

Abstract
Human cytomegalovirus (CMV) is a latent and persistent virus whose proliferation increases morbidity and mortality of immune-compromised individuals. The current anti-CMV therapeutics targeting the viral DNA polymerase or the major immediate-early (MIE) gene locus are somewhat effective at limiting CMV-associated disease. However, due to low bioavailability, severe toxicity, and the development of drug resistant CMV strains following prolonged treatment, current anti-CMV therapeutics are insufficient. To help address this shortfall, we established a high-content assay to identify inhibitors targeting CMV entry and the early steps of infection. The infection of primary human fibroblasts with a variant of the CMV laboratory strain AD169 expressing a chimeric IE2-yellow fluorescence protein (YFP) (AD169IE2-YFP) provided the basis for the high-content assay. The localization of IE2-YFP to the nucleus shortly following an AD169IE2-YFP infection induced a robust fluorescent signal that was quantified using confocal microscopy. The assay was optimized to achieve outstanding assay fitness and high Z′ scores. We then screened a bioactive chemical library consisting of 2080 compounds and identified hit compounds based on the decrease of fluorescence signal from IE2-YFP nuclear expression. The hit compounds likely target various cellular processes involved in the early steps of infection including capsid transport, chromatin remodeling, and viral gene expression. Extensive secondary assays confirmed the ability of a hit compound, convallatoxin, to inhibit infection of both laboratory and clinical CMV strains and limit virus proliferation. Collectively, the data demonstrate that we have established a robust high-content screen to identify compounds that limit the early steps of the CMV life cycle, and that novel inhibitors of early infection events may serve as viable CMV therapeutics.
Keywords: Human cytomegalovirus, high throughput, therapeutics, small-molecule screen, antivirals, cardiac glycoside
Introduction
Human Cytomegalovirus (CMV) is the prototypic member of the Betaherpesvirinae subfamily. The coevolution of CMV with humans over the past millions of years has led to the development of complex and elegant methods of immune evasion that have allowed the virus to infect a majority of the human population. Indeed, CMV causes latent and persistent infections in 60-90% of individuals (Mocarski ES, 2007).
While most CMV carriers are asymptomatic, virus proliferation significantly increases morbidity and/or mortality in immunocompromised individuals. This includes newborns, organ transplant recipients, AIDS patients, and the elderly. CMV is clearly a major medical problem affecting a significant number of individuals. To date, very few drugs have been developed for either prophylactic, pre-emptive, or direct treatment of CMV disease (ganciclovir, foscarnet, cidofovir, fomivirsen, and high-dose acyclovir) (Mercorelli et al., 2011). Although these drugs have some efficacy against CMV infection (Steininger, 2007), toxicity, drug-drug interactions, and drug resistance are common limitations (Chou et al., 2008). With the exception of fomivirsen, all approved anti-CMV therapeutic compounds target the viral DNA polymerase (Razonable, 2011) leading to high toxicity due to off–target effects on the host DNA polymerase. Therefore, more potent and diverse anti-CMV therapies are clearly warranted.
The early stage of a CMV infection includes virus attachment and entry followed promptly by expression of the immediate early gene products. The Major Immediate Early transcript (MIE) is generated shortly following infection from spliced products creating the IE1 and IE2 gene products. IE2 is a transactivator for early viral proteins (Cherrington and Mocarski, 1989; Meier and Stinski, 1997) that can complex with host factors of the cellular transcription machinery. IE1 stimulates a variety of viral and cellular promoters through its ability to interact with multiple transcriptional regulators, disrupt repressive nuclear architecture, and work synergistically with IE2 to activate viral promoters (Meier and Stinski, 1997). These are essential viral gene products whose expression is a critical first step for transcription of the CMV genome. Therefore, assessment of IE gene expression can serve as a rapid proxy for the measurement of CMV infection. We and others have utilized CMV virus strains encoding fluorescent chimeric proteins to measure IE expression in infected cells (Gardner et al., 2013; Straschewski et al., 2010).
In this study, we employed a CMV strain that expresses a yellow fluorescent protein (YFP), fused to the IE2 transcript (AD169IE2-YFP), to develop a sensitive and specific assay to identify inhibitors of virus infection. The assay, which is cell-based and multiparametric, represents a high-content approach to identify antiviral compounds (Gasparri, 2009). Based on fluorescent protein expression in CMV-infected fibroblasts, we identified and validated a panel of small molecule compounds that block diverse, early events of CMV infection in vitro. Secondary assays confirmed the inhibition of viral gene expression when cells were treated with the hit compounds. Additionally we show that our lead compound, the cardiac glycoside convallatoxin, was capable of limiting CMV proliferation and production of infectious virus. Together, we present a rapid, high-content platform for identifying agents that target the early events of a CMV infection.
Materials and Methods
Cell lines, antibodies, viruses and chemicals
Human lung fibroblasts (MRC5) (ATCC, Manassas, VA) were cultured in complete Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 U of penicillin-streptomycin/ml and 100 U of HEPES/ml at 37°C in a humidified incubator with 5% CO2. CMV AD169IE2-YFP and TB40/EFLAG-YFP were propagated as previously described (Gardner et al., 2013). Infectious virus yield was assayed on human fibroblasts by median tissue culture infective dose (TCID50). The TCID50 results were used to estimate infectious particles/ml to infect cells at the desired multiplicity of infection (MOI). Cycloheximide was purchased from MP Biomedicals, Solon, Ohio (Cat. No. 100183).
High-content screen
MRC5 cells were seeded and treated the following day for one hour with controls and samples. Virus was added to the cells and adsorbed for 1 hour at 37°C, spin-infected at 1200 rpm for one hour at room temperature and returned to 37°C incubator. At 16 hours post infection (hpi), cells were fixed with 4% Paraformaldehyde (PFA) for 30 minutes at 4°C. Cells were stained with 25μg/mL Hoechst in phosphate buffered saline (PBS) with 1% bovine serum albumin (BSA) and 0.1% saponin for one hour at RT. Cells were then washed twice with PBS and images were collected using Channel 1 (excitation at 405 nm; emission detection at 442-447 nm) and Channel 2 (excitation at 488 nm; emission detection at 502-548 nm) to detect DAPI and YFP respectively on a Molecular Devices ImageXpress Ultra (IXU) plate-scanning confocal microscope. Positive cells were identified based on the following criteria: primary stain (nuclei) between 6-21μm and at least 4500 gray levels above local background and secondary stain (IE2-YFP) was only present in the nucleus, between 6-21μm and at least 1000 gray levels above background. MetaXpress software (Molecular Devices, Sunnyvale, CA) was used to calculate the fluorescent intensity (W2 stained average intensity/well). These values were utilized to determine Robust Z-scores (Zhang et al., 1999). The median fluorescent intensity value was calculated for each plate (excluding controls), and used to determine the % fluorescent intensity of each well.
Optimal cell plating density, positive control concentration and MOI were determined by pilot assays testing the following ranges for each parameter: 500-3000 cells/well, cycloheximide concentrations of 0.5-50μg/ul and MOI 0.5-15. The Z′ factor as described in (Zhang et al., 1999) was used to evaluate assay fitness using the equation Z′ = 1-[(3σc+ + 3σc-)/(|μc+ - μc-|)]. The ideal conditions were determined to be 3000 cells/well at an MOI of 0.5, with a positive control (cycloheximide) concentration of 9.8μM (10mg/mL) (samples measured in quadruplicate).
The bioactive chemical library at the Mount Sinai Integrated Screening Core was purchased from Microsource Discovery (Gaylordsville, CT) and consisted of 2,080 total compounds. Aurora 384 black well plates (Brooks Automation, Chelmsford, MA) were seeded with 3,000 cells/well and maintained at 37°C, 5% CO2 overnight. The following day the respective chemical compound source plate (final 5.6μM) and controls (cycloheximide, final 9.8μM) were transferred by pin tool (V & P Scientific) into previously-seeded plates (in duplicate) and incubated for one hour at 37°C. Cells were then mock-infected or infected with AD169IE2-YFP at an MOI of 0.5 for one hour at 37°C and then spin-infected at 1200 rpm for one hour at RT. Plates were then returned to the incubator and fixed and stained using Hoechst reagent (25μg/ml) at 16 hours post infection. Each library source plate was screened in duplicate. Images of four fields/well were acquired by the IXU plate-scanning confocal microscope. Image segmentation and analysis was carried out using MetaXpress.
Determining half maximal effective concentration (EC50) of hit compounds
MRC5 cells (10,000 in 100μL) were plated in each well of a 96-well plate (Greiner, Monroe, NC). The following day, media was replaced with DMEM with varying concentrations (0-5μM) of the compounds (final 100μL) in sextuplicate (1hr @ 37°C) and then cells were infected with AD169IE2-YFP (MOI:3). Following a one-hour inoculation cells were washed with media and then fresh media without drug was added back to the wells. At 18 hpi, the plates were analyzed with an Acumen eX3 cytometer for the number of IE2-YFP positive cells/well based on IE2-YFP fluorescent intensity/well (Gardner et al., 2013). Fluorescent emission above 2 standard deviations of background was registered as positive signal. Any fluorescent signal larger than 5μm, smaller than 300μm, and separated from any other emission by at least 0.5μm in both x and y axes was classified as an “event”. Using DMSO treated cells (0.1%) infected with AD169IE2-YFP as 100% infection, the percent infection of cells pre-treated with increasing amounts of compound was determined. For TB40/EFLAG-YFP infections, ARPE-19 cells were plated (10,000 in 100uL) as above. The following day, media was replaced with DMEM with varying concentrations (0-10nM) of convallatoxin (final 100μL) in sextuplicate and then cells were infected with TB40/EFLAG-YFP. At 120hpi, plates were analyzed with an Acumen eX3 cytometer as above. The EC50 values were calculated using Prism 5′s nonlinear fit log (inhibitor) vs. response (three parameters) analysis of the average % infection values.
Determining half maximal toxicity concentration (CC50)
MRC5 or ARPE-19 cells (10,000 in 100μL) were plated in each well of a 96-well plate. The following day, media was replaced with DMEM with varying concentrations (0-5μM) of the compounds (final 100μL) in sextuplicate. After 19 hours, NucGreen dead 488 (NucGreen) (Life Technologies, Grand Island, NY) was directly added to three out of six wells per condition (10 min at RT). (NucGreen) is a dye that is impermeable to living cells but not dead cells, that emits a fluorescent signal when bound to DNA. The number of NucGreen-positive cells was determined by measuring the fluorescent intensity/well using a fluorescent plate reader. To determine total number of cells/well, cells were fixed with 4% paraformaldehyde in PBS for up to 2 hrs, replaced with PBS (100μL) and NucGreen was added to the wells for 10 min. The number of NucGreen-positive cells were determined by measuring the fluorescent intensity detected by the plate reader. The cell toxicity of the compound-treated cells was calculated into percent cell viability using total number of NucGreen-positive cells as 100% compared to the number of NucGreen-positive cells from compound-treated cells. The CC50 values were calculated using Prism5's nonlinear fit log(agonist) vs. response (three parameters) analysis of these averages and extrapolation to the concentration that would produce cell toxicity of 50% above media alone.
Analysis of viral protein expression in infected cells
MRC5 cells treated with DMSO only (0.1%), podofilox (5-50nM), convallatoxin (5-50nM), or colchicine (10-500nM) (1hr at 37 °C) were infected with CMV AD169 (MOI:3) and washed 1× with DMEM. Total cell lysates from uninfected and infected cells 20 hpi were subjected to SDS-PAGE followed by immunoblot analysis using anti-CMV US11 antibody (Baker and Tortorella, 2007) and an anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Chemicon, Billerica, MA). Representative data is included from three independent experiments.
Multi-cycle Infection Studies
MRC5 cells (10,000 in 100μL) were plated in each well of two 96-well plates (Greiner, Cat No. 655180). In one plate the following day, the media was replaced with DMEM containing convallatoxin (0, 5, 10, or 50nM) in sextuplicate followed immediately by either infection with AD169IE2-YFP (MOI: 0.5) or addition of media alone. On the other plate, cells were either infected with AD169IE2-YFP (MOI:0.5) or mock infected for 36 hours before replacement of media with convallatoxin (0, 5, 10, or 50nM) in sextuplicate. The plates were analyzed for up to 168 hpi for IE2-YFP positive cells or NucGreen-positive cells using a fluorescent plate reader. The percent cell viability of compound-treated cells was determined as described above.
Plaque reduction assay
MRC5 cells were seeded in duplicate with DMEM at a density of 5×104 cells/well in a 24-well plate (BD-Falcon, Franklin Lakes, NJ). The next day cells were treated with 0.1% DMSO, 12μM ganciclovir (APP pharmaceuticals, Schaumberg, IL), or 5nM convallatoxin for one hour prior to infection with AD169 WT (MOI 0.002) (1hr at 37 °C). Following infection, cells were washed 2× with DMEM and wells were refilled with 3% DMEM containing DMSO, ganciclovir or convallatoxin and supplemented with 10μg/mL Cytogam (CSL Behring, King of Prussia, PA). At 5dpi, cells were fixed in 4% PFA (20 minutes at 4 °C), and stained with Giemsa (Harleco, EMD Millipore) (1hr at 37 °C). Wells were washed with dH20 5× and plaques were blind counted using phase contrast microscopy.
Analysis of fluorescence expression and infectious virus production in a multi-cycle replication
MRC5 cells (10,000 in 100uL) were plated in a 96-well plate. The following day cells were pretreated for 1 hour with DMEM containing DMSO (0.1%), ganciclovir (12μM), or convallatoxin (50nM). Following drug pretreatment, cells were infected with AD169IE2-YFP (MOI:0.5). (1hr at 37°C). At 24hpi, wells were aspirated and washed 1× with DMEM. Wells were then filled with 100uL of fresh DMEM without any compound. Beginning at 24hpi, cells were analyzed in 48-hour intervals for YFP+ cells using the Acumen eX3 cytometer. Immediately following fluorescence readings, cell supernatant was removed and snap frozen. At the conclusion of the 9-day timecourse, MRC5 cells were seeded in DMEM at a density of 5×104 cells/well in 24-well plates. The following day, serial dilution of supernatants were adsorbed onto fibroblasts (1 hr at 37°C). Cells were washed 2× with DMEM and then replaced with 3% DMEM supplemented with 10μg/mL of Cytogam. The infected cells were then cultured for 10 days at which time they were fixed with 4% PFA. Plaques were stained with Giemsa and counted using phase contrast microscopy.
Results
Characterization of AD169IE2-YFP -infected fibroblasts
The CMV immediate early (IE) gene products IE1 and IE2 direct expression of downstream lytic genes and perform key functions in producing infectious virions within infected cells (Meier and Stinski, 1997). Thus, expression of the immediate-early gene IE2 in the nucleus was utilized as the readout to quantify CMV infection. A CMV strain of AD169 engineered to express a viral chimeric protein consisting of the IE2 protein product and yellow fluorescent protein (YFP) (AD169IE2-YFP) was employed to visualize and quantify virus-infected nuclei. Previous studies indicated that the AD169IE2-YFP demonstrated similar growth kinetics to the parental AD169 strain (Gardner et al., 2013). Our initial experiment examined mock-infected and AD169IE2-YFP-infected human lung fibroblasts (MRC5) (multiplicity of infection (MOI): 3). We examined protein expression of immediate early genes by immunoblot (Figure 1). Consistent with published data (Gardner et al., 2013; Teng et al., 2012), both IE1 and IE2-YFP proteins were detected as early as 6 hours post-infection (hpi) and through 24hpi (Figure 1a, lanes 2-4 and 6-8). Mock-infected cells (Figure 1A, lanes, 1 and 5) validated the specific detection of viral gene products and the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) immunoblot confirmed equivalent protein loading (Figure 1A, lanes 9-12). To confirm that the IE2-YFP was a fluorescent species, AD169IE2-YFP-infected MRC5 cells were analyzed by fluorescence microscopy at 24hpi to ensure accumulation of the chimeric protein. Analysis of mock- and virus-infected cells demonstrated an exclusive fluorescence signal localized to the nucleus of ∼50% of virus-infected cells (Figure 1B), a result consistent with previous published results (DeMeritt et al., 2006; Straschewski et al., 2010). Together, the data confirmed the expression of IE2-YFP in AD169IE2-YFP-infected cells as fluorescent species.
Figure 1. AD169IE2-YFP-infected cells express IE2-YFP as an immediate-early protein.

(A) Mock (lanes 1, 5, and 9) and AD169IE2-YFP-infected (lanes 2-4, 6-8, 10-12) (MOI:5) MRC5 cells were harvested up to 24 hpi and subjected to immunoblot analysis for CMV IE2 (lanes 1-4), CMV IE1 (lanes 5-8), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (lanes 9-12) polypeptides. The respective polypeptides and relative molecular mass markers are indicated. (B) Mock- and AD169IE2-YFP-infected MRC5 cells (MOI:3) were stained with Hoechst reagent (blue) and analyzed by fluorescent microscopy (×20) at 24hpi. Both the Hoechst reagent and IE2-YFP (green) are localized to the nucleus and the overlay highlights the virus-infected cells.
AD169IE2-YFP-based high-content screen
We next established AD169IE2-YFP-infection conditions to be used in screening for inhibitors to the early steps of CMV infection. The initial experiment examined the fluorescent intensity and assay fitness of virus-infected cells with varying cell number and multiplicity of infection (Figure 2). A previous study determined that 16hpi of AD169IE2-YFP-infected cells was the optimal time to quantify a virus infection (Teng et al., 2012) and was used for the current study. Varying numbers of MRC5 cells (500, 1500, and 3000 cells/well in a 384-well plate) infected with a range of MOIs (2, 5, 7.5, 10, and 15) were subjected to analysis by confocal microscopy for stained average intensity (Figure 2A). Spinoculation was used at all MOIs in order to enhance infection levels. The fluorescence intensity of the virus-infected cells increased with cell number and MOI. The largest increase in fluorescence intensity was observed when 3,000 cells/well were infected with increasing MOIs (Figure 2A, gray bars). In contrast, the fluorescence intensity from cells plated at 500 cells/well only slightly increased at higher MOIs indicating that the infection conditions reached saturation (Figure 2A, white bars). The fluorescence intensity values from non-infected cells (as background values) were utilized to calculate assay fitness by the Z′ factor (Zhang et al., 1999) (Figure 2B). All of the infection conditions yielded Z′ factor values >0.5 indicating a robust assay.
Figure 2. Optimized conditions for an AD169IE2-YFP-based high-content screen.
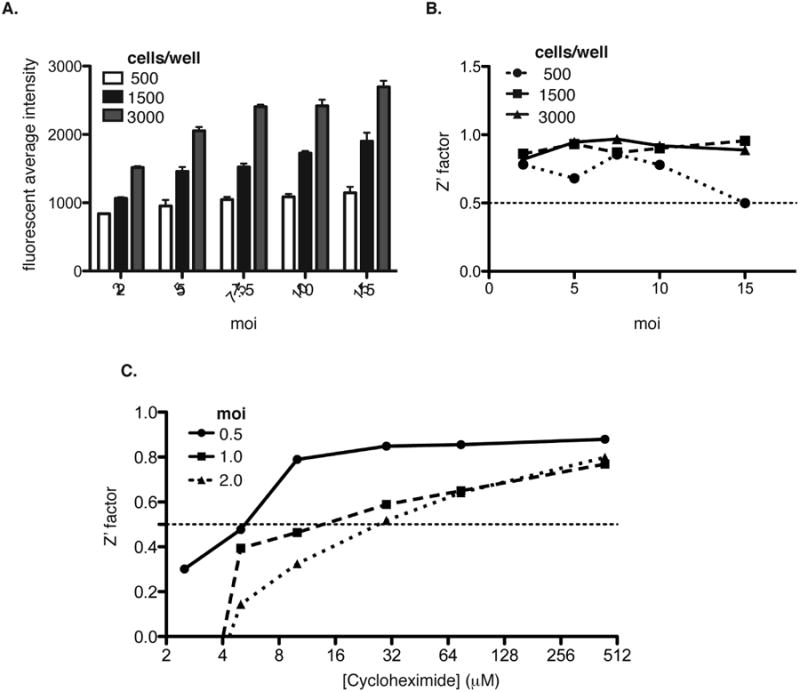
The fluorescent average intensity (A) was determined from 500 (white bars), 1500 (black bars), and 3000 (gray bars) MRC5 cells/well infected with AD169IE2-YFP at MOIs 2, 5, 7.5, 10 and 15. Error bars represent standard error of the mean of samples measured in quadruplicate. (B) The fluorescent intensity values were used to calculate the Z′ factor values for infection conditions of (A). (C) Z′ factor values were calculated using the fluorescence intensity values from MRC5 cells infected with AD169IE2-YFP at MOIs 0.5, 1.0, and 2.0 and treated with various concentrations (0-512 μM) of cycloheximide.
To further optimize conditions for the high-content screen, we examined the concentration of the translation inhibitor cycloheximide as a positive control to block IE2-YFP expression. MRC5 cells (3000 cells/well) infected with AD169IE2-YFP (MOI: 0.5, 1, or 2) were exposed to varying concentrations of cycloheximide (0-512 μM) one hour prior to infection (Figure 2C). The fluorescent intensity from the virus-infected cells and cycloheximide-treated cells were utilized to calculate the Z′ factor for all of the conditions. Strikingly, an MOI of 0.5 provided an excellent Z′ factor (∼0.8) at lower concentrations of cycloheximide (8-64 μM). In contrast, MOIs of 1 and 2 reached Z′ factor values >0.5 at cycloheximide concentrations >32μM. The results suggest that cycloheximide treatment can effectively block the expression of IE2-YFP in AD169IE2-YFP-infected cells. Thus, an MOI of 0.5 with cycloheximide >10μM as a control provide excellent conditions to identify anti-CMV probes.
High-content screen for identifying inhibitors of a CMV infection
Using optimized infection and analysis conditions for AD169IE2-YFP-infected MRC5 cells, a bioactive chemical library (2080 compounds) was screened in a high-content assay to identify inhibitors that block a CMV infection (Figure 3A and 3B). The Robust Z score was calculated for each compound and plotted based on chemical ID number (Chung et al., 2008). By applying the 3-sigma rule, we identified eighteen compounds that reduced YFP fluorescent intensity beyond the normal distribution (Figure 3A). To ensure that the putative hits were accurate, we determined YFP intensity as a distinct parameter to determine its value as an appropriate readout. The YFP mean fluorescent intensity (MFI)/well was calculated as percent YFP fluorescence intensity using the median YFP MFI from each plate (excluding controls) as 100% and plotted based on chemical ID number (Figure 3B). The collective data from the bioactive collection revealed high parity in relative fluorescence levels between all plates, and that >98% of the compounds did not decrease the levels of YFP MFI. Of the initial eighteen compounds, twelve decreased YFP intensity >70%, which was chosen as a stringent cut-off in order to identify high-quality hit compounds to examine in secondary assays. This level corresponded to a hit rate of 0.58% (Figures 3B and Table 1). Given the timing of the assay, it was expected that compounds that reduce YFP expression will target an early step of the viral infectious cycle by interfering with attachment, fusion, intracellular trafficking of the capsid to the nucleus, transcription, or translation.
Figure 3. High-content screen for identifying CMV inhibitors.
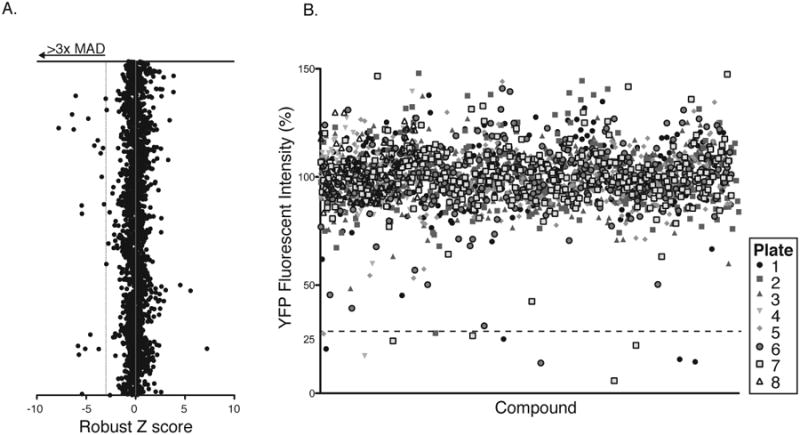
(A) The Robust Z-score was calculated from the fluorescence intensity MRC5 cells infected with AD169IE2-YFP-infected fibroblasts pretreated for each compound (X axis) and plotted against the chemical ID number (Y axis). Eighteen compounds reduced YFP expression >3 median absolute deviations beyond the mean score (dotted line). (B) Average YFP mean fluorescent intensity from MRC5 cells infected with AD169IE2-YFP-infected fibroblasts pretreated for each compound (2 replicates) was calculated into %YFP fluorescence intensity using DMSO treated cells as 100%. The %YFP fluorescence (Y axis) was plotted based on chemical ID number (X axis). Symbols represent the library plate that corresponds to each compound. The dotted line indicates the selected hit cutoff of 70% inhibition (30% infection).
Table 1. Hit compounds from bioactive compound library.
| Structure | Compound | Characterized Function | Robust Z Score | YFP Intensity (%) | Group |
|---|---|---|---|---|---|
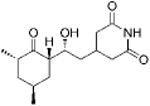
|
Cycloheximide | Translation inhibition | -6.34 | 11.2 +/- 1.8 | Control |
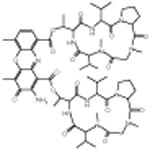
|
Actinomycin D | Protein synthesis inhibition | -7.8 | 5.8 +/- 0.11 | 1 |
|
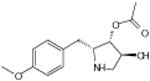
Anisomycin | -5.11 | 25.0 +/- 0.52 | ||
|
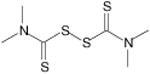
Thiram | Pesticide and Fungicide | -6.64 | 14.0 +/- 0.29 | 2 |
|
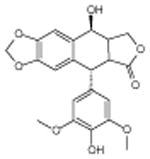
4′-demethyl-epipodophyllotoxin | Alters DNA topoisomerase activity | -5.75 | 15.7 +/- 0.31 | 3 |
|
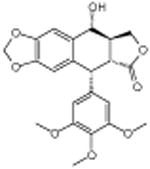
Podofilox | -6.45 | 14.5 +/- 0.31 | ||
|
β-peltatin | Disrupts microtubule | -5.43 | 20.5 +/- 0.41 | 4 |
|
Podophyllotoxin acetate | -6.44 | 22.2 +/- 0.42 | ||
|
Colchicine | -5.44 | 17.3 +/- 0.34 | ||
|
Vinblastine sulfate | Disrupts microtubule | -6.27 | 24.2 +/- 0.48 | 5 |
|
Vincristine sulfate | -6.07 | 26.6 +/- 0.53 | ||
|
Convallatoxin | Cardiac glycoside | -4.59 | 27.8 +/- 0.87 | 6 |
|
Anthothecol | Anti malarial | -5.47 | 27.5 +/- 0.55 | 7 |
The hit compounds identified from the high-content screen represent a collection of inhibitors that could target diverse steps of the entry pathway according to their previously characterized functions (Table 1). The chemicals from Group 1, actinomycin and anisomycin are general inhibitors of protein synthesis (Grollman, 1967). The Group 2 compound, thiram, functions as a pesticide and fungicide inducing cellular toxicity likely from altering cellular metabolism (Cereser et al., 2001b). 4′ Demethylepipodophyllotoxin (Group 3) targets DNA topoisomerase. Groups 4 and 5 consist of compounds that alter microtubule polymerization. Group 4 hit compounds are derived from podophyllotoxin and group 5 compounds are microtubule inhibitors from other classes. Convallatoxin (Group 6) is a cardiac glycoside (Babula et al., 2013) that alters the Na+/K+ ATPase. Finally, anthothecol (Group 7) was previously characterized to have anti-malarial activity functioning through an unknown mechanism (Lee et al., 2008). Taken together, we have developed a robust high-content screen for the identification of numerous CMV inhibitors targeting diverse steps of virus infection.
Determination of efficacy and toxicity of hit compounds
We next determined the half maximal effective concentration (EC50) and half maximal cytotoxicity concentration (CC50) of a compound from each group (Table 1). MRC5 cells untreated or in DMSO (to control for DMSO-content of compound-treated cells) were infected with CMV AD169IE2-YFP (MOI: 3) in a 96 well plate and subsequently analyzed for IE2-YFP fluorescence intensity for up to 18hpi using an Acumen eX3 cytometer (Figure 4A). Consistent with published results (Gardner et al., 2013), the IE2-YFP positive cells (YFP+ cells) increased over time, briefly peaked at 12hpi, and reached a stable plateau by 15hpi. Importantly, cells infected in the presence of DMSO displayed lower infection levels than media possibly due to modulation of cellular integrity or metabolism. Thus, subsequent assays were normalized to cells infected in the presence of DMSO. Together, the data support the utilization of fluorescent cytometry of CMV AD169IE2-YFP-infected fibroblasts as a robust method to measure the efficacy of hit compounds to inhibit a CMV infection.
Figure 4. Determination of EC50 and CC50 values of representative hit compounds.
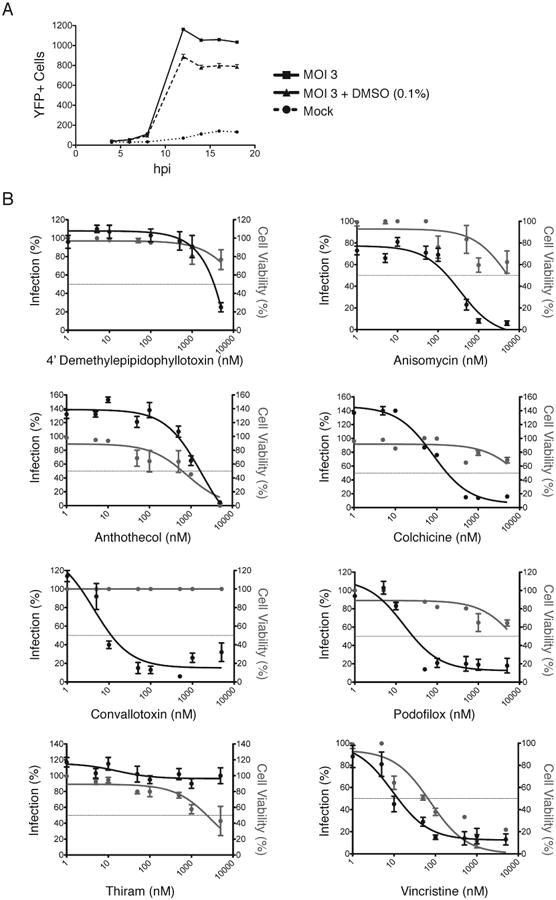
(A) Mock and AD169IE2-YFP-infected MRC5 cells (MOI:3) treated with or without DMSO were analyzed for fluorescence signal using an Acumen eX3 cytometer. The number of YFP+ cells/well was plotted over the course of infection (18hpi). (B) MRC5 cells treated with DMSO and increasing amounts of hit compounds (0-5 μM) were infected with AD169IE2-YFP (MOI:3) and subjected to analysis using an Acumen eX3 cytometer for YFP+ cells. The % infection (left axis, black lines, filled circles) was determined based on the YFP+ cells from virus infected cells treated using DMSO treated cells as 100% infection. The intranuclear cell staining using NucGreen fluorescent cell dye (GFP + cells) was utilized to determine cell viability (right axis, gray lines and gray circles) with DMSO treated cells as 100% cell viability. Error bars represent standard error of the mean.
MRC5 cells pretreated for one hour with DMSO or increasing concentrations (0-5μM) of representative hit compounds 4′demethylepipodophyllotoxin, anisomycin, anthothecol, colchicine, thiram, convallatoxin, podofilox, and vincristine were infected with CMV AD169IE2-YFP and analyzed for YFP+ cells at 18hpi due to the consistent fluorescent signal observed by 15hpi (Figure 4B). Using the number of YFP+ cells from AD169IE2-YFP-infected cells treated with DMSO as 100% infection, the percent infection of AD169IE2-YFP-infected cells was plotted versus the concentration of respective hit compounds (Figure 4B, black lines, filled circles). Consistent with the data from the primary screen (Figure 3), all of the compounds, excluding thiram, resulted in a <20% infection at the maximum concentration tested. Thiram-induced inhibition of a CMV infection was inconsistent with the primary screening results and was subsequently excluded as a hit compound. Using the % infection values, we determined a range of EC50 values of 0.01μM for convallatoxin and vincristine to 3.4μM for 4′ demethylepipodophyllotoxin (Figure 4B and Table 2) with most compounds yielding EC50 values < 0.2μM.
Table 2. Hit compound efficacy.
| Compound | EC50 (μM) | CC50 (μM) | Selective Ratio |
|---|---|---|---|
| 4-demethyl-epipodophyllotoxin | 3.42 | >5 | >1.5 |
| Anisomycin | 0.17 | >5 | >29 |
| Anthothecol | 1.68 | 0.7 | 0.4 |
| Colchicine | 0.18 | >5 | >28 |
| Convallatoxin | 0.01 | >5 | >454 |
| Podofilox | 0.03 | >5 | >192 |
| Thiram | >5 | 2.2 | <0.4 |
| Vincristine sulfate | 0.01 | 0.04 | 3.4 |
Next, the half maximal cytotoxicity concentration (CC50) of the hit compounds was determined using the cell impermeant dye NucGreen (Life Technologies) that measures cell permeability, an irreversible change defining cell viability. MRC5 cells treated with DMSO or increasing concentrations (0-5μM) of hit compounds 4′ demethylepipodophyllotoxin, anisomycin, anthothecol, colchicine, convallatoxin, podofilox, thiram, and vincristine were stained with NucGreen both before and after fixing the cells in 4% paraformaldehyde. Using cells treated with DMSO as comparison, the percentage of non-permeable (viable) cells was determined for the hit compounds (Figure 4B, gray lines and filled circles). Five of the eight hit compounds had greater than 50% cell viability at the highest concentration tested (5μM). Remarkably, the translational inhibitor anisomycin had a high CC50 value (>5μM) likely due to the cell type and prolonged doubling time of cells (12-24hrs). The least toxic was convallatoxin, with 100% viability at 5μM; the most toxic was vincristine, with a CC50 of 0.04μM (Figure 4B and Table 2). The CC50 values reported here are similar to previously published results in diverse cell systems using different toxicity assays (Cereser et al., 2001a; Chen et al., 2004; Davenport et al., 2014; Li et al., 2012; Pan et al., 2013; Tran et al., 2012; Yadav et al., 2014).
The EC50 and CC50 values of the eight compounds tested were used to determine a selective ratio (CC50/EC50) to quantify the difference between the toxic and effective doses of the compounds (Table 2). The baseline of an effective selective ratio is dependent on the stringency of the assays used to determine the EC50 and CC50 values. Given that the translation inhibitor anisomycin has a selective ratio of ∼30 and is toxic upon prolonged treatment, we would consider 4′-demethylepipidophyllotoxin, anthothecol, colchicine, thiram, and vincristine with selective ratios <30 to have limited specificity in inhibiting a CMV infection and a narrow therapeutic index (2013). Yet, hit compounds convallatoxin and podofilox, have high selective ratios of 454 and 192, respectively, suggesting they are highly effective against a CMV infection.
Validation of selective hit compound to inhibit viral gene expression
Immediate early gene expression is a requirement of early and late gene expression. To verify that our hit compounds also inhibited expression of these later gene classes, AD169-WT-infected (MOI: 3) cells pretreated with podofilox, convallatoxin, and colchicine were assayed for expression of an early gene product at concentrations spanning the previously calculated EC50 values. Cells were harvested at 20hpi and analyzed for expression of CMV early protein US11 (Figure 5). Total cell lysates from the mock-infected cells or infected cells pretreated with DMSO or selective hit compounds were subjected to immunoblot analysis using antibodies against CMV US11 (Figure 5, lanes 1-11) and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Figure 5, lanes 12-22). As expected, US11 protein was detected in virus-infected cells treated with DMSO (Figure 5, lane 2). Consistent with the AD169IE2-YFP infection data (Figure 4B), lower levels of US11 were observed in infected cells treated with 50 or 500nM of respective compound (Figure 5A, lanes 3, 6 and 9). Strikingly, convallatoxin treatment blocked the expression of US11 at all concentrations (Figure 5, lanes 6-8). Complete inhibition at 5nM was remarkable, as the EC50 was found to be 10nM (Table 2). This may be due to the fact that a reduction in IE gene expression dramatically impacts subsequent early gene expression. Taken together, these data confirm the efficacy of the high quality hit compounds to limit a CMV infection.
Figure 5. Validation of selective hit compounds to inhibit a CMV infection.

MRC5 cells pre-treated with DMSO, podofilox (5-50nM), convallatoxin (5-50nM), or colchicine (10-500nM) were infected with AD169-WT (MOI:3) and total cell lysates were resolved on a SDS-polyacrylamide gel (12%). Mock (lanes 1 and 12) and infected (lanes 2-11, 13-22) cells were harvested at 20 hpi and subjected to immunoblot analysis for the early CMV gene product US11 (lanes 1-11) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (lanes 12-22). The respective polypeptides and relative molecular mass markers are indicated.
Convallatoxin inhibits infection of a clinical CMV strain
To confirm the anti-CMV property of the lead compound, convallatoxin, we utilized a reporter virus derived from a clinical strain, TB40/EFLAG-YFP, previously engineered and characterized in our lab (Noriega et al., 2014). The TB40/EFLAG-YFP strain substitutes a chimeric FLAG-YFP sequence for the US28 gene, which is expressed with early kinetics. ARPE-19 epithelial cells treated with DMSO were infected with CMV TB40/EFLAG-YFP (MOI: 20) in a 96-well plate and subsequently analyzed for FLAG-YFP fluorescence intensity for up to 7dpi using an Acumen eX3 cytometer (Figure 6A). The YFP+ cells increased beginning at day 2, reaching their highest levels at 6 dpi and thereby defining infection conditions.
Figure 6. Determination of convallatoxin antiviral activity using a clinical infection model.
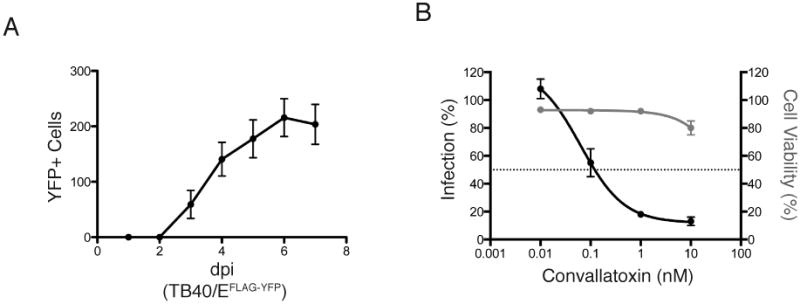
(A). ARPE-19 epithelial cells were infected with the clinical CMV strain TB40/EFLAG-YFP (MOI:20). Fluorescence signal was measured using an Acumen eX3 cytometer. The number of YFP+ cells/well was plotted over the course of infection (up to 7dpi.) (B) ARPE-19 epithelial cells treated with DMSO and increasing amounts of convallatoxin (0-10 nM) were infected with TB40/EFLAG-YFP (MOI:20) and analyzed for YFP+ cells. The % infection (left axis, black lines, filled circles) was determined based on the YFP+ cells using DMSO-treated cells as 100% infection. The intranuclear cell staining using NucGreen fluorescent cell dye (GFP + cells) was utilized to determine cell viability (right axis, gray lines and gray circles) with DMSO treated cells as 100% cell viability. Error bars represent standard error of the mean.
Following establishment of infection conditions, ARPE-19 cells were pretreated for one hour with increasing concentrations (0-10nM) of convallatoxin and then infected for up to 120 hours, at which time the number of YFP+ cells was used to determine percent infection (Figure 6B, black line). Cells treated with convallatoxin demonstrated a significant decrease in YFP+ cells, with an EC50 value of 0.1nM. NucGreen was used to measure viability of ARPE-19 cells (Figure 6B, gray line). Convallatoxin treatment had a minimal effect on cellular viability, with a slight drop in cell viability to approximately 80% at the highest concentration tested (10nM) (Figure 6B, gray line). Interestingly, the EC50 value of convallatoxin treatment of TB40/EFLAG-YFP-infected MRC5 cells was similar to AD169 infection (data not shown). The lower EC50 and CC50 values of TB40/EFLAG-YFP-infected ARPE cells suggest that these cells are more sensitive to convallatoxin treatment and demonstrate the effectiveness of an early-step inhibitor as a potential antiviral.
Convallatoxin limits CMV proliferation
We further characterized convallatoxin-mediated inhibition of CMV entry and subsequent proliferation. AD169IE2-YFP-infected MRC5 cells treated with 0, 5, 10, or 50nM convallatoxin at either 0 or 36 hours post-infection were analyzed for YFP+ cells using a fluorescent plate reader for up to 136hpi (Figure 7A). When cells were treated with DMSO at either 0 or 36hpi, the number of YFP+ cells slowly increased during the first 36 hours and then quickly peaked at approximately 96hpi (Figure 7A, filled circles). Convallatoxin treatment reduced the number of YFP+ cells in a dose-dependent manner compared to DMSO-treated cells when added simultaneously with virus (0hpi) (Top panel). By 100hpi, cells treated with 5nM convallatoxin displayed approximately half of the fluorescent expression of the DMSO-treated cells likely due to the lower MOI and increased drug exposure time. Strikingly, convallatoxin treatment of infected cells at 36hpi also resulted in the significant decrease of YFP+ cells (Figure 7A, bottom panel) consistent with the inhibition of IE2-YFP expression.
Figure 7. Analysis of CMV infection following long-term convallatoxin treatment.
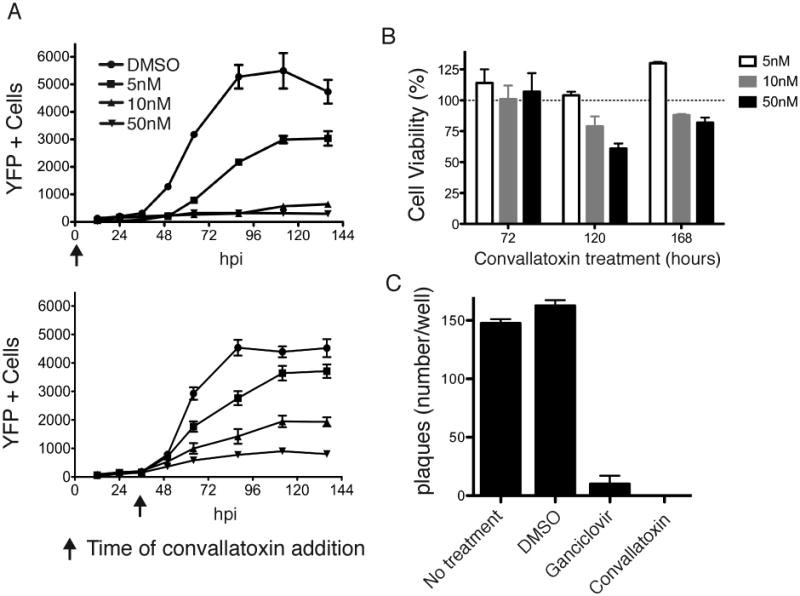
(A) The number of YFP+ cells (left axis, black lines) were analyzed from MRC5 cells treated with convallatoxin (5, 10, or 50nM) at time of infection (top panel) or 36 hpi (bottom panel) for up to 136hpi. (B) MRC5 cells were exposed to 5, 10, or 50nM convallatoxin for up to 168 hours. Using the total number of cells treated with DMSO as 100% cell viability, the % cell viability of convallatoxin-treated MRC5 cells was plotted for 72, 120, and 168 hpi. Error bars represent standard error of the mean. (C) MRC5 cells untreated or pretreated with DMSO, ganciclovir (12μM), or convallatoxin (5nM) were infected with AD169-WT (MOI: 0.002). The cells were subjected to a viral plaque assay 5 dpi. Error bars represent standard error of the mean.
To ensure that the decrease in YFP+ cells was not due to convallatoxin-induced toxicity during prolonged treatment, uninfected MRC5 fibroblasts treated with 5, 10, or 50nM convallatoxin were stained with NucGreen and analyzed for number of YFP+ cells using a fluorescent cytometer. The number of YFP+ cells in convallatoxin-treated wells compared to total number of cells/well was used to calculate percent cell viability (Figure 7B). While sensitivity to intranuclear cell staining in the presence of 10nM and 50nM convallatoxin demonstrated limited toxicity at 120 hours post treatment (79% and 61%, respectively) and 168 hours post treatment (91% and 82%, respectively), 5nM convallatoxin had no detectable effect throughout the timecourse. Thus, the hit compound convallatoxin can inhibit initial CMV infection as well as mitigate the progression of an established CMV infection in human fibroblasts at concentrations that cause minimal cellular toxicity.
Finally, to assess the effect of convallatoxin treatment on cell-cell CMV dissemination, we employed a plaque-reduction assay to compare the effect of convallatoxin treatment to cells treated with the anti-CMV compound, ganciclovir. MRC5 cells were untreated or pre-incubated (1hr) with DMSO, convallatoxin (5nM), or ganciclovir (12μM, an efficacious concentration for limiting viral replication in vitro (Shigeta et al., 1991)) before infection with AD169IE2-YFP (MOI: 0.002). At 5 dpi, the number of viral plaques per well was calculated (Figure 7C). As expected, similar number of viral plaques were observed in the untreated and DMSO-treated cells. Remarkably, while DMSO-treated cells displayed an average of 163 plaques/well, both ganciclovir and convallatoxin-treated cells showed a nearly complete reduction in viral plaques (10 and 0 plaques, respectively). The data demonstrate that convallatoxin is able to prevent cell-to-cell spread during a CMV infection.
Convallatoxin limits CMV production upon short-term treatment
Can short-term exposure of convallatoxin to virus-infected cells limit viral production? To address this question, we measured the fluorescence levels of AD169IE2-YFP-infected MRC5 cells (MOI: 0.5) together with the titer of productive virus released over 9 days post-infection from cells pretreated for one hour with DMSO, convallatoxin or ganciclovir, and left in the presence of each respective compound for 24 hpi. The number of YFP+ cells was calculated as percent infection compared to DMSO-treated cells (Figure 8A). As expected, ganciclovir-treated cells exhibited only a small decrease in YFP fluorescence (Figure 8A, triangles). In contrast, convallatoxin-treated cells displayed a significant decrease in fluorescence (3, 5 and 7 dpi) with a slight increase at later time points (Figure 8A, squares). These results suggest that inhibition of initial steps of infection has a prolonged affect on viral gene expression.
Figure 8. Convallatoxin treatment limits CMV production.
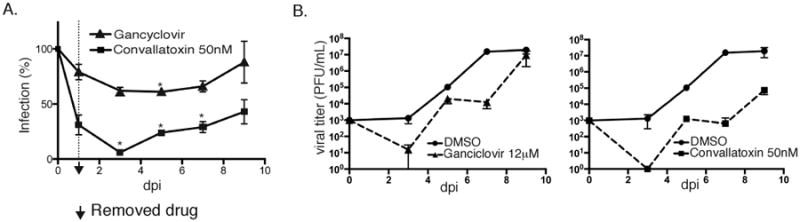
MRC5 cells were pretreated with DMSO, 12μM ganciclovir, or 5nM convallatoxin and then infected with AD169IE2-YFP (MOI:0.5). At 1dpi, each respective compound was removed and the cell culture media was replaced. YFP+ cells were measured in 48-hour intervals using an Acumen eX3 cytometer and then supernatant was harvested for titration of infectious extracellular virus using a standard plaque assay. (A) The % infection of ganciclovir-treated cells (filled triangles) and convallatoxin-treated cells (filled squares) was calculated throughout the 9-day timecourse. (B) Day 3, 5, 7, and 9 supernatants from the ganciclovir-treated infection (filled triangles, dotted line) (left panel) or the convallatoxin-treated infection (filled squares, dotted line) (right panel), were plotted together with supernatant from the DMSO-treated infection (filled circles, solid line) (both panels). Error bars represent standard error of the mean.
The supernatant of the virus-infected cells was isolated and infectious progeny were measured using a standard plaque assay (Figure 8B). As expected, ganciclovir treatment slightly decreased the viral titer given the short exposure time of the drug (Figure 8c, triangle, dotted line). Yet, convallatoxin treatment resulted in delayed viral growth kinetics throughout the time course (Figure 8B, square, dotted line) with an ∼10 fold decrease in virus titer when compared to ganciclovir treatment. Taken together, the data demonstrate the ability of convallatoxin to limit the production of infectious extracellular virus during a CMV infection after short-term treatment.
Discussion
The initial phase of a CMV infection requires multiple virus-host interactions. These events, which are critical for viral entry and gene expression, may offer opportunities for therapeutic intervention. Thus, we utilized the AD169 strain of CMV expressing a chimeric IE2-YFP protein, AD169IE2-YFP (Figure 1) to identify novel compounds that affect the early steps of a CMV infection. The high-content screening of a small chemical library identified compounds that block IE2 expression. The steps at which the hit compounds act provide a detailed portrayal of the series of virus-host interactions that lead to initial viral gene expression during CMV infection (Figure 9). Importantly, the assay identified hit compounds that have been characterized to alter cellular processes directly or indirectly involved in a CMV infection (Table 1). The targeting of cellular processes would contrast from current anti-CMV drugs, which specifically target viral gene products by inhibiting the viral DNA polymerase, or interfering with immediate early gene transcription through inhibition of the Major Immediate-Early gene locus (Mercorelli et al., 2011).
Figure 9. Potential steps of the CMV infectious cycle targeted by hit compounds.

Viral attachment and fusion to the cellular membrane is followed by release of the CMV capsid and tegument proteins into the cytoplasm. The capsid travels along cellular microtubule networks, which are sensitive to the Group 4 and 5 hit compounds (colchicine, podophyllotoxin, podofilox, vinblastine, vincristine, and β–peltatin). At the nuclear membrane, the capsid is uncoated and viral DNA is deposited into the nucleus where host chromatin modifications permit the expression of viral DNA. This step is likely targeted by the group 3 hit compound, 4′ dimethyl-epipodophyllotoxin. Viral gene transcription produces mRNA transcripts that are translated in the cytoplasm and blocked by the group 1 compounds (actinomycin D and anisomycin). Convallatoxin (Group 6) and anthothecol (Group 7) may function either directly or indirectly at any points preceding immediate early gene expression. Following the expression of early and late viral genes, newly synthesized viral genomes are packaged into viral capsid and then undergo two rounds of envelopment accompanied by packaging tegument proteins. Finally, an infectious virion buds from the infected cell into the extracellular space.
CMV infection is a multi-step process that involves viral binding to the cell surface followed by viral membrane fusion, microtubule-mediated transport of the capsid to the nucleus and viral gene expression (Figure 9). The HCS identified specific hit compounds that disrupt many of these steps. While protein synthesis inhibitors such as actinomycin D and anisomycin were identified as hits, not every protein synthesis inhibitor contained in the compound library was identified as a hit. For example, cells treated with the compound emetine (Jimenez et al., 1977) reduced YFP intensity by only 41%, well above our cutoff at the 5μM screening dose.
The hit compounds colchicine and vincristine are well-characterized microtubule inhibitors (Jordan et al., 1998) that were identified to block CMV infection (Table 1, group 5) and reduced infection when added up to 4hpi (data not shown). Consistent with these findings, additional well-characterized microtubule inhibitors limit CMV infection (Ogawa-Goto et al., 2003); however, not all microtubule inhibitors in the screen were identified as hits. Noscapine (Landen et al., 2002) did not inhibit CMV infection (YFP Intensity 105.6%), The podophyllotoxin derivatives podofilox and beta-peltatin bind to the same site on the tubulin monomer as colchicine (Cortese et al., 1977) although they are described as having distinct microtubule depolymerization characteristics (Desbene and Giorgi-Renault, 2002). Viral DNA deposition in the host nucleus causes significant chromatin rearrangement, permitting viral gene expression and replication. Such arrangements require host enzymes, such as DNA topoisomerase II (Benson and Huang, 1988). Thus, the hit compound 4′demethylepipodophyllotoxin (group 3) which inhibits DNA topoisomerase II (Huang et al., 1992; Wozniak and Ross, 1983) likely prevents remodeling of the viral genome. Of note, not all topoisomerase inhibitors in the library were scored as hit compounds, for example doxorubicin (Fornari et al., 1994) treated cells still had YFP intensity of 64%. Further characterization of these compounds will provide a more comprehensive understanding of the early steps of a CMV infectious cycle.
Convallatoxin (group 6), an efficacious compound identified in the screen is characterized as a cardiac glycoside belonging to a family of compounds including ouabain, digitalis and digoxin (Yancy et al., 2013). Ouabain and digoxin have been described to inhibit virus infection through targeting immediate-early gene expression but are considerably more toxic than convallatoxin (Kapoor et al., 2012). Interestingly, cardiac glycosides ouabain and peruvoside were included in the compound library tested, but they were excluded as hit compounds because they did not reduce YFP expression >70% (data not shown).
There is no direct evidence that cardiac glycosides inhibit CMV infection by interfering with a viral protein. One study demonstrates that cardiac glycosides act by preventing the upregulation of ether-a-go-go-related gene potassium channel 1 (Kapoor et al., 2012), suggesting it functions by targeting a transcription event. Another possible mechanism of viral inhibition is through manipulation of the Src kinase pathway by reducing the interaction between Src kinase and the Na+/K+ ATPase, leading to disregulated Src activity (Reinhard et al., 2013). Src kinase may be important for CMV entry into monocytes where the gH/gL/UL128-UL131 pentamer is critical for mediating endocytic entry while the role in fibroblasts, where CMV entry does not require the pentameric complex, is less clear (Nogalski et al., 2013). Indeed in our experiments, the potency of convallatoxin is enhanced roughly 100 fold in the context of an infection of epithelial cells with the clinically-derived reporter strain TB40/E (Figure 6B) compared to infection of fibroblasts with our AD169-based reporter virus (Figure 4B). In the future, efforts to create a convallatoxin-resistant CMV strain should be conducted in order to dissect the nature of the drug's ability to inhibit CMV infection and distinguish between a viral or cellular target.
Given that the development of viral resistance is a limitation of many CMV therapeutics, interfering with cellular components involved in CMV infection may offer a distinct strategy that is less susceptible to viral adaptation. For example, data have recently emerged on the utility of leflunomide treatment in cases of ganciclovir-resistant CMV infection. Leflunomide, an immune-suppresive compound marketed for treatment of rheumatoid arthritis, is thought to interfere with pyrimidine synthesis and phosphorylation of tyrosine kinases involved in B- and T- cell activity (Chacko and John, 2012). Thus, rather than inhibiting viral DNA synthesis, leflunomide likely imparts an antiviral activity by interfering with cellular processes.
A drawback to targeting a cellular process to prevent or ameliorate CMV infection is the potential toxicity accompanied with prolonged treatment. The determination of a selective ratio provides some insight in comparing the efficacy of the compound to its toxicity when conducting in vitro studies. When characterizing a compound as a potential drug, the therapeutic index (Muller and Milton, 2012) is a more accurate value to define the effectiveness of the compound based on the severity of the disease and the threshold of toxicity (e.g. lethal dose vs. presence of adverse but not life-threatening side effects). Cytomegalovirus infection causes a wide range of symptoms, and the therapeutic index needed to prevent vision loss from CMV would be different from the dose used as prophylaxis in a pregnant woman. While our selective hit compound convallatoxin possessed a high selective ratio during a 16-hour infection (Figure 4B and Table 2), it displayed minute toxicity in longer timecourse experiments (Figure 7B). Future experiments will determine whether toxicity of hit compounds that target initial CMV infection events can extend to the longer time points necessary for treatment of chronic CMV infection or long-term prophylaxis.
Several new anti-CMV compounds are presently in clinical development (Boeckh and Geballe, 2011). These include an additional viral polymerase inhibitor (CMX001 or brincidofovir), a potential terminase inhibitor (AIC246 or letermovir), as well as a UL97 kinase inhibitor (maribavir). Additionally, the licensed drug imatinib shows some promise as an anti-CMV agent (Boeckh and Geballe, 2011). While these new compounds offer benefits over current anti CMV treatment such as improved potency and limited toxicity, they require further validation and may ultimately have drawbacks that limit their efficacy (Schubert et al., 2013; Winston et al., 2012). This and the continued relevance of drug-resistant CMV disease (Boivin et al., 2012) warrant the continued identification of anti-CMV compounds that can offer improvements over current standard of care.
In conclusion, a high-content screen identified a panel of bioactive compounds that interfere with diverse, early events in CMV infection. One of the hit compounds, convallatoxin, demonstrated that an early-event inhibitor can limit viral infection and proliferation. The data described here support the development of early-event inhibitors as future CMV therapeutics.
Highlights.
A novel, high-content assay was developed to identify anti-CMV therapeutics
The assay relies on a reporter CMV strain, AD169IE2-YFP
The assay was thoroughly optimized to achieve outstanding assay fitness
Several efficacious compounds were identified from a library of >2000 compounds
Extensive secondary assays confirmed the antiviral activity of the lead compounds
Acknowledgments
This work was supported in part by the NIH grants DA035190 and AI101820 and the CTSA grant UL1TR000067. T.J.G. is a predoctoral trainee supported in part by an American Heart Association pre-doctoral fellowship. T.C. is also sponsored in part by a USPHS Institutional Research Training Award T32-AI07647
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- U.S.F.a.D. Administration., editor. Code of Federal Regulations Subchapter D, Drugs for Human Use, Part 320. 2013. Bioavailability and bioequivalence requirements. [Google Scholar]
- Babula P, Masarik M, Adam V, Provaznik I, Kizek R. From Na+/K+-ATPase and cardiac glycosides to cytotoxicity and cancer treatment. Anti-cancer agents in medicinal chemistry. 2013;13:1069–1087. doi: 10.2174/18715206113139990304. [DOI] [PubMed] [Google Scholar]
- Baker BM, Tortorella D. Dislocation of an endoplasmic reticulum membrane glycoprotein involves the formation of partially dislocated ubiquitinated polypeptides. The Journal of biological chemistry. 2007;282:26845–26856. doi: 10.1074/jbc.M704315200. [DOI] [PubMed] [Google Scholar]
- Benson JD, Huang ES. Two specific topoisomerase II inhibitors prevent replication of human cytomegalovirus DNA: an implied role in replication of the viral genome. Journal of virology. 1988;62:4797–4800. doi: 10.1128/jvi.62.12.4797-4800.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckh M, Geballe AP. Cytomegalovirus: pathogen, paradigm, and puzzle. The Journal of clinical investigation. 2011;121:1673–1680. doi: 10.1172/JCI45449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin G, Goyette N, Farhan M, Ives J, Elston R. Incidence of cytomegalovirus UL97 and UL54 amino acid substitutions detected after 100 or 200 days of valganciclovir prophylaxis. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology. 2012;53:208–213. doi: 10.1016/j.jcv.2011.12.019. [DOI] [PubMed] [Google Scholar]
- Cereser C, Boget S, Parvaz P, Revol A. An evaluation of thiram toxicity on cultured human skin fibroblasts. Toxicology. 2001a;162:89–101. doi: 10.1016/s0300-483x(01)00347-x. [DOI] [PubMed] [Google Scholar]
- Cereser C, Boget S, Parvaz P, Revol A. Thiram-induced cytotoxicity is accompanied by a rapid and drastic oxidation of reduced glutathione with consecutive lipid peroxidation and cell death. Toxicology. 2001b;163:153–162. doi: 10.1016/s0300-483x(01)00401-2. [DOI] [PubMed] [Google Scholar]
- Chacko B, John GT. Leflunomide for cytomegalovirus: bench to bedside. Transplant infectious disease : an official journal of the Transplantation Society. 2012;14:111–120. doi: 10.1111/j.1399-3062.2011.00682.x. [DOI] [PubMed] [Google Scholar]
- Chen SW, Tian X, Tu YQ. Synthesis and cytotoxic activity of novel derivatives of 4′-demethylepipodophyllotoxin. Bioorganic & medicinal chemistry letters. 2004;14:5063–5066. doi: 10.1016/j.bmcl.2004.07.094. [DOI] [PubMed] [Google Scholar]
- Cherrington JM, Mocarski ES. Human cytomegalovirus ie1 transactivates the alpha promoter-enhancer via an 18-base-pair repeat element. Journal of virology. 1989;63:1435–1440. doi: 10.1128/jvi.63.3.1435-1440.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S, Marousek G, Li S, Weinberg A. Contrasting drug resistance phenotypes resulting from cytomegalovirus DNA polymerase mutations at the same exonuclease locus. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology. 2008;43:107–109. doi: 10.1016/j.jcv.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung N, Zhang XD, Kreamer A, Locco L, Kuan PF, Bartz S, Linsley PS, Ferrer M, Strulovici B. Median absolute deviation to improve hit selection for genome-scale RNAi screens. Journal of biomolecular screening. 2008;13:149–158. doi: 10.1177/1087057107312035. [DOI] [PubMed] [Google Scholar]
- Cortese F, Bhattacharyya B, Wolff J. Podophyllotoxin as a probe for the colchicine binding site of tubulin. The Journal of biological chemistry. 1977;252:1134–1140. [PubMed] [Google Scholar]
- Davenport J, Balch M, Galam L, Girgis A, Hall J, Blagg BS, Matts RL. High-throughput screen of natural product libraries for hsp90 inhibitors. Biology. 2014;3:101–138. doi: 10.3390/biology3010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMeritt IB, Podduturi JP, Tilley AM, Nogalski MT, Yurochko AD. Prolonged activation of NF-kappaB by human cytomegalovirus promotes efficient viral replication and late gene expression. Virology. 2006;346:15–31. doi: 10.1016/j.virol.2005.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbene S, Giorgi-Renault S. Drugs that inhibit tubulin polymerization: the particular case of podophyllotoxin and analogues. Current medicinal chemistry Anti-cancer agents. 2002;2:71–90. doi: 10.2174/1568011023354353. [DOI] [PubMed] [Google Scholar]
- Fornari FA, Randolph JK, Yalowich JC, Ritke MK, Gewirtz DA. Interference by doxorubicin with DNA unwinding in MCF-7 breast tumor cells. Molecular pharmacology. 1994;45:649–656. [PubMed] [Google Scholar]
- Gardner TJ, Bolovan-Fritts C, Teng MW, Redmann V, Kraus TA, Sperling R, Moran T, Britt W, Weinberger LS, Tortorella D. Development of a high-throughput assay to measure the neutralization capability of anti-cytomegalovirus antibodies. Clinical and vaccine immunology : CVI. 2013;20:540–550. doi: 10.1128/CVI.00644-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparri F. An overview of cell phenotypes in HCS: limitations and advantages. Expert opinion on drug discovery. 2009;4:643–657. doi: 10.1517/17460440902992870. [DOI] [PubMed] [Google Scholar]
- Grollman AP. Inhibitors of protein biosynthesis. II. Mode of action of anisomycin. The Journal of biological chemistry. 1967;242:3226–3233. [PubMed] [Google Scholar]
- Huang ES, Benson JD, Huong SM, Wilson B, van der Horst C. Irreversible inhibition of human cytomegalovirus replication by topoisomerase II inhibitor, etoposide: a new strategy for the treatment of human cytomegalovirus infection. Antiviral research. 1992;17:17–32. doi: 10.1016/0166-3542(92)90087-l. [DOI] [PubMed] [Google Scholar]
- Jimenez A, Carrasco L, Vazquez D. Enzymic and nonenzymic translocation by yeast polysomes. Site of action of a number of inhibitors. Biochemistry. 1977;16:4727–4730. doi: 10.1021/bi00640a030. [DOI] [PubMed] [Google Scholar]
- Jordan A, Hadfield JA, Lawrence NJ, McGown AT. Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. Medicinal research reviews. 1998;18:259–296. doi: 10.1002/(sici)1098-1128(199807)18:4<259::aid-med3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Cai H, Forman M, He R, Shamay M, Arav-Boger R. Human cytomegalovirus inhibition by cardiac glycosides: evidence for involvement of the HERG gene. Antimicrobial agents and chemotherapy. 2012;56:4891–4899. doi: 10.1128/AAC.00898-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landen JW, Lang R, McMahon SJ, Rusan NM, Yvon AM, Adams AW, Sorcinelli MD, Campbell R, Bonaccorsi P, Ansel JC, Archer DR, Wadsworth P, Armstrong CA, Joshi HC. Noscapine alters microtubule dynamics in living cells and inhibits the progression of melanoma. Cancer research. 2002;62:4109–4114. [PubMed] [Google Scholar]
- Lee SE, Kim MR, Kim JH, Takeoka GR, Kim TW, Park BS. Antimalarial activity of anthothecol derived from Khaya anthotheca (Meliaceae) Phytomedicine : international journal of phytotherapy and phytopharmacology. 2008;15:533–535. doi: 10.1016/j.phymed.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Li JY, Huang JY, Li M, Zhang H, Xing B, Chen G, Wei D, Gu PY, Hu WX. Anisomycin induces glioma cell death via down-regulation of PP2A catalytic subunit in vitro. Acta pharmacologica Sinica. 2012;33:935–940. doi: 10.1038/aps.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JL, Stinski MF. Effect of a modulator deletion on transcription of the human cytomegalovirus major immediate-early genes in infected undifferentiated and differentiated cells. Journal of virology. 1997;71:1246–1255. doi: 10.1128/jvi.71.2.1246-1255.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercorelli B, Lembo D, Palu G, Loregian A. Early inhibitors of human cytomegalovirus: state-of-art and therapeutic perspectives. Pharmacology & therapeutics. 2011;131:309–329. doi: 10.1016/j.pharmthera.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocarski ES, T S, Pass RF. Cytomegaloviruses. In: K DM, M P, editors. Fields Virology. Wolters Kluwer Health/Lippincott Williams & Wilkins; Philadelphia: 2007. 2007. [Google Scholar]
- Muller PY, Milton MN. The determination and interpretation of the therapeutic index in drug development. Nature reviews Drug discovery. 2012;11:751–761. doi: 10.1038/nrd3801. [DOI] [PubMed] [Google Scholar]
- Nogalski MT, Chan GC, Stevenson EV, Collins-McMillen DK, Yurochko AD. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS pathogens. 2013;9:e1003463. doi: 10.1371/journal.ppat.1003463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noriega VM, Gardner TJ, Redmann V, Bongers G, Lira SA, Tortorella D. Human cytomegalovirus US28 facilitates cell-to-cell viral dissemination. Viruses. 2014;6:1202–1218. doi: 10.3390/v6031202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa-Goto K, Tanaka K, Gibson W, Moriishi E, Miura Y, Kurata T, Irie S, Sata T. Microtubule network facilitates nuclear targeting of human cytomegalovirus capsid. Journal of virology. 2003;77:8541–8547. doi: 10.1128/JVI.77.15.8541-8547.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan T, Huang B, Zhang W, Gabos S, Huang DY, Devendran V. Cytotoxicity assessment based on the AUC50 using multi-concentration time-dependent cellular response curves. Analytica chimica acta. 2013;764:44–52. doi: 10.1016/j.aca.2012.12.047. [DOI] [PubMed] [Google Scholar]
- Razonable RR. Antiviral drugs for viruses other than human immunodeficiency virus. Mayo Clinic proceedings. 2011;86:1009–1026. doi: 10.4065/mcp.2011.0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard L, Tidow H, Clausen MJ, Nissen P. Na(+),K (+)-ATPase as a docking station: protein-protein complexes of the Na(+),K (+)-ATPase. Cellular and molecular life sciences : CMLS. 2013;70:205–222. doi: 10.1007/s00018-012-1039-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert A, Ehlert K, Schuler-Luettmann S, Gentner E, Mertens T, Michel D. Fast selection of maribavir resistant cytomegalovirus in a bone marrow transplant recipient. BMC infectious diseases. 2013;13:330. doi: 10.1186/1471-2334-13-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeta S, Konno K, Baba M, Yokota T, De Clercq E. Comparative inhibitory effects of nucleoside analogues on different clinical isolates of human cytomegalovirus in vitro. The Journal of infectious diseases. 1991;163:270–275. doi: 10.1093/infdis/163.2.270. [DOI] [PubMed] [Google Scholar]
- Steininger C. Novel therapies for cytomegalovirus disease. Recent patents on anti-infective drug discovery. 2007;2:53–72. doi: 10.2174/157489107779561634. [DOI] [PubMed] [Google Scholar]
- Straschewski S, Warmer M, Frascaroli G, Hohenberg H, Mertens T, Winkler M. Human cytomegaloviruses expressing yellow fluorescent fusion proteins-- characterization and use in antiviral screening. PloS one. 2010;5:e9174. doi: 10.1371/journal.pone.0009174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng MW, Bolovan-Fritts C, Dar RD, Womack A, Simpson ML, Shenk T, Weinberger LS. An endogenous accelerator for viral gene expression confers a fitness advantage. Cell. 2012;151:1569–1580. doi: 10.1016/j.cell.2012.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TD, Pham NB, Fechner G, Zencak D, Vu HT, Hooper JN, Quinn RJ. Cytotoxic cyclic depsipeptides from the Australian marine sponge Neamphius huxleyi. Journal of natural products. 2012;75:2200–2208. doi: 10.1021/np3006474. [DOI] [PubMed] [Google Scholar]
- Winston DJ, Saliba F, Blumberg E, Abouljoud M, Garcia-Diaz JB, Goss JA, Clough L, Avery R, Limaye AP, Ericzon BG, Navasa M, Troisi RI, Chen H, Villano SA, Uknis ME. Efficacy and safety of maribavir dosed at 100 mg orally twice daily for the prevention of cytomegalovirus disease in liver transplant recipients: a randomized, double-blind, multicenter controlled trial. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2012;12:3021–3030. doi: 10.1111/j.1600-6143.2012.04231.x. [DOI] [PubMed] [Google Scholar]
- Wozniak AJ, Ross WE. DNA damage as a basis for 4′-demethylepipodophyllotoxin-9-(4,6-O-ethylidene-beta-D-glucopyranoside) (etoposide) cytotoxicity. Cancer research. 1983;43:120–124. [PubMed] [Google Scholar]
- Yadav R, Kumar D, Kumari A, Yadav SK. Encapsulation of podophyllotoxin and etoposide in biodegradable poly-D,L-lactide nanoparticles improved their anticancer activity. Journal of microencapsulation. 2014;31:211–219. doi: 10.3109/02652048.2013.834988. [DOI] [PubMed] [Google Scholar]
- Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128:e240–327. doi: 10.1161/CIR.0b013e31829e8776. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. Journal of biomolecular screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]