

Abstract
An alternative to the canonical insulin signalling pathway for glucose transport is muscle contraction/exercise. Mechanical stress is an integrated part of the muscle contraction/relaxation cycle, and passive stretch stimulates muscle glucose transport. However, the signalling mechanism regulating stretch-stimulated glucose transport is not well understood. We recently reported that the actin cytoskeleton regulating GTPase, Rac1, was activated in mouse muscle in response to stretching. Rac1 is a regulator of contraction- and insulin-stimulated glucose transport, however, its role in stretch-stimulated glucose transport and signalling is unknown. We therefore investigated whether stretch-induced glucose transport in skeletal muscle required Rac1 and the actin cytoskeleton. We used muscle-specific inducible Rac1 knockout mice as well as pharmacological inhibitors of Rac1 and the actin cytoskeleton in isolated soleus and extensor digitorum longus muscles. In addition, the role of Rac1 in contraction-stimulated glucose transport during conditions without mechanical load on the muscles was evaluated in loosely hanging muscles and muscles in which cross-bridge formation was blocked by the myosin ATPase inhibitors BTS and Blebbistatin. Knockout as well as pharmacological inhibition of Rac1 reduced stretch-stimulated glucose transport by 30–50% in soleus and extensor digitorum longus muscle. The actin depolymerizing agent latrunculin B similarly decreased glucose transport in response to stretching by 40–50%. Rac1 inhibition reduced contraction-stimulated glucose transport by 30–40% in tension developing muscle but did not affect contraction-stimulated glucose transport in muscles in which force development was prevented. Our findings suggest that Rac1 and the actin cytoskeleton regulate stretch-stimulated glucose transport and that Rac1 is a required part of the mechanical stress-component of the contraction-stimulus to glucose transport in skeletal muscle.
Key points
Rac1 regulates stretch-stimulated (i.e. mechanical stress) glucose transport in muscle.
Actin depolymerization decreases stretch-induced glucose transport in skeletal muscle.
Rac1 is a required part of the mechanical stress-component of the contraction-stimulus to glucose transport in skeletal muscle.
Introduction
Mechanical stress is an integrated part of muscle contraction and exercise stimulus. Mechanical stress may be necessary to fully activate glucose transport because the prevention of tension development reduced contraction-stimulated glucose transport (Blair et al. 2009; Ihlemann et al. 1999; Jensen et al. 2014b) and mechanical stress-induced signalling (Jensen et al. 2014b). Furthermore, passive stretching of incubated rodent muscle activates glucose transport (Ihlemann et al. 1999; Sakamoto et al. 2003; Ito et al. 2006; Chambers et al. 2009; Jensen et al. 2014b). However, the mediators of stretch-stimulated glucose transport remain unknown.
A novel candidate is the small Rho family GTPase Rac1, which has been implicated in the Akt-dependent (Nozaki et al. 2013; Takenaka et al. 2014) or Akt-independent (JeBailey et al. 2007; Chiu et al. 2011; Sylow et al. 2013a; Sylow et al. 2014a) regulation of insulin-induced glucose transport and, more recently, contraction-stimulated (Sylow et al. 2013b) glucose transport. Interestingly, a number of cell culture studies suggest that Rac1 can be activated by mechanical stress (Kawamura et al. 2003; Poh et al. 2009; Boccafoschi et al. 2011). Interestingly, passive stretching of incubated rodent muscles also increase Rac1 GTP loading (i.e. activation) in skeletal muscle (Zhou et al. 2007; Sylow et al. 2013b). Stretch-signalling to glucose transport is independent of insulin signalling and AMP activated protein kinase (AMPK) (Chambers et al. 2009; Jensen et al. 2014b). Because AMPK signalling and mechanical stress likely represent independent signals to stimulate glucose transport, and Rac1 is activated by passive stretching, Rac1 is an excellent candidate for regulating stretch-stimulated glucose transport.
Rac1-dependent reorganization of the actin cytoske-leton is necessary for insulin-stimulated glucose transport in cultured muscle cells (JeBailey et al. 2004) and pharma-cological disruption of the actin cytoskeleton decreases insulin- and contraction-stimulated glucose transport ex vivo in mouse muscles (Brozinick et al. 2004; Brozinick et al. 2007; Chiu et al. 2011; Sylow et al. 2013a; Sylow et al. 2013b; Sylow et al. 2014a). However, to our knowledge, the requirement for Rac1 and the actin cytoskeleton in the regulation of stretch-stimulated glucose transport in skeletal muscle has not been investigated.
The present study aimed to investigate whether stretch-induced glucose transport in mouse skeletal muscle relies on Rac1 and an intact actin cytoskeleton. Furthermore, we investigated downstream signalling in response to stretch after Rac1 inhibition or Rac1 knockout (KO) and actin cytoskeleton depolymerization. In addition, the role of Rac1 in contraction-stimulated glucose transport during conditions without mechanical stress on the muscles was evaluated. We hypothesized that stretch-stimulated glucose transport relies on the activation of Rac1 and an intact actin cytoskeleton and that Rac1 regulates glucose transport in response to contraction via mechanical stress-activated signals.
Methods
Ethical approval
All experiments were approved by the Danish Animal Experimental Inspectorate and complied with the European Convention for the Protection of Vertebrate Animals Used for Experiments and Other Scientific Purposes.
Animals
A total of 76 female C57BL/6 mice (Taconic, Lille Skensved, Denmark), aged 12–16 weeks, were used for all inhibitor experiments.
Tetracycline-inducible muscle-specific Rac1 KO mice
Twelve inducible muscle-specific Rac1 KO mice were obtained as described previously (Sylow et al. 2013b). In brief, Rac1 floxed mice (Chrostek et al. 2006) were crossed with mice containing a tetracycline-controlled transactivator coupled to the human skeletal muscle actin promoter, which drives the muscle-specific expression of the Cre recombinase (Rao & Monks, 2009). Mice were back-crossed until N5 (96.9% congenic on a C57BL/6 background). Control wild-type (WT) mice were littermates carrying either the Cre recombinase or the Rac1 flox on one or both alleles. Rac1 KO mice were homozygous for Rac1 flox and either homozygous or heterozygous for the Cre recombinase. Rac1 KO was induced at 10–14 weeks of age by adding the tetracycline analogue doxycycline to the drinking water (1 g l−1; Sigma-Aldrich, St Louis, MO, USA) for 3 weeks (control mice also received doxycycline) followed by a washout period of 3 weeks. All animals were maintained under a 12:12 h light/dark cycle and received standard rodent chow diet (Altromin no. 1324; Chr. Pedersen, Copenhagen, Denmark) and water ad libitum.
Muscle incubations
Soleus and extensor digitorum longus (EDL) muscles were dissected from 2 h fasted, anaesthetized [pentobarbital sodium; 6 mg (100 g body weight)–1] mice and were suspended at resting tension (2–4 mN) in incubation chambers (Multi Myograph system; Danish Myo-Technology, Aarhus, Denmark) in Krebs–Ringer–Henseleit buffer with 2 mm pyruvate and 8 mm mannitol at 30°C, as described previously (Jensen et al. 2007). For inhibitor experiments, the muscles were pre-incubated for 1 h in Krebs–Ringer–Henseleit buffer with Rac1 Inhibitor II (Rac1 InhibII; 15 μm; Calbiochem, San Diego, CA, USA), 50 μm N-benzyl-p-toluene sulphonamide (BTS, B3082; TCI Europe NV, Zwijndrecht, Belgium) and 75 μm Blebbistatin (B0560; Sigma-Aldrich), or 45 min with latrunculin B (5 μm; Cytoskeleton, Inc., Denver CO, USA), or 40 min with cytochalasin B (50 μm; Sigma-Aldrich) as indicated, or a corresponding amount of DMSO as vehicle control. After the pre-incubation period, muscles were stimulated with passive stretching by mechanically increasing the distance between one end of the muscle and the other until the measured force production reached 100–130 mN for 15 min. As the force development declined (the amount of force produced by muscles during electrically-induced contraction) during the stimulation period, the distance between the muscle ends was continuously adjusted. Contractions were induced by electrical stimulation every 15 s with 2 s trains of 0.2 ms pulses delivered at 100 Hz (∼35 V) for 12 min. This corresponded to a net stimulation time of 0.27%.
2-Deoxyglucose (2DG) transport
2DG transport was measured with 1 mm 2DG during the last 10 min of the stimulation period using 3H 2DG (0.6 μCi ml−1) and 14C mannitol (0.18 μCi ml−1) tracers as described previously (Jensen et al. 2007).
Muscle analysis
Immediately after stretch or contraction stimulation, muscle tissue was quickly washed in ice cold saline and carefully dried and frozen in liquid nitrogen and stored at –80°C. Tissue was homogenized 2 × 30 s at 30 Hz using a Tissuelyser II (Qiagen, Valencia, CA, USA) in 50 mm Hepes (pH 7.5), 150 mm NaCl, 20 mm sodium pyrophosphate, 20 mm β-glycerophosphate, 10 mm NaF, 2 mm sodium orthovanadate, 2 mm EDTA, 1% NP-40, 10% glycerol, 2 mm phenylmethanesulfonyl fluoride, 1 mm MgCl2, 1 mm CaCl2, 10 μg ml−1 leupeptin, 10 μg ml−1 aprotinin and 3 mm benzamidine. After end-over-end rotation for 20 min, lysate supernatants were collected by centrifugation (10,000 g) for 15 min at 4°C.
Immunoblotting
Lysate protein concentrations were measured using the bicinchoninic acid method, with BSA standards (Pierce, Rockford, IL, USA) and bicinchoninic acid assay reagents (Pierce) in triplicates. Total protein and phosphorylation levels of relevant proteins were determined by standard immunoblotting techniques, with equal amounts of pro-tein loaded per well. The primary antibodies used were p-p38 MAPKThr180/Tyr182, actin, PAK1, p-PAK1/2Thr423/402 (Cell Signaling Technology, Beverly, MA, USA) and Rac1 (Cytoskeleton, Inc.). Polyvinylidene difluoride membr-anes (Immobilon Transfer Membrane; Millipore, Billerica, MA, USA) were blocked for 30 min in Tris-buffered saline-Tween 20 plus 2% skim milk or 5% BSA protein, at room temperature. Membranes were incubated with primary antibodies overnight at 4°C, followed by incuba-tion with horseradish peroxidase-conjugated secondary antibody (Dako, Glostrup, Denmark) for 1 h at room temperature. Bands were visualized using the ChemiDoc MP Imaging System (Bio-Rad, Hercules, CA, USA) plus enhanced chemiluminescence (ECL+; Amersham Biosciences, Little Chalfont, UK).
Statistical analysis
The results are reported as the mean ± sem. Statistical testing was performed using paired t tests or two-way repeated measurements ANOVA as appropriate. Tukey's post hoc test was performed when ANOVA revealed signi-ficant interaction. Statistical evaluation was performed using Sigmaplot, version 11.0 (Systat Software Inc., Chicago, IL, USA). P < 0.05 was considered statistically significant.
Results
Pharmacological Rac1 inhibition reduces stretch-induced glucose transport
To investigate the involvement of Rac1 in stretch-induced glucose transport, we incubated soleus and EDL muscles with or without the Rac1 inhibitor, Inhibitor II. We recently reported that the Rac1 Inhibitor II reduces insulin- and electrically-induced contraction- but not AICAR-stimulated glucose transport; it further inhibits downstream signalling of Rac1 at a concentration of 15 μm (Sylow et al. 2013a, 2014a). At that concentration, the Rac1 Inhibitor II reduced the stretch-induced approximately three-fold increment in glucose transport by ∼30% and ∼50% in soleus and EDL muscles, respectively (Fig. 1A). These findings suggest that Rac1 is an important regulator of glucose transport induced by mechanical stress in skeletal muscle.
Figure 1.
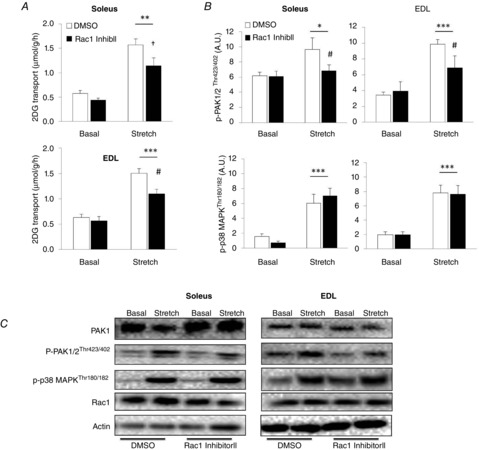
Stretch-stimulated glucose transport is partially regulated by Rac1
A, bar graphs showing stretch-stimulated 2DG transport in soleus and EDL muscles incubated for 1 h with 15 μm Rac1 Inhibitor II or a corresponding amount of DMSO (n = 7–8). B, bar graphs showing quantifications of p-PAK1/2Thr423/402 and p-p38 MAPKThr180/182 immunoblots (n = 7–8). C, Representative blots of PAK1, p-PAK1/2Thr423/402, p-p38 MAPKThr180/182, Rac1 and actin. *P < 0.05; **P < 0.01; ***P < 0.001: significant effect of stretch stimulation. †P < 0.05: significant effect of inhibitor (main effect). #P < 0.05: significant interaction between stretch and inhibitor. Values are the mean ± sem.
Passive stretching increases phosphorylation of the downstream target of Rac1: PAK1/2
We have previously reported that the binding of Rac1 to GTP increased in response to stretching (Sylow et al. 2013b). In kidney derived BHK cells, Rac1 regulates actin cytoskeleton dynamics by facilitating the phosphorylation of its downstream target, PAK1/2 (Edwards et al. 1999). GTP-bound Rac1 binds to PAK1/2 and relieves it from its auto-inhibitory domain and thereby promotes the phosphorylation of PAK1 on Thr423 and PAK2 on Thr402 (referred to as p-PAK1/2Thr423/402). p-PAK1/2Thr423/402 increased by ∼30% in soleus and ∼120% in EDL muscles in response to passive stretching (Fig. 1B and C). Rac1 Inhibitor II abolished this response in soleus and reduced it by ∼60% in EDL. p-p38 MAPKThr180/Tyr182 was unaffected by Rac1 Inhibitor II and increased by ∼200% in both soleus and EDL in response to stretching. There were no increases in p-AMPKThr172, p-ACCSer221 or p-AS160 (using the phospho Akt substrate antibody; data not shown), suggesting that AMPK- or Akt- mediated signalling does not regulate stretch-stimulated glucose transport in skeletal muscle.
Stretch-stimulated glucose transport is reduced in Rac1 KO mouse muscle
To further confirm the involvement of Rac1 in stretch-induced glucose transport, we analysed glucose transport and downstream signalling in inducible muscle-specific Rac1 KO mice. These mice have the advantage over conventional KO models in that the genetic modification is performed in adult mice, minimizing the risk of compensation for the missing gene. In soleus and EDL muscles, Rac1 content was decreased by ∼75% and
∼85%, respectively compared to WT littermate controls (Fig.2A and D). Stretching increased glucose transport by ∼100% in WT mice and similar to pharmacological Rac1 inhibition, stretch-stimulated glucose transport was reduced by ∼40% in soleus and by ∼30% in EDL KO muscle (Fig. 2B). As previously reported (Sylow et al. 2013a, 2013b), Rac1 KO resulted in an increased PAK1 protein content in both soleus and EDL muscles (Fig. 2A and D). Therefore, analysis of PAK signalling at site Thr423/402 was related to the total amount of PAK1. We found that stretch-stimulated p-PAK1/2Thr423/402 increased ∼150% in soleus muscle of WT but not Rac1 KO mice. In EDL, the stretch-induced ∼200% increase in p-PAK1/2Thr423/402 was blocked by Rac1 KO (Fig. 2C and D). Stretch-induced p-p38 MAPKThr180/Tyr182 was not different between genotypes. These data indicate a significant role for Rac1 in the regulation of stretch-induced glucose transport in skeletal muscle and show that PAK1/2 but not p38 MAPK is regulated downstream of Rac1.
Figure 2.
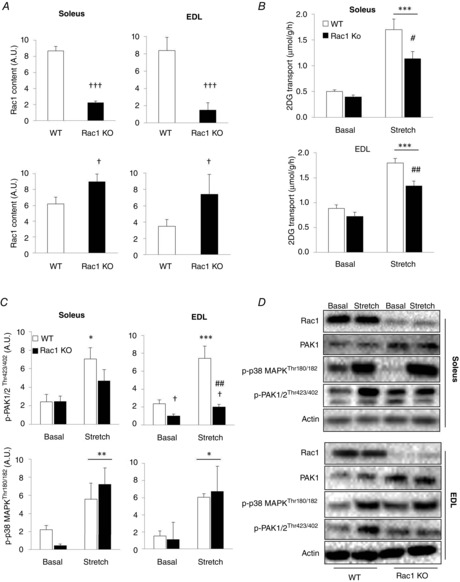
PAK but not p38 MAPK stretch signalling is reduced Rac1 KO muscle
A, bar graphs showing quantifications of immunoblots of Rac1 and PAK1 in soleus and EDL muscle from inducible muscle-specific Rac1 KO or WT littermates. (n = 5–7) B, stretch-stimulated 2DG transport in soleus and EDL from Rac1 KO or WT littermates (n = 5–7). C, bar graphs showing quantifications of immunoblots of p-PAK1/2Thr423/402 (related to total PAK1) and p-p38 MAPKThr180/182 in response to stretching in soleus and EDL muscle from muscle-specific Rac1 KO or WT littermates (n = 5–7). D, representative blots of PAK1, p-PAK1/2Thr423/402, p-p38 MAPKThr180/182, Rac1 and actin. *P < 0.05; **P < 0.01; ***P < 0.001: significant effect of stretch stimulation. #P < 0.05; ##P < 0.01): significant interaction between stretch and genotype. †P < 0.05; †††P < 0.001: significant effect of genotype is indicated. Values are the mean ± sem.
The actin cytoskeleton regulates stretch-stimulated glucose transport
Rac1 is a major regulator of the actin cytoskeleton in L6 muscle cells in response to insulin (JeBailey et al. 2004, 2007) and a functional actin cytoskeleton may also be necessary for insulin and muscle contraction to normally regulate glucose transport in mature skeletal muscle (Brozinick et al. 2004; Sylow et al. 2013b, 2014a). However, whether the actin cytoskeleton is required for stretch-stimulated glucose transport is not known. Accordingly, we incubated soleus and EDL muscles with the actin depolymerizing agent, latrunculin B, and analysed stretch-stimulated glucose transport. Latrunculin B significantly reduced stretch-stimulated glucose transport by ∼50% in soleus and ∼40% in EDL muscle (Fig. 3A). These findings suggest that, similar to insulin and contraction, an intact actin cytoskeleton is required for the normal regulation of stretch-induced glucose transport.
Figure 3.
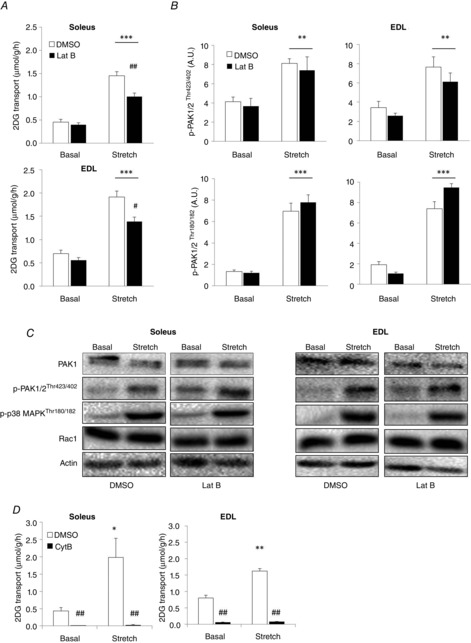
Stretch-stimulated glucose transport is inhibited by actin depolymerization
A, bar graphs showing stretch-stimulated 2DG transport in soleus and EDL muscles incubated for 45 min with 5 μm latrunculin B (Lat B) or a corresponding amount of DMSO (n = 8–10). B, bar graphs showing quantifications of immunoblots of p-PAK1/2Thr423/402 and p-p38 MAPKThr180/182 (n = 8–10). C, representative blots of PAK1, p-PAK1/2Thr423/402, p-p38 MAPKThr180/182, Rac1 and actin. All blots are from the same gel (n = 8–10). D, stretch-stimulated 2DG transport in soleus and EDL muscles incubated for 40 min with 50 μm cytochalasin B (Cyt B) or a corresponding amount of DMSO (n = 3–4). *P < 0.05; **P < 0.01; ***P < 0.001: significant effect of stretch stimulation. #P < 0.05; ##P < 0.01: significant interaction between stretch and inhibitors. Values are the mean ± sem.
Disruption of the actin cytoskeleton does not affect intracellular signalling during stretching
The actin cytoskeleton has been proposed to act as a scaffolding structure for signalling proteins in response to growth factor stimulation (Khayat et al. 2000; Peyrollier et al. 2000) and thus could participate in the transduction of extracellular-mediated signals to Rac1 and other signalling proteins (Zhou et al. 2007; Constantin, 2014). However, we did not detect any decreased activation of PAK1/2 or p38 MAPK proteins after latrunculin B treatment, suggesting that the actin cytoskeleton is not essential for stretch-activated signalling in skeletal muscle (Fig. 3B and C). Rather, the actin cytoskeleton may facilitate the docking and fusion of GLUT4 vesicles to allow glucose to enter the cell. To confirm that passive stretching of skeletal muscle relies on glucose transporter proteins and is not an indirect consequence of damaged or leaky plasma membranes, we applied the glucose transporter inhibitor cytochalasin B to our passive stretch model. Cytochalasin B binds to GLUT4 and GLUT1 (Hellwig & Joost, 1991) and thereby inhibits all glucose transport into the muscle cells that is facilitated by glucose transporters (Klip & Paquet, 1990). Should stretch-induced glucose transport occur via a glucose transporter independent mechanism, stretch would increase 2DG transport despite the inhibition of glucose transporters. Cytochalasin B completely blocked stretch-stimulated 2DG transport (Fig. 3D), showing that passive stretching stimulates glucose transport via regulation of glucose transporter proteins.
Rac1 regulates contraction-stimulated glucose transport but only during conditions of force development
Muscle contraction, which potently increases glucose transport, is accompanied by mechanical stress acting on the muscle. The mechanical stress-component of muscle contraction is necessary to fully activate glucose transport because the prevention of tension development reduced contraction-stimulated glucose transport (Ihlemann et al. 1999; Blair et al. 2009; Jensen et al. 2014b). Based on our findings showing that Rac1 regulates both contraction- (Sylow et al. 2013b) and stretch-stimulated glucose transport (present study), we hypothesized that Rac1 is part of the signal by which mechanical stress increases glucose transport during muscle contraction. To test this, we used Rac1 inhibitor II during conditions of muscle contraction with maximal or no force development. We used two previously published methods to disrupt tension development during electrically-induced contraction: (i) loosely hanging muscles in which resting length of the muscles was adjusted to achieve no force output (Ihlemann et al. 1999) and (ii) pre-treatment with the myosin heavy chain ATPase inhibitors BTS and Blebbistatin, which prevent cross-bridge formation (Blair et al. 2009; Jensen et al. 2014b). Loosely hanging soleus and EDL muscles displayed ∼70% reduced glucose transport compared to muscles in which maximal tension development was recorded (Fig. 4A). The residual 40% (soleus) and 50% (EDL) increase in glucose transport in loosely hanging muscles was not affected by the inhibition of Rac1. In agreement with these results, BTS/Blebbistatin treatment, which inhibited force production by >95%, reduced contraction-stimulated glucose transport by 65% and 55% in soleus and EDL, respectively. The inhibition of Rac1 in tension developing muscles reduced glucose transport by 20–40% in soleus and EDL muscles. However, the Rac1 Inhibitor II in combination with BTS/Blebbistatin did not affect the residual 50% and 70% increase in glucose transport in non-force developing soleus or EDL muscle, respectively (Fig. 2B). These findings suggest that Rac1 is partially required for contraction-stimulated glucose transport because it is a component of the mechanical stress-signal to glucose transport.
Figure 4.
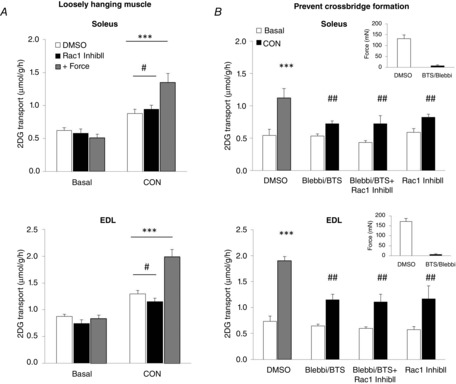
Glucose transport in contracting non-tension developing muscle is not regulated by Rac1
A, bar graphs showing electrically-induced contraction-stimulated 2DG transport in tension developing (+ Force) or loosely hanging (white and black bars) soleus and EDL muscles incubated for 1 h with 15 μm Rac1 Inhibitor II (Rac1 InhibII) or a corresponding amount of DMSO (n = 6). B, bar graphs showing electrically-induced contraction-stimulated 2DG transport in soleus and EDL muscles incubated for 1 h with 15 μm Rac1 Inhibitor II ± 75 μm BTS/50 μm Blebbistatin (Blebbi) or a corresponding amount of DMSO. Inserts show the effect of BTS/Blebbi on initial force production during contraction (n = 5–8). *P < 0.05; **P < 0.01; ***P < 0.001: significant effect of contraction. #P < 0.05; ##P < 0.01: significant interaction between contraction and inhibitors. Values are the mean ± sem.
Discussion
In the present study, we report that Rac1 and a func-tional actin cytoskeleton are necessary for the normal regulation of stretch-induced glucose transport in skeletal muscle. In addition, we found that Rac1 regulated contrac-tion-stimulated glucose transport only when force deve-lopment was allowed, suggesting that Rac1 specifically mediated the mechanical stress-component of con-traction-stimulated glucose transport.
Muscle stretching was first reported to increase glucose transport almost 30 years ago (Shoji, 1986) but the underlying mechanism remains not well described. Mechanical stress is sensed by a number of proteins at the plasma membrane, including the dystrophin glycoprotein complex, ion channels, integrins and focal adhesions. We previously reported that passive stretching of mouse muscle activated Rac1 (Sylow et al. 2013b) and, in the present study, we show that stretching also increased phosphorylation of the downstream target of Rac1: PAK1/2. These findings are in agreement with earlier studies showing that Rac1 co-localized with PAK in response to passive stretching in skeletal muscle (Zhou et al. 2007). The mechanism by which Rac1 regulates glucose transport in response to stretching remains to be clarified. In response to insulin, Rac1 facilitates GLUT4 translocation in L6 myotubes via reorganization of the cortical actin cytoskeleton (JeBailey et al. 2004). The occurrence of cortical actin remodelling in mature skeletal muscle and its role in stretch-stimulated glucose transport needs further investigation. However, based on the sensitivity to latrunculin B, muscle stretching could require an intact actin cytoskeleton to stimulate glucose transport, similar to the regulation of insulin-stimulated GLUT4 translocation and glucose transport in cell culture (JeBailey et al. 2004, 2007). It is therefore tempting to hypothesize that Rac1 regulates actin cytoskeleton dynamics and thereby mediates stretch-stimulated glucose transport.
Our overall working hypothesis is that muscle contraction facilitates glucose transport via two distinct pathways. This includes a metabolic signal via AMPK and a mechanical signal. By contrast, our data do not support a major role for proteins activated exclusively by sarcoplasmic reticulum Ca2+ release (Jensen et al. 2014b). The findings of the present study build on this hypothesis because stretch-stimulated glucose transport partly depended on Rac1, whereas Rac1 inhibition failed to suppress glucose transport when force production was compromised by either suboptimal sarcomere length or myosin ATPase inhibition. This suggests that Rac1 is part of an AMPK-independent, stretch-signal to glucose transport during contraction. The independence of metabolic stress/AMPK and mechanical stress/Rac1 in regulating muscle glucose transport is further supported by a number of previous findings because (i) passive stretch for 15 min activates Rac1 but not AMPK signalling (Chambers et al. 2009; Jensen et al. 2014b); (ii) AICAR activates AMPK but not Rac1 signalling (Sylow et al. 2013b); (iii) kinase-dead AMPK expression does not prevent exercise-induced Rac1 signalling (Sylow et al. 2013b); (iv) Rac1 inhibition does not inhibit AMPK signalling (Sylow et al. 2013b); (v) both AMPK and Rac1 activation appear to be sufficient to increase glucose transport (Chiu et al. 2013; Lai et al. 2014); and (f) combined AICAR and passive stretch-stimulation mimic the contraction glucose transport response and this response displays additivity with insulin but not contraction-stimulation (Jensen et al. 2014b).
A major unresolved question is whether and how the numerous proteins and mechanisms, previously shown to be required for glucose transport stimulation in muscle, fit into our AMPK and mechanical stress-model (Jensen et al. 2014a; Sylow et al. 2014b). For some molecules, their sensitivity to the AMPK activator AICAR might lend a clue as to their involvement in AMPK signalling to glucose transport. Hence, the inhibition of putative glucose transport regulators such as CaMKII, nitric oxide synthase and reactive oxygen species does not appear to inhibit AICAR (AMPK)-stimulated glucose transport (Stephens et al. 2004; Wright et al. 2004; Merry et al. 2010), although not all studies support this (Fryer et al. 2000; Sandstrom et al. 2006). Given that these molecules are not AMPK-dependent, their involvement in glucose transport-regulation could occur via mechanical stress, as shown in the present study for Rac1. Interestingly, all of the above mentioned molecules appear to be induced by mechanical stress (Kitajima et al. 2011; Ito et al. 2013) and a role of free oxygen radicals has already been suggested for stretch-stimulated glucose transport (Chambers et al. 2009). The involvement of these putative candidates in the regulation of stretch- and contraction-stimulated glucose transport needs further investigation.
The glucose transport-response to stretch was only partially reduced by Rac1 inhibition or by the actin depolymerizing agent, latrunculin B. If the possibility is considered that glucose transport in muscle in response to insulin (Brozinick et al. 2004; Sylow et al. 2013a, 2014a), contraction (Sylow et al. 2013b) and stretching (present study) is partially dependent on Rac1 and filamentous actin, then what is the residual Rac1 and actin-independent component? The residual glucose transport might reflect an increase in intrinsic GLUT4 activity (Furtado et al. 2002; Huang et al. 2002), the contribution from other glucose transporter isoforms in muscle (Purcell et al. 2011), the regulation of fusion of already docked GLUT4 vesicles (Bai et al. 2007) or perhaps the inhibition of GLUT4 endocytosis rather than exocytosis (Foley et al. 2011). Indeed, in isolated ex vivo incubated muscles, the rate-limiting step for glucose uptake is probably transport (Hansen et al. 1994) and GLUT4 appears to account for the majority of this transport (Zisman et al. 2000). In the present study, we did not directly measure GLUT4 translocation and it is possible that inhibiting the actin cytoskeleton and its regulators has a greater effect on GLUT4 movement than glucose transport. In support of this, we have observed that inducible Rac1 KO abolishes GLUT4 translocation but only partially reduces glucose uptake in tibialis anterior muscle in response to exercise (L. Sylow, unpublished data). Similarly, insulin-stimulated GLUT4 translocation was almost completely prevented in conventional Rac1 KO (Ueda et al. 2010) compared to the modest reduction in insulin-stimulated glucose transport in the inducible Rac1 KO mouse muscle (Sylow et al. 2013a). Thus, it is possible that the GLUT4 translocation-response is Rac1 and/or actin-dependent, and yet the result is only a partial reduction in glucose transport.
Previous studies in vascular smooth muscle cells reported that p38 MAPK is activated downstream of Rac1 in response to cyclic strain stress (Li et al. 2000; Qi et al. 2010). p38 MAPK has been suggested to play an essential role in stretch-stimulated glucose transport because a pharmacological inhibitor of p38 MAPK abolished glucose transport during passive stretching (Chambers et al. 2009), although that inhibitor was reported to directly inhibit intrinsic GLUT4 activity (Ribe et al. 2005). In addition, another p38 MAPK inhibitor, VX-702, did not reduce stretch-stimulated glucose transport in mouse muscles despite a complete blockade of p38 MAPK signalling, making the exact contribution of p38 MAPK unclear (Jensen et al. 2014b). Whatever the role of p38 MAPK in glucose transport regulation, the findings of the present study show that Rac1 signals independently of p38 MAPK because stretch-induced glucose transport was decreased by Rac1 Inhibitor II and Rac1 KO, despite normal p38 MAPKThr180/Tyr182 phosphorylation.
In conclusion, we show that Rac1 and an intact actin cytoskeleton are critical for the normal regulation of stretch-stimulated glucose transport in skeletal muscle. In addition, Rac1 is necessary for the normal regulation of contraction-stimulated glucose transport but only during conditions of force development. These findings suggest that Rac1 regulates the mechanical stress-component of contraction-stimulated glucose transport, possibly via the actin cytoskeleton.
Acknowledgments
We acknowledge the skilled technical assistance of Betina Bolmgren and Irene Bech Nielsen (Molecular Physiology Group, Denmark). Rac1 floxed mice were a kind gift from Cord Brakebusch (Biomedical Institute, BRIC, University of Copenhagen, Denmark). Tetracycline-activated Cre mice were a kind gift from Ashley Monks (Department of Psychology, University of Toronto Mississauga, CA, USA).
Glossary
Abbreviations
- AMPK
AMP activated protein kinase
- 2DG
2-deoxyglucose
- EDL
extensor digitorum longus
- KO
knockout
- WT
wild-type
Additional information
Competing interests
The authors declare that there are no competing interests.
Author contributions
L.S. and T.E.J. designed the study. L.S. conducted the experiments, performed the laboratory analysis and wrote the manuscript. T.E.J., M.K., L.L.V.M and E.A.R. conducted the experiments. All authors revised and approved the final version of the manuscript submitted for publication. T.E.J. is the guarantor of this work and, as such, has full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding
The study was supported by grants from, the Danish Independent Research Council/Medicine and Science, the Novo Nordisk Foundation, the Lundbeck Foundation and the University of Copenhagen Excellence Program 2016 and the UNIK – Food Fitness and Pharma.
References
- Bai L, Wang Y, Fan J, Chen Y, Ji W, Qu A, Xu P, James DE. Xu T. Dissecting multiple steps of GLUT4 trafficking and identifying the sites of insulin action. Cell Metab. 2007;5:47–57. doi: 10.1016/j.cmet.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Blair DR, Funai K, Schweitzer GG. Cartee GD. A myosin II ATPase inhibitor reduces force production, glucose transport, and phosphorylation of AMPK and TBC1D1 in electrically stimulated rat skeletal muscle. Am J Physiol Endocrinol Metab. 2009;296:E993–E1002. doi: 10.1152/ajpendo.91003.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccafoschi F, Mosca C, Bosetti M. Cannas M. The role of mechanical stretching in the activation and localization of adhesion proteins and related intracellular molecules. J Cell Biochem. 2011;112:1403–1409. doi: 10.1002/jcb.23056. [DOI] [PubMed] [Google Scholar]
- Brozinick JT, Jr, Berkemeier BA. Elmendorf JS. “Actin”g on GLUT4: membrane & cytoskeletal components of insulin action. Curr Diabetes Rev. 2007;3:111–122. doi: 10.2174/157339907780598199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozinick JT, Jr, Hawkins ED, Strawbridge AB. Elmendorf JS. Disruption of cortical actin in skeletal muscle demonstrates an essential role of the cytoskeleton in glucose transporter 4 translocation in insulin-sensitive tissues. J Biol Chem. 2004;279:40699–40706. doi: 10.1074/jbc.M402697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers MA, Moylan JS, Smith JD, Goodyear LJ. Reid MB. Stretch-stimulated glucose uptake in skeletal muscle is mediated by reactive oxygen species and p38 MAP-kinase. J Physiol. 2009;587:3363–3373. doi: 10.1113/jphysiol.2008.165639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu TT, Jensen TE, Sylow L, Richter EA. Klip A. Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cell Signal. 2011;23:1546–1554. doi: 10.1016/j.cellsig.2011.05.022. [DOI] [PubMed] [Google Scholar]
- Chiu TT, Sun Y, Koshkina A. Klip A. Rac-1 superactivation triggers insulin-independent glucose transporter 4 (GLUT4) translocation that bypasses signaling defects exerted by c-Jun N-terminal kinase (JNK)- and ceramide-induced insulin resistance. J Biol Chem. 2013;288:17520–17531. doi: 10.1074/jbc.M113.467647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrostek A, Wu X, Quondamatteo F, Hu R, Sanecka A, Niemann C, Langbein L, Haase I. Brakebusch C. Rac1 is crucial for hair follicle integrity but is not essential for maintenance of the epidermis. Mol Cell Biol. 2006;26:6957–6970. doi: 10.1128/MCB.00075-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim Biophys Acta. 2014;1838:635–642. doi: 10.1016/j.bbamem.2013.08.023. [DOI] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM. Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- Foley K, Boguslavsky S. Klip A. Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry. 2011;50:3048–3061. doi: 10.1021/bi2000356. [DOI] [PubMed] [Google Scholar]
- Fryer LG, Hajduch E, Rencurel F, Salt IP, Hundal HS, Hardie DG. Carling D. Activation of glucose transport by AMP-activated protein kinase via stimulation of nitric oxide synthase. Diabetes. 2000;49:1978–1985. doi: 10.2337/diabetes.49.12.1978. [DOI] [PubMed] [Google Scholar]
- Furtado LM, Somwar R, Sweeney G, Niu W. Klip A. Activation of the glucose transporter GLUT4 by insulin. Biochem Cell Biol. 2002;80:569–578. doi: 10.1139/o02-156. [DOI] [PubMed] [Google Scholar]
- Hansen PA, Gulve EA. Holloszy JO. Suitability of 2-deoxyglucose for in vitro measurement of glucose transport activity in skeletal muscle. J Appl Physiol. 1994;76:979–985. doi: 10.1152/jappl.1994.76.2.979. [DOI] [PubMed] [Google Scholar]
- Hellwig B. Joost HG. Differentiation of erythrocyte-(GLUT1), liver-(GLUT2), and adipocyte-type (GLUT4) glucose transporters by binding of the inhibitory ligands cytochalasin B, forskolin, dipyridamole, and isobutylmethylxanthine. Mol Pharmacol. 1991;40:383–389. [PubMed] [Google Scholar]
- Huang C, Somwar R, Patel N, Niu W, Torok D. Klip A. Sustained exposure of L6 myotubes to high glucose and insulin decreases insulin-stimulated GLUT4 translocation but upregulates GLUT4 activity. Diabetes. 2002;51:2090–2098. doi: 10.2337/diabetes.51.7.2090. [DOI] [PubMed] [Google Scholar]
- Ihlemann J, Ploug T, Hellsten Y. Galbo H. Effect of tension on contraction-induced glucose transport in rat skeletal muscle. Am J Physiol Endocrinol Metab. 1999;277:E208–E214. doi: 10.1152/ajpendo.1999.277.2.E208. [DOI] [PubMed] [Google Scholar]
- Ito N, Ruegg UT, Kudo A, Miyagoe-Suzuki Y. Takeda S. Activation of calcium signaling through Trpv1 by nNOS and peroxynitrite as a key trigger of skeletal muscle hypertrophy. Nat Med. 2013;19:101–106. doi: 10.1038/nm.3019. [DOI] [PubMed] [Google Scholar]
- Ito Y, Obara K, Ikeda R, Ishii M, Tanabe Y, Ishikawa T. Nakayama K. Passive stretching produces Akt- and MAPK-dependent augmentations of GLUT4 translocation and glucose uptake in skeletal muscles of mice. Pfluger's Arch. 2006;451:803–813. doi: 10.1007/s00424-005-1512-5. [DOI] [PubMed] [Google Scholar]
- JeBailey L, Rudich A, Huang X, Di Ciano-Oliveira C, Kapus A. Klip A. Skeletal muscle cells and adipocytes differ in their reliance on TC10 and Rac for insulin-induced actin remodeling. Mol Endocrinol. 2004;18:359–372. doi: 10.1210/me.2003-0294. [DOI] [PubMed] [Google Scholar]
- JeBailey L, Wanono O, Niu W, Roessler J, Rudich A. Klip A. Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes. 2007;56:394–403. doi: 10.2337/db06-0823. [DOI] [PubMed] [Google Scholar]
- Jensen TE, Angin Y, Sylow L. Richter EA. Is contraction-stimulated glucose transport feedforward regulated by Ca2+ Exp Physiol. 2014;99:1562–1568. doi: 10.1113/expphysiol.2014.081679. [DOI] [PubMed] [Google Scholar]
- Jensen TE, Rose AJ, Jorgensen SB, Brandt N, Schjerling P, Wojtaszewski JF. Richter EA. Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. Am J Physiol Endocrinol Metab. 2007;292:E1308–E1317. doi: 10.1152/ajpendo.00456.2006. [DOI] [PubMed] [Google Scholar]
- Jensen TE, Sylow L, Rose AJ, Madsen AB, Angin Y, Maarbjerg SJ. Richter EA. Contraction-stimulated glucose transport in muscle is controlled by AMPK and mechanical stress but not sarcoplasmatic reticulum Ca2+ release. Molecular Metabolism. 2014b;3:742–753. doi: 10.1016/j.molmet.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura S, Miyamoto S. Brown JH. Initiation and transduction of stretch-induced RhoA and Rac1 activation through caveolae: cytoskeletal regulation of ERK translocation. J Biol Chem. 2003;278:31111–31117. doi: 10.1074/jbc.M300725200. [DOI] [PubMed] [Google Scholar]
- Khayat ZA, Tong P, Yaworsky K, Bloch RJ. Klip A. Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J Cell Sci. 2000;113(Pt 2):279–290. doi: 10.1242/jcs.113.2.279. [DOI] [PubMed] [Google Scholar]
- Kitajima N, Watanabe K, Morimoto S, Sato Y, Kiyonaka S, Hoshijima M, Ikeda Y, Nakaya M, Ide T, Mori Y, Kurose H. Nishida M. TRPC3-mediated Ca2+ influx contributes to Rac1-mediated production of reactive oxygen species in MLP-deficient mouse hearts. Biochem Biophys Res Commun. 2011;409:108–113. doi: 10.1016/j.bbrc.2011.04.124. [DOI] [PubMed] [Google Scholar]
- Klip A. Paquet MR. Glucose transport and glucose transporters in muscle and their metabolic regulation. Diabetes Care. 1990;13:228–243. doi: 10.2337/diacare.13.3.228. [DOI] [PubMed] [Google Scholar]
- Lai YC, Kviklyte S, Vertommen D, Lantier L, Foretz M, Viollet B, Hallen S. Rider MH. A small-molecule benzimidazole derivative that potently activates AMPK to increase glucose transport in skeletal muscle: comparison with effects of contraction and other AMPK activators. Biochem J. 2014;460:363–375. doi: 10.1042/BJ20131673. [DOI] [PubMed] [Google Scholar]
- Li C, Hu Y, Sturm G, Wick G. Xu Q. Ras/Rac-Dependent activation of p38 mitogen-activated protein kinases in smooth muscle cells stimulated by cyclic strain stress. Arterioscler Thromb Vasc Biol. 2000;20:E1–E9. doi: 10.1161/01.atv.20.3.e1. [DOI] [PubMed] [Google Scholar]
- Merry TL, Steinberg GR, Lynch GS. McConell GK. Skeletal muscle glucose uptake during contraction is regulated by nitric oxide and ROS independently of AMPK. Am J Physiol Endocrinol Metab. 2010;298:E577–E585. doi: 10.1152/ajpendo.00239.2009. [DOI] [PubMed] [Google Scholar]
- Nozaki S, Takeda T, Kitaura T, Takenaka N, Kataoka T. Satoh T. Akt2 regulates Rac1 activity in the insulin-dependent signaling pathway leading to GLUT4 translocation to the plasma membrane in skeletal muscle cells. Cell Signal. 2013;25:1361–1371. doi: 10.1016/j.cellsig.2013.02.023. [DOI] [PubMed] [Google Scholar]
- Peyrollier K, Hajduch E, Gray A, Litherland GJ, Prescott AR, Leslie NR. Hundal HS. A role for the actin cytoskeleton in the hormonal and growth-factor-mediated activation of protein kinase B. Biochem J. 2000;352(Pt 3):617–622. [PMC free article] [PubMed] [Google Scholar]
- Poh YC, Na S, Chowdhury F, Ouyang M, Wang Y. Wang N. Rapid activation of Rac GTPase in living cells by force is independent of Src. PLoS One. 2009;4:e7886. doi: 10.1371/journal.pone.0007886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SH, Aerni-Flessner LB, Willcockson AR, Diggs-Andrews KA, Fisher SJ. Moley KH. Improved insulin sensitivity by GLUT12 overexpression in mice. Diabetes. 2011;60:1478–1482. doi: 10.2337/db11-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi YX, Qu MJ, Yan ZQ, Zhao D, Jiang XH, Shen BR. Jiang ZL. Cyclic strain modulates migration and proliferation of vascular smooth muscle cells via Rho-GDIalpha, Rac1, and p38 pathway. J Cell Biochem. 2010;109:906–914. doi: 10.1002/jcb.22465. [DOI] [PubMed] [Google Scholar]
- Rao P. Monks DA. A tetracycline-inducible and skeletal muscle-specific Cre recombinase transgenic mouse. Dev Neurobiol. 2009;69:401–406. doi: 10.1002/dneu.20714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribe D, Yang J, Patel S, Koumanov F, Cushman SW. Holman GD. Endofacial competitive inhibition of glucose transporter-4 intrinsic activity by the mitogen-activated protein kinase inhibitor SB203580. Endocrinology. 2005;146:1713–1717. doi: 10.1210/en.2004-1294. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Aschenbach WG, Hirshman MF. Goodyear LJ. Akt signaling in skeletal muscle: regulation by exercise and passive stretch. Am J Physiol Endocrinol Metab. 2003;285:E1081–E1088. doi: 10.1152/ajpendo.00228.2003. [DOI] [PubMed] [Google Scholar]
- Sandstrom ME, Zhang SJ, Bruton J, Silva JP, Reid MB, Westerblad H. Katz A. Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. J Physiol. 2006;575:251–262. doi: 10.1113/jphysiol.2006.110601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji S( Effects of stretch and starvation on glucose uptake of rat soleus and extensor digitorum longus muscles. Muscle Nerve. 1986;9:144–147. doi: 10.1002/mus.880090206. [DOI] [PubMed] [Google Scholar]
- Stephens TJ, Canny BJ, Snow RJ. McConell GK. 5'-aminoimidazole-4-carboxyamide-ribonucleoside-activated glucose transport is not prevented by nitric oxide synthase inhibition in rat isolated skeletal muscle. Clin Exp Pharmacol Physiol. 2004;31:419–423. doi: 10.1111/j.1440-1681.2004.04014.x. [DOI] [PubMed] [Google Scholar]
- Sylow L, Jensen TE, Kleinert M, Hojlund K, Kiens B, Wojtaszewski J, Prats C, Schjerling P. Richter EA. Rac1 signaling is required for insulin-stimulated glucose uptake and is dysregulated in insulin resistant murine and human skeletal muscle. Diabetes. 2013a;62((6)):1865–1875. doi: 10.2337/db12-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylow L, Jensen TE, Kleinert M, Mouatt JR, Maarbjerg SJ, Jeppesen J, Prats C, Chiu TT, Boguslavsky S, Klip A, Schjerling P. Richter EA. Rac1 is a novel regulator of contraction-stimulated glucose uptake in skeletal muscle. Diabetes. 2013b;62:1139–1151. doi: 10.2337/db12-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylow L, Kleinert M, Pehmoller C, Prats C, Chiu TT, Klip A, Richter EA. Jensen TE. Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cell Signal. 2014a;26:323–331. doi: 10.1016/j.cellsig.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Sylow L, Moller LL, Kleinert M, Richter EA. Jensen TE. Rac1- A novel regulator of contraction-stimulated glucose uptake in skeletal muscle. Exp Physiol. 2014b;99:1574–1580. doi: 10.1113/expphysiol.2014.079194. [DOI] [PubMed] [Google Scholar]
- Takenaka N, Yasuda N, Nihata Y, Hosooka T, Noguchi T, Aiba A. Satoh T. Role of the guanine nucleotide exchange factor in Akt2-mediated plasma membrane translocation of GLUT4 in insulin-stimulated skeletal muscle. Cell Signal. 2014;26:2460–2469. doi: 10.1016/j.cellsig.2014.07.002. [DOI] [PubMed] [Google Scholar]
- Ueda S, Kitazawa S, Ishida K, Nishikawa Y, Matsui M, Matsumoto H, Aoki T, Nozaki S, Takeda T, Tamori Y, Aiba A, Kahn CR, Kataoka T. Satoh T. Crucial role of the small GTPase Rac1 in insulin-stimulated translocation of glucose transporter 4 to the mouse skeletal muscle sarcolemma. FASEB J. 2010;24:2254–2261. doi: 10.1096/fj.09-137380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DC, Hucker KA, Holloszy JO. Han DH. Ca2+ and AMPK both mediate stimulation of glucose transport by muscle contractions. Diabetes. 2004;53:330–335. doi: 10.2337/diabetes.53.2.330. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Jiang D, Thomason DB. Jarrett HW. Laminin-induced activation of Rac1 and JNKp46 is initiated by Src family kinases and mimics the effects of skeletal muscle contraction. Biochemistry. 2007;46:14907–14916. doi: 10.1021/bi701384k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB, Wojtaszewski JF, Hirshman MF, Virkamaki A, Goodyear LJ, Kahn CR. Kahn BB. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med. 2000;6:924–928. doi: 10.1038/78693. [DOI] [PubMed] [Google Scholar]