
- Liou J, Fivaz M, Inoue T. Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA. 2007;104:9301–9306. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE., Jr Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez E, Jardin I, Berna-Erro A, Bermejo N, Salido GM, Sage SO, Rosado JA. Redondo PC. STIM1 tyrosine-phosphorylation is required for STIM1-Orai1 association in human platelets. Cell Signal. 2012;24:1315–1322. doi: 10.1016/j.cellsig.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Luik RM, Wang B, Prakriya M, Wu MM. Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454:538–542. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji SS, Parker NJ, Williams RT, Van Stekelenburg L, Pearson RB, Dziadek M. Smith PJ. STIM1: a novel phosphoprotein located at the cell surface. Biochim Biophys Acta. 2000;1481:147–155. doi: 10.1016/s0167-4838(00)00105-9. [DOI] [PubMed] [Google Scholar]
- Mignen O, Thompson JL. Shuttleworth TJ. Reciprocal regulation of capacitative and arachidonate-regulated noncapacitative Ca2+ entry pathways. J Biol Chem. 2001;276:35676–35683. doi: 10.1074/jbc.M105626200. [DOI] [PubMed] [Google Scholar]
- Mignen O, Thompson JL. Shuttleworth TJ. Calcineurin directs the reciprocal regulation of calcium entry pathways in nonexcitable cells. J Biol Chem. 2003;278:40088–40096. doi: 10.1074/jbc.M306365200. [DOI] [PubMed] [Google Scholar]
- Mignen O, Thompson JL. Shuttleworth TJ. Arachidonate-regulated Ca2+-selective (ARC) channel activity is modulated by phosphorylation and involves an A-kinase anchoring protein. J Physiol. 2005;567:787–798. doi: 10.1113/jphysiol.2005.090209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignen O, Thompson JL. Shuttleworth TJ. STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store depletion or translocation to the plasma membrane. J Physiol. 2007;579:703–715. doi: 10.1113/jphysiol.2006.122432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignen O, Thompson JL. Shuttleworth TJ. Both Orai1 and Orai3 are essential components of the arachidonate-regulated Ca2+-selective (ARC) channels. J Physiol. 2008;586:185–195. doi: 10.1113/jphysiol.2007.146258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignen O, Thompson JL, Yule DI. Shuttleworth TJ. Agonist activation of arachidonate-regulated Ca2+-selective (ARC) channels in murine parotid and pancreatic acinar cells. J Physiol. 2005;564:791–801. doi: 10.1113/jphysiol.2005.085704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero M, Garcia-Sancho J. Alvarez J. Phosphorylation down-regulates the store-operated Ca2+ entry pathway of human neutrophils. J Biol Chem. 1994;269:3963–3967. [PubMed] [Google Scholar]
- Murphy CT, Bullock AJ. Westwick J. A role for protein phosphorylation in modulating Ca2+ elevation in rabbit platelets treated with thapsigargin. Biochem J. 1996;313:83–89. doi: 10.1042/bj3130083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Gomez LL. Dell'Acqua ML. Imaging kinase-AKAP79-phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J Cell Biol. 2003;160:101–112. doi: 10.1083/jcb.200209127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB. Penner R. Depletion-activated calcium current is inhibited by protein kinase in RBL-2H3 cells. Proc Natl Acad Sci U S A. 1995;92:7907–7911. doi: 10.1073/pnas.92.17.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE. Lewis RS. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo-Guisado E, Campbell DG, Deak M, Alvarez-Barrientos A, Morrice NA, Alvarez IS, Alessi DR. Martin-Romero FJ. Phosphorylation of STIM1 at ERK1/2 target sites modulates store-operated calcium entry. J Cell Sci. 2010;123:3084–3093. doi: 10.1242/jcs.067215. [DOI] [PubMed] [Google Scholar]
- Rice LV, Bax HJ, Russell LJ, Barrett VJ, Walton SE, Deakin AM, Thomson SA, Lucas F, Solari R, House D. Begg M. Characterization of selective calcium-release activated calcium channel blockers in mast cells and T-cells from human, rat, mouse and guinea-pig preparations. Eur J Pharmacol. 2013;704:49–57. doi: 10.1016/j.ejphar.2013.02.022. [DOI] [PubMed] [Google Scholar]
- Sakai T. Ambudkar IS. Role for protein phosphatase in the regulation of Ca2+ influx in parotid gland acinar cells. Am J Physiol Cell Physiol. 1996;271:C284–C294. doi: 10.1152/ajpcell.1996.271.1.C284. [DOI] [PubMed] [Google Scholar]
- Sekar RB. Periasamy A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J Cell Biol. 2003;160:629–633. doi: 10.1083/jcb.200210140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan JT, Gilmore RC, Watson MJ, Archer CB. Tarran R. 17β-Estradiol inhibits phosphorylation of stromal interaction molecule 1 (STIM1) protein: implication for store-operated calcium entry and chronic lung diseases. J Biol Chem. 2013;288:33509–33518. doi: 10.1074/jbc.M113.486662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuttleworth TJ. Arachidonic acid activates the noncapacitative entry of Ca2+ during [Ca2+]i oscillations. J Biol Chem. 1996;271:21720–21725. doi: 10.1074/jbc.271.36.21720. [DOI] [PubMed] [Google Scholar]
- Smyth JT, Petranka JG, Boyles RR, DeHaven WI, Fukushima M, Johnson KL, Williams JG. Putney JW., Jr Phosphorylation of STIM1 underlies suppression of store-operated calcium entry during mitosis. Nat Cell Biol. 2009;11:1465–1472. doi: 10.1038/ncb1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopulos PB, Schindl R, Fahrner M, Zheng L, Gasmi-Seabrook GM, Muik M, Romanin C. Ikura M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat Commun. 2013;4:2963. doi: 10.1038/ncomms3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JL. Shuttleworth TJ. Orai channel-dependent activation of phospholipase C-δ: a novel mechanism for the effects of calcium entry on calcium oscillations. J Physiol. 2011;589:5057–5069. doi: 10.1113/jphysiol.2011.214437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JL. Shuttleworth TJ. A plasma membrane-targeted cytosolic domain of STIM1 selectively activates ARC channels, an arachidonate-regulated store-independent Orai channel. Channels. 2012;6:370–378. doi: 10.4161/chan.21947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JL. Shuttleworth TJ. Molecular basis of activation of the arachidonate-regulated Ca2+ (ARC) channel, a store-independent Orai channel, by plasma membrane STIM1. J Physiol. 2013;591:3507–3523. doi: 10.1113/jphysiol.2013.256784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojyo Y, Tanimura A. Matsumoto Y. Suppression of capacitative Ca2+ entry by serine/threonine phosphatase inhibitors in rat parotid acinar cells. Jpn J Pharmacol. 1995;69:381–389. doi: 10.1254/jjp.69.381. [DOI] [PubMed] [Google Scholar]
- Wu MM, Buchanan J, Luik RM. Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 2006;174:803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Jin H, Cai X, Li S. Shen Y. Structural and mechanistic insights into the activation of stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci U S A. 2012;109:5657–5662. doi: 10.1073/pnas.1118947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino T, Ishikawa J, Ohga K, Morokata T, Takezawa R, Morio H, Okada Y, Honda K. Yamada T. YM-58483, a selective CRAC channel inhibitor, prevents antigen-induced airway eosinophilia and late phase asthmatic responses via Th2 cytokine inhibition in animal models. Eur J Pharmacol. 2007;560:225–233. doi: 10.1016/j.ejphar.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Yu F, Sun L. Machaca K. Orai1 internalization and STIM1 clustering inhibition modulate SOCE inactivation during meiosis. Proc Natl Acad Sci U S A. 2009;106:17401–17406. doi: 10.1073/pnas.0904651106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA. Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]