

Abstract
The role of interleukin-6 (IL-6) in hyperthermia and heat stroke is poorly understood. Plasma IL-6 is elevated following hyperthermia in animals and humans, and IL-6 knockout mice are more intolerant of severe hyperthermia. We evaluated the effect of IL-6 supplementation on organ injury following severe hyperthermia exposure in anaesthetized mice. Two hours prior to hyperthermia, mice were treated with 0.6 μg intraperitoneal IL-6, or identical volumes of saline in controls. Mice were anaesthetized, gavaged with FITC–dextran for measures of gastrointestinal permeability, and exposed to incremental (0.5°C every 30 min) increases in temperature. Heating stopped when maximum core temperature (Tc) of 42.4°C was attained (Tc,max). The mice recovered at room temperature (≈22°C) for 30 or 120 min, at which time plasma and tissues were collected. IL-6-treated mice, on average, required ≈25 min longer to attain Tc,max. Injury and swelling of the villi in the duodenum was present in untreated mice after 30 min of recovery. These changes were blocked by IL-6 treatment. IL-6 also reduced gastrointestinal permeability, assayed by the accumulation of FITC–dextran in plasma. Plasma cytokines were also attenuated in IL-6-treated animals, including significant reductions in TNFα, MCP-1 (CXCL2), RANTES (CCL5) and KC (CCL5). The results demonstrate that IL-6 has a protective influence on the pattern of physiological responses to severe hyperthermia, suggesting that early endogenous expression of IL-6 may provide a protection from the development of organ damage and inflammation.
Key points
Heat stroke afflicts thousands of humans each year, worldwide.
The immune system responds to hyperthermia exposure resulting in heat stroke by producing an array of immunological proteins, such as interleukin-6 (IL-6). However, the physiological functions of IL-6 and other cytokines in hyperthermia are poorly understood.
We hypothesized that IL-6 plays a protective role in conditions of heat stroke. To test this, we gave small IL-6 supplements to mice prior to exposing them to hot environments sufficient to induce conditions of heat stroke.
Pretreatment with IL-6 resulted in improved ability to withstand heat exposure in anaesthetized mice, it protected the intestine from injury, reducing the permeability of the intestinal barrier, and it attenuated the release of other cytokines involved in inflammation.
The results support the hypothesis that IL-6 is a ‘physiological stress hormone’ that plays an important role in survival during acute life-threatening conditions such as heat stroke.
Introduction
Heat stroke (HS) afflicts thousands of people every year, across all walks of life. It has a mortality of 5–50%, depending on the population being studied, but there are also long term complications (Dematte et al. 1998; Bouchama ´ Knochel, 2002; Davido et al. 2006; Argaud et al. 2007). During exposure to high environmental temperatures that can result in HS, one of the earliest and most robust cytokines seen in the circulation in mammals is interleukin-6 (IL-6) (Robins et al. 1995; Bouchama et al. 2005b; Welc et al. 2013a). IL-6 has been found to have many different functions that fall under categories of both pro- and anti-inflammatory activity, wound healing, cell survival signalling and metabolic control, and has been shown to have a number of functional roles in acute and chronic illnesses. For example, it is the chief hormone regulating the release of acute phase proteins from the liver, which is critical for surviving invasion of pathogens or in responding to injury or physical stress (Heinrich et al. 1990; Heath et al. 1993; Nemeth et al. 2004). In models of acute, life-threatening illnesses, removing the entire IL-6 response by knockout or antibody treatment causes markedly increased mortality rates. These conditions include sepsis (Barton ´ Jackson, 1993; Leon et al. 1998; Wang et al. 2001), acute pancreatitis (Cuzzocrea et al. 2002), liver failure (Cressman et al. 1996) and HS (Leon, 2007). In further support of a role for IL-6 as a protective hormone, supplementation in the early stages of haemorrhagic shock results in increased survival (Alten et al. 2008), prevention of circulatory collapse (Alten et al. 2008), reductions in lung injury (Moran et al. 2009) and decreased liver injury (Moran et al. 2008). One of the mechanisms by which IL-6 may function in these acute settings is as a mediator of pre- or post-conditioning through activation of the receptor-mediated JAK/STAT3 signalling pathway, a powerful mechanism promoting cell survival (Dawn et al. 2004; Matsumoto et al. 2006; Sakata et al. 2012).
Based on the observations above, we hypothesized that IL-6 production normally emerging early in the response to hyperthermia is part of a stress-induced cytokine response (Welc et al. 2013a) and plays a protective preconditioning role, promoting organism survival. To explore the influences of early IL-6 signalling in the outcomes of HS, in early experiments we tested if IL-6 pretreatment had an effect on the ability to resist elevations in core temperature (Tc) in response to a preprogrammed elevation in environmental temperature (Tenv). Surprisingly, we found that in anaesthetized animals treated with IL-6, Tc did not increase as fast as in untreated animals. We then wished to test whether IL-6 pretreatment affected the onset of organ injury or the timing of the immune system responses to hyperthermia. However, to compare animals with the same total heat exposure, we redesigned the experiment so that the core temperature profile in both treated and untreated mice remained the same. Results demonstrate that in response to exposure to the same Tc profile resulting in conditions of HS, IL-6 pretreatment has potent protective influences on the organism, reducing intestinal barrier dysfunction and injury and attenuating the circulating cytokine responses that are characteristic of extreme hyperthermia exposures.
Methods
Mice and IL-6 supplementation
All animal protocols were approved by the University of Florida Institutional Animal Care and Use Committee. Male, wild-type C57BL/6 mice (119 total) were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and housed at the University of Florida on a 12/12 h light–dark cycle with the dark cycle between 19.00 and 07.00 h until the day of the experiment. The temperature and humidity of the rooms was maintained at 19–23°C and approximately 40–60% relative humidity. Animals were brought from the animal facility to the laboratory the afternoon before the experiment in a new cage in order to adapt to the laboratory and to recover from the stress of transport. They were maintained on the same 12/12 h light–dark cycle at 19–23°C and 40–60% relative humidity in the laboratory. Experiments were performed the next day, during daytime hours with experiments beginning between at 07.00 and 12.00 h. The ages of the mice ranged between 8 and 16 weeks; weights are reported for individual groups in the Results. The animals were provided food and water ad libitum prior to anaesthesia. The mice were weighed and anaesthetized by i.p. injection with 50 mg kg−1 pentobarbital (Nembutal) diluted in sterile saline (10%). Pentobarbital supplements of 0.05 ml were given throughout the study, as needed. As no surgery was performed on the animals throughout the protocols, the level of anaesthesia was kept at a low level, just sufficient to eliminate toe withdrawal reflexes. Mice receiving IL-6 supplementation were administered 0.6 μg recombinant mouse IL-6 in 100 μl sterile saline by i.p. injection, without anaesthesia, 2 h prior to initiation of the HS protocols. The dosage of IL-6 was based on the pharmacokinetic experiments of i.p. IL-6 injections in mice (Peters et al. 1996). These studies predicted that a 0.6 μg i.p. dose would result in a peak plasma dose of ≈200–300 pg ml−1 by 1 h and this level would be maintained for approximately 4 h. The plasma target of 200–300 pg ml−1 was based on known concentrations of plasma IL-6 during the early recovery period after passive heat stroke in unanaesthetized mice (Leon et al. 2006). Sham control mice received an identical i.p. injection of 100 μl sterile saline.
HS protocols
Two different HS protocols were implemented. In the first, referred to as the ‘Tenv clamp’, the animals were placed supine in a modified plexiglass mouse cage with a plexiglass lid that could be quickly removed for anaesthesia supplementation without greatly affecting the mouse’s temperature exposure (Fig. 1A). A custom-made servo heating system, consisting of a high capacity variable blower and a heat exchange element made of copper pipe, was used to manipulate the Tenv of the cage. Baffles on both ends of the cage ensured a laminar flow and a uniform temperature distribution. The sides of the cage were insulated and the transparent top was double-paned. Tenv was regulated by a servo-controlled, proportional, integral and differential controller (Digisense, Cole-Parmer, Vernon Hills, IL, USA). Rectal temperature was monitored using a 2 mm rectal thermistor (YSI, Yellow Springs, OH, USA). The anaesthetized mice were brought to a steady state Tc of 36°C using the temperature controller and then were started on a programmed temperature profile in which Tenv was first brought to 39.5°C, over 30 min, and then elevated 0.5°C every 30 min until Tc attained a maximum value of 42.4°C (Tc,max), a temperature corresponding to moderate HS in unanaesthetized mice (Leon et al. 2005). After reaching Tc,max, the animals were removed from the chamber and allowed to recover at room temperature for 30 min, reaching approximately 35–36°C. All experiments were matched with anaesthetized sham controls in which Tc was maintained at 37°C (night-time active Tc for mice (Leon et al. 2005)) during equivalent periods.
Figure 1. Experimental set-up.
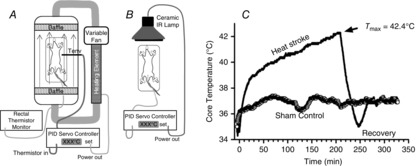
A, environmental temperature clamp (Tenv clamp) apparatus for inducing HS in anaesthetized mice. This was used to measure the ability to resist rising Tenv. B, core temperature clamp (Tc clamp) apparatus, used to force Tc to a set temperature profile. C, a typical Tc profile of an HS experiment using apparatus in B vs. a typical tracing of a sham control mouse.
A second anaesthetized HS model was developed to measure variables such as intestinal permeability, intestinal injury and cytokine expression, all of which are time-dependent. As will be shown in detail in the Results, IL-6 pretreatment had drastic effects on the length of time that it took a given animal to reach the targeted Tc,max, when exposed to the same Tenv profile. Therefore, we developed a second method that clamped Tc to a preset, programmed time course, thus fixing the time–temperature profile during hyperthermia for all mice (Fig. 1C). We called this second approach the ‘Tc clamp’ method. Briefly, the environmental cage was removed and the animals were placed under a long wavelength ceramic infrared lamp with radiant energy emitted at 4–14 μm wavelength. The heating protocol was identical to the Tenv clamp method in terms of the incremental changes in temperature that were controlled at each stage. This resulted in a relatively constant thermal load of ≈110°C min, calculated as time × (rectal temp. − 40.4°C) (Hubbard et al. 1977). After acquiring Tc,max at ≈200 min, the animals were allowed to recover for either 30 min or 2 h, with the controller Tc set point maintained at 36°C (Fig. 1C). Results were compared against sham controls that were studied over the same durations of anaesthesia and monitoring.
Measurement of intestinal permeability and blood sampling
For gastrointestinal permeability measurements, just after induction of anaesthesia, a 4 kD FITC–dextran (FD4) solution (20.8 mM) was gently gavaged into the lower oesophagus at a dose of 10 μl g−1 bodyweight. This method was originally based on the approach of Lambert et al. (2002) in rats and was adapted for mice by Oliver et al. (2012). The animals were held upright for ∼10 s and then placed in the temperature controlling system in the supine position. At the end of the study, the mice were given a strong supplemental dose of pentobarbital, and a 0.5–1 ml blood sample was obtained under anaesthesia by transthoracic stick into a 1 ml, EDTA-loaded syringe. The animals were then killed by thoracotomy and removal of the heart from the great vessels, under anaesthesia. The plasma sample was immediately spun down in a refrigerated centrifuge and plasma pipetted off from the buffy coat. The samples were aliquoted and frozen at −80 °C until FITC concentrations within the plasma samples were measured using standard spectrofluorometry and calibrated to FITC standards. Fresh plasma from naïve control animals was used as a negative blank control. Aliquots of plasma were frozen for multiplex cytokine measurements.
Histological analyses of intestinal samples
The entire intestines were removed immediately following blood withdrawal. The small intestines were then rinsed and the mesentery removed so that they could be straightened, limiting mechanical stress on the tissue. The entire intestine was then placed in 4% formalin for fixation. At a later time, 1 cm transverse sections of the duodenum (immediately following the pyloric sphincter), the jejunum (half way between the pyloric sphincter and ileocaecal junction) and the ileum (immediately preceding the ileocaecal junction) were cut out and used for histological assessment. Tissues were then processed by the University of Florida Molecular Pathology Core Lab, for paraffin embedding, cutting 4 μm thin sections, mounting on slides and staining with haematoxylin–eosin (H´E). To analyse villus injury in an unbiased way, the slides were blindly scored for injury by two independent readers using methods as described previously (Novosad et al. 2013). These methods are based on the original approach of Chiu et al. (1970) and range from 0 to 5, with 0 = normal mucosal villi, 1 = subepithelial space at the villus tips, 2 = extension of the subepithelial space with moderate lifting, 3 = massive epithelial lifting down the sides of the villi, some tips denuded, 4 = denuded villi, dilated capillaries and 5 = disintegration of the lamina propria. In addition, villus height and width were measured using calibrated microscope image analysis, also as previously described (Novosad et al. 2013). Specifically, every fourth villus was graded and measured until a total of 10 villus measurements and injury grades were made for each section of intestine in each animal.
Cytokine measurements
Plasma cytokines and chemokines were qualitatively and quantitatively evaluated, utilizing the MILLIPLEX XMAP Mouse Cytokine/Chemokine-Premixed 22-Plex Assays (Millipore, Billerica, MA, USA). The soluble mediators, granulocyte colony stimulating factor (G-CSF), granulocyte macrophage colony stimulating factor (GM-CSF), interferon-gamma (IFN-γ), interleukin 10 (IL-10), IL-12 (p70), IL-13, IL-15, IL-17, IL-1a, IL-1ß, IL-2, IL-4, IL-5, IL-6, IL-7, IL-9, interferon gamma inducing protein 10 (IP-10), keratinocyte-derived cytokine (KC), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1 alpha (MIP-1α), RANTES (regulation on activation normal T cell expressed and secreted) and tumour necrosis factor alpha (TNF-α), were probed for according to the manufacturer’s protocols. Detailed methods for multiplex sample preparation and analysis for our laboratory were previously reported (Welc et al. 2012). Concentrations (pg ml–1) were determined using a standard curve, five-parameter logistics and Milliplex analyst (Viagene, Tampa, FL, USA).
Statistics
The treatments within each model were randomized such that control animals and experimental animals were interspersed. Only animals that survived the entire anaesthesia, heating and recovery protocol were used for data analysis (50–63% survival rate of the anaesthetized animals through the entire protocol). For non-skewed data, central tendency and variance were expressed as means ± SEM or SD as appropriate; skewed data were expressed as medians ±25–75% quartiles (only the bottom 25% were graphed for clarity). No outliers were removed. For comparisons of parametric data (intestinal histology), a two-way ANOVA was performed with heat and IL-6 as the factors of interest with significant differences between groups tested with least squares contrasts (SAS JMP software). Multi-group comparisons for non-parametric data used Kruskal–Wallis ANOVA followed by the post hoc Steel–Dwas test for comparison of all groups (SAS JMP). For fold changes in multiplex cytokine/chemokine measurements, significant differences were determined by the sign test; bias due to multiple sampling for the sign test was estimated using the Benjamini–Hochberg procedure for calculation of the maximum false discovery rate (FDR) (Benjamini ´ Hochberg, 1995).
Results
Response to changing environmental temperature
Using the ‘Tenv clamp’ method, animals that were pretreated with IL-6 withstood longer periods of environmental heat exposure before reaching the target core temperature of 42.4°C (Fig. 2). Specifically, it required ≈35% longer for the IL-6-treated animals to complete the entire protocol prior to the recovery period. Furthermore, on average, IL-6-treated mice withstood core temperatures above 40°C for 41% longer than sham-treated mice, before reaching their target Tc,max. Therefore, IL-6 pretreatment resulted in delayed rise in Tc in response to a fixed pattern of elevation in Tenv. The peak Tenv at the Tc,max end point was 41.0 ± 0.1°C for control animals and was not significantly different from IL-6-treated animals, 41.4 ± 0.4°C (P = 0.1). There were no differences in body weights between groups in this experiment: 25.8 ± 1.8 vs. 24.2 ± 2.0 g (mean±SD) in HS vs. HS + IL-6, respectively.
Figure 2. Using the ‘Tenv clamp’ method the duration that animals could withstand core temperature elevations above 40°C and the duration of the total heating and recovery period were significantly elevated in IL-6-treated animals.
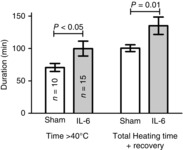
Values are means ± SEM. Significance was determined by a two-sample t-test. Results are only from animals that survived the protocol: 60% in untreated HS, 63% in IL-6-treated HS.
Intestinal barrier function
The ‘Tc clamp’ method was used for these and all remaining experiments, ensuring that the time elapsed from the beginning of heat exposure to Tc,max was fixed at ≈200 min and thus keeping the thermal load and time elapsed for diffusion of FD4 from intestine to plasma constant. As shown in Fig. 3, 30 min after Tc,max, although there were apparent elevations in plasma FD4 in both untreated and IL-6-treated mice exposed to HS, only the IL-6-treated mice reached statistical significance at this time point. However, at 120 min after Tc,max, there were significant elevations in plasma FD4 in untreated control HS mice, while plasma FD4 levels in IL-6-treated HS mice had returned to baseline levels. There were no statistical differences in body weight between any of the groups. For animals killed at the 30 min time window: untreated controls, 29.0 ± 3.2 g; IL-6-treated controls, 27.2 ± 3.5 g; untreated HS, 29.6 ± 1.1 g; IL-6-treated HS, 27.1 ± 2.6 g. For animals killed at the 120 min window: untreated controls, 28.3 ± 1.2 g; IL-6-treated controls, 27.6 ± 1.2 g; untreated HS, 29.2 ± 2.9 g; IL-6-treated HS, 27.6 ± 1.2 g.
Figure 3. Effects of HS on the permeability of the intestine to high molecular weight FD4 gavaged into the gastrointestinal lumen.
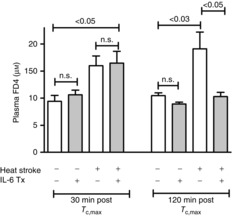
The perpendicular brackets on the ends of the lines below the P values are directly over the corresponding groups that were found to be signficantly different by post hoc comparisons. Measurements were taken at 30 and 120 min after Tc,max. All experiments used the Tc clamp method. Shaded bars: IL-6 treatment. Values are means ± SEM, n = 5–7 per group. Signficance levels were from non-parametric tests following Kruskal–Wallis ANOVA (Steel–Dwass).
Intestinal injury and morphology
Most of the histological evidence of altered villus structure and injury was identified in the duodenum, with minor changes in the jejunum. As shown in representative images in Fig. 4, in untreated HS mice 30 min after Tc,max, intestinal villi in the duodenum exhibited marked damage, with loss of the integrity of the epithelial lining, apparent swelling and villus shortening (Fig. 4B). In contrast, in IL-6-treated HS mice, although villus morphology was changed, the villi were relatively protected from injury (Fig. 4C). The changes in morphology in this group were largely due to shortened villus lengths. As shown in Fig. 5, at 30 min after Tc,max, the sham-treated HS mice exhibited approximately a 2-fold elevation in the average villi injury scores in the duodenum, which was statistically higher than that observed in the IL-6-treated HS mice (Fig. 5A). This injury was largely resolved by 120 min after Tc,max (Fig. 5D). At that time point, the differences in the extent of injury between IL-6 and sham-treated HS animals were abrogated. Villus height was reduced in response to HS at both 30 and 120 min time points in duodenum and jejunum, and there were no effects of IL-6 treatment in either region (Fig. 5D and E). Finally, villus width, an indicator of swelling or contraction, was significantly elevated at 30 min after Tc,max in control HS mice in the duodenum, but not in IL-6-treated HS mice (Fig. 5C), whereby at 120 min villus width had returned to roughly baseline levels or below in both experimental groups (Fig. 5D).
Figure 4. Typical H´E stained sections of duodenal region of the intestine.
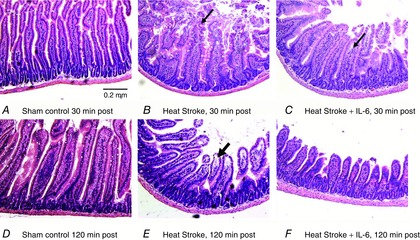
A and D, sham control animals at 30 and 120 min time points equivalent to HS durations. B, HS animals after 30 min of recovery. Arrow highlights typical area of swelling and epithelial disorganization. E, typical intestine from HS mice after 120 min of recovery. Note the shortened villi and the continued epithelial lifting from the lamina propria. C, duodenum from HS animals treated with IL-6 after 30 min of recovery. Note the more intact villi but with some residual epithelial lifting from the lamina propria. F, intestine of HS plus IL-6 treatment showing shortened villi but without clear evidence of continuing injury. For individual images, contrast, brightness and colour were adjusted within PowerPoint.
Figure 5. Effects of HS and IL-6 treatment on intestinal injury and villus dimensions.
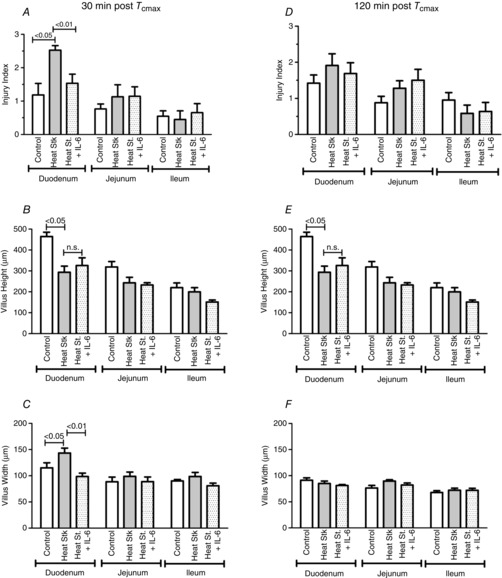
HS with IL-6 treatment (dotted fill), HS with no IL-6 (grey fill), sham-timed control (open). A and D, injury scores; B and E, villus height; and C and F, villus width. Values are means ± SEM; n = 5–7 animals per group. Significance levels represent least squares contrasts following two-way ANOVA.
Circulating cytokines
HS significantly altered the circulating cytokine profile. Because of the complexity of the responses at the two time points, two different quantitative analyses were performed. Initially, a non-parametric test (Kruskal–Wallis ANOVA) and post hoc analyses were used to compare absolute concentration levels of specific cytokines measured within each time period between IL-6-treated and the sham HS mice (Fig. 6). The particular groups of cytokines represented in Fig. 6 are only those that were significantly different at the overall ANOVA level. At 30 min after Tc,max, cytokines that are directly or indirectly involved in innate immunity, (IL-6, IL-10, TNFα, IL-12(P70)) were significantly elevated by HS. In addition, several chemokines, including KC (CXCL1), IP-10 (CXCL10) and MCP-1 (CCL2), were also significantly elevated.
Figure 6. Cytokine and chemokine protein expression in the circulation at 30 and 120 min after Tc,max, compared to sham controls.
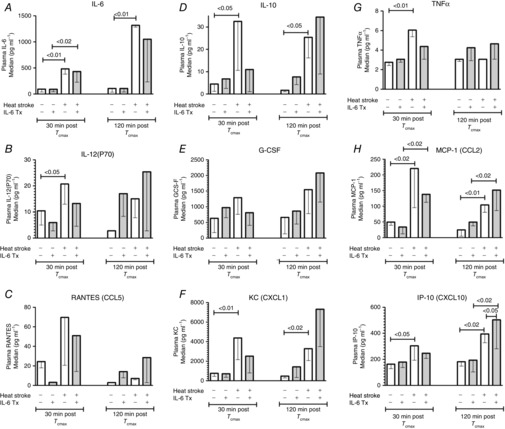
Shaded bars represent IL-6-treated animals; open bars represent either controls or untreated HS animals; n = 6–13 animals per group. Bar heights represent medians; hanging bracket represents the location of the bottom 25% quartile. Significance levels are from non-parametric Kruskal–Wallis followed by Steel–Dwass post hoc tests.
Interestingly, pretreatment of HS animals with IL-6 resulted in a general reduction in the expression of many cytokines, although direct comparisons between IL-6-treated and untreated groups in HS were not often statistically different. In most cases, however, the HS responses in the IL-6 group were also not statistically different from control. These include TNFα, IL-10, IL-12(P70), KC (CXCL1) and IP-10(CXCX10). At 120 min after Tc,max, IL-6, IL-10, MCP-1, KC and IP-10 were still significantly elevated compared to control mice. There was a general trend for IL-6 pretreatment to induce global upregulation of cytokines and chemokines at 120 min after Tc,max but this only reached statistical significance for IP-10 (Fig. 6L).
To reduce the effects of control group variance, a second analytical approach was used to look exclusively at the influence of IL-6 treatment on fold changes of expression from the average cytokine measurements in the corresponding untreated HS conditions (Fig. 7). Only specific cytokines that showed statistically significant treatment effects in Fig. 6 were included in this simpler analysis. Thirty minutes after Tc,max, IL-6, TNFα, KC, MCP-1 and RANTES were all significantly reduced by pretreatment with IL-6, while at 120 min after Tc,max no statistically significant effects of IL-6 treatment were observed for any cytokine (data not shown). All of these P values fell within an FDR of <0.15, meaning that it is highly unlikely that our conclusions are biased due to multiple sampling of cytokines (Benjamini ´ Hochberg, 1995).
Figure 7. Secondary analyses of cytokine responses to IL-6 treatment during HS.
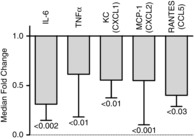
Values are medians–(bottom 25% quartile) calculated as a fold change of IL-6-treated HS over the mean value in untreated HS. P values are from a Wilcoxon test. The maximum FDR for all of these samples was < 0.15.
Discussion
Exogenous IL-6 administration, given prior to HS, resulted in a delay in the rise of core temperature during exposure to increasing environmental temperatures. This suggests that IL-6 is having either a direct or an indirect global impact on the physiology of heat exchange in severe hyperthermia. In addition, IL-6 prevented well-known outcomes of severe heat exposure; specifically, it decreased injury to the duodenum and reduced the loss of intestinal permeability (Hall et al. 2001; Oliver et al. 2012). The global cytokine response to hyperthermia was also significantly dampened following HS, an effect that influenced the expression of nearly every cytokine or chemokine that was responsive to heat. Overall, the data are consistent with the concept that the early emergence of circulating IL-6 during conditions of severe hyperthermia exposure may function as a stress response hormone that works via multiple mechanisms to reduce organ injury and elevate chances for survival.
Possible mechanisms of altered responses to environmental temperature
As mice in this study were anaesthetized, they would be unable to regulate temperature around a set point (Refinetti ´ Carlisle, 1988). Therefore, any effect of IL-6 treatment on the ability to resist elevations in core temperature is not likely to involve inhibition of the influence of pyrogens on set point control, particularly because IL-6 is itself known as a pyrogen (LeMay et al. 1990). It appears more likely that the effects of IL-6 involve support of some aspect of cardiovascular function, thus delaying the rapid rise in core temperature that occurs at the end stages of HS, where cardiovascular collapse results in an inability to dissipate heat (Hall et al. 2001). There are precedents for this effect on the cardiovascular system. For example, in severe haemorrhagic shock, IL-6 supplementation prevents circulatory collapse during reperfusion and reduces mortality 5-fold (Alten et al. 2008). Although the mechanisms are not entirely clear, the effect is STAT3-dependent. It is well known that IL-6, via JAK/STAT3 signalling, can have a potent preconditioning effect on the heart, protecting it from ischaemia-reperfusion injury (Dawn et al. 2004; Smart et al. 2006; Boengler et al. 2008). Secondly, attenuation of hyperthermia-induced cytokines such as TNFα could protect vascular control or cardiac function in HS (Meldrum, 1998). Thirdly, IL-6 plays a very important role in activating the acute phase response of the liver (Heinrich et al. 1990; Heath et al. 1993; Conley et al. 2005; Tiberio et al. 2006). Pretreatment of IL-6 could therefore provide an umbrella of protection for the cardiovascular system, via activation and release of proteins that reduce oxidant stress, inhibit protease activity, remove reactive metals from circulation and reduce vascular leakage, thus protecting multiple organ systems in life threatening illness (Heinrich et al. 1990; Nemeth et al. 2004; Conley et al. 2005).
Possible mechanisms for IL-6 protection of the intestine
There are two main mechanisms for intestinal injury in HS. Hyperthermia alone is a potent cause of gastrointestinal epithelial cell injury (Dokladny et al. 2006; Novosad et al. 2013), but whole body hyperthermia also leads to a reduction in splanchnic blood flow, resulting in intestinal ischaemia (Hall et al. 1999). In many acute settings, IL-6 has been shown to be protective of the intestinal epithelium in ischaemia. For example, IL-6 treatment inhibits intestinal epithelial apoptosis, causes cell proliferation, lengthening of the villi and promotion of intestinal cell recovery following ischaemia-reperfusion injury (Jin et al. 2008). In transplant studies, IL-6 infusions 2 h before transplant have also been shown to prevent intestinal ischaemia-reperfusion injury (Kimizuka et al. 2004). Similar protective roles on the liver have been identified following ischaemia-reperfusion or haemorrhage (Camargo et al. 1997; Arikan et al. 2006). In conditions of compromised intestinal barriers, IL-6 protects the colonic barrier by stimulation of keratin isoform expression, which contributes to cytoskeletal elements important in maintaining barrier integrity (Wang et al. 2007). IL-6 infusion in humans to ≈35 pg ml−1 also results in acute elevations in splanchnic blood flow (Lyngso et al. 2002), which might be a mechanism for IL-6-induced protection from the intestinal ischaemia associated with HS.
Damage to the villi along the intestinal lining initiates a programmed wound healing response that includes villus contraction, followed by sloughing off of the villus tip and proliferation and migration of new epithelium, a complex process called ‘restitution’ (Blikslager et al. 2007; Derikx et al. 2008). These events are well underway within 30 min of an ischaemic injury (Derikx et al. 2008). A loss of villus length was evident in this study 30 min after reaching Tc,max (Fig. 5B). Interestingly, although no significant evidence of injury was present in the IL-6-treated mice at this time, the average villus length was already shortened to the same extent as in the untreated mice, and remained shortened through 120 min of recovery (Fig. 5E). This may mean that IL-6 either did not prevent the physical contraction of the villus structure or that IL-6 pretreatment accelerated the process of restitution. However, the role of IL-6 in restitution of the villus structure after injury is not yet understood and there are reports that it does and does not play a significant role in this process (Jin et al. 2008; Grivennikov et al. 2009; Pickert et al. 2009) For example, it is known that STAT3 signalling, the most important downstream mediator of IL-6 receptor signalling, is important in the restitution and healing process (Jin et al. 2008), but the direct involvement of IL-6 versus other STAT3-activating cytokines such as IL-22 or IL-10 is still not clear. It may be that the most important influence of IL-6 is simply the inhibition of apoptosis, which is a relatively well-established influence of IL-6 signalling in the gastrointestinal tract and other organ systems (Jin et al. 2008; Grivennikov et al. 2009).
Finally, the reduction in intestinal permeability observed in IL-6 pretreated animals may have arisen from the effect of IL-6 on reducing the circulating levels of inflammatory cytokines such as TNFα (Fig. 6G). TNFα has a potent impact on permeability to large molecular weight molecules in the small intestine by opening the cytoskeletal components of the epithelial tight junctions (Turner, 2000; Marchiando et al. 2010). In fact, although TNFα actions are generally considered to relate to local inflammation in the mucosal lining, infusion of TNFα into the bloodstream is often used as a model of intestinal barrier dysfunction (Marchiando et al. 2010). The exact role of TNFα in heat stroke progression is not entirely known. Much of the TNFα signal may be dampened by a large upregulation of soluble receptors for TNFα in heat stroke (Bouchama et al. 2005a; Blaha et al. 2009), which probably play a protective role (Van Zee et al. 1992). Recent studies in TNF receptor II knockouts suggest that TNFα plays a complex role in thermoregulation and has indirect influences on the acute phase response (Leon et al. 2013)
Possible mechanisms for IL-6 reduction of inflammatory cytokines
The generalized reduction in circulating cytokine expression observed in the first 30 min of recovery may reflect the well-known anti-inflammatory actions of IL-6 on the expression of other inflammatory and anti-inflammatory cytokines (Tilg et al. 1994; Xing et al. 1998; Starkie et al. 2003; Steensberg et al. 2003). However, it is difficult to conceive that this would be an across-the-board effect, observed in essentially all cytokines within the set that were up-regulated by heat (see Fig. 6). A more likely scenario is that the response reflects secondary influence from protection of the intestinal barrier. That is, by preventing endotoxin or other pro-inflammatory mediators (Chang et al. 2012) from being released from the intestinal lumen, there would understandably be less overall stimulation of the innate immune response during HS via a reduction in the activation of toll like receptors (TLRs) on inflammatory cells and other tissues. However, an alternative mechanism could reflect the overall effect of IL-6 preconditioning on cell survival or the effect of the acute phase protein response on reduction of organ injury. Both of these effects of IL-6 would presumably reduce the net release of damage-associated molecular patterns, which can also activate cytokine production through TLR activation in HS (Kawai ´ Akira, 2010; Dehbi et al. 2012).
The effects of pretreatment with IL-6 appeared to influence soluble mediator expression in the early but not the later stages of recovery. At the 120 min window of recovery, the cytokines in general were unaffected by IL-6 pretreatment, and in some cases the effect showed a trend toward rebound of cytokine expression or possibly overexpression. Exactly what this means in terms of the processes of recovery from HS, ongoing injury and the mechanisms of tissue repair is unclear, but a critical feature of protection may involve the timing of early IL-6 signalling.
Critique of the experimental approach
Our use of anaesthetized animals rather than conscious animals for these studies was based on our need to perform gavage in the mice prior to study. Our early experience in conscious animals, submitted to hyperthermia conditions resulting in HS, made it clear that the responses of mice are very sensitive to psychological stress. Gavage in awake animals is very stressful and therefore we avoided gavaging until after the animals were anaesthetized. Secondly, conscious animals generally exhibit differing time courses of response to elevated Tenv (Leon et al. 2006) and this presents some problems with respect to interpretation of permeability measurements and the timing of expression of soluble cytokines and other mediators. By clamping the Tc profile to a set time, we had complete control of the hyperthermia stimulus and the timing of sample collection. The data presented here provide a basis to now test these effects in conscious animals, which are likely to have more variability in responses.
An interesting outcome of the experiment was that we never saw an elevation in circulating IL-6 after i.p. IL-6 infusion, at the time windows we studied. Based on pharmacokinetic studies of i.p. IL-6 injections, we had expected to elevate IL-6 approximately 200–300 pg (Peters et al. 1996). However, even in control mice, we could not measure any elevations of IL-6 at any time point. This means that our signal was probably very small and transient or that it was quickly bound to soluble receptors, which in our hands results in lowered sensitivity to antibodies for IL-6 used in multiplex technology. Another consideration is that in some cell types, IL-6 receptor activation has been shown to inhibit endogenous IL-6 transcription and secretion via a negative feedback loop involving suppressor for cytokine signalling-3 (SOCS-3) (Yan et al. 2010). Therefore, inhibition of endogenous IL-6 could have offset the effects of supplementation. However, our consistent results, across a variety of different measurements, ensure that we had biological activity of our IL-6 injections. Another possibility is that the IL-6 injected into the peritoneum could have triggered a different immune response by activating resident lymphocytes or macrophages in the peritoneum to produce mediators that subsequently suppressed cytokine signalling or induced protective pathways.
The source of the large elevation in endogenous IL-6 in HS is not known. Hyperthermia is a significant stimulus for IL-6 production and release from skeletal muscle (Welc et al. 2012), and hyperthermia further amplifies stimuli common in HS, such as responses to endotoxin and catecholamines (unpublished data). Because skeletal muscle makes up a very large proportion of body mass, it would seem likely that muscle has some role in contributing to plasma IL-6 levels in these conditions. In contrast, in inflammatory cells and fibroblasts hyperthermia suppresses IL-6 production from inflammatory mediators (Ostberg et al. 2000; Takii et al. 2010). However, many other types of parenchymal cells secrete IL-6 in inflammation, including adipose tissue (Mohamed-Ali et al. 2001), intestinal epithelium (Meyer et al. 1994) and cardiac muscle (Jeron et al. 2003), but how these are affected by hyperthermia is unknown. As most parenchymal tissues and inflammatory cells have the capacity to produce IL-6 under a variety of conditions common in HS, it is very difficult to be certain of the source of secretion in such a complex pathological setting.
We used an approach towards measuring intestinal permeability that does not take into consideration the loss of the FD4 into the urine or in peripheral vascular beds during the course of the experiment. In many cases on autopsy, using UV light, we could clearly see FD4 in the urine of the animals and sometimes beneath the skin. We also routinely looked for distribution of FD4 down the small intestine at autopsy, and by the end of the experiment it was always distributed well throughout the small intestine. Therefore, the FD4 in the blood at any time point is likely to reflect a balance of the rate at which the FD4 is entering the bloodstream from the intestine and the rate at which it is being eliminated into the urine or into other leaky vascular beds. This is a plausible explanation for how FD4 measurements could even go down with IL-6 treatment at the 120 min time point (Fig. 3). Despite these limitations, we felt that the clear presence of protection from morphological injury in the IL-6-treated animals verified that the approach reliably provided a measure of intestinal barrier function.
Glossary
Abbreviations
- FD4
4 kD FITC–dextran for permeability measurements
- FDR
false discovery rate due to multiple sampling
- G-CSF
granulocyte colony stimulating factor
- GM-CSF
granulocyte macrophage colony stimulating factor
- HS
heat stroke
- IFNγ
interferon-gamma
- IL-10
interleukin-10
- IL-12(p70)
interleukin-10-p70, the active heterodimer of IL-12
- IL-13
interleukin 13
- IL-15
interleukin 15
- IL-17
interleukin 17
- IL-2
interleukin-2
- IL-4
interleukin-4
- IL-5
interleukin-5
- IL-6
interleukin-6
- IL-7
interleukin-7
- IL-9
interleukin 9
- IP-10
interferon gamma inducing protein 10, same as CXCL10
- JAK/STAT3
an IL-6 receptor signalling complex containing ‘Janus kinase’ and ‘signal transducer and activator of transcription-3’
- KC
keratinocyte-derived cytokine, same as CXCL1
- MCP-1
monocyte chemoattractant protein-1, same as CCl2
- MIP-1α
macrophage inflammatory protein-1 alpha, same as CCL3
- RANTES
regulation on activation normal T cell expressed and secreted, same as CCL5
- Tc
core temperature
- Tc,max
maximum core temperature achieved during hyperthermia
- Tenv
environmental temperature
- TNFα
tumour necrosis factor alpha
Additional information
Competing interests
The authors have no conflict of interest for any portion of this study.
Author contributions
All experiments were performed in the Department of Applied Physiology and Kinesiology at the University of Florida. N.A.P. performed the majority of animal experiments and contributed to the design of the study and first draft of the manuscript. S.S.W. assisted on some of the analytical tests, contributed to the design of the study and was involved in interpretation and statistical analysis of data. He played a major role in editing the manuscript. S.M.W. performed the oversight and analyses of all cytokine measurements and contributed to writing and reviewing the manuscript. M.A.K. performed experimental procedures and analyses, provided image analysis and careful editing of the manuscript. T.L.C. designed the project, performed some animal experiments, statistical analysis and graphics, and wrote and edited the last drafts of the manuscript.
Funding
This work was supported by the American Heart Association GRNT7990119 (T.L.C.) and the BK and Betty Stevens Endowment from the University of Florida (T.L.C.). N.P. was the recipient of the 2012 American Physiological Society Military Physiology Award for this research.
Translational perspective
One implication of these experiments is that some form of low dose IL-6 supplement could have potential as a therapeutic approach for acute life-threatening conditions such as HS. The practical timing of such a supplement, however, would be difficult to perform clinically because one would have to know that conditions of HS or similar acute problems are imminent. It is possible that supplements later in the course of heat exposure or at higher doses would be equally or more effective. In experimental haemorrhagic shock, for example, therapeutic benefits have been studied when IL-6 injections are given intravenously, at the beginning of the resuscitation period (Alten et al. 2008). In passive HS, endogenous IL-6 remains at a low level immediately after Tc,max (Leon et al. 2006; Welc et al. 2013a), so intervention, with i.v. IL-6 at this time point may be of some benefit. Because we used i.p. injection, with considerable delays in protein absorption in the gut (Peters et al. 1996), it would have been difficult to time the IL-6 application accurately to specifically hit this target.
A deeper implication of these experimental results is that they provide a framework for greater clarity with respect to the consequences of early IL-6 stimulation and release in real life situations. For example, consider active military combatants or athletes exposed to hot and humid environments. They have repeated periods of activity and then inactivity, when core temperature and presumably IL-6 signalling are likely to go through cycles. For example, exercise alone (Steensberg et al. 2000) or hyperthermia alone (Welc et al. 2013b), in the absence of HS, are independent stimuli for elevating IL-6 in the circulation. It is possible that these early signalling events induce a kind of natural ‘preconditioning’ endocrine signal that results in changes in the physiology of heat exchange and an attenuation of the severe consequences of organ injury and vascular collapse that accompanies HS. As IL-6 signalling has inherent feedback loops that turn STAT3 signalling off over time (Kubo et al. 2003), the timing and sequence of events that naturally occur and how they influence the protective aspects of IL-6 may be a key determinant in understanding which athlete, which military combatant or child left in a hot car succumbs to HS and under what real life circumstances.
References
- Alten JA, Moran A, Tsimelzon AI, Mastrangelo MA, Hilsenbeck SG, Poli V. Tweardy DJ. Prevention of hypovolemic circulatory collapse by IL-6 activated Stat3. PLoS One. 2008;3:e1605. doi: 10.1371/journal.pone.0001605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaud L, Ferry T, Le QH, Marfisi A, Ciorba D, Achache P, Ducluzeau R. Robert D. Short- and long-term outcomes of heatstroke following the 2003 heat wave in Lyon, France. Arch Intern Med. 2007;167:2177–2183. doi: 10.1001/archinte.167.20.ioi70147. [DOI] [PubMed] [Google Scholar]
- Arikan AA, Yu B, Mastrangelo MAA. Tweardy DJ. Interleukin-6 treatment reverses apoptosis and blunts susceptibility to intraperitoneal bacterial challenge following hemorrhagic shock. Crit Care Med. 2006;34:771–777. doi: 10.1097/01.ccm.0000201901.30292.c2. [DOI] [PubMed] [Google Scholar]
- Barton BE. Jackson JV. Protective role of interleukin 6 in the lipopolysaccharide–galactosamine septic shock model. Infect Immun. 1993;61:1496–1499. doi: 10.1128/iai.61.4.1496-1499.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y. Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995;57:289–300. [Google Scholar]
- Blaha MD, Helwig BG. Leon LR. Plasma soluble cytokine receptor levels are elevated during heat stroke recovery in mice. FASEB J. 2009;23:1026. [Google Scholar]
- Blikslager AT, Moeser AJ, Gookin JL, Jones SL. Odle J. Restoration of barrier function in injured intestinal mucosa. Physiol Rev. 2007;87:545–564. doi: 10.1152/physrev.00012.2006. [DOI] [PubMed] [Google Scholar]
- Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G. Schulz R. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther. 2008;120:172–185. doi: 10.1016/j.pharmthera.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Bouchama A. Knochel JP. Heat stroke. N Engl J Med. 2002;346:1978–1988. doi: 10.1056/NEJMra011089. [DOI] [PubMed] [Google Scholar]
- Bouchama A, Ollivier V, Roberts G, Al Mohanna F, de Prost D, Eldali A, Saussereau E, El-Sayed R. Chollet-Martin S. Experimental heatstroke in baboon: analysis of the systemic inflammatory response. Shock. 2005a;24:332–335. doi: 10.1097/01.shk.0000180620.44435.9c. [DOI] [PubMed] [Google Scholar]
- Bouchama A, Roberts G, Al Mohanna F, El-Sayed R, Lach B, Chollet-Martin S, Ollivier V, Al Baradei R, Loualich A, Nakeeb S, Eldali A. de Prost D. Inflammatory, hemostatic, and clinical changes in a baboon experimental model for heatstroke. J Appl Physiol (1985) 2005b;98:697–705. doi: 10.1152/japplphysiol.00461.2004. [DOI] [PubMed] [Google Scholar]
- Camargo CA, Jr, Madden JF, Gao W, Selvan RS. Clavien PA. Interleukin-6 protects liver against warm ischemia/reperfusion injury and promotes hepatocyte proliferation in the rodent. Hepatology. 1997;26:1513–1520. doi: 10.1002/hep.510260619. [DOI] [PubMed] [Google Scholar]
- Chang M, Kistler EB. Schmid-Schonbein GW. Disruption of the mucosal barrier during gut ischemia allows entry of digestive enzymes into the intestinal wall. Shock. 2012;37:297–305. doi: 10.1097/SHK.0b013e318240b59b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CJ, McArdle AH, Brown R, Scott HJ. Gurd FN. Intestinal mucosal lesion in low-flow states. I. A morphological, hemodynamic, and metabolic reappraisal. Arch Surg. 1970;101:478–483. doi: 10.1001/archsurg.1970.01340280030009. [DOI] [PubMed] [Google Scholar]
- Conley L, Geurs TL. Levin LA. Transcriptional regulation of ceruloplasmin by an IL-6 response element pathway. Brain Res Mol Brain Res. 2005;139:235–241. doi: 10.1016/j.molbrainres.2005.05.027. [DOI] [PubMed] [Google Scholar]
- Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V. Taub R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274:1379–1383. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Mazzon E, Dugo L, Centorrino T, Ciccolo A, McDonald MC, de Sarro A, Caputi AP. Thiemermann C. Absence of endogenous interleukin-6 enhances the inflammatory response during acute pancreatitis induced by cerulein in mice. Cytokine. 2002;18:274–285. doi: 10.1006/cyto.2002.0883. [DOI] [PubMed] [Google Scholar]
- Davido A, Patzak A, Dart T, Sadier MP, Meraud P, Masmoudi R, Sembach N. Cao TH. Risk factors for heat related death during the August 2003 heat wave in Paris, France, in patients evaluated at the emergency department of the Hôpital Européen Georges Pompidou. Emerg Med J. 2006;23:515–518. doi: 10.1136/emj.2005.028290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawn B, Xuan YT, Guo Y, Rezazadeh A, Stein AB, Hunt G, Wu WJ, Tan W. Bolli R. IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc Res. 2004;64:61–71. doi: 10.1016/j.cardiores.2004.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehbi M, Uzzaman T, Baturcam E, Eldali A, Ventura W. Bouchama A. Toll-like receptor 4 and high-mobility group box 1 are critical mediators of tissue injury and survival in a mouse model for heatstroke. PLoS One. 2012;7:e44100. doi: 10.1371/journal.pone.0044100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dematte JE, O’Mara K, Buescher J, Whitney CG, Forsythe S, McNamee T, Adiga RB. Ndukwu IM. Near-fatal heat stroke during the 1995 heat wave in Chicago. Ann Intern Med. 1998;129:173–181. doi: 10.7326/0003-4819-129-3-199808010-00001. [DOI] [PubMed] [Google Scholar]
- Derikx JP, Matthijsen RA, de Bruine AP, van Bijnen AA, Heineman E, van Dam RM, Dejong CH. Buurman WA. Rapid reversal of human intestinal ischemia-reperfusion induced damage by shedding of injured enterocytes and reepithelialisation. PLoS One. 2008;3:e3428. doi: 10.1371/journal.pone.0003428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokladny K, Moseley PL. Ma TY. Physiologically relevant increase in temperature causes an increase in intestinal epithelial tight junction permeability. Am J Physiol Gastrointest Liver Physiol. 2006;290:G204–212. doi: 10.1152/ajpgi.00401.2005. [DOI] [PubMed] [Google Scholar]
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L. Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DM, Baumgardner KR, Oberley TD. Gisolfi CV. Splanchnic tissues undergo hypoxic stress during whole body hyperthermia. Am J Physiol. 1999;276:G1195–G1203. doi: 10.1152/ajpgi.1999.276.5.G1195. [DOI] [PubMed] [Google Scholar]
- Hall DM, Buettner GR, Oberley LW, Xu L, Matthes RD. Gisolfi CV. Mechanisms of circulatory and intestinal barrier dysfunction during whole body hyperthermia. Am J Physiol Heart Circ Physiol. 2001;280:H509–H521. doi: 10.1152/ajpheart.2001.280.2.H509. [DOI] [PubMed] [Google Scholar]
- Heath DI, Cruickshank A, Gudgeon M, Jehanli A, Shenkin A. Imrie CW. Role of interleukin-6 in mediating the acute phase protein response and potential as an early means of severity assessment in acute pancreatitis. Gut. 1993;34:41–45. doi: 10.1136/gut.34.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich PC, Castell JV. Andus T. Interleukin-6 and the acute phase response. Bio J. 1990;265:621–636. doi: 10.1042/bj2650621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard RW, Bowers WD, Matthew WT, Curtis FC, Criss RE, Sheldon GM. Ratteree JW. Rat model of acute heatstroke mortality. J Appl Physiol. 1977;42:809–816. doi: 10.1152/jappl.1977.42.6.809. [DOI] [PubMed] [Google Scholar]
- Jeron A, Kaiser T, Straub RH, Weil J, Riegger GA. Muders F. Myocardial IL-6 regulation by neurohormones–an in vitro superfusion study. Brain Behav Immun. 2003;17:245–250. doi: 10.1016/s0889-1591(03)00053-9. [DOI] [PubMed] [Google Scholar]
- Jin X, Zimmers TA, Zhang Z, Pierce RH. Koniaris LG. Interleukin-6 is an important in vivo inhibitor of intestinal epithelial cell death in mice. Gut. 2008;59:186–196. doi: 10.1136/gut.2008.151175. [DOI] [PubMed] [Google Scholar]
- Kawai T. Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Kimizuka K, Nakao A, Nalesnik MA, Demetris AJ, Uchiyama T, Ruppert K, Fink MP, Stolz DB. Murase N. Exogenous IL-6 inhibits acute inflammatory responses and prevents ischemia/reperfusion injury after intestinal transplantation. Am J Transplant. 2004;4:482–494. doi: 10.1111/j.1600-6143.2004.00368.x. [DOI] [PubMed] [Google Scholar]
- Kubo M, Hanada T. Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol. 2003;4:1169–1176. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]
- Lambert GP, Gisolfi CV, Berg DJ, Moseley PL, Oberley LW. Kregel KC. Hyperthermia-induced intestinal permeability and the role of oxidative and nitrosative stress. J Appl Physiol. 2002;92:1750–1761. doi: 10.1152/japplphysiol.00787.2001. [DOI] [PubMed] [Google Scholar]
- LeMay LG, Vander AJ. Kluger MJ. Role of interleukin 6 in fever in rats. Am J Physiol. 1990;258:R798–R803. doi: 10.1152/ajpregu.1990.258.3.R798. [DOI] [PubMed] [Google Scholar]
- Leon LR. Heat stroke and cytokines. Prog Brain Res. 2007;162:481–524. doi: 10.1016/S0079-6123(06)62024-4. [DOI] [PubMed] [Google Scholar]
- Leon LR, Blaha MD. DuBose DA. Time course of cytokine, corticosterone, and tissue injury responses in mice during heat strain recovery. J Appl Physiol. 2006;100:1400–1409. doi: 10.1152/japplphysiol.01040.2005. [DOI] [PubMed] [Google Scholar]
- Leon LR, Dineen S, Blaha MD, Rodriguez-Fernandez M. Clarke DC. Attenuated thermoregulatory, metabolic, and liver acute phase protein response to heat stroke in TNF receptor knockout mice. Am J Physiol Regul Integr Comp Physiol. 2013;305:R1421–R1432. doi: 10.1152/ajpregu.00127.2013. [DOI] [PubMed] [Google Scholar]
- Leon LR, DuBose DA. Mason CW. Heat stress induces a biphasic thermoregulatory response in mice. Am J Physiol Regul Integr Comp Physiol. 2005;288:R197–R204. doi: 10.1152/ajpregu.00046.2004. [DOI] [PubMed] [Google Scholar]
- Leon LR, White AA. Kluger MJ. Role of IL-6 and TNF in thermoregulation and survival during sepsis in mice. Am J Physiol. 1998;275:R269–R277. doi: 10.1152/ajpregu.1998.275.1.R269. [DOI] [PubMed] [Google Scholar]
- Lyngso D, Simonsen L. Bulow J. Metabolic effects of interleukin-6 in human splanchnic and adipose tissue. J Physiol. 2002;543:379–386. doi: 10.1113/jphysiol.2002.021022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR, 2nd, Raleigh DR, Guan Y, Watson AJ, Montrose MH. Turner JR. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. 2010;189:111–126. doi: 10.1083/jcb.200902153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, O’Malley K, Efron PA, Burger C, McAuliffe PF, Scumpia PO, Uchida T, Tschoeke SK, Fujita S, Moldawer LL, Hemming AW. Foley DP. Interleukin-6 and STAT3 protect the liver from hepatic ischemia and reperfusion injury during ischemic preconditioning. Surgery. 2006;140:793–802. doi: 10.1016/j.surg.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Meldrum DR. Tumor necrosis factor in the heart. Am J Physiol. 1998;274:R577–R595. doi: 10.1152/ajpregu.1998.274.3.R577. [DOI] [PubMed] [Google Scholar]
- Meyer TA, Noguchi Y, Ogle CK, Tiao G, Wang JJ, Fischer JE. Hasselgren PO. Endotoxin stimulates interleukin-6 production in intestinal epithelial cells. A synergistic effect with prostaglandin E2. Arch Surg. 1994;129:1290–1294. doi: 10.1001/archsurg.1994.01420360080010. discussion 1294–1295. [DOI] [PubMed] [Google Scholar]
- Mohamed-Ali V, Flower L, Sethi J, Hotamisligil G, Gray R, Humphries SE, York DA. Pinkney J. β-Adrenergic regulation of IL-6 release from adipose tissue: in vivo and in vitro studies. J Clin Endocrinol Metab. 2001;86:5864–5869. doi: 10.1210/jcem.86.12.8104. [DOI] [PubMed] [Google Scholar]
- Moran A, Akcan Arikan A, Mastrangelo MA, Wu Y, Yu B, Poli V. Tweardy DJ. Prevention of trauma and hemorrhagic shock-mediated liver apoptosis by activation of Stat3α. Int J Clin Exp Med. 2008;1:213–247. [PMC free article] [PubMed] [Google Scholar]
- Moran A, Tsimelzon AI, Mastrangelo MA, Wu Y, Yu B, Hilsenbeck SG, Poli V. Tweardy DJ. Prevention of trauma/hemorrhagic shock-induced lung apoptosis by IL-6-mediated activation of Stat3. Clin Transl Sci. 2009;2:41–49. doi: 10.1111/j.1752-8062.2008.00076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK. Ganz T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–1276. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novosad VL, Richards JL, Phillips NA, King MA. Clanton TL. Regional susceptibility to stress-induced intestinal injury in the mouse. Am J Physiol Gastrointest Liver Physiol. 2013;305:G418–G426. doi: 10.1152/ajpgi.00166.2013. [DOI] [PubMed] [Google Scholar]
- Oliver SR, Phillips NA, Novosad VL, Bakos MP, Talbert EE. Clanton TL. Hyperthermia induces injury to the intestinal mucosa in the mouse: evidence for an oxidative stress mechanism. Am J Physiol Regul Integr Comp Physiol. 2012;302:R845–R853. doi: 10.1152/ajpregu.00595.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostberg JR, Taylor SL, Baumann H. Repasky EA. Regulatory effects of fever-range whole-body hyperthermia on the LPS-induced acute inflammatory response. J Leukoc Biol. 2000;68:815–820. [PubMed] [Google Scholar]
- Peters M, Jacobs S, Ehlers M, Vollmer P, Mullberg J, Wolf E, Brem G, Meyer zum Buschenfelde KH. Rose-John S. The function of the soluble interleukin 6 (IL-6) receptor in vivo: sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongat-ion of the plasma half-life of IL-6. J Exp Med. 1996;183:1399–1406. doi: 10.1084/jem.183.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S, Ouyang W, Neurath MF. Becker C. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refinetti R. Carlisle HJ. Thermoregulation during pentobarbital and ketamine anesthesia in rats. J Physiol (Paris) 1988;83:300–303. [PubMed] [Google Scholar]
- Robins HI, Kutz M, Wiedemann GJ, Katschinski DM, Paul D, Grosen E, Tiggelaar CL, Spriggs D, Gillis W. d’Oleire F. Cytokine induction by 41.8 degrees C whole body hyperthermia. Cancer Lett. 1995;97:195–201. doi: 10.1016/0304-3835(95)03976-4. [DOI] [PubMed] [Google Scholar]
- Sakata H, Narasimhan P, Niizuma K, Maier CM, Wakai T. Chan PH. Interleukin 6-preconditioned neural stem cells reduce ischaemic injury in stroke mice. Brain. 2012;135:3298–3310. doi: 10.1093/brain/aws259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart N, Mojet MH, Latchman DS, Marber MS, Duchen MR. Heads RJ. IL-6 induces PI 3-kinase and nitric oxide-dependent protection and preserves mitochondrial function in cardiomyocytes. Cardiovasc Res. 2006;69:164–177. doi: 10.1016/j.cardiores.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Starkie R, Ostrowski SR, Jauffred S, Febbraio M. Pedersen BK. Exercise and IL-6 infusion inhibit endotoxin-induced TNF-α production in humans. FASEB J. 2003;17:884–886. doi: 10.1096/fj.02-0670fje. [DOI] [PubMed] [Google Scholar]
- Steensberg A, Fischer CP, Keller C, Moller K. Pedersen BK. IL-6 enhances plasma IL-1ra, IL-10, and cortisol in humans. Am J Physiol Endocrinol Metab. 2003;285:E433–E437. doi: 10.1152/ajpendo.00074.2003. [DOI] [PubMed] [Google Scholar]
- Steensberg A, van Hall G, Osada T, Sacchetti M, Saltin B. Klarlund Pedersen B. Production of interleukin-6 in contracting human skeletal muscles can account for the exercise-induced increase in plasma interleukin-6. J Physiol. 2000;529:237–242. doi: 10.1111/j.1469-7793.2000.00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takii R, Inouye S, Fujimoto M, Nakamura T, Shinkawa T, Prakasam R, Tan K, Hayashida N, Ichikawa H, Hai T. Nakai A. Heat shock transcription factor 1 inhibits expression of IL-6 through activating transcription factor 3. J Immunol. 2010;184:1041–1048. doi: 10.4049/jimmunol.0902579. [DOI] [PubMed] [Google Scholar]
- Tiberio L, Tiberio GA, Bardella L, Cervi E, Cerea K, Dreano M, Garotta G, Fra A, Montani N, Ferrari-Bravo A, Callea F, Grigolato P, Giulini SM. Schiaffonati L. Mechanisms of interleukin-6 protection against ischemia-reperfusion injury in rat liver. Cytokine. 2006;34:131–142. doi: 10.1016/j.cyto.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Tilg H, Trehu E, Atkins MB, Dinarello CA. Mier JW. Interleukin-6 (IL-6) as an anti-inflammatory cytokine: induction of circulating IL-1 receptor antagonist and soluble tumor necrosis factor receptor p55. Blood. 1994;83:113–118. [PubMed] [Google Scholar]
- Turner JR( ‘Putting the squeeze’ on the tight junction: understanding cytoskeletal regulation. Semin Cell Dev Biol. 2000;11:301–308. doi: 10.1006/scdb.2000.0180. [DOI] [PubMed] [Google Scholar]
- van Zee KJ, Kohno T, Fischer E, Rock CS, Moldawer LL. Lowry SF. Tumor necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumor necrosis factor α in vitro and in vivo. Proc Nat Acad Sci U S A. 1992;89:4845–4849. doi: 10.1073/pnas.89.11.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Fang CH. Hasselgren PO. Intestinal permeability is reduced and IL-10 levels are increased in septic IL-6 knockout mice. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1013–R1023. doi: 10.1152/ajpregu.2001.281.3.R1013. [DOI] [PubMed] [Google Scholar]
- Wang L, Srinivasan S, Theiss AL, Merlin D. Sitaraman SV. Interleukin-6 induces keratin expression in intestinal epithelial cells: potential role of dertin-8 in interleukin-6-induced barrier function alterations. J Biol Chem. 2007;282:8219–8277. doi: 10.1074/jbc.M604068200. [DOI] [PubMed] [Google Scholar]
- Welc SS, Clanton TL, Dineen SM. Leon LR. Heat stroke activates a stress-induced cytokine response in skeletal muscle. J Appl Physiol (1985) 2013a;115:1126–1137. doi: 10.1152/japplphysiol.00636.2013. [DOI] [PubMed] [Google Scholar]
- Welc SS, Judge AR. Clanton TL. Skeletal muscle interleukin-6 regulation in hyperthermia. Am J Physiol Cell Physiol. 2013b;305:C406–C413. doi: 10.1152/ajpcell.00084.2013. [DOI] [PubMed] [Google Scholar]
- Welc SS, Oca-Cossio J, Phillips NA, Wallet SM. Clanton TL. Hyperthermia increases interleukin-6 in mouse skeletal muscle. Am J Physiol Cell Physiol. 2012;303:C455–C466. doi: 10.1152/ajpcell.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF. Achong MK. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101:311–320. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Cao J, Wu M, Zhang W, Jiang T, Yoshimura A. Gao H. Suppressor of cytokine signaling 3 inhibits LPS-induced IL-6 expression in osteoblasts by suppressing CCAAT/enhancer-binding protein β activity. J Biolog Chem. 2010;285:37227–37239. doi: 10.1074/jbc.M110.132084. [DOI] [PMC free article] [PubMed] [Google Scholar]