

Abstract
Protein disulfide isomerases (PDI) are eukaryotic oxidoreductases that catalyze the formation and rearrangement of disulfide bonds during folding of substrate proteins. Structurally, PDI enzymes share as a common feature the presence of at least one active thioredoxin-like domain. PDI enzymes are also involved in holding, refolding, and degradation of unfolded or misfolded proteins during stressful conditions. The EhPDI enzyme (a 38 kDa polypeptide with two active thioredoxin-like domains) has been used as a model to gain insights into protein folding and disulfide bond formation in E. histolytica. Here, we performed a functional complementation assay, using a ΔdsbC mutant of E. coli, to test whether EhPDI exhibits isomerase activity in vivo. Our preliminary results showed that EhPDI exhibits isomerase activity; however, further mutagenic analysis revealed significant differences in the functional role of each thioredoxin-like domain. Additional studies confirmed that EhPDI protects heat-labile enzymes against thermal inactivation, extending our knowledge about its chaperone-like activity. The characterization of EhPDI, as an oxidative folding catalyst with chaperone-like function, represents the initial step to dissect the molecular mechanisms involved in protein folding in E. histolytica.
1. Introduction
In eukaryotic cells, folding and posttranslational modifications of proteins are the primary function of the endoplasmic reticulum (ER) [1]. Formation of disulfide bonds, a common modification observed in several secretory proteins, takes place mainly in that compartment [1, 2]. Almost all organisms have a set of proteins involved in folding; however, the cellular and molecular details of this process have been elucidated only in a few model systems, such as the yeast ER and the bacterial periplasmic compartment [3, 4].
Protein disulfide isomerases (PDI) are eukaryotic oxidoreductases that catalyze the formation and rearrangement of disulfide bonds during folding of substrate proteins [5]. Structurally, PDI enzymes share as a common feature the presence of at least one active thioredoxin-like domain. Some organisms, such as yeast and mammals, have a family of PDI homologues that exhibit distinct domain organization and function [6–8].
Under physiological conditions, the cellular mechanisms that respond to proteotoxic stress remain in an inactive state; however, under stressful conditions, several response mechanisms are triggered to restore proteome stability, but if these fail, the apoptotic pathways are activated, leading ultimately to cell death [9, 10]. In addition to assisting oxidative folding of nascent polypeptides, PDI enzymes are also involved in holding, refolding, and degradation of unfolded or misfolded proteins under stressful conditions [10]. Furthermore, since the blocking of PDI activity could lead to protein misfolding, prolonged proteotoxic stress and apoptosis [11, 12] considering PDI as a therapeutic target to stop the progression of some diseases seem plausible [13, 14].
Human amebiasis, the parasitic infection caused by the protozoan Entamoeba histolytica, is a prevalent infection in developing countries [15]. Interestingly, the continued secretion of proteins, including virulence factors, is well recognized as the primary feature of this parasite [16]. Moreover, protein folding and correct disulfide bond formation are essential for secreted virulence factors, such as the Gal/GalNAc-inhibitable lectin [17] and the pore-forming peptide A (amoebapore A) [18].
The E. histolytica genome has 11 genes encoding PDI homologues [19]. From these, the enzyme named EhPDI (a 38 kDa polypeptide with two active thioredoxin-like domains) has been used as a model to study protein folding and disulfide bond formation in E. histolytica. By using in vitro assays and standard substrates (such as insulin, lysozyme, and ribonuclease A), we have confirmed that EhPDI exhibits the distinctive oxidoreductase activities (reductase, oxidase, and isomerase) as well as the typical chaperone-like function (suppression of polypeptide aggregation) [20, 21]. Only the oxidase activity has been demonstrated in vivo, through functional complementation of the dsbA mutation in E. coli [22].
Here, to test whether EhPDI exhibits isomerase activity in vivo, we performed a functional complementation assay using the ΔdsbC mutant of E. coli as a model and the defective expression of the periplasmic protein AppA as the phenotype. The acid phosphatase-phytase enzyme (AppA) has three consecutive disulfide bonds and one nonconsecutive that renders it dependent on DsbC [23]. Our preliminary results showed that EhPDI exhibits isomerase activity; however, further mutagenic analysis revealed significant differences in the functional role of each thioredoxin-like domain. Finally, additional studies confirmed that EhPDI protects two heat-labile enzymes, α-glucosidase and NdeI endonuclease, against thermal inactivation, extending our knowledge about its chaperone-like activity.
2. Materials and Methods
2.1. Materials
DNA amplification reagents and DNA purification kits were from Qiagen (Valencia, CA). Bacterial media were from Becton Dickinson (Franklin Lakes, NJ). Electrophoresis reagents were from Bio-Rad (Hercules, CA). Endonucleases and other enzymes were from New England Biolabs (Ipswich, MA). Other biochemicals were from Sigma-Aldrich (St. Louis, MO), otherwise mentioned in the text. All reagents used were analytical or molecular biology grade.
2.2. Bacterial Strains, Plasmids, and Growth Conditions
Escherichia coli strains and plasmids used in this study are listed in Table 1. Bacterial cultures were grown in LB medium at 37°C, with appropriate antibiotics (ampicillin at 150 μg/mL and chloramphenicol at 15 μg/mL). Recombinant plasmids were constructed by using standard molecular cloning protocols.
Table 1.
Strains and plasmids used in this study.
| Strains or plasmids | Relevant genotype or features | Source or reference |
|---|---|---|
| Strains | ||
| XL1-Blue MRF′ | Δ (mcrA)183 Δ (mcrCB-hsdSMR-mrr)173 endA1 supE44 thi-1 recA1 gyrA96 relA1 lac [F′ proAB lacI q ZΔM15 Tn10 (Tet R)] | Stratagene |
| Shuffle Express | fhuA2 [lon] ompT ahpC gal λatt::pNEB3-r1-cDsbC (Spec R, lacI q)ΔtrxB sulA11 R (mcr-73::miniTn10– Tet S)2 [dcm] R (zgb-210::Tn10– Tet S) endA1 Δgor Δ (mcrC-mrr)114::IS10 | NEB1 |
| BW25113 | F − Δ (araD-araB)567 ΔlacZ4787(::rrnB-3) λ − rph-1 Δ (rhaD-rhaB)568 hsdR514 | CGSC2
[54] |
| JW2861-1 | BW25113 dsbC744::kan | GCSC2
[54] |
| Plasmid | ||
| pBAD33 | Arabinose regulation, p15A origin, CmR | ATCC3
[55] |
| pBAD-AppA | pBAD33-based, periplasmic AppA | This study |
| pBluescript SK- | Lactose regulation, ColE1 origin, AmpR | Stratagene |
| pBAppA | pBluescript-based, periplasmic AppA | This study |
| pBPelB-EhPDI | pBluescript-based, periplasmic EhPDI (wild type) | [22] |
| pBRM05 | pBluescript-based, periplasmic EhPDISS/CC (C44S; C47S) | [20] |
| pBRM06 | pBluescript-based, periplasmic EhPDICC/SS (C160S; C163S) | [20] |
| pBRM15 | pBluescript-based, periplasmic EhPDISS/SS (C44S; C47S; C160S; C163S) | [20] |
| pQE30 | Lactose regulation, ColE1 origin, AmpR | Qiagen |
| pQHPDI | pQE30-based, recombinant EhPDI (wild type) | [20] |
| pQRM05 | pQE30-based, recombinant EhPDISS/CC (C44S; C47S) | This study |
| pQRM06 | pQE30-based, recombinant EhPDICC/SS (C160S; C163S) | This study |
| pQRM15 | pQE30-based, recombinant EhPDISS/SS (C44S; C47S; C160S; C163S) | This study |
1New England Biolabs; 2Coli Genetic Stock Center; 3American Type Culture Collection.
2.2.1. Construction of pBAD-AppA Plasmid
Full-length sequence of the bacterial appA gene was amplified from genomic DNA (XL1-Blue MRF′ strain), using the synthetic oligonucleotides EcAppAF (5′-cgc gcg gaa ttc ATG AAA AGC GGA AAC ATA TCG-3′) and EcAppAR (5′-cgc gcg tct aga TTA CAA ACT GCA CGC CGG TAT-3′) as primers. The PCR product was then digested with EcoRI and XbaI endonucleases and cloned into the EcoRI-XbaI sites of pBluescript SK-, yielding the pBAppA plasmid. To obtain the pBAD-AppA plasmid, a site for EcoRV (located immediately upstream of the EcoRI) was used to get a restriction fragment from EcoRV-XbaI sites, which was then subcloned into SmaI-XbaI sites of pBAD33. The appA gene was confirmed by DNA sequencing.
2.2.2. Construction of the pQRM05, pQRM06, and pQRM15 Plasmids
The EhPDI gene variants (with Cys to Ser substitutions) were amplified from its corresponding pBluescript-based plasmid (Table 1), using the synthetic oligonucleotides EhPDIp38F (5′-cat cac gga tcc GCT GAT GTA GTA TCA TTA AAT C-3′) and M13FW (5′-GTA AAA CGA CGG CCA GTG-3′) as primers. Then, PCR products were digested with BamHI and HindIII endonucleases and subcloned into the same sites of pQE30 (in frame with the sequence encoding the N-terminal hexahistidine tag). The EhPDI gene variants were confirmed by DNA sequencing.
2.3. DsbC Complementation and AppA Activity Assay
2.3.1. Periplasmic Expression of AppA and Coexpression with EhPDI
E. coli strains BW25113 (wild type) and JW2861-1 (ΔdsbC mutant) were transformed with pBAD33 (as control) or pBAD-AppA. Stable transformants were cultured in LB medium, supplemented with chloramphenicol, and the periplasmic expression of AppA was induced with 0.2% arabinose. Bacterial cell pellets (from 1 mL) were obtained by centrifugation (2 min at 10,000 rpm).
E. coli strain JW2861-1 harboring pBAD-AppA was transformed with pBluescript-based plasmids expressing EhPDI variants (Table 1). The plasmid pBluescript SK- was used as a control. Stable cotransformants were cultured in LB medium, supplemented with ampicillin and chloramphenicol, and the periplasmic coexpression of AppA and EhPDI was induced with 0.2% arabinose and 1 mM IPTG. Bacterial cell pellets were obtained as before.
2.3.2. Acid Phosphatase Activity Assay
The acid phosphatase activity was determined by a colorimetric assay [23]. Bacterial cell pellets were resuspended in glycine buffer (0.25 M; pH 2.5) and adjusted to 0.3–0.6 units of A 600 per mL. Then, 20 μL aliquots were further diluted with 80 μL of the same buffer and mixed with 100 μL of 50 mM p-nitrophenyl phosphate. After 15 min of incubation at 37°C, reactions were stopped by adding 1 mL of 1.2 N NaOH. Immediately, supernatants were separated by centrifugation (5 min at 14,500 rpm) and the released p-nitrophenolate was quantified by measuring the A 420. Light scattering by cellular debris was also considered (recording the A 550). The acid phosphatase activity was expressed in Miller units [23, 24].
2.4. Purification of Recombinant EhPDI Enzymes
E. coli strain Shuffle Express was transformed with pQE30-based plasmids expressing recombinant EhPDI enzyme variants (see Table 1). Stable transformants were cultured in LB medium, supplemented with ampicillin, and protein expression was induced with 0.1 mM IPTG. Bacterial cells (from 100 mL) were harvested and lysed under native conditions, using the CelLytic B Plus Kit (Sigma-Aldrich). From the soluble fraction, recombinant proteins were purified by Ni-affinity chromatography (The QIAexpressionist, Qiagen). Eluate fractions were analyzed by SDS-PAGE and those containing more than 95% of pure protein were pooled and concentrated/desalted by ultrafiltration, using a Microsep UF Spin Filter (Pall Co.). Protein concentration was determined by performing the BCA colorimetric assay (Sigma-Aldrich), using BSA as standard.
2.5. Oxidative Refolding Assay
Oxidative refolding of denatured-reduced ribonuclease A (drRNAse) by recombinant amebic PDI enzymes was assayed by following a reported protocol [25]. Refolding was achieved by diluting drRNAse (7.8 μM) into a reaction buffer (2 mM GSH, 0.4 mM GSSG, 100 mm Tris-HCl, pH 8.0) containing 5 μM of amebic PDI enzymes and 4.5 mM of cCMP (RNAse substrate). The reactivation of RNAse was followed for 60 min by recording the absorbance at 296 nm. Active RNAse (μM) was calculated from the first derivative of the absorbance over time and corrected for the depletion of the substrate and the formation of the product (CMP, RNAse inhibitor) [25]. The isomerase activity was determined from the linear increase of active RNAse over time (μM/min), after the lag phase (which reflects the oxidase activity, min−1).
2.6. Disulfide Reductase Assay
Disulfide reduction of bovine insulin catalyzed by recombinant amebic PDI enzymes was assayed according to a standard turbidity method [21, 26]. Recombinant enzymes (2 μM final) were added to a reaction buffer (2 mM EDTA and 100 mM HEPES; pH 7.0) containing bovine insulin (100 μM final). Disulfide reduction was started by adding DTT (0.3 mM final) and followed for 90 min by recording the A 650 every 5 min. Reductase activity was determined from the linear increase of absorbance over time after the lag phase (A 650/min2) [27].
2.7. Chaperone Activity Assays
Chaperone-like activity of EhPDI was evaluated by performing a protection against thermal inactivation assay of two heat-labile enzymes: α-glucosidase [28, 29] and NdeI endonuclease [30, 31].
2.7.1. Thermal Inactivation of α-Glucosidase
Different concentrations of EhPDI (0–5 μM final) were added to a reaction buffer (50 mM KH2PO4; pH 6.8) containing yeast α-glucosidase (16 μg/mL final). Thermal inactivation was performed by incubating at 43°C for 60 min (a control without treatment was carried out for each concentration). Then, reactions were cooled on ice for 1 min and the aggregated protein was separated by centrifugation (5 min at 14,500 rpm). α-glucosidase activity was determined by diluting a 40 μL aliquot of the supernatant with 160 μL of reaction buffer containing 0.125 mM of reduced glutathione and 1.25 mM of p-nitrophenyl-α-D-glucopyranoside. After 20 min of incubation at room temperature, reactions were stopped by adding 50 μL of 0.5 M Na2CO3. The released p-nitrophenolate was quantified by measuring the absorbance at 415 nm. The α-glucosidase activity (AG) was defined by the increase in absorbance over time (A 415/min). For each concentration of EhPDI, the percentage of protection against thermal inactivation (chaperone-like activity) was determined by using the following equation: protection (%) = (AG T/AG U) × 100, where AG T represents the remaining α-glucosidase activity after thermal treatment (60 min at 43°C), while the untreated enzyme is represented by AG U.
2.7.2. Thermal Inactivation of NdeI Endonuclease
Different concentrations of EhPDI (0–2 μM final) were added to a reaction buffer (75 mM potassium acetate, 30 mM Tris-acetate, 15 mM magnesium acetate, 1.5 mM DTT; pH 7.9) containing NdeI endonuclease (1 U/μL final). Thermal inactivation was performed by incubating at 50°C for 30 min (control reactions without incubation were carried out). Then, the reactions were cooled on ice for 2 min and briefly centrifuged (to collect a 10 μL volume). Endonucleolytic activity was determined by adding 5 μL of a plasmid solution, containing 0.1 μg of pUC19 and 0.1 mg of BSA. After 2 hours of incubation at 37°C, standard agarose gel electrophoresis and ethidium bromide staining were used to analyze the restriction fragments. The NdeI endonuclease activity was defined by the relative amount of linearized plasmid, estimated by digital densitometry. For each concentration of EhPDI, the percentage of protection against thermal inactivation (chaperone-like activity) was determined by using the following equation: protection (%) = [(EN T − C2)/(C1 − C2)] × 100, where EN T represents the remaining NdeI endonuclease activity after thermal treatment (30 min at 50°C), while C1 (with NdeI) and C2 (without NdeI) correspond to the control reactions without treatment.
2.8. Statistical Analysis
Unless otherwise mentioned, activity data were from three independent experiments and are represented as mean ± standard error. All statistical analysis were performed using Prism v.5 (GraphPad Software, San Diego, CA). Unpaired t-test was used for routine comparison of data sets. P values less than 0.05 were considered statistically significant.
3. Results and Discussion
3.1. EhPDI Exhibits In Vivo Isomerase Activity
To study the functional activities of eukaryotic PDI enzymes in vivo, yeast and bacterial cells have been successfully used to complement phenotypes associated with defective formation of disulfide bonds [32, 33]. In E. coli cells, the oxidative folding of polypeptides is carried out in the periplasmic compartment and performed by the Dsb proteins: oxidation and isomerization of disulfide bonds are catalyzed by DsbA and DsbC, respectively [34].
The DsbC protein is particularly notable since it shares structural and functional similarities with eukaryotic PDI enzymes [35]. In fact, its functional role as disulfide isomerase has been studied using eukaryotic multidisulfide proteins as substrates [35, 36]. Four physiological substrates of DsbC have been identified so far: AppA [23], RcsF [37], MepA, and RNAse I [38]; from these, in vivo studies using AppA as substrate protein showed that DsbC plays an important role during folding of proteins with nonconsecutive disulfide bonds [23]. Then, to test whether EhPDI exhibits disulfide isomerase activity in vivo, we performed a functional complementation assay using the ΔdsbC mutant of E. coli as a model and the defective periplasmic expression of AppA as the phenotype.
Initially, the absence of acid phosphatase activity (as background) was confirmed, indicating that the chromosomal appA gene was not induced under our experimental conditions (data not shown). Then, pBAD-AppA (Table 1) was used to transform both wild type and ΔdsbC mutant strains. Stable transformants were cultured and AppA expression was induced properly. As noted in Figure 1, a high level of acid phosphatase activity was detected in the wild type strain, 9595 ± 593 Miller units, whereas a low level of activity was observed in the ΔdsbC mutant (1593 ± 70 Miller units), confirming the DsbC-dependence of AppA (Figure 1). Then, pBPelB-EhPDI (Table 1) was used to transform the ΔdsbC/pBAD-AppA strain, using a two-plasmid system [39]. Stable cotransformants were cultured and protein expression was induced accordingly. Periplasmic expression of EhPDI was confirmed by immunoblot (data not shown). Interestingly, a significant increase in acid phosphatase activity was detected (5151 ± 344 Miller units), suggesting that the correct disulfide bond formation of AppA was assisted by the isomerase activity of EhPDI.
Figure 1.
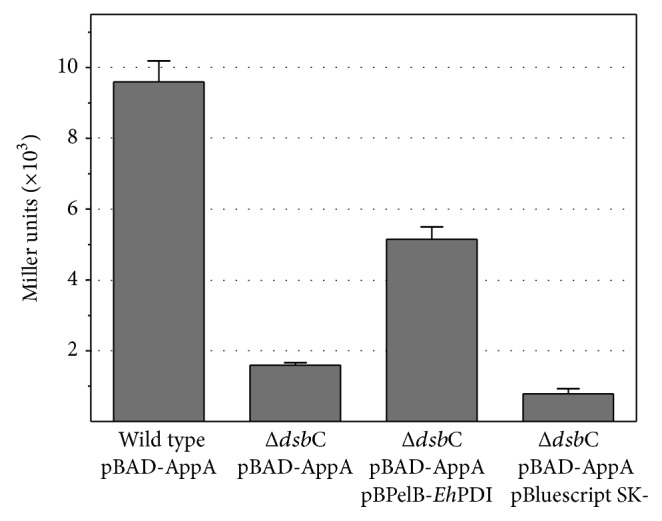
Acid phosphatase activity when AppA was expressed in the wild type and ΔdsbC mutant strains, as well as when it was coexpressed with EhPDI in the ΔdsbC mutant strain. The activity (expressed in Miller units) is shown on the left. The plasmids used for transfection of E. coli are also indicated.
3.2. Isomerase Activity of EhPDI Is Limited and Dependent on Its Reductase Activity
The active site of the thioredoxin-like domains from DsbA, DsbC, and PDI enzymes is characterized by the presence of the motif CXXC, where the cysteine residues play an important role in the enzymatic activity [40, 41]. EhPDI contains two thioredoxin-like domains (referred to as N- and C-Trx, resp.; both having the motif CGHC) that are essential for its in vivo oxidase activity [20]. To test whether both domains contribute to the isomerase activity, we carried out mutagenic analysis followed by a functional complementation assay.
The pBluescript-based plasmids expressing EhPDI variants (Table 1) were used to transform the ΔdsbC/pBAD-AppA strain. Stable cotransformants were cultured and protein expression was induced accordingly. Periplasmic expression of EhPDI variants was confirmed by immunoblot (data not shown). As indicated in Table 2, the isomerase activity of EhPDI is dependent on its CGHC active sites, since a complete loss of the AppA activity was observed when the variant having both domains inactivated was coexpressed (EhPDISS/SS). Also, low AppA activity was detected when each domain was tested without the background of the other (EhPDISS/CC and EhPDICC/SS); furthermore, the slight difference observed between these two variants can be explained by considering that the thioredoxin-like domains are not equivalent with regard to the isomerase activity [33, 42]. However, it is important to take into account the cellular features of the bacterial model to better understand the role of EhPDI as an isomerase in vivo.
Table 2.
In vivo isomerase activity of EhPDI enzyme variants.
| Enzyme variant | Acid phosphatase (AppA)∗ activity in Miller units (%)¶ |
|---|---|
| EhPDISS/SS | 732 ± 74 (−1) |
| EhPDISS/CC | 1086 ± 136 (7) |
| EhPDICC/SS | 1646 ± 230 (20) |
∗The data was expressed as ± standard error (n = 6).
¶Under the background of ΔdsbC/pBAD-AppA, normalization was performed considering the mean activity of pBPelB-EhPDI as maximal (A max = 5151) and pBluescript SK- as minimal (A min = 780). The percentage (%) was calculated as [(A − A min)/(A max − A min)] × 100, where A represents the activity of the enzyme variant.
In the periplasmic compartment, substrate proteins with misoxidized disulfide bonds are shuffled to properly oxidized states by two mechanisms: (1) the isomerase pathway, where DsbC acts on the substrate as reductase-oxidase, and (2) the reductase pathway, where DsbC simply acts as reductase, allowing DsbA another chance to correctly oxidize the substrate [43]. In addition, when the ΔdsbC mutant of E. coli was complemented with the protein TrxP from Bacteroides fragilis (a periplasmic reductase with poor isomerase activity), a fully restored AppA activity was observed, indicating that the disulfide bond isomerization of this substrate is accomplished mainly through the reductase pathway [43]. Hence, it is reasonable to think that the low AppA activity detected when the ΔdsbC mutant of E. coli was complemented with any of the variants (Table 2) suggests that the isomerase function of EhPDI is dependent on its reductase activity. To test this, we performed two in vitro activity assays, oxidative refolding of ribonuclease and reduction of insulin, using purified recombinant enzymes, that is, EhPDI variants.
As indicated in Table 3, the wild type (EhPDI) and variants (EhPDISS/CC and EhPDICC/SS) showed comparable oxidative refolding capabilities. In contrast, significant differences were observed in their reductive activities: EhPDICC/SS retained about 60%, whilst EhPDISS/CC roughly retained 20%. These results confirmed that EhPDI is dependent on its reductase activity to function as oxidoreductase in vivo and in vitro.
Table 3.
In vitro activities of purified EhPDI enzyme variants.
| Enzyme | RNAse A oxidative refolding∗†¶ |
Insulin reduction∗†¶
[×10−6 A 650/min2] (%) |
|
|---|---|---|---|
| Oxidase [×10−6 min−1] (%) |
Isomerase [×10−6 μM/min] (%) |
||
| EhPDI | 513 ± 16 (100) | 39 ± 1 (100) | 109 ± 11 (100) |
| EhPDISS/CC | 551 ± 16 (107) | 33 ± 1 (85) | 22 ± 2 (20) |
| EhPDICC/SS | 517 ± 13 (101) | 31 ± 1 (79) | 65 ± 5 (60) |
∗The data was expressed as mean ± standard error (n = 3).
†The activity of the enzyme variant EhPDISS/SS was not statistically significant, as compared with the reaction performed in the absence of enzyme.
¶The activity ratio (%) was calculated as (variant/wild type) × 100.
3.3. EhPDI Protects Proteins against Thermal-Induced Aggregation
The chaperone-like function of PDI enzymes is determined by their ability to protect misfolded/unfolded substrate proteins against thermal-induced aggregation and to assist refolding [44, 45]. This function is essential in order for PDI to act as an efficient folding catalyst, since it allows access to buried thiols and disulfide bonds in the substrates and prevents nonspecific interactions between partially folded intermediates [1]. Typically, the chaperone-like function has been studied in vitro by measuring the ability to prevent protein aggregation induced by different physical or chemical conditions, such as temperature or denaturants [5, 46].
We have already reported that EhPDI exhibits chaperone-like function by showing its ability to prevent the DTT-induced aggregation of the B chain of insulin [21]. Although this assay was a simple approach to test the chaperone-like function of EhPDI, the low molecular mass of the substrate (3.4 kDa) represents a limitation of this assay, since it offers a restricted number of contact sites to form stable complexes [47]. Hence, to gain further insights regarding the chaperone-like function of EhPDI, we performed additional in vitro assays to test its ability to prevent thermal-induced aggregation, using as substrates two heat-labile enzymes: α-glucosidase and NdeI endonuclease.
As shown in Figures 2 and 3, the chaperone-like function of EhPDI was dose-dependent, since an increment of activity was observed as a result of augmenting its concentration. Moreover, to estimate its chaperone-like ability, the half-maximal effective concentration (EC50) and Hill slope were calculated by fitting the data to a model of one specific binding site with a variable slope. Interestingly, the apparent values obtained for α-glucosidase (EC50 = 3.0 ± 0.4 μM; Hill slope = 1.0) and NdeI endonuclease (EC50 = 0.26 ± 0.06 μM; Hill slope = 2.4) suggest that EhPDI exhibits differences in substrate specificity and affinity [48].
Figure 2.

Protection of thermal inactivation of α-glucosidase assisted by EhPDI. Relative chaperone-like activity (%) of recombinant EhPDI.
Figure 3.
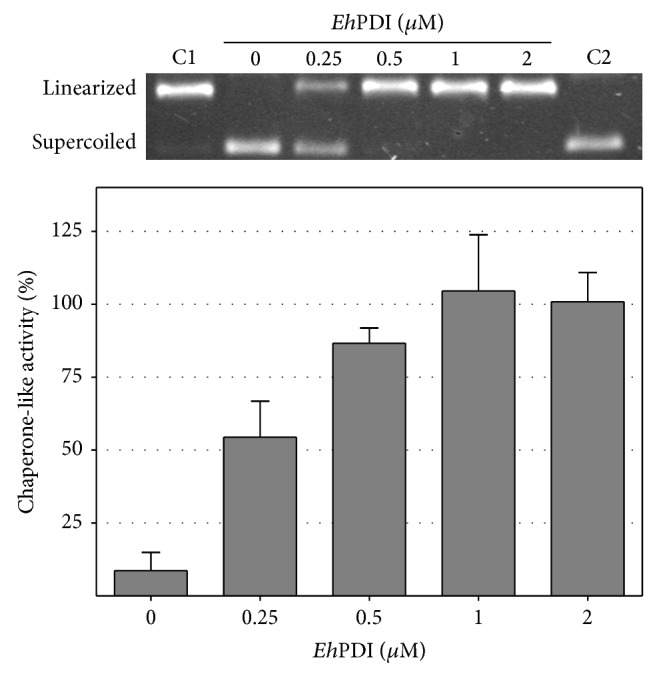
Protection of thermal inactivation of NdeI endonuclease assisted by EhPDI. Relative chaperone-like activity (%) of recombinant EhPDI. Upper panel: agarose gel indicating the relative mobility of the linearized and supercoiled plasmid (C1 and C2); also, concentrations of EhPDI are indicated.
Although EhPDI does not have a substrate-binding b′-domain (a-a′-D) as the mammalian homologue (a-b-b′-a′-c) [49], the notion that some other domains (e.g., a′ or D) might be involved in the chaperone activity is evident. This idea is supported by the results of two previous reports: (i) ERp46, which lacks a b′-domain, is able to bind peptides through its catalytic domains (a0, a, and a′) [50] and (ii) the D-domain of ERp29 contains a discrete and conserved substrate-binding site [51].
3.4. Closing Remarks
Although little is known about the E. histolytica mechanisms that act in response to proteotoxic stress [52], the upregulation of genes encoding typical chaperones (such as Hsp70 and Hsp90) in a response to thermal stress suggests that it contains the cellular machinery necessary to preserve and restore the stability of the proteome [53]. So, the identification and characterization of EhPDI as a folding catalyst with chaperone-like activity represents an additional step to dissect the molecular mechanisms involved in both protein folding and proteotoxic stress in E. histolytica. Hence, it is conceivable to suppose that inhibition of EhPDI could lead to an increase in protein misfolding, promoting a sustained proteotoxic stress, eventually inducing apoptosis and, thus, preventing infection by this parasite.
Acknowledgments
This work was supported in part by Grants from PROMEP (NPTC-103.5/11/3713 to Rosa E. Mares), UABC (CGPI-CI17/3878 to Samuel G. Meléndez-López), and CONACYT (CB-2010/01/155714 and SSA/IMSS/ISSSTE-2011/01/161544 to Marco A. Ramos). Rosa E. Mares, Samuel G. Meléndez-López, and Marco A. Ramos are National Researchers (SNI-CONACYT, Mexico) and members of the Biological-Pharmaceutical Academic Group (Health Sciences, UABC).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Imaoka S. Chemical stress on protein disulfide isomerases and inhibition of their functions. International Review of Cell and Molecular Biology. 2011;290:121–166. doi: 10.1016/B978-0-12-386037-8.00003-X. [DOI] [PubMed] [Google Scholar]
- 2.Inaba K. Structural basis of protein disulfide bond generation in the cell. Genes to Cells. 2010;15(9):935–943. doi: 10.1111/j.1365-2443.2010.01434.x. [DOI] [PubMed] [Google Scholar]
- 3.Sevier C. S., Kaiser C. A. Conservation and diversity of the cellular disulfide bond formation pathways. Antioxidants & Redox Signaling. 2006;8(5-6):797–811. doi: 10.1089/ars.2006.8.797. [DOI] [PubMed] [Google Scholar]
- 4.Denoncin K., Collet J.-F. Disulfide bond formation in the bacterial periplasm: major achievements and challenges ahead. Antioxidants and Redox Signaling. 2013;19(1):63–71. doi: 10.1089/ars.2012.4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu S., Sankar S., Neamati N. Protein disulfide isomerase: a promising target for cancer therapy. Drug Discovery Today. 2014;19(3):222–240. doi: 10.1016/j.drudis.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 6.Hatahet F., Ruddock L. W. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxidants and Redox Signaling. 2009;11(11):2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 7.Selles B., Jacquot J.-P., Rouhier N. Comparative genomic study of protein disulfide isomerases from photosynthetic organisms. Genomics. 2011;97(1):37–50. doi: 10.1016/j.ygeno.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Galligan J. J., Petersen D. R. The human protein disulfide isomerase gene family. Human Genomics. 2012;6, article 6 doi: 10.1186/1479-7364-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urra H., Dufey E., Lisbona F., Rojas-Rivera D., Hetz C. When ER stress reaches a dead end. Biochimica et Biophysica Acta: Molecular Cell Research. 2013;1833(12):3507–3517. doi: 10.1016/j.bbamcr.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 10.Niforou K., Cheimonidou C., Trougakos I. P. Molecular chaperones and proteostasis regulation during redox imbalance. Redox Biology. 2014;2(1):323–332. doi: 10.1016/j.redox.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee S., Min Kim S., Dotimas J., et al. Thioredoxin-interacting protein regulates protein disulfide isomerases and endoplasmic reticulum stress. EMBO Molecular Medicine. 2014;6(6):732–743. doi: 10.15252/emmm.201302561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muller C., Bandemer J., Vindis C., et al. Protein disulfide isomerase modification and inhibition contribute to ER stress and apoptosis induced by oxidized low density lipoproteins. Antioxidants and Redox Signaling. 2013;18(7):731–742. doi: 10.1089/ars.2012.4577. [DOI] [PubMed] [Google Scholar]
- 13.Jasuja R., Passam F. H., Kennedy D. R., et al. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. The Journal of Clinical Investigation. 2012;122(6):2104–2113. doi: 10.1172/JCI61228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu S., Butkevich A. N., Yamada R., et al. Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(40):16348–16353. doi: 10.1073/pnas.1205226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ximénez C., Morán P., Rojas L., Valadez A., Gómez A. Reassessment of the epidemiology of amebiasis: state of the art. Infection, Genetics and Evolution. 2009;9(6):1023–1032. doi: 10.1016/j.meegid.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Olivos-García A., Saavedra E., Ramos-Martínez E., Nequiz M., Pérez-Tamayo R. Molecular nature of virulence in Entamoeba histolytica . Infection, Genetics and Evolution. 2009;9(6):1033–1037. doi: 10.1016/j.meegid.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Barroso L., Abhyankar M., Noor Z., et al. Expression, purification, and evaluation of recombinant LecA as a candidate for an amebic colitis vaccine. Vaccine. 2014;32(10):1218–1224. doi: 10.1016/j.vaccine.2013.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hecht O., van Nuland N. A., Schleinkofer K., et al. Solution structure of the pore-forming protein of Entamoeba histolytica . Journal of Biological Chemistry. 2004;279(17):17834–17841. doi: 10.1074/jbc.M312978200. [DOI] [PubMed] [Google Scholar]
- 19.Ramos M. A., Mares R. E., Magaña P. D., Ortega J. E., Cornejo-Bravo J. M. In silico identification of the protein disulfide isomerase family from a protozoan parasite. Computational Biology and Chemistry. 2008;32(1):66–70. doi: 10.1016/j.compbiolchem.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Mares R. E., Magaña P. D., Meléndez-López S. G., Licea A. F., Cornejo-Bravo J. M., Ramos M. A. Oxidative folding and reductive activities of EhPDI, a protein disulfide isomerase from Entamoeba histolytica . Parasitology International. 2009;58(3):311–313. doi: 10.1016/j.parint.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 21.Ramos M. A., Mares R. E., Magaña P. D., Rivas I. D., Meléndez-López S. G. Entamoeba histolytica: biochemical characterization of a protein disulfide isomerase. Experimental Parasitology. 2011;128(1):76–81. doi: 10.1016/j.exppara.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Ramos M. A., Sanchez-Lopez R., Mares R. E., Olvera F., Alagón A. Identification of an Entamoeba histolytica gene encoding a protein disulfide isomerase that functionally complements the dsbA mutation in Escherichia coli . Molecular and Biochemical Parasitology. 2005;143(2):236–240. doi: 10.1016/j.molbiopara.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 23.Berkmen M., Boyd D., Beckwith J. The nonconsecutive disulfide bond of Escherichia coli phytase (AppA) renders it dependent on the protein-disulfide isomerase, DsbC. The Journal of Biological Chemistry. 2005;280(12):11387–11394. doi: 10.1074/jbc.M411774200. [DOI] [PubMed] [Google Scholar]
- 24.Miller J. H. A Short Course in Bacterial Genetics. Cold Spring Harbor, NY, USA: CSHL Press; 1992. [Google Scholar]
- 25.Wilkinson B., Xiao R., Gilbert H. F. A structural bisulfide of yeast protein-disulfide isomerase destabilizes the active site disulfide of the N-terminal thioredoxin domain. Journal of Biological Chemistry. 2005;280(12):11483–11487. doi: 10.1074/jbc.M414203200. [DOI] [PubMed] [Google Scholar]
- 26.Holmgren A. Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. The Journal of Biological Chemistry. 1979;254(19):9627–9632. [PubMed] [Google Scholar]
- 27.Martinez-Galisteo E., Padilla C. A., Garcia-Alfonso C., Lopez-Barea J., Barcena J. A. Purification and properties of bovine thioredoxin system. Biochimie. 1993;75(9):803–809. doi: 10.1016/0300-9084(93)90131-B. [DOI] [PubMed] [Google Scholar]
- 28.Stromer T., Ehrnsperger M., Gaestel M., Buchner J. Analysis of the interaction of small heat shock proteins with unfolding proteins. The Journal of Biological Chemistry. 2003;278(20):18015–18021. doi: 10.1074/jbc.M301640200. [DOI] [PubMed] [Google Scholar]
- 29.Kern R., Malki A., Holmgren A., Richarme G. Chaperone properties of Escherichia coli thioredoxin and thioredoxin reductase. Biochemical Journal. 2003;371(3):965–972. doi: 10.1042/BJ20030093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suragani M., Aadinarayana V. D., Pinjari A. B., et al. Human resistin, a proinflammatory cytokine, shows chaperone-like activity. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(51):20467–20472. doi: 10.1073/pnas.1306145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hess J. F., FitzGerald P. G. Protection of a restriction enzyme from heat inactivation by [alpha]-crystallin. Molecular Vision. 1998;4(article 29) [PubMed] [Google Scholar]
- 32.Kimura T., Hosoda Y., Kitamura Y., Nakamura H., Horibe T., Kikuchi M. Functional differences between human and yeast protein disulfide isomerase family proteins. Biochemical and Biophysical Research Communications. 2004;320(2):359–365. doi: 10.1016/j.bbrc.2004.05.178. [DOI] [PubMed] [Google Scholar]
- 33.Stafford S. J., Lund P. A. Mutagenic studies on human protein disulfide isomerase by complementation of Escherichia colidsbA and dsbC mutants. FEBS Letters. 2000;466(2-3):317–322. doi: 10.1016/S0014-5793(99)01728-7. [DOI] [PubMed] [Google Scholar]
- 34.Rietsch A., Belin D., Martin N., Beckwith J. An in vivo pathway for disulfide bond isomerization in Escherichia coli . Proceedings of the National Academy of Sciences of the United States of America. 1996;93(23):13048–13053. doi: 10.1073/pnas.93.23.13048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rietsch A., Bessette P., Georgiou G., Beckwith J. Reduction of the periplasmic disulfide bond isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. Journal of Bacteriology. 1997;179(21):6602–6608. doi: 10.1128/jb.179.21.6602-6608.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bessette P. H., Qiu J., Bardwell J. C. A., Swartz J. R., Georgiou G. Effect of sequences of the active-site dipeptides of DsbA and DsbC on in vivo folding of multidisulfide proteins in Escherichia coli . Journal of Bacteriology. 2001;183(3):980–988. doi: 10.1128/JB.183.3.980-988.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leverrier P., Declercq J.-P., Denoncin K., et al. Crystal structure of the outer membrane protein RcsF, a new substrate for the periplasmic protein-disulfide isomerase DsbC. The Journal of Biological Chemistry. 2011;286(19):16734–16742. doi: 10.1074/jbc.M111.224865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hiniker A., Bardwell J. C. A. In vivo substrate specificity of periplasmic disulfide oxidoreductases. Journal of Biological Chemistry. 2004;279(13):12967–12973. doi: 10.1074/jbc.M311391200. [DOI] [PubMed] [Google Scholar]
- 39.Skowronek K., Kasprzak A. A. A two-plasmid system for independent genetic manipulation of subunits of homodimeric proteins and selective isolation of chimeric dimers. Analytical Biochemistry. 2002;300(2):185–191. doi: 10.1006/abio.2001.5456. [DOI] [PubMed] [Google Scholar]
- 40.Kadokura H., Beckwith J. Mechanisms of oxidative protein folding in the bacterial cell envelope. Antioxidants and Redox Signaling. 2010;13(8):1231–1246. doi: 10.1089/ars.2010.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karala A. R., Lappi A. K., Ruddock L. W. Modulation of an active-site cysteine pka allows pdi to act as a catalyst of both disulfide bond formation and isomerization. Journal of Molecular Biology. 2010;396(4):883–892. doi: 10.1016/j.jmb.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 42.Lyles M. M., Gilber H. F. Mutations in the thioredoxin sites of protein disulfide isomerase reveal functional nonequivalence of the N- and C-terminal domains. Journal of Biological Chemistry. 1994;269(49):30946–30952. [PubMed] [Google Scholar]
- 43.Shouldice S. R., Cho S.-H., Boyd D., et al. In vivo oxidative protein folding can be facilitated by oxidation-reduction cycling. Molecular Microbiology. 2010;75(1):13–28. doi: 10.1111/j.1365-2958.2009.06952.x. [DOI] [PubMed] [Google Scholar]
- 44.Taylor M., Burress H., Banerjee T., et al. Substrate-induced unfolding of protein disulfide isomerase displaces the cholera toxin A1 subunit from its holotoxin. PLoS Pathogens. 2014;10(2) doi: 10.1371/journal.ppat.1003925.e1003925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tian R., Li S.-J., Wang D.-L., Zhao Z., Liu Y., He R.-Q. The acidic C-terminal domain stabilizes the chaperone function of protein disulfide isomerase. Journal of Biological Chemistry. 2004;279(47):48830–48835. doi: 10.1074/jbc.M407076200. [DOI] [PubMed] [Google Scholar]
- 46.Chaudhuri R., Cheng Y., Middaugh C. R., Volkin D. B. High-throughput biophysical analysis of protein therapeutics to examine interrelationships between aggregate formation and conformational stability. AAPS Journal. 2014;16(1):48–64. doi: 10.1208/s12248-013-9539-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fu X.-M., Zhu B. T. Human pancreas-specific protein disulfide-isomerase (PDIp) can function as a chaperone independently of its enzymatic activity by forming stable complexes with denatured substrate proteins. Biochemical Journal. 2010;429(1):157–169. doi: 10.1042/BJ20091954. [DOI] [PubMed] [Google Scholar]
- 48.Irvine A. G., Wallis A. K., Sanghera N., et al. Protein disulfide-isomerase interacts with a substrate protein at all stages along its folding pathway. PLoS ONE. 2014;9(1) doi: 10.1371/journal.pone.0082511.e82511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bryne L. J., Sidhu A., Wallis A. K., et al. Mapping of the ligand-binding site on the b' domain of human PDI: interaction with peptide ligands and the x-linker region. Biochemical Journal. 2009;423(2):209–217. doi: 10.1042/BJ20090565. [DOI] [PubMed] [Google Scholar]
- 50.Funkner A., Parthier C., Schutkowski M., et al. Peptide binding by catalytic domains of the protein disulfide isomerase-related protein ERp46. Journal of Molecular Biology. 2013;425(8):1340–1362. doi: 10.1016/j.jmb.2013.01.029. [DOI] [PubMed] [Google Scholar]
- 51.Lippert U., Diao D., Barak N. N., Ferrari D. M. Conserved structural and functional properties of D-domain containing redox-active and -inactive protein disulfide isomerase-related protein chaperones. The Journal of Biological Chemistry. 2007;282(15):11213–11220. doi: 10.1074/jbc.M604440200. [DOI] [PubMed] [Google Scholar]
- 52.Santi-Rocca J., Smith S., Weber C., et al. Endoplasmic reticulum stress-sensing mechanism is activated in Entamoeba histolytica upon treatment with nitric oxide. PLoS ONE. 2012;7(2) doi: 10.1371/journal.pone.0031777.e31777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weber C., Guigon G., Bouchier C., et al. Stress by heat shock induces massive down regulation of genes and allows differential allelic expression of the Gal/GalNAc lectin in Entamoeba histolytica . Eukaryotic Cell. 2006;5(5):871–875. doi: 10.1128/EC.5.5.871-875.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baba T., Ara T., Hasegawa M., et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular Systems Biology. 2006;2 doi: 10.1038/msb4100050.2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guzman L.-M., Belin D., Carson M. J., Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose P(BAD) promoter. Journal of Bacteriology. 1995;177(14):4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]