

Abstract
Patients with chronic kidney disease (CKD) have a higher risk of developing cardiovascular diseases. Their vascular endothelium is dysfunctional, among other things, because it is permanently exposed to uraemic toxins, several of which, mostly protein-bound compounds such as indoxyl sulfate (IS) and p-cresyl sulphate, having poor clearance by conventional dialysis, induce endothelial toxicity. However, the molecular mechanism by which uraemic toxins regulate early stages of endothelial dysfunction remains unclear. Recent studies have demonstrated the important role of integrin-linked kinase (ILK) in the maintenance of endothelial integrity. In this study, we investigate the involvement of ILK in the mechanism underlying vascular endothelial damage that occurs in uraemia. First, we show that incubation of EA.hy926 cells with human uraemic serum from CKD patients upregulates ILK activity. This ILK activation also occurs when the cells are exposed to IS (25–100 μg ml−1), p-cresol (10–100 μg ml−1) or both combined, compared to human serum control. Next, we observed that high doses of both toxins together induce a slight decrease in cell proliferation and increase apoptosis and reactive oxygen species production. Interestingly, these toxic effects displayed a strong increase when the ILK protein is knocked down by small interfering RNA, even at low doses of uraemic toxins. Abrogation of AKT has demonstrated the ILK/AKT signalling pathway involved in these processes. This study has demonstrated the implication of ILK in the protection against endothelial cell damage induced by uraemic toxins, a molecular mechanism that could play a protective role in the early stages of endothelial dysfunction observed in uraemic patients.
Key points
Patients with chronic kidney disease have a higher risk of developing cardiovascular diseases than the general population. Their vascular endothelium is dysfunctional, among other things, because it is permanently exposed to uraemic toxins, several of which have poor clearance by conventional dialysis.
Recent studies have demonstrated the important role of integrin-linked kinase (ILK) in the maintenance of endothelial integrity and in this study we investigate the involvement of ILK in the mechanism underlying vascular endothelial damage that occurs in uraemia.
For the first time, we demonstrate the implication of ILK in the protection against endothelial cell damage (inhibition of proliferation, toxicity, oxidative stress and programed cell death) induced by uraemic serum from chronic kidney disease patients and uraemic toxins.
This molecular mechanism may have clinical relevance because it highlights the importance of maintaining high levels of ILK activity to help preserve endothelial integrity, at least in early stages of chronic kidney disease.
Introduction
Patients with chronic kidney disease (CKD) are at higher risk of cardiovascular diseases than the general population (Wheeler, 1996; Parfrey & Foley, 1999). This cannot be explained only by the high prevalence of traditional cardiovascular risk factors such as hypertension, hyperlipidaemias, diabetes, smoking or left ventricular hypertrophy. Thus, the possible contribution of other factors, such as endothelial dysfunction, has been studied in recent years (Passauer et al. 2000). Maintaining the functional integrity of the endothelium is important in the prevention of vascular diseases (Ross, 1993). Under physiological conditions, endothelial lesions can be repaired by proliferation and migration of different reparative cells and also by circulating endothelial populations such as endothelial progenitor cells (EPCs) (Sabatier et al. 2009; Rabelink et al. 2010). Chronic exposure to vascular risk factors alters the regulatory properties of the endothelium, which progresses toward a pro-inflammatory pattern, senescence and apoptosis (Ross, 1993). In fact, there is a body of evidence indicating that endothelial dysfunction begins early in the progression of CKD, especially as a result of an imbalance between endothelial damage and repair (Stam et al. 2006; Jourde-Chiche et al. 2011).
CKD often evolves to chronic renal failure. In the final stages, a clinical situation in which the internal environment is completely altered, known as uraemia, is reached (Rodriguez-Puyol & Praga, 1998). The endothelium of patients with CKD is therefore permanently exposed to uraemic toxins. A uraemic environment promotes endothelial damage, suggesting an involvement of uraemia-specific factors in this process (Jourde-Chiche et al. 2011). Uraemic toxins are classified into three groups: water-soluble molecules with low molecular weight such as urea, middle molecules and protein-bound uraemic toxins (Vanholder et al. 2003). The last named are poorly eliminated by conventional haemodialysis (Vanholder et al. 2003; Jourde-Chiche et al. 2009). Several uraemic toxins, mostly protein-bound compounds such as indoxyl sulfate (IS) and p-cresyl sulfate, the main circulating form of p-cresol (pc), have specific endothelial toxicity (Dou et al. 2004; Meijers et al. 2009; Adelibieke et al. 2013) and have recently emerged as the likely pathogens inducing endothelial dysfunction in CKD (Günthner et al. 2009; Liabeuf et al. 2010). In this regard, it has been observed that removing uraemic toxins improves endothelial function and repair by decreasing endothelial microparticles and increasing EPCs (Ramírez et al. 2002; De Groot et al. 2004).
However, the mechanisms by which increased uraemia might influence endothelial cells, and especially the early responses of endothelial cell injury, are still not well understood.
Endothelial cell dysfunction is a consequence of a different expression pattern of several intracellular signalling pathways. In the cardiovascular system, integrins are the molecules that propagate signals from outside the cell by binding to the extracellular matrix and the cytoskeleton (Alenghat & Ingber, 2002). In this way, integrin linked kinase (ILK) is a key component of the integrin signalling complex that functions as an intracellular scaffold molecule and as a kinase that links the cell-adhesion receptors, integrins and growth factors to the actin cytoskeleton and to a range of signalling pathways (Hannigan et al. 1996; Wu & Dedhar, 2001; Legate et al. 2006). ILK-mediated signalling proteins include protein kinase B (PKB/AKT), glycogen synthase kinase 3β (GSK-3β) and mitogen-activated protein kinases (MAPKs) (Boulter & Van Obberghen-Schilling, 2006). ILK regulates signalling pathways for cell adhesion-mediated cell survival (anoikis), apoptosis, proliferation and mitosis, migration, invasion, and vascularization and tumour angiogenesis (Hannigan et al. 2011).
It has been observed that endothelial ILK plays a critical role in vascular vessel integrity and angiogenesis, is essential for extracellular matrix and endothelial cell interaction, confers key survival signals to mature endothelium and regulates the recruitment and survival of EPCs (Friedrich et al. 2004; Kaneko et al. 2004; Cho et al. 2005). However, despite its importance, the involvement of ILK in endothelial dysfunction in the CKD pathophysiological context has not been analysed in depth. Therefore, the present study has been designed to investigate if the ILK protein is involved in uraemic serum or uraemic toxin-induced endothelial cell damage. We clearly demonstrate that the initial response of endothelial cells to uraemic serum from patients with CKD or uraemic toxins involves ILK/AKT pathway activation and suggest a relevant role of this molecular mechanism of protection against endothelial cell damage induced by uraemic toxins in its early stages.
Methods
Ethical approval
The project was approved by the Ethical Committee of Renal Research of the Spanish biobank network (National Register Number 0000931). Informed consent was obtained in writing from all patients after institutional approval. The studies conformed to the standards set by the latest revision of the Declaration of Helsinki.
Drugs and other reagents
Uraemic solutes pc and IS were obtained from Sigma Chemical Company (St Louis, MO, USA). Primary antibodies against ILK, GSK-3β, P-GSK-3β, AKT and P-AKT were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-rabbit secondary antibody was purchased from Dako (Barcelona, Spain) and anti-goat secondary antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All antibody dilutions were 1:500 or 1:5000. Enhanced chemiluminescent reagent and X-ray films were from Thermo Fisher Scientific (Waltham, MA, USA). 5-Bromo-2′-deoxy-uridine (BrdU) was from Roche (Meyland, France). Fluorescein isothiocyanate (FITC)-conjugated anti-BrdU was from Becton-Dickinson (San José, CA, USA). Specific Signal-Silencer AKT small interfering RNA (siRNA), targets AKT1 and AKT2 and the GSK-3 fusion protein were purchased from Cell Signaling Technology. Silencer negative control #1 siRNA was from Ambion, Inc. (Austin, TX, USA), and ILK-specific siRNA was from Bionova Científica (Barcelona, Spain). ReliaBLOT Block and the ReliaBLOT HRP Conjugated were from Bethyl Laboratories, Inc. (Montgomery, TX, USA). The fluorescent probe dichlorodihydrofluorescein-diacetate (H2DCFDA) was from Molecular Probes (Life Technologies, Paisley, UK). Antibiotics, lipofectamine, opti-MEM and RPMI medium were from Invitrogen (Life Technologies). Dulbecco's modified Eagle's medium and fetal bovine serum (FBS) were from Lonza (Basel, Switzerland). All reagents were prepared in DMSO so that the final concentration was <0.1%. Other antibodies and reagents were from Sigma Chemical Company.
Human uraemic serum
Serum samples were obtained from the Spanish biobank network (National Register Number 0000931). Twenty-six CKD stage 4–5 patients undergoing conservative treatment (14 females, 12 men) with a mean age of 73.7 years (range: 45–79 years) were selected. Renal function was calculated per the Modification of Diet in Renal Disease 7 (MDR-D7) protocol, and creatinine clearance (ClCr) was 21.3 ± 5.1 ml min–1 (range: 14–24). The criterion for patient selection was: absence of inflammatory disease, acute or chronic infection, autoimmune diseases, hepatic insufficiency, diabetes or malignancy; and non-smokers. Serum samples were frozen at −80°C until analysis. To minimize differences between patients, serum samples were pooled for the experiments (uraemic serum). Human autologous serum (AB serum; BioWhittaker, Walkersville, MD, USA) was used as control (normal serum).
Uraemic toxins
Uraemic toxins were tested at concentrations in the uraemic range. p-Cresol was diluted from a stock solution prepared in methanol and IS was diluted from stock solutions prepared in water. Solutes were diluted at least 1:500 in culture medium to reach mean uraemic concentrations. Uraemic solutes were compared with their respective controls (methanol or water).
Cell culture and treatments
The permanent human endothelial cell line (EA.hy926), established by fusion of human umbilical vein endothelial cells (HUVECs) and the adenocarcinoma epithelial cell line A549 (Edgell et al. 1983), was obtained from ATCC. Cells were maintained in culture medium supplemented with 20 mm l-glutamine and antibiotics (penicillin, 100 U ml−1; streptomycin, 100 mg ml−1) and 10% fetal bovine serum. Cultures from passages 2–10 were used. HUVECs were obtained from umbilical cord vein by collagenase digestion, as previously described (Dou et al. 2004). For the experiments, subconfluent cells were serum-deprived for 24 h and incubated with the different treatments at variable concentrations and times (see figure legends).
Western blot analysis
Cell samples were lysated in a solution containing 150 mm NaCl, 10 mm Tris-HCl (pH 7.4), 5 mm EDTA, 1% deoxycholic acid, 0.1% SDS, 1% Triton X-100 and protease inhibitors 1 mm phenylmethylsulphonylfluoride, 10 mg ml−1 aprotinin, 2 mg ml−1 leupeptin and the phosphatase inhibitor 0.2 mm NaVO4. Cell proteins (30–40 μg) were run in an 8–10% SDS–polyacrylamide gel, transferred onto a nitrocellulose membrane (Trans-Blot Transfer Medium, Bio-Rad, CA, USA) and incubated overnight at 4°C with specific antibodies as previously described (Gonzalez-Ramos et al. 2013). This incubation was followed by a second incubation with a peroxidase-conjugated secondary antibody and immunoreactive products were detected by chemiluminescence using the enhanced chemiluminescent Western blotting detection reagents (Amersham Biosciences, Amersham, UK) following the protocol provided by the manufacturer.
Endothelial cell proliferation assay by BrdU incorporation
Endothelial cell proliferation was assessed by BrdU incorporation into cellular DNA according to the manufacturer's instructions (see below). After treatments, BrdU was added for 2 h. The level of BrdU cell incorporation was detected in labelling cells with propidium iodine (PI) and FITC-conjugated anti-BrdU monoclonal antibodies, analysed on a FACScan flow cytometer (Becton-Dickinson), at 488 nm.
Endothelial cell proliferation assay by proliferating cell nuclear antigen (PCNA) expression
The expression of PCNA was measured by flow cytometry and confocal immunofluorescence, according to the manufacturer's instructions (see below).
For flow cytometry, adherent cells were collected after treatment, washed with PBS and fixed with 70% ice-cold ethanol (2 h, −20°C). Cells were washed twice with ice-cold staining buffer (PBS with 1% FBS, 0,09% NaN3) and incubated with 20 μl FITC-conjugated anti-PCNA antibody or FITC isotype control (Becton-Dickinson) for 30 min, in the dark. Cells were washed twice and, after cellular DNA staining with 5 ng ml−1 PI dissolved in PBS, they were analysed on a FACScan flow cytometer. FITC fluorescence signal was measured in PI-negative cells and PCNA expression level was estimated using the mean fluorescence intensity of the cell population. Data were analysed with WinMDI software (Scripps Research Institute, La Jolla, CA, USA).
Confocal immunofluorescence experiments were performed in cells fixed with ice cold methanol, using a monoclonal antibody against PCNA (1:100, Proliferating Cell Nuclear Antigen, PC10, Santa Cruz Biotechnology) and a secondary antibody, Alexa Fluor 647 goat-anti-mouse IgG (1:300, Life Technologies). Cell nuclei were identified by staining with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma). Samples were analysed using a LEICA TCS-SP5 confocal micro-scope (Leica Microsystems, Wetzlar, Germany). Pictures were obtained and fluorescence intensity was measured by densitometry using ImageJ software (http://rsbweb.nih.gov/ij/).
Endothelial viability assay
The effect of uraemic serum or solutes on cell viability was determined by Trypan Blue exclusion. In brief, EA.hy926 cells cultured on six-well culture plates were incubated for 24 or 48 h with treatments or with their respective controls. Endothelial cells were then detached with trypsin-EDTA solution, Trypan Blue solution was added and the percentage of cells excluding Trypan Blue was determined.
Metabolic activity assay
After treatment, 3-[4,5-dimethylthiazol-2-yl]-2,5- diphenyltetrazolium bromide (MTT) stock solution in PBS was added to the culture (final concentration 0.5 mg ml−1) and cells were incubated (2.5 h, 37°C). Formazan crystals were dissolved in 0.1 m HCl in isopropanol, and absorbance was measured on a microplate reader (test wavelength 570 nm, reference wavelength 690 nm).
Quantification of sub-G1 DNA content by flow cytometry
Quantification of apoptotic cells was carried out by measurement of the fraction of cells with sub-G1 DNA content by flow cytometry analysis after PI staining. Adherent and floating cells were collected after treatment, washed with ice-cold PBS, and fixed with 70% ice-cold ethanol (30 min, 4°C). Cells were washed twice with PBS and treated with RNAse (1 mg ml−1; 30 min, 37°C). After staining of cellular DNA with 5 ng ml−1 PI dissolved in PBS, cells were analysed on a FACScan flow cytometer. Percentages of cells in different cell cycle phases were calculated from DNA histograms. Cells with sub-G1 DNA content were considered apoptotic.
Assessment of apoptosis by Annexin V and PI staining
Apoptosis was also detected using an Annexin V-FITC Apoptosis Detection Kit (Calbiochem, La Jolla, CA, USA). Briefly, cells were double stained with annexin V-FITC and PI, following manufacturer's instructions. Staining was measured by flow cytometry on the FACScan and the distribution of cells was analysed using CellQuest software (Becton-Dickinson). Data from 10,000 cells were collected for each data file. Early apoptosis was defined as Annexin V-FITC-positive and PI-negative cells, and late apoptosis was defined as double positive Annexin V-FITC+/PI+ staining.
siRNA transfection
To deplete expression of AKT or ILK protein by specific siRNA, oligonucleotides cells were seeded 24 h before transfection. Cells were transfected in 1 ml of opti-MEM with 100 nm siRNA or Silencer-negative control (Scrambled RNA) using LipofectAMINE. After 8 h incubation with the RNA complex, 1 ml of medium containing FBS was added. The cells were treated as indicated.
ILK activity measurement
ILK activity in human mesangial cells extracts was determined using an immunoprecipitation in vitro kinase assay (Del Nogal et al. 2012). Briefly, 300 μg of extracted cell proteins were immunoprecipitated with an anti-ILK antibody. GSK-3β fusion protein was used as the ILK substrate and ATP was the phosphate donor. Proteins were run on 12% SDS-PAGE gels. Both ReliaBLOT Block and the ReliaBLOT HRP Conjugated were used for membrane blotting and antibody incubation following the protocol provided by the manufacturer (Bethyl Laboratories). Immunoreactive products were detected as previously described and, after autoradiography, the phosphorylated GSK-3β protein was quantified by densitometric analysis with Scion Image software.
Reactive oxygen species (ROS) detection
ROS production was determined by two methods.
First, as described by González-Ramos et al. (2012), H2DCFDA (5 mm) loaded cells were detached by trypsinization after treatments and cellular fluorescence intensity was measured on a FACScan flow cytometer with a 488 nm argon laser. For each analysis, 10,000 events were recorded and cell viability was controlled with PI. ROS production was estimated using the mean fluorescence intensity of the cell population. Data were analysed with WinMDI software (Scripps Research Institute).
In addition, ROS production was measured by fluorescence confocal microscopy, according to the manufacturer's instructions. After treatments, cells were washed twice and loaded with 5 μm CellROX Deep Red Reagent fluorogenic probe (Molecular Probes, Life Technologies), by incubating them at 37°C for 1 h. The medium was removed and cells were washed three times with PBS. Samples were analysed using a LEICA TCS-SP5 confocal microscope (Leica Microsystems) at excitation/emission of 640/665 nm (Deep Red). Pictures were obtained and fluorescence intensity was measured by densitometry using ImageJ software (http://rsbweb.nih.gov/ij/).
Data analysis and statistical procedures
All values are presented as mean ± standard error of the mean (SEM). Experiments were repeated a minimum of three times. Comparisons between groups were performed by using non-parametric statistics. A P value of < 0.05 was considered significant.
Results
Uraemic serum and uraemic toxins increase ILK activity in endothelial cells
First, we tested the effect of uraemic serum on ILK expression levels or activation by performing dose and time–response experiments on EA.hy926 endothelial cells. As shown in Fig. 1A and B, uraemic serum induced an increase in the ILK downstream effector GSK-3β phosphorylation levels at serine-9, compared to cells exposed to normal human serum control medium, without changes in ILK expression levels. This increase was significant from exposure to 2.5% serum and 2 h of incubation. Given that most previous in vitro studies have been carried out in HUVECs, we confirmed this finding by incubating the cells with different percentages of serum for 24 h. We observed the same effect on GSK-3β phosphorylation in a dose-dependent manner, with no changes observed in ILK cellular content (Fig. 1C). Thus, we decided to use the EA.hy926 endothelial cell line as a cellular model. Next, we tested the ability of two uraemic retention solutes, IS and pc, to increase ILK activity in EA.hy926 cells. As shown in Fig. 2A and B, IS (25–100 μg ml−1) and pc (10–100 μg ml−1) (acting as a surrogate of the main in vivo metabolite, p-cresyl sulfate) induced both rapid and sustained GSK-3β phosphorylation when they were added to cells growing in normal human serum (2.5%). Moreover, the exposure of cells to a combination of both toxins at low concentration (IS (25 μg ml−1) plus pc (10 μg ml−1)) or high concentration (IS plus pc (100 μg ml−1 each)) induced an additive effect in GSK-3β phosphorylation after 2–24 h of treatment. All these effects were dose- and time-dependent and reached maximum at high doses of both toxins together (Fig. 3A), without changes in ILK content. Furthermore, because GSK-3β protein may be targeted by other kinases, the ability of uraemic toxins alone or combined to increase specific ILK activity was tested by in vitro analysis of immunoprecipitated ILK activity, measured as capacity to phosphorylate GSK-3β fusion protein (Fig. 3B). In addition, we confirmed that the increased phosphorylation of GSK-3β is ILK-dependent because it is completely abolished when ILK is knocked down by ILK siRNA, as measured by Western blotting (Fig. 3C). Thereby, these results strongly confirmed the specific ILK activity.
Figure 1. Uraemic serum up-regulates ILK activity in EA.hy926 cells and HUVECs.
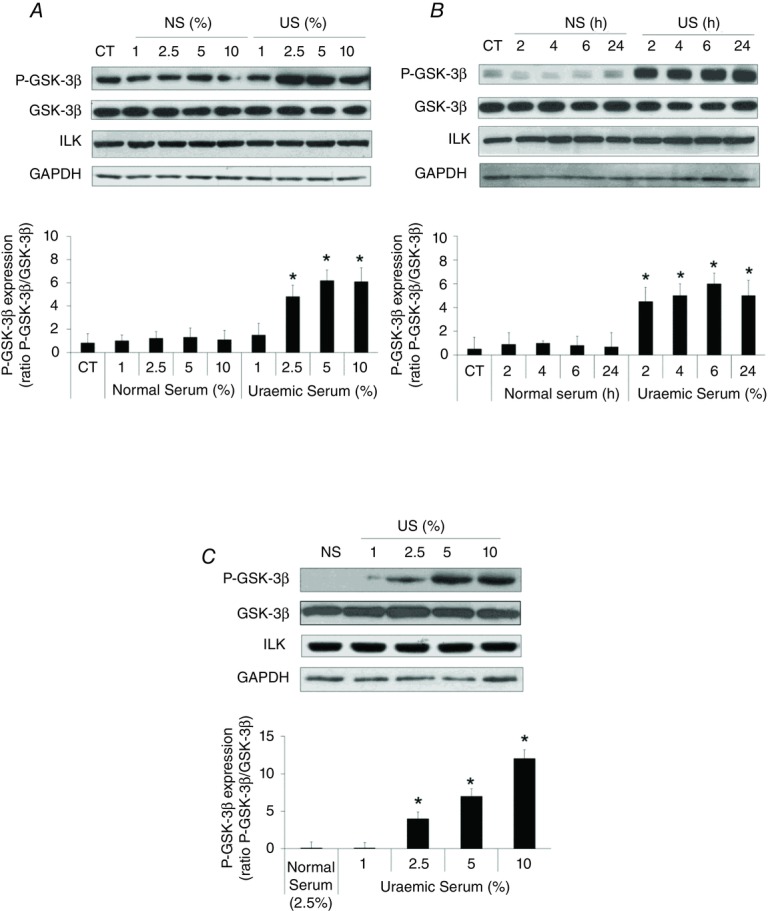
A and B, EA.hy926 cells were incubated in medium supplemented with different normal serum (NS) or uraemic serum (US) concentrations for 24 h (A) or with 2.5% NS or US during different times (B). C, HUVECs were incubated in medium supplemented with 2.5% NS or 1–10% US for 24 h. Representative Western blots of phosphorylated GSK-3β in the serine-9 residue (P-GSK-3β) or ILK are shown. Total GSK-3β or GAPDH levels were determined as their respective endogenous controls. Bars represent the normalized densitometric analysis of the blots against the endogenous control values. All the values are represented as mean ± SEM of five independent experiments. *P < 0.05 vs. NS (CT serum-depleted cells, 24 h).
Figure 2. Uraemic toxins increase ILK activity in EA.hy926 cells.
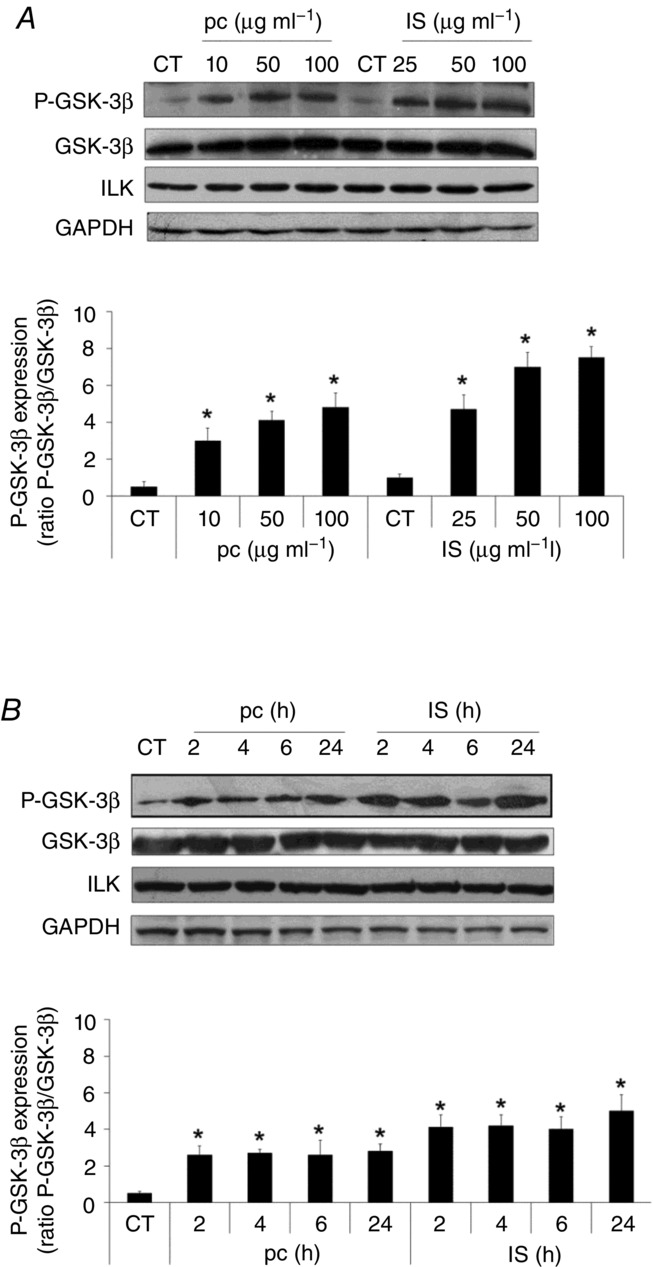
Cells were incubated in medium supplemented with 2.5% normal serum (NS) plus p-cresol (pc; 10, 50 or 100 μg ml−1) or indoxyl sulfate (IS; 10, 25, 50 and 100 μg ml−1) for 24 h (A) or with 2.5% NS plus pc (10 μg ml−1) or IS (25 μg ml−1) for different times (B). Representative Western blots of phosphorylated GSK-3β in the serine-9 residue (P-GSK-3β) or ILK are shown. Total GSK-3β or GAPDH levels were determined as their respective endogenous control. Bars represent the normalized densitometric analysis of the blots against the endogenous control values. All the values are represented as mean ± SEM of five independent experiments. *P < 0.05 vs. control (CT; 2.5% NS, 24 h).
Figure 3. Combination of uraemic toxins increases ILK activity in EA.hy926 cells.
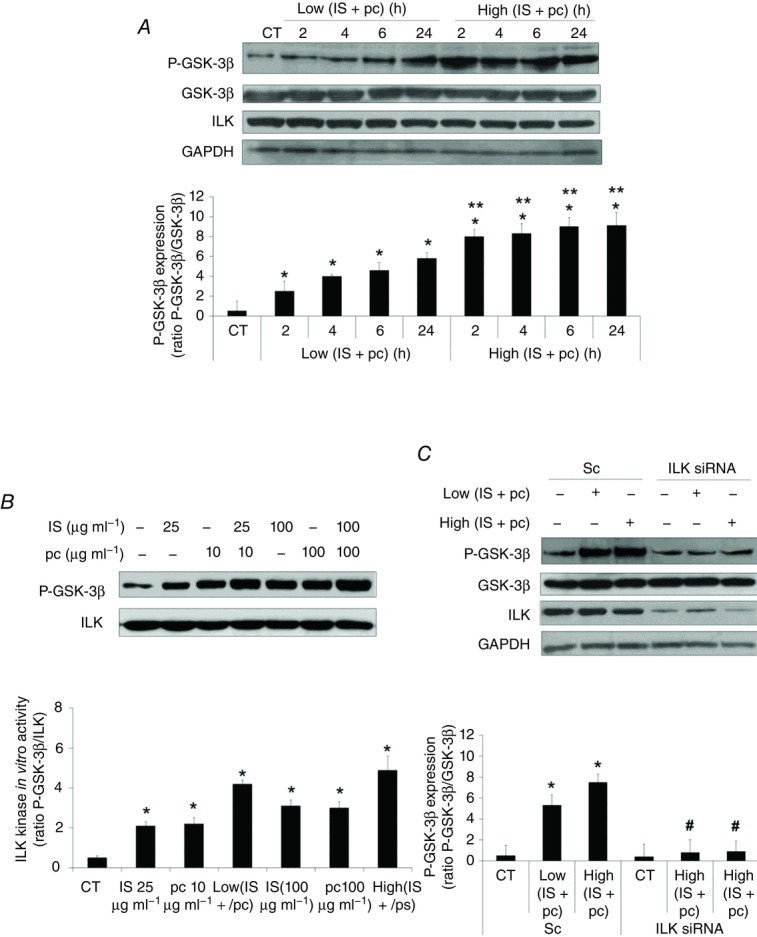
A, cells were incubated in medium supplemented with 2.5% normal serum (NS) plus a combination of low concentrations of indoxyl sulfate (IS; 25 μg ml−1) and p-cresol (pc; 10 μg ml−1) (Low IS + pc) or plus a combination of high concentrations of IS (100 μg ml−1) and pc (100 μg ml−1) (High IS + pc) for different times. Representative Western blots of phosphorylated GSK-3β in the serine-9 residue (P-GSK-3β) or ILK are shown. Total GSK-3β or GAPDH levels were determined as endogenous control. B, cells were incubated in medium supplemented with 2.5% NS plus IS (25 or 100 μg ml−1), pc (10 or 100 μg ml−1), Low (IS + pc) or High (IS + pc), for 24 h. In vitro kinase activity of ILK was determined in cell lysates, by immunoprecipitation of ILK followed by incubation with a fixed amount of exogenous GSK-3 protein-fusion as substrate. Levels of xogenous GSK-3 protein phosphorylation in the serine-9 residue (P-GSK-3β) were measured by Western blot and equal ILK loading was confirmed. C, cells were depleted of ILK with specific siRNA (100 nm) and treated afterwards as in A for 24 h. Representative Western blots of phosphorylated GSK-3β in the serine-9 residue (P-GSK-3β) or ILK are shown. Total GSK-3β or GAPDH levels were determined as endogenous control. Bars represent the normalized densitometric analysis of the blots against the endogenous control (A and C) or ILK (B) values. All values are represented as mean ± SEM of six independent experiments. *P < 0.05 vs. control (CT; 2.5% NS, 24 h), **P < 0.05 vs. Low (IS + pc), #P < 0.05 vs. Sc.
ILK protein plays a protective role in uraemic toxin-dependent induction of cell proliferation and viability decrease and apoptosis increase of EA.hy926 endothelial cells
The effect of uraemic serum in cell proliferation was analysed by BrdU incorporation into cellular DNA. Incubation with uraemic serum (2.5%, 24 and 48 h) decreased cellular proliferation compared with normal serum (Fig. 4A). Next, to determine the specific biological activity of uraemic toxins, EA.hy926 cells were exposed to different concentrations of IS and pc alone or combined for 24 h. As shown in Fig. 4B, only exposure of cells to high doses of IS or pc, or both together, significantly decreased cell proliferation. Interestingly, the cells displayed a strong decrease in cell proliferation when the ILK protein was knocked down by siRNA at low doses of IS and pc (Fig. 4C), something that also occurred at high doses (data not shown). Furthermore, we confirmed that the effect of uraemic toxins on proliferation is specifically affected by ILK siRNA, by a second method, measuring the expression of the S-phase marker PCNA both by flow cytometry and confocal immunofluorescence (Fig. 4D and E). To investigate whether uraemic serum or uraemic toxins would affect endothelial cell viability, the effect of these compounds was determined by Tripan Blue exclusion. Again, after 24 or 48 h, endothelial cell viability was impaired by the uraemic serum (Fig. 5A). Likewise, cell viability was significantly reduced in cells incubated with high doses of IS or pc or both together for 24 h (Fig. 5B). The effect of ILK was confirmed in cells abrogated by specific ILK siRNA, in which cell proliferation and viability determined using the MTT assay were significantly reduced at low doses of IS and pc (Fig. 5C) as well as at high doses (data not shown). Next, the percentage of hypodiploid apoptotic cells was analysed by flow cytometry. Apoptosis levels produced by uraemic serum were low (Fig. 6A) and IS, pc or both together induced a slight increase in the percentage of apoptotic cells, at either 24 h (Fig. 6B) or 48 h (data not shown), with increased cytotoxicity at higher concentrations and combinations of toxins (Fig. 6B). ILK depletion substantially increased the apoptosis induced by low doses of uraemic toxins (Fig. 6C). As expected, this increase was also significant at high doses (data not shown). Taken together, these data suggest that upregulation of the ILK pathway by uraemic toxins activates a mechanism of protection against a decrease in cell proliferation and viability and increase in apoptosis induced by IS and pc in EA.hy926 cells.
Figure 4. Role of ILK in the EA.hy926 cell proliferation decrease induced by uraemic toxins.
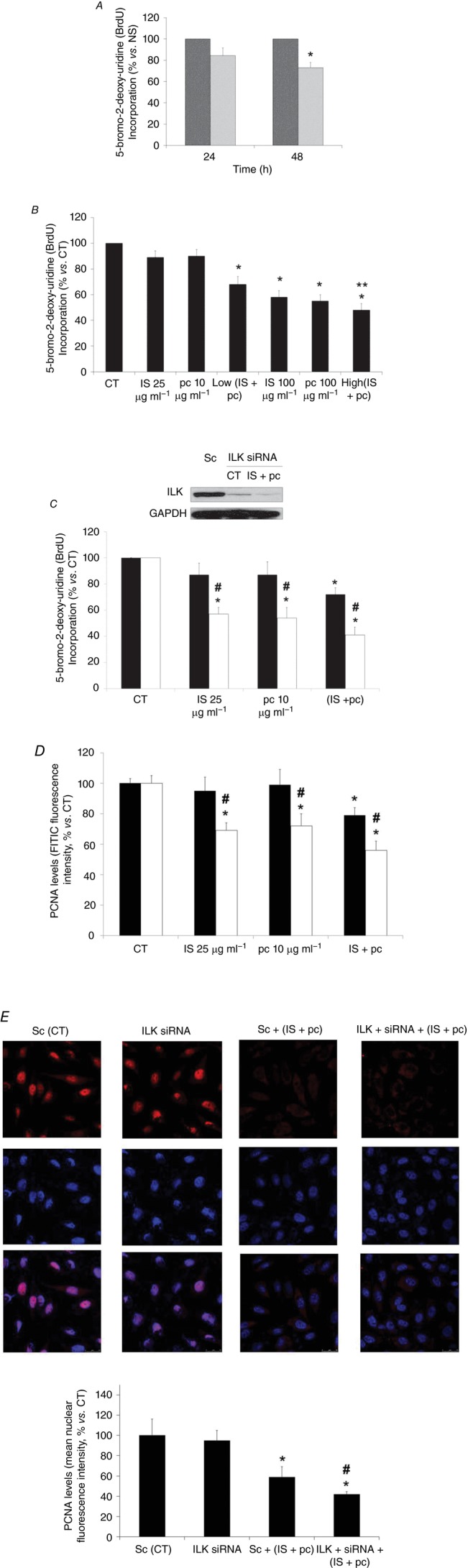
A, cells were incubated in medium supplemented with normal serum (NS, dark grey bars) (10%) or uraemic serum (light grey bars) (10%) for 24 and 48 h. B, cells were incubated in medium supplemented with 2.5% NS plus indoxyl sulfate (IS; 25 or 100 μg ml−1), p-cresol (pc; 10 or 100 μg ml−1), a combination of low concentrations of IS (25 μg ml−1) and pc (10 μg ml−1) (Low IS + pc) or plus a combination of high concentrations of IS (100 μg ml−1) and pc (100 μg ml−1) (High IS + pc) for 24 h. **P < 0.05 vs. Low (IS + pc). C and D, cells were depleted of ILK with specific siRNA (open bars) (100 nm) and treated afterwards with IS (25 μg ml−1), pc (10 μg ml−1) or both, for 24 h. Scrambled RNA (Sc) (filled bars) was used as control. After incubation, endothelial cell proliferation was measured by cytometric analysis of BrdU incorporation (C) or by cytometric analysis of PCNA expression levels, measured by fluorescence intensity of labelled cells with an FITC-conjugated antibody (D) and calculated as percentage of fluorescence intensity versus control (CT; 2.5% NS, 24 h). Data are expressed as mean ± SEM of six independent experiments. *P < 0.05 vs. control (CT; 2.5% NS, 24 h); #P < 0.05 vs. Sc. Representative Western blot of total ILK levels to check its depletion, and GAPDH levels as endogenous control are shown in C. E, after incubation as in C and D, nuclear PCNA expression (red) was determined by confocal microscopy. Nuclei were stained with DAPI (blue). A representative experiment is shown. Bar graphs represent the densitometric analysis of the fluorescence of 20 cells of six independent experiments. The results are expressed as a percentage of nuclear fluorescence intensity versus scrambled untreated control (Sc (CT); 2.5% NS, 24 h) and are the mean ± SEM of six different experiments. *P < 0.05 vs. Sc (CT); #P < 0.05 vs. Sc + (IS +pc).
Figure 5. Role of ILK in the EA.hy926 cell viability decrease induced by uraemic toxins.
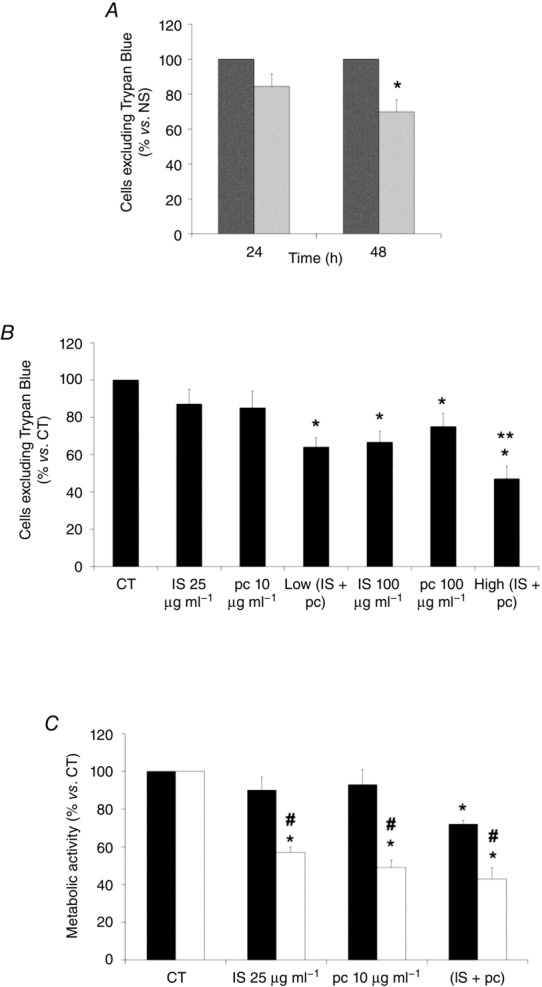
A, cells were incubated in medium supplemented with normal serum (NS, dark grey bars) (10%) or uraemic serum (light grey bars) (10%) for 24 and 48 h. B, cells were incubated in medium supplemented with 2.5% NS plus indoxyl sulfate (IS; 25 or 100 μg ml−1), p-cresol (pc; 10 or 100 μg ml−1), a combination of low concentrations of IS (25 μg ml−1) and pc (10 μg ml−1) (Low IS + pc) or plus a combination of high concentrations of IS (100 μg ml−1) and pc (100 μg ml−1) (High IS + pc) for 24 h. After incubation, endothelial cell viability was determined by Trypan Blue exclusion. **P < 0.05 vs. Low (IS + pc). C, cells were depleted of ILK with specific siRNA (open bars) (100 nm) and treated afterwards with IS (25 μg ml−1), pc (10 μg ml−1) or both, for 24 h. Scrambled RNA (Sc) (filled bars) was used as control. After incubation, endothelial cell viability was measured by MTT assay. Data are expressed as mean ± SEM of six independent experiments. *P < 0.05 vs. control (CT; 2.5% NS, 24 h); #P < 0.05 vs. Sc.
Figure 6. Role of ILK in the EA.hy926 cell uraemic toxin-induced apoptosis.
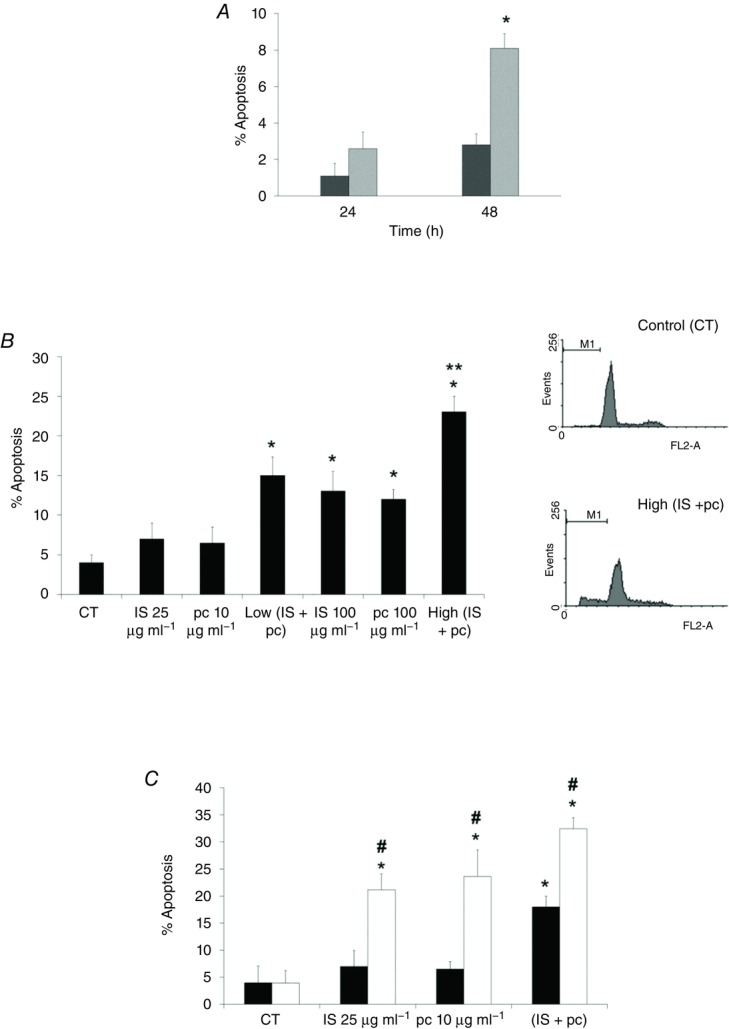
A, cells were incubated in medium supplemented with normal serum (NS, dark grey bars) (10%) or uraemic serum (light grey bars) (10%) for 24 and 48 h. B, cells were incubated in medium supplemented with 2.5% NS plus indoxyl sulfate (IS; 25 or 100 μg ml−1), p-cresol (pc; 10 or 100 μg ml−1), a combination of low concentrations of IS (25 μg ml−1) and pc (10 μg ml−1) (Low IS + pc) or plus a combination of high concentrations of IS (100 μg ml−1) and pc (100 μg ml−1) (High IS + pc) for 24 h. **P < 0.05 vs. Low (IS + pc). C, cells were depleted of ILK with specific siRNA (open bars) (100 nm) and treated afterwards with IS (25 μg ml−1), pc (10 μg ml−1) or both, for 24 h. Scrambled RNA (Sc) (filled bars) was used as control. After incubation, apoptosis was determined in PI-stained cells and analysed by flow cytometry. Representative graphs are shown. Data are expressed as mean ± SEM of seven independent experiments. *P < 0.05 vs. control (CT; 2.5% NS, 24 h); #P < 0.05 vs. Sc.
ILK protects against both apoptosis and ROS production induced by uraemic toxins via activation of the AKT protein pathway
Next, because ILK has been shown to directly activate AKT through phosphorylation on Ser473 (Persad et al. 2001), to analyse the mechanism by which ILK protects against apoptosis induction by uraemic toxins, we tested the possible effect of AKT. As shown in Fig. 7A, a significant increase in AKT phosphorylation at Ser473 was induced by uraemic toxins, and ILK depletion completely blocked the increase in AKT phosphorylation, demonstrating the importance of ILK as an upstream mediator of this effect. AKT knockdown by specific siRNA promoted an increase of apoptosis in uraemic toxin-treated cells, measured by flow cytometry as the percentage of the sub-G1 hypodiploid cell population (Fig. 7B) and verified by determining the level of phosphatidyl serine exposed outside the cell membrane by labelling with Annexin V-FITC (Fig. 7C). These findings support the involvement of this protein in the protective effect of ILK from the uraemic toxin-induced apoptosis. Finally, we tested whether ROS were augmented, as previously described for HUVECs (Dou et al. 2007; Carracedo et al. 2013). Uremic serum (Fig. 8A) and uraemic toxins (Fig. 8B–D) produced a moderate increase in ROS production, which significantly increased when ILK (Fig. 8B and C) or AKT (Fig. 8B and D) proteins were abrogated by specific siRNAs, even in cells that were treated with low doses of uraemic toxins.
Figure 7. AKT activation is involved in ILK protection against uraemic toxin-induced apoptosis of EA.hy926 cells.
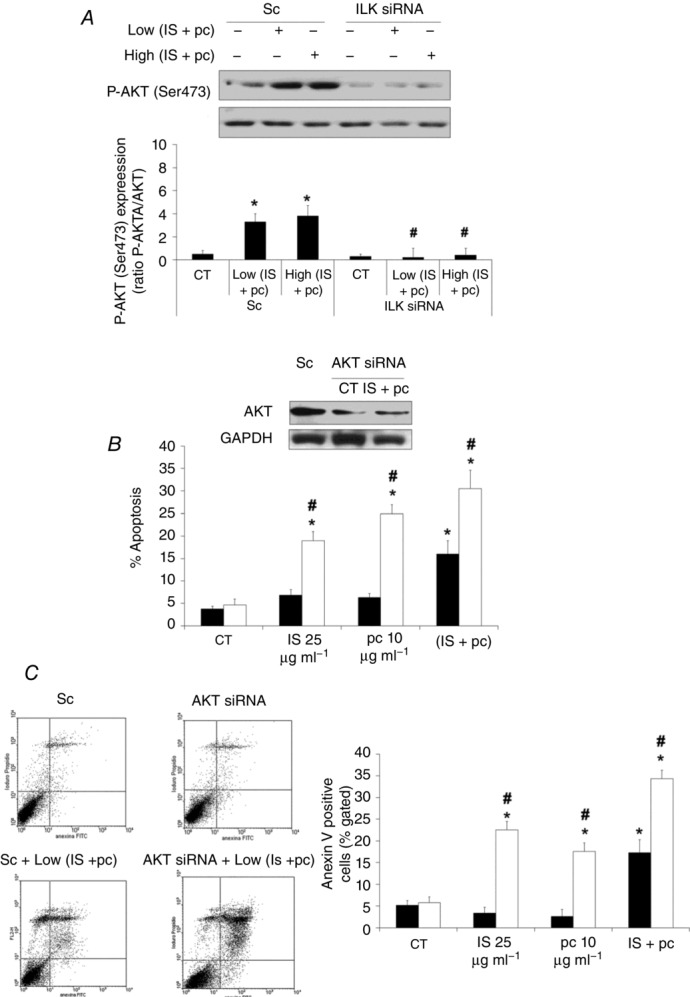
A, cells were depleted of ILK with specific siRNA (100 nm) and afterwards incubated in medium supplemented with 2.5% normal serum (NS) plus a combination of low concentrations of indoxyl sulfate (25 μg ml−1) and p-cresol (10 μg ml−1) (Low IS + pc) or plus a combination of high concentrations of IS (100 μg ml−1) and pc (100 μg ml−1) (High IS + pc) for 24 h. Scrambled RNA (Sc) was used as control. Representative Western blots of phosphorylated AKT in the serine-473 residue (P-AKT (Ser473)) and total AKT as control are shown. Bars represent the normalized densitometric analysis of blots against control values. B and C, cells were depleted of AKT with specific siRNA (open bars) (100 nm) and treated afterwards with IS (25 μg ml−1), pc (10 μg ml−1) or both, for 24 h. Scrambled RNA (Sc) (filled bars) was used as control. After incubation, apoptosis was determined in PI-stained cells and analysed by flow cytometry (B) or cells were stained with annexin V-FITC and PI, followed by flow cytometry analysis (C). Some results are shown by representative dot plots. Data are expressed as mean ± SEM of five independent experiments. *P < 0.05 vs. control (CT; 2.5% NS, 24 h); #P < 0.05 vs. Sc.
Figure 8. ILK and AKT protect EA.hy926 cells from uraemic toxin-mediated ROS production.
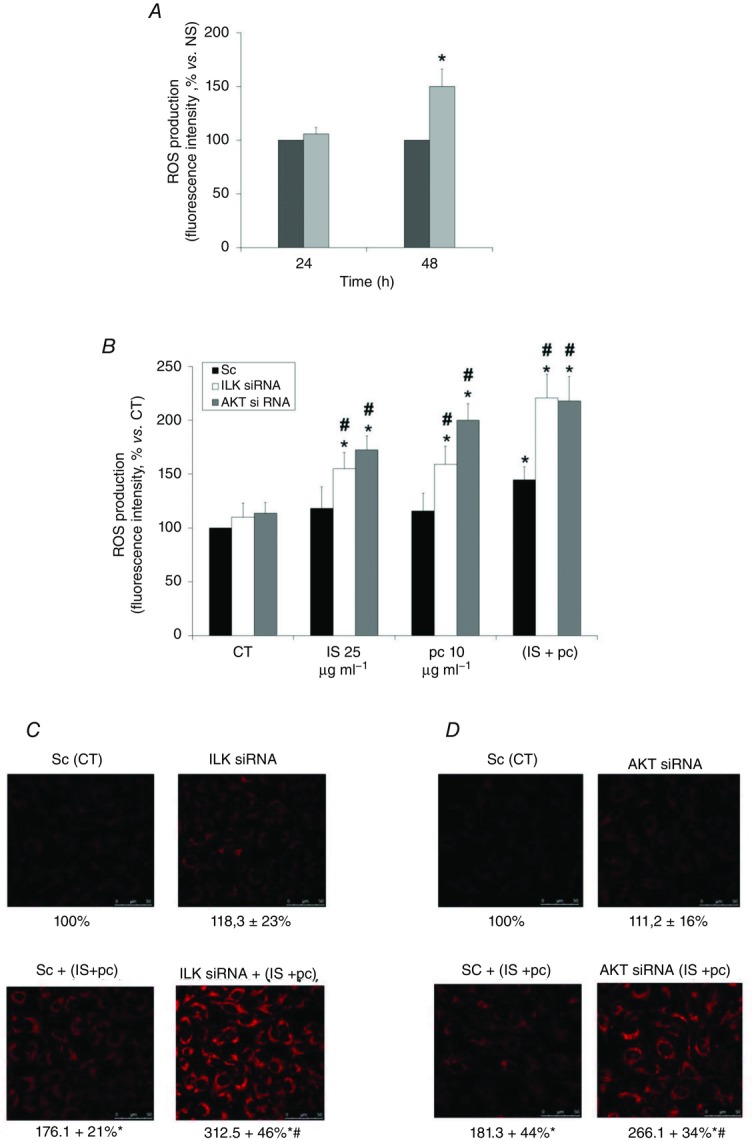
A, cells were incubated in medium supplemented with normal serum (NS, dark grey bars) (10%) or uraemic serum (light grey bars) (10%) for 24 and 48 h. After incubation, intracellular ROS production was determined by flow cytometry using 5 mm H2DCFDA probe, calculated as the percentage of fluorescence intensity versus normal serum. *P < 0.05 vs. NS. B, cells were depleted of ILK (open bars) or AKT (light grey bars) with specific siRNA (100 nm) and afterwards were incubated in medium supplemented with 2.5% NS plus indoxyl sulfate (IS; 25 or 100 μg ml−1), p-cresol (pc; 10 or 100 μg ml−1) or a combination of low concentrations of IS (25 μg ml−1) and pc (10 μg ml−1) (IS + pc) for 24 h. Scrambled RNA (Sc) (filled bars) was used as control. After incubation, intracellular ROS production was determined by flow cytometry using 5 mm H2DCFDA probe, calculated as the percentage of fluorescence intensity versus control (CT; 2.5% NS, 24 h). Data are expressed as mean ± SEM of five independent experiments. *P < 0.05 vs. control; #P < 0.05 vs. Sc. C and D, cells were depleted of ILK (C) or AKT (D) with specific siRNA (100 nm) and afterwards they were incubated in medium supplemented with 2.5% NS and a combination of low concentrations of IS (25 μg ml−1) and pc (10 μg ml−1) (IS + pc) for 24 h. Scrambled RNA (Sc) was used as control. After incubation, intracellular ROS production was determined by confocal microscopy, using 5 μm CellROX Deep Red Reagent fluorogenic probe. A representative experiment is shown. Data represent the densitometric analysis of the fluorescence of 20 cells of six independent experiments. The results are expressed as a percentage of scrambled untreated control and are the mean ± SEM of the mean from six different experiments. *P < 0.05 vs. Sc (CT); #P < 0.05 vs. Sc + (IS +pc).
The present results suggest that in early stages of CKD, ILK activation protects against apoptosis and ROS production induced by uraemic toxins via an ILK/AKT signalling pathway.
Discussion
The main finding of this study has been ILK as a potential mediator that prevents early stages of endothelial damage induced by uraemic toxins in CKD. The ILK/AKT pathway protects endothelial cells against decreases in cell proliferation and viability and increases in apoptosis and ROS production induced by uraemic toxins. Therefore, the maintenance of high levels of ILK activity appears as a possible therapeutic target in the treatment of CKD.
Several studies have indicated the contribution of uraemic toxins present in the serum of CKD patients to endothelial dysfunction, although the mechanisms involved remain obscure (Dou et al. 2004; Tumur & Niwa, 2009; Adelibieke et al. 2013).
Our first approach was to investigate the effect of ILK in serum derived from patients with CKD on endothelial cells. Interestingly, with uraemic serum we observed a dose- and time-dependent increase in ILK activity. There is a body of evidence indicating that ILK plays a critical role in vascular development through integrin–matrix interactions and endothelial cell survival (Friedrich et al. 2004; Kaneko et al. 2004; Cho et al. 2005) and, in addition, transgenic mouse models demonstrate the important role of ILK in vascular development (Boulter & Van Obberghen-Schilling, 2006; Kogata et al. 2009). Moreover, our group has recently demonstrated the relevance of ILK function in the regulation of endothelial nitric oxide production and vasomotor tone (Herranz et al. 2012). The present results support that ILK activation induced by uraemic serum may mediate protection against endothelial damage in CKD.
Moreover, the uraemic serum induced a slight decrease in cell proliferation and viability and increased cellular apoptosis, compared to normal serum. Differences between our and previously obtained results may be explained because, to our knowledge, all previous studies used HUVECs (Serradell et al. 2003; Zafeiropoulou et al. 2012; Carracedo et al. 2013). Furthermore, some authors used increased levels or longer times of uraemic serum exposition, with no apoptosis, accelerating the proliferation observed (Serradell et al. 2003). Other reports provided similar information by comparing endothelial cell toxicity of uraemic serum derived from patients before or after haemodialysis (Zafeiropoulou et al. 2012). Furthermore, we observed an increased ROS production, as previously reported (Carracedo et al. 2013).
Next, we investigated the direct participation of uraemic toxins in endothelial cell damage induced by uraemic serum. We evaluated the effect of two protein-bound uraemic toxins, IS and pc (acting as a surrogate of p-cresyl sulphate (p-CS)), which have specific endothelial toxicity (Dou et al. 2004; Meijers et al. 2009; Zhu et al. 2012). The accumulation of these compounds represents a serious clinical problem in CKD patients, as their removal by conventional haemodialysis is low because they do not diffuse through the dialysis membrane (Jourde-Chiche et al. 2009). Based on previous reports, the concentrations of the uraemic solutes used in this study (1–100 μg ml−1) are in the range of those reported in CKD (Duranton et al. 2012; Zhu et al. 2012). It has been observed that in CKD patients, serum levels of IS and pc increased by approximately 50- to 90-fold (Niwa et al. 1988; Vanholder et al. 2003) and10-fold (De Smet et al. 1998), respectively. In this regard, Itoh et al. (2012) measured uraemic toxins in haemodialysis patients by liquid chromatography/tandem mass spectrometry. The authors observed that the total serum levels of IS and p-CS in healthy volunteers were 0.5 ± 0.1 and 2.2 ± 0.9 μg ml−1 and in serum samples obtained from 45 haemodialysis patients were 29.9 ± 1.8 and 37.1 ± 2.8 μg ml−1, respectively. In addition, a recent study from the European Uremic Toxin Work Group has reported that total serum levels of IS and p-CS in the haemodialysis population reached up to 236 and 105 μg ml−1, respectively (Vanholder et al. 2003; Duranton et al. 2012). In respect to the values in patients with non-dialysis CKD, of which there is little information, an analysis of 10 CKD Stage 5 patients found 16% of patients had values >46 μg ml−1 p-CS (Eloot et al. 2011) and another study with 70 stable CKD patients (stages 3–5) showed IS concentrations >4.9 μg ml−1 in 55% of patients (Lin et al. 2012). Moreover, higher levels of IS and p-CS in CKD Stage 1–5 progressors versus non-progressors were observed (Wu et al. 2011). In addition, we explored the additive effect of the combination of both toxins, finding synergistic effects when tested together, as previously observed in p-CS combined with the p-cresylglucoronide (Meert et al. 2012), the other conjugate of pc, in in vitro assays, in contrast to other experimental in vivo findings (Pletinck et al. 2013).
Our results with both uraemic toxins clearly demonstrated a quick and significant increase in ILK activity and suggested this kinase is involved in the protection against many forms of endothelial damage observed: a reduction in cell proliferation and viability corresponded to an increase in apoptosis levels. Furthermore, uraemic toxins induced endothelial oxidative stress, reflected by increased ROS production (Dou et al. 2007; Tumur & Niwa, 2009; Adelibieke et al. 2012; Itoh et al. 2012). We found a slight increase in ROS production in cells exposed to high concentrations or both uraemic toxins combined. In addition, we rejected the possibility that these observed ROS increases were the causes of ILK activity upregulation, as we previously described in H2O2-stimulated human mesangial cells (Gonzalez-Ramos et al. 2013), as preincubation of cells with N-acetylcysteine, an antioxidant that reduces ROS production, did not modify increases in ILK activity induced by uraemic toxins (data not shown).
Interestingly, even different concentrations of toxins or both toxins combined may induce differential effects in the cells, reaching a maximum at high concentrations or when exposed to toxins combined. ILK was involved in the protection against endothelial damage, as demonstrated by the abrogation by specific siRNAs, which in turn significantly increased these effects, most notably at low concentrations of toxins. It is interesting that low concentrations of uraemic toxins did not produce endothelial damage, which could suggest that the activation of ILK may be sufficient to protect endothelium from uraemia in early stages of disease development. In advanced stages of the disease, with high concentrations of all these retention solutes, activation of ILK was not sufficient to prevent endothelial damage, probably due to the intervention of other factors induced by uraemia, especially pro-inflammatory ones, often related to ROS increases, both in vivo (Pletinck et al. 2013) and in vitro (Tumur & Niwa, 2009; Tumur et al. 2010; Caballo et al. 2012). This may have clinical relevance, because it highlights the importance of maintaining high levels of ILK activity to help preserve endothelial integrity, at least in early stages of CKD.
Finally, we investigated the mechanisms underlying ILK-mediated survival after endothelial cell stimulation by uraemic toxins. According to previous results from our group and others, ILK phosphorylates AKT in serine 473, a mechanism implicated in integrin-mediated survival and gene expression regulation (Vivanco & Sawyers, 2002; Edwards et al. 2005). Here, the implication of AKT was demonstrated by the increase in both apoptosis and ROS production in AKT-depleted cells. One possible explanation for the mechanism implicated could be activation of NFκB (Tan et al. 2002), a pivotal transcription factor that is active in uraemia and is mainly involved in the production of ROS, inflammation and apoptosis (Masai et al. 2010; Tumur et al. 2010; Carracedo et al. 2013; Poveda et al. 2013). In addition, IS has been reported to increase NAD(P)H oxidase activity and strongly decrease levels of intracellular glutathione, the most active non-enzymatic antioxidant in endothelial cells (Dou et al. 2007). In contrast to our findings, Adelibieke et al. (2013) observed that IS inhibits erythropoietin-induced HUVEC survival and proliferation, as well as their anti-apoptosis function trough suppression of AKT phosphorylation. Considering the data previously discussed above, a possible putative model of uraemic toxin-induced ILK-mediated survival in EA.hy926 endothelial cells can be envisaged (Fig. 9).
Figure 9. Schematic representation of the mechanisms involved in the protective effect of the ILK/AKT pathway in uraemic toxin-induced apoptosis and ROS production of EA.hy926 cells.
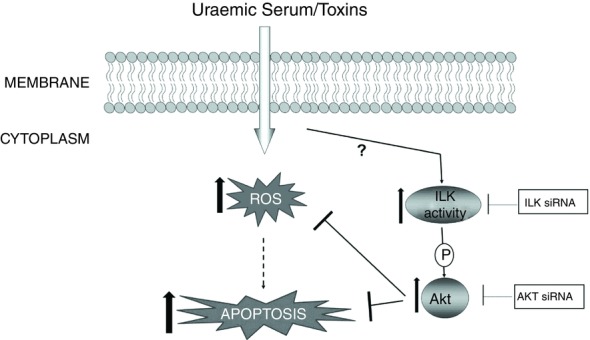
The added uraemic toxins upregulate the ILK/AKT pathway, which provides a survival signal protecting endothelial cells from decreases in proliferation and increases in apoptosis and ROS production induced by uraemic toxins. The points of inhibition by the different agents used in this study are shown.
Acknowledgments
None declared.
Translational perspective
Uraemic retention solutes and their potential toxicity have received attention as putative promoters of the augmented cardiovascular diseases and mortality in CKD. Because CKD is a silent pathology, often diagnosed when it is irreversible, current therapy strategies aim to delay clinical manifestations until the final stages of the disease. In this sense, understanding the primary responses of the endothelium to a cytotoxic uraemic environment is of major importance for the design of efficient therapeutic strategies.
The present work tried to elucidate the role of ILK in upregulation of endothelial cell damage induced by uraemic toxins, as an attempt to understand the molecular mechanism underlying the effect of uraemic toxins in the early stages of endothelial dysfunction. We found the ILK/AKT signalling pathway as a potential survival mediator, and this has been the first study, to our knowledge, that has demonstrated the implication of ILK activation as a possible intracellular protective mechanism that is activated in response to cell damage caused by uraemic toxins.
This may have clinical relevance, because it highlights the importance of maintaining high levels of ILK activity to help preserve endothelial integrity, at least in the early stages of CKD.
Glossary
Abbreviations
- BrdU
5-bromo-2′-deoxy-uridine
- CKD
chronic kidney disease
- EA.hy926
human endothelial cell line
- FBS
fetal bovine serum
- GSK-3β
glycogen synthase kinase 3β
- H2DCFDA
dichlorodihydrofluorescein-diacetate
- HUVEC
human umbilical vein endothelial cell
- ILK
integrin-linked kinase
- IS
indoxyl sulphate
- MTT
(3-[4,5-dimethylthiazol-2-yl]-2,5- diphenyltetrazolium bromide
- NS
normal serum
- pc
p-cresol
- p-CS
p-cresyl sulphate
- PCNA
proliferating cell nuclear antigen
- PI
propidium iodide
- PKB/AKT
protein kinase B
- ROS
reactive oxygen species
- siRNA
small interfering RNA
- US
uraemic serum
Additional information
Competing interests
The authors declare that there are no competing interests
Author contributions
L.C., M.R.-P. and D.R.-P. performed the statistical analyses and drafted the paper. A.G.-J., A.L. and L.C. carried out cell culture and treatments, proliferation, toxicity and apoptosis assays, siRNA transfections, determination of in vitro ILK activity, Western blot analysis and measurements of ROS levels. J.C. and R.R.-C. provided intellectual input. D.R.-P., M.R.-P., R.R.-C. and L.C. conceived the experiments. All authors read and approved the final paper.
Funding
This study was supported by grant ISCIII-RETIC (REDinREN/RD06/0016/0002), by a grant from Fundación Mutua Madrileña (FMM2011-001) to L.C., by a grant from Ministerio de Educación (SAF2010–16198) to M.R.P., by a grant from Fondo de Investigaciones Sanitarias (FISSPI11/01630) to D.R.P. and by Instituto de Investigaciones Sanitarias Reina Sofía (IRSIN) and Fundación Renal Iñigo Álvarez de Toledo (FRIAT).
References
- Adelibieke Y, Shimizu H, Muteliefu G, Bolati D. Niwa T. Indoxyl sulfate induces endothelial cell senescence by increasing reactive oxygen species production and p53 activity. J Ren Nutr. 2012;22:86–89. doi: 10.1053/j.jrn.2011.10.027. [DOI] [PubMed] [Google Scholar]
- Adelibieke Y, Shimizu H, Saito S, Mironova R. Niwa T. Indoxyl sulfate counteracts endothelial effects of erythropoietin through suppression of Akt phosphorylation. Circ J. 2013;77:1326–1336. doi: 10.1253/circj.cj-12-0884. [DOI] [PubMed] [Google Scholar]
- Alenghat FJ. Ingber DE. Mechanotransduction: all signals point to cytoskeleton, matrix and integrins. Sci STKE. 2002;119:1–4. doi: 10.1126/stke.2002.119.pe6. [DOI] [PubMed] [Google Scholar]
- Boulter E. Van Obberghen-Schilling E. Integrin-linked kinase and its partners: a modular platform regulating cell-matrix adhesion dynamics and cytoskeletal organization. Eur J Cell Biol. 2006;85:255–263. doi: 10.1016/j.ejcb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Caballo C, Palomo M, Cases A, Galán AM, Molina P, Vera M, Bosch X, Escolar G. Diaz-Ricart M. NFκB in the development of endothelial activation and damage in uremia: an in vitro approach. PLoS ONE. 2012;7:e43374. doi: 10.1371/journal.pone.0043374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo J, Buendía P, Merino A, Soriano S, Esquivias E, Martín-Malo A, Aljama P. Ramírez R. Cellular senescence determines endothelial cell damage induced by uremia. Exp Gerontol. 2013;48:766–773. doi: 10.1016/j.exger.2013.04.004. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Youn SW, Cheon SI, Kim TY, Hur J, Zhang SY, Lee SP, Park KW, Lee MM, Choi YS, Park YB, Kim HS. Regulation of endothelial cell and endothelial progenitor cell survival and vasculogenesis by integrin-linked kinase. Arterioscler Thromb Vasc Biol. 2005;25:1154–1160. doi: 10.1161/01.ATV.0000164312.20008.93. [DOI] [PubMed] [Google Scholar]
- De Groot K, Bahlmann FH, Sowa J, et al. Uremia causes endothelial progenitor cell deficiency. Kidney Int. 2004;66:641–646. doi: 10.1111/j.1523-1755.2004.00784.x. [DOI] [PubMed] [Google Scholar]
- Del Nogal M, Luengo A, Olmos G, Lasa M, Rodriguez-Puyol D, Rodriguez-Puyol M. Calleros L. Balance between apoptosis or survival induced by changes in extracellular-matrix composition in human mesangial cells: a key role for ILK-NFκB pathway. Apoptosis. 2012;17:1261–74. doi: 10.1007/s10495-012-0769-3. [DOI] [PubMed] [Google Scholar]
- De Smet R, David F, Sandra P, Van Kaer J, Lesaffer G, Dhondt A, Lameire N, Vanholder R. A sensitive HPLC method for the quantification of free and total p-cresol in patients with chronic renal failure. Clin Chim Acta. 1998;278:1–21. doi: 10.1016/s0009-8981(98)00124-7. [DOI] [PubMed] [Google Scholar]
- Dou L, Bertrand E, Cerini C, Faure V, Sampol J, Vanholder R, Berland Y. Brunet P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004;65:442–451. doi: 10.1111/j.1523-1755.2004.00399.x. [DOI] [PubMed] [Google Scholar]
- Dou L, Jourde-Chiche N, Faure V, Cerini C, Berland Y, Dignat-George F. Brunet P. The uremic solute indoxyl sulphate induces oxidative stress in endothelial cells. J Thromb Haemost. 2007;5:1302–1308. doi: 10.1111/j.1538-7836.2007.02540.x. [DOI] [PubMed] [Google Scholar]
- Duranton F, Cohen G, De Smet R, Rodriguez M, Jankowski J, Vanholder R. Argiles A. European Uremic Toxin Work Group: Normal and pathologic concentrations of uremic toxins. J Am Soc Nephrol. 2012;23:1258–1270. doi: 10.1681/ASN.2011121175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgell CJ, McDonald CC. Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci USA. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards LA, Thiessen B, Dragowska WH, Daynard T, Bally MB. Dedhar S. Inhibition of ILK in PTEN-mutant human glioblastomas inhibits PKB/Akt activation, induces apoptosis, and delays tumor growth. Oncogene. 2005;24:3596–605. doi: 10.1038/sj.onc.1208427. [DOI] [PubMed] [Google Scholar]
- Eloot S, Schepers E, Barreto DV, Barreto FC, Liabeuf S, Van Biesen W, Verbeke F, Glorieux G, Choukroun G, Massy Z, Vanholder R. Estimated glomerular filtration rate is a poor predictor of concentration for a broad range of uremic toxins. Clin J Am Soc Nephrol. 2011;6:1266–1273. doi: 10.2215/CJN.09981110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich EB, Liu E, Sinha S, Cook S, Milstone DS, MacRae CA, Mariotti M, Kuhlencordt PJ, Force T, Rosenzweig A, St-Arnaud R, Dedhar S, Gerszten RE. Integrin-linked kinase regulates endothelial cell survival and vascular development. Mol Cell Biol. 2004;24:8134–8144. doi: 10.1128/MCB.24.18.8134-8144.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Ramos M, de Frutos S, Griera M, Luengo A, Olmos G, Rodriguez-Puyol D, Calleros Rodriguez-Puyol M. Integrin-linked kinase mediates the hydrogen peroxide-dependent transforming growth factor-β1 up-regulation. Free Radic Biol Med. 2013;61C:416–427. doi: 10.1016/j.freeradbiomed.2013.04.029. [DOI] [PubMed] [Google Scholar]
- González-Ramos M, Mora I, de Frutos S, Garesse R, Rodríguez-Puyol M, Olmos G. Rodríguez-Puyol D. Intracellular redox equilibrium is essential for the constitutive expression of AP-1 dependent genes in resting cells: studies on TGF-β1 regulation. Int J Biochem Cell Biol. 2012;44:963–971. doi: 10.1016/j.biocel.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Günthner T, Jankowski V, Kretschmer A, Nierhaus M. Giet M, Zidek W, Jankowski J. Endothelium and vascular smooth muscle cells in the context of uremia. Semin Dial. 2009;22:428–432. doi: 10.1111/j.1525-139X.2009.00594.x. van der. [DOI] [PubMed] [Google Scholar]
- Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC, Dedhar S. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin linked protein kinase. Nature. 1996;379:91–96. doi: 10.1038/379091a0. [DOI] [PubMed] [Google Scholar]
- Hannigan GE, McDonald PC, Walsh MP. Dedhar S. Integrin-linked kinase: not so ‘pseudo’ after all. Oncogene. 2011;30:4375–4385. doi: 10.1038/onc.2011.177. [DOI] [PubMed] [Google Scholar]
- Herranz B, Marquez S, Guijarro B, Aracil E, Aicart-Ramos C, Rodriguez-Crespo I, Serrano I, Rodríguez-Puyol M, Zaragoza C, Saura M. Integrin-linked kinase regulates vasomotor function by preventing endothelial nitric oxide synthase uncoupling: role in atherosclerosis. Circ Res. 2012;110:439–449. doi: 10.1161/CIRCRESAHA.111.253948. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Ezawa A, Kikuchi K, Tsuruta K. Niwa T. Protein-bound uremic toxins in hemodialysis patients measured by liquid chromatography/tandem mass spectrometry and their effects on endothelial ROS production. Anal Bioanal Chem. 2012;403:1841–1850. doi: 10.1007/s00216-012-5929-3. [DOI] [PubMed] [Google Scholar]
- Jourde-Chiche N, Dou L, Cerini C, Dignat-George F. Brunet P. Vascular incompetence in dialysis patients—protein-bound uremic toxins and endothelial dysfunction. Semin Dial. 2011;24:327–337. doi: 10.1111/j.1525-139X.2011.00925.x. [DOI] [PubMed] [Google Scholar]
- Jourde-Chiche N, Dou L, Cerini C, Dignat-George F, Vanholder R. Brunet P. Protein-bound toxins – update 2009. Semin Dial. 2009;22:334–339. doi: 10.1111/j.1525-139X.2009.00576.x. [DOI] [PubMed] [Google Scholar]
- Kaneko Y, Kitazato K. Basaki Y. Integrin-linked kinase regulates vascular morphogenesis induced by vascular endothelial growth factor. J Cell Sci. 2004;117:407–415. doi: 10.1242/jcs.00871. [DOI] [PubMed] [Google Scholar]
- Kogata N, Tribe RM, Fassler R, Way M. Adams RH. Integrin-linked kinase controls vascular wall formation by negatively regulating Rho/ ROCK-mediated vascular smooth muscle cell contraction. Genes Dev. 2009;23:2278–2283. doi: 10.1101/gad.535409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legate KR, Montañez E, Kudlacek O. Fässler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol. 2006;7:20–31. doi: 10.1038/nrm1789. [DOI] [PubMed] [Google Scholar]
- Liabeuf S, Barreto DV, Barreto FC, Meert N, Glorieux G, Schepers E, Temmar M, Choukroun G, Vanholder R, Massy ZA European Uraemic Toxin Work Group. European Uraemic Toxin Work Group (EUTox): free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol Dial Transplant. 2010;25:1183–1189. doi: 10.1093/ndt/gfp592. [DOI] [PubMed] [Google Scholar]
- Lin CJ, Liu HL, Pan CF, Chuang CK, Jayakumar T, Wang TJ, Chen HH, Wu CJ. Indoxyl sulfate predicts cardiovascular disease and renal function deterioration in advanced chronic kidney disease. Arch Med Res. 2012;43:451–456. doi: 10.1016/j.arcmed.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Masai N, Tatebe J, Yoshino G, Morita T. Indoxyl sulfate stimulates monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells by inducing oxidative stress through activation of the NADPH oxidase-nuclear factor-κB pathway. Circ J. 2010;74:2216–24. doi: 10.1253/circj.cj-10-0117. [DOI] [PubMed] [Google Scholar]
- Meert N, Schepers E. Glorieux G. Novel method for simultaneous determination of p-cresylsulphate and p-cresylglucuronide: clinical data and pathophysiological implications. Nephrol Dial Transplant. 2012;27:2388–2396. doi: 10.1093/ndt/gfr672. [DOI] [PubMed] [Google Scholar]
- Meijers BK, Van Kerckhoven S, Verbeke K, Dehaen W, Vanrenterghem Y, Hoylaerts MF, Evenepoel P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am J Kidney Dis. 2009;54:891–901. doi: 10.1053/j.ajkd.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Niwa T, Takeda N, Tatematsu A. Maeda K. Accumulation of indoxyl sulfate, an inhibitor of drug-binding, in uremic serum as demonstrated by internal-surface reversed-phase liquid chromatography. Clin Chem. 1988;34:2264–2267. [PubMed] [Google Scholar]
- Parfrey PS. Foley RN. The clinical epidemiology of cardiac disease in chronic renal failure. J Am Soc Nephrol. 1999;10:1606–1615. doi: 10.1681/ASN.V1071606. [DOI] [PubMed] [Google Scholar]
- Passauer J, Büssemaker E, Range U, Plug M. Gross P. Evidence in vivo showing increase of baseline nitric oxide generation and impairment of edothelin-dependent vasodilatation in normotensive patients on chronic hemodialysis. J Am Soc Nephrol. 2000;11:1726–1734. doi: 10.1681/ASN.V1191726. [DOI] [PubMed] [Google Scholar]
- Persad S, Attwell S, Gray V, Mawji N, Deng JT, Leung D, Yan J, Sangher J, Walsh MP. Dedhar S. Regulation of protein kinase B/Akt-serine-473 phosphorylation by integrin linked kinase (ILK): critical roles for kinase activity and amino acids arginine-211 and serine-343. J Biol Chem. 2001;276:27462–27469. doi: 10.1074/jbc.M102940200. [DOI] [PubMed] [Google Scholar]
- Pletinck A, Glorieux G, Schepers E, Cohen G, Gondouin B, Van Landschoot M, Eloot S, Rops A, Van de Voorde J, De Vriese A, van der Vlag J, Brunet P, Van Biesen W, Vanholder R. Protein-bound uremic toxins stimulate crosstalk between leukocytes and vessel wall. J Am Soc Nephrol. 2013;24:1981–1994. doi: 10.1681/ASN.2012030281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poveda J, Sanchez-Niño MD, Glorieux G, Sanz AB, Egido J, Vanholder R. Ortiz A. p-Cresyl sulphate has pro-inflammatory and cytotoxic actions on human proximal tubular epithelial cells. Nephrol Dial Transplant. 2013;29:56–64. doi: 10.1093/ndt/gft367. [DOI] [PubMed] [Google Scholar]
- Rabelink TJ, de Boer HC. van Zonneveld AJ. Endothelial activation and circulating markers of endothelial activation in kidney disease. Nat Rev Nephrol. 2010;6:404–414. doi: 10.1038/nrneph.2010.65. [DOI] [PubMed] [Google Scholar]
- Ramirez R, Martin-Malo A. Aljama P. Inflammation and hemodiafiltration. Contrib Nephrol. 2002;158:210–215. doi: 10.1159/000107252. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Puyol D. Praga M. Causas de insuficiencia renal crónica y sus mecanismos de protección. In: Lamas-Peláez S, editor; Hernando-Avendaño L, Aljama-García P, Arias-Rodríguez M, Caramelo-Díaz C, Egido-de los Ríos J, editors. Nefrología Clínica. 1st edn. Madrid: Panamericana; 1998. pp. 535–546. [Google Scholar]
- Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- Sabatier F, Camoin-Jau L, Anfosso F, Sampol J. Dignat-George F. Circulating endothelial cells, microparticles and progenitors: key players towards the definition of vascular competence. J Cell Mol Med. 2009;13:454–471. doi: 10.1111/j.1582-4934.2008.00639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serradell M, Díaz-Ricart M, Cases A, Petriz J, Ordinas A. Escolar G. Uraemic medium accelerates proliferation but does not induce apoptosis of endothelial cells in culture. Nephrol Dial Transplant. 2003;18:1079–1085. doi: 10.1093/ndt/gfg161. [DOI] [PubMed] [Google Scholar]
- Stam F, van Guldener C, Becker A, Dekker JM, Heine RJ, Bouter LM, Stehouwer CD. Endothelial dysfunction contributes to renal function associated cardiovascular mortality in a population with mild renal insufficiency: the Hoorn study. J Am Soc Nephrol. 2006;17:537–545. doi: 10.1681/ASN.2005080834. [DOI] [PubMed] [Google Scholar]
- Tan C, Mui A. Dedhar S. Integrin-linked kinase regulates inducible nitric oxide synthase and cyclooxygenase-2 expression in an NF-κB-dependent manner. J Biol Chem. 2002;277:3109–3116. doi: 10.1074/jbc.M108673200. [DOI] [PubMed] [Google Scholar]
- Tumur Z. Niwa T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am J Nephrol. 2009;9:551–557. doi: 10.1159/000191468. [DOI] [PubMed] [Google Scholar]
- Tumur Z, Shimizu H, Enomoto A, Miyazaki H. Niwa T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-κB activation. Am J Nephrol. 2010;31:435–441. doi: 10.1159/000299798. [DOI] [PubMed] [Google Scholar]
- Vanholder R, De Smet R, Glorieux G, Argilés A, Baurmeister U, Brunet P, Clark W, Cohen G, De Deyn PP, Deppisch R, Descamps-Latscha B, Henle T, Jörres A, Lemke HD, Massy ZA, Passlick-Deetjen J, Rodriguez M, Stegmayr B, Stenvinkel P, Tetta C, Wanner C, Zidek W European Uremic Toxin Work Group (EUTox) European Uremic Toxin Work Group (EUTox): Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 2003;63:1934–1943. doi: 10.1046/j.1523-1755.2003.00924.x. [DOI] [PubMed] [Google Scholar]
- Vanholder R, Schepers E, Pletinck A, Neirynck N, Glorieux G. An update on protein bound uremic retention solutes. J Ren Nutr. 2012;22:90–94. doi: 10.1053/j.jrn.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Wheeler DC. Cardiovascular disease in patients with chronic renal failure. Lancet. 1996;348:1673–1674. doi: 10.1016/S0140-6736(05)65816-3. [DOI] [PubMed] [Google Scholar]
- Wu C. Dedhar S. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J Cell Biol. 2001;155:505–510. doi: 10.1083/jcb.200108077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu IW, Hsu KH, Lee CC, Sun CY, Hsu HJ, Tsai CJ, Tzen CY, Wang YC, Lin CY, Wu MS. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol Dial Transplant. 2011;26:938–947. doi: 10.1093/ndt/gfq580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafeiropoulou K, Bita T, Polykratis A, Karabina S. Vlachojannis J. Hemodialysis removes uremic toxins that alter the biological actions of endothelial cells. PLoS ONE. 2012;7:e30975. doi: 10.1371/journal.pone.0030975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JZ, Zhang J, Yang K, Du R, Jing YJ, Lu L. Zhang RY. P-cresol, but not p-cresylsulphate, disrupts endothelial progenitor cell function in vitro. Nephrol Dial Transplant. 2012;27:4323–4330. doi: 10.1093/ndt/gfs382. [DOI] [PubMed] [Google Scholar]