

Abstract
Streptococcus pneumoniae is a common nasopharyngeal resident in healthy people but, at the same time, one of the major causes of infectious diseases such as pneumonia, meningitis, and sepsis. The shift from commensal to pathogen and its interaction with host cells are poorly understood. One of the major limitations for research on pneumococcal-host interactions is the lack of suitable tools for live-cell imaging. To address this issue, we developed a generally applicable strategy to create genetically stable, highly fluorescent bacteria. Our strategy relies on fusing superfolder green fluorescent protein (GFP) or a far-red fluorescent protein (RFP) to the abundant histone-like protein HlpA. Due to efficient translation and limited cellular diffusion of these fusions, the cells are 25-fold brighter than those of the currently best available imaging S. pneumoniae strain. These novel bright pneumococcal strains are fully virulent, and the GFP reporter can be used for in situ imaging in mouse tissue. We used our reporter strains to study the effect of the polysaccharide capsule, a major pneumococcal virulence factor, on different stages of infection. By dual-color live-cell imaging experiments, we show that unencapsulated pneumococci adhere significantly better to human lung epithelial cells than encapsulated strains, in line with previous data obtained by classical approaches. We also confirm with live-cell imaging that the capsule protects pneumococci from neutrophil phagocytosis, demonstrating the versatility and usability of our reporters. The described imaging tools will pave the way for live-cell imaging of pneumococcal infection and help further understanding of the mechanisms of pneumococcal pathogenesis.
INTRODUCTION
Streptococcus pneumoniae is a major cause of morbidity and mortality worldwide, and pneumococcal infections (e.g., pneumonia, septicemia, and meningitis) kill more than 1 million people every year (1). Pneumococci are also quiescent colonizers of the upper respiratory tract, particularly in children, but little is known about the mechanisms underlying the transition from commensal to pathogen. It is therefore of crucial importance to understand the entire pneumococcal pathogenesis cycle in detail.
The polysaccharide capsule covering the cell surface is the most central virulence factor of S. pneumoniae. The involvement of the capsule in pneumococcal pathogenesis has been appreciated since Griffith in 1928 (2) published his famous transformation experiment with rough and smooth strains of S. pneumoniae. Today, it is known that the bulky capsule, which is either negatively charged or neutral, contributes to pathogenesis by protecting pneumococci against the human immune system. For example, the capsule hinders phagocytosis and inhibits complement activity (3–6). Over 90 different pneumococcal serotypes have been identified to date (7), and the different serotypes vary in how well they protect the bacteria against phagocytosis (6). Furthermore, the amount of capsule differs between bacteria, and it has been shown, for example, that strains with thinner capsule adhere better than strains with thick capsule during the initial nasopharyngeal colonization (8). While the capsule is an important virulence factor, molecular epidemiology studies have also shown that nontypeable (unencapsulated) strains are abundant within human populations and act as “hubs” for recombination between pneumococci, driving antibiotic resistance and serotype switching (9). Direct observations of encapsulated and unencapsulated pneumococci in live host-pathogen assays are lacking, and it thus remains unclear how much the capsule contributes to the virulence cycle.
Most of the knowledge concerning pneumococcal interactions with host cells and host tissue we have today has been obtained in vitro by biochemical or immunological assays and in vivo by traditional postinfection plating and CFU counts, as well as by electron microscopy of fixed samples of clinical isolates of S. pneumoniae. To further extend our knowledge about pneumococcal pathogenicity, the key method would be the possibility of imaging interactions between bacteria and host cells in real time. Such a technique will provide understanding of the localization and dynamics of S. pneumoniae during the course of infection and in that manner unravel factors important for the infection process. It will also open up the possibilities to study the role of the capsule in isogenic pneumococci during host attachment and immune evasion. Imaging of bacteria interacting with host cells and host tissue requires labeling to discriminate the bacteria from other cells and the surroundings. In vivo imaging of S. pneumoniae is typically done today using immunofluorescence, where antibodies bound to fluorescent dyes are used to target S. pneumoniae (10, 11), or by live/dead staining (12). However, these techniques do not permit imaging of live cells. Alternatively, bacteria can be stained in vitro prior to the experiment using membrane-permeable fluorescent dyes (13–15). This permits live-cell imaging, but the method is limited by the potential toxicity of the dyes and dilution of the fluorescent signal over time due to either secretion or cell division.
A better solution for live imaging is therefore to use strains that express fluorescence or bioluminescence. In vivo imaging with bioluminescent luciferase (lux) reporters has been used to follow the course of infection of S. pneumoniae in mice (16–19). This is a powerful strategy that allows monitoring of the infection in real time using in vivo imaging systems (IVIS). One of the limitations using this approach is that rather high concentrations of bacteria are required for detection, and single-cell detection is not possible (20). Another important aspect is that luciferase signals, which depend on the expression of five genes (luxCDABE), are emitted only from metabolically active cells. This may be an advantage since only living cells are detected; on the other hand, lux reporters cannot thus be detected after fixation and embedding of animal tissues. The method of choice would therefore be to have strains expressing fluorescent proteins, yet there are only very few examples of such imaging of S. pneumoniae published. These examples include the work of Kadioglu et al. (21), who studied pneumococcal invasion of bronco-epithelial cells in mice, and Ribes et al. (22), who imaged pneumococcal interactions with murine microglial cells. In both of these cases, the S. pneumoniae strains contained a green fluorescent protein (GFP) expressed from a multicopy plasmid. The likely reason for limited use of these strains is the lack of a homogenous and sufficiently bright fluorescent signal being emitted from the pneumococcal cells.
Here, we present bright fluorescent and genetically stable strains of S. pneumoniae constructed using a generally applicable strategy. We show that these fluorescent strains are fully virulent in a mouse model and that they are highly suitable for live imaging of bacterium-host cell interactions. By comparing a wild-type encapsulated strain with an unencapsulated mutant, we show that the polysaccharide capsule protects pneumococci against human neutrophils, but at the same time we show that the encapsulated strain is less efficient in adhering to human epithelial cells. This provides further evidence for the role of the capsule in pneumococcal infection and confirms that, in vivo, capsule production and display must be tightly controlled to provide successful colonization of S. pneumoniae within the human body.
MATERIALS AND METHODS
Bacterial growth conditions and transformation.
S. pneumoniae was grown in liquid casein-based medium with yeast extract (C+Y medium) (23) at 37°C and plated in Columbia agar (Oxoid, Basingstoke, United Kingdom) supplemented with 2% (vol/vol) defibrinated sheep blood (Johnny Rottier, Kloosterzade, The Netherlands). For selection, 4.5 μg/ml chloramphenicol or 250 μg/ml kanamycin was added to the plates. Strains and oligonucleotides are listed in Tables 1 and 2, respectively.
TABLE 1.
S. pneumoniae strains used in this study
| Strain | Descriptiona | Reference or source |
|---|---|---|
| D39 | Serotype 2, encapsulated | 41 |
| JWV500 | D39 hlpA-gfp_Camr | This study |
| MK119 | D39 hlpA_hlpA-rfp_Camr | 28 |
| MK127 | JWV500 Δcps2E::Kanr | This study |
| MK128 | MK119 Δcps2E::Kanr | This study |
| MK147 | D39 hlpA_gfp_Camr | This study |
| P92 | D39 pGFP1 (multicopy GFPmut3 gene) | 21 |
Transcriptional and translational fusions are indicated by an underscore and a hyphen, respectively.
TABLE 2.
Oligonucleotides used in this study
| Oligonucleotide name | Sequence (5′→ 3′)a |
|---|---|
| cam-F+BamHI+SpeI | GCGTGGATCCACTAGTAGGAGGCATATCAAATGAACTTTA |
| cam-R+BlpI | AGCTGCTCAGCTTATAAAAGCCAGTCATTAGG |
| gfp-dsm-link-F+BamHI | CGATGGATCCGGATCTGGTGGAGAAGCTGCAGCTAAAGGATCAAAAGGAGAAGAGCTGTTCACAGG |
| sPG12_camR+NotI | ACGTGCGGCCGCTTATAAAAGCCAGTCATTAG |
| hlpA-up-F | AACAAGTCAGCCACCTGTAG |
| hlpA-up-R+BamHI | CTGCGGATCCTTTAACAGCGTCTTTAAGAGCTTTACCAGC |
| hlpA-down-F+NotI | GACGCGGCCGCACTCAGTCTTTAAAAAGCCTATTGTAT |
| hlpA-down-R | CGTGGCTGACGATAATGAGG |
| hlpA-R-SphI | CGCGCATGCAGACTGATTATTTAACAGCGTC |
| gfp(dsm)_F_rbshlpA_SphI | CGCGCATGCTGGAGGAATCATTAACATGTCAAAAGGAGAAGAGCTGTTCACAGG |
Restriction sites are underlined.
For transformation, S. pneumoniae was grown in C+Y medium (pH 6.8) at 37°C until the optical density at 600 nm (OD600) reached 0.1; then 100 ng/ml synthetic competence-stimulating peptide 1 (CSP-1) was added, and cells were incubated for 12 min at 37°C to activate the transformation machinery. DNA was added to the activated cells, and a 20-min incubation at 30°C followed. Cells were subsequently diluted 10 times in fresh C+Y medium and incubated for 1.5 h at 37°C. Transformants were selected by plating in Columbia blood agar containing the appropriate antibiotics.
Growth curves were monitored using 96-wells plates in a Tecan Infinite 200 PRO microtiter plate reader essentially as described before (24).
Construction of bacterial strains. (i) JWV500 (PhlpA-hlpA-gfp_Camr).
JWV500 (PhlpA-hlpA-gfp_Camr) (in the genotype, the hyphen indicates a translational fusion, whereas the underscore indicates a transcriptional fusion) expresses the histone-like protein HlpA with a superfolder fused to its C-terminal end from the hlpA locus (Fig. 1A). A domain-breaking linker (RGSGGEAAAKAGTS) was inserted between HlpA and superfolder GFP (sfGFP) to give structural flexibility (25). A fragment containing a chloramphenicol resistance gene (cat) was amplified from genomic DNA of strain sPG6 (26) with primers cam-F+BamHI+SpeI and cam-R+BlpI and ligated into the SpeI and BlpI sites of plasmid pKB01 harboring the sfgfp from Bacillus subtilis [pKB01_sfgfp(Bs)] (27) to obtain plasmid pJWV503 with cat located downstream of sfgfp(Bs). The sfgfp(Bs)-cat fragment was then amplified from pJWV503 using primers GFP_DSM-link-F+BamHI (linker sequence introduced in this primer) and sPG12-cam-R. hlpA and the upstream region (hlpA-up) were amplified from genomic DNA of S. pneumoniae D39 using primers hlpA-up-F and hlpA-up-R+BamHI. The region downstream of hlpA (hlpA-down) was amplified from genomic DNA of D39 using primers hlpA-down-F+NotI and hlpA-down-R. The hlpA-up fragment was then cut with BamHI, the sfgfp(Bs)-cam fragment was cut with BamHI and NotI, and the hlpA-down fragment was cut with NotI. The three fragments were ligated and transformed into S. pneumoniae D39. Transformants were selected on Columbia blood agar with chloramphenicol. Correct transformants were verified by PCR and sequencing.
FIG 1.

Generating bright fluorescent pneumococci. (A) Schematic representation of the conserved chromosomal locus of hlpA (SPD_0997) and the reporter strains expressing hlpA-gfp and hlpA-rfp. The chloramphenicol acetyltransferase (cat) gene provides easy selection of chloramphenicol-resistant transformants. The hlpA-promoter (PhlpA) and the transcriptional terminators (lollipops) are indicated. Note that we were not able to generate a strain with hlpA-rfp as the only copy of hlpA, indicating that this fusion is not functional (28). (B) Time-lapse fluorescence microscopy of strains JWV500 and MK119, showing that HlpA-GFP and HlpA-RFP are stably expressed throughout growth and division. Images are overlays of fluorescence signal and phase-contrast micrographs. Scale bar, 5 μm. (C) Growth curves of cells grown in C+Y medium at 37°C showing that growth of the modified S. pneumoniae strains JWV500 and MK119 is similar to that of the wild-type strain D39 in vitro. Averages of three replicates are shown. Error bars show standard deviations.
(ii) MK119 (PhlpA-hlpA_hlpA-rfp_Camr).
Construction of strain MK119 is described elsewhere (28). This strain contains the gene hlpA fused to the far-red fluorescent protein (RFP) mKate2 (hlpA-mKate2; here called hlpA-rfp) and a chloramphenicol resistance gene immediately downstream of the native hlpA gene. The same ribosomal binding site is present upstream of both versions of hlpA.
(iii) MK127 (PhlpA-hlpA-gfp_Camr Δcps2E::Kanr) and MK128 (PhlpA-hlpA_hlpA-rfp_Camr Δcps2E::Kanr).
The strains MK127 and MK128, containing a deletion mutation in the capsule locus, were made by transforming the strains JWV500 and MK119, respectively, with a PCR product causing replacement of the gene cps2E (encoding a glucose phosphotransferase which initiates capsule synthesis) with a kanamycin resistance gene (M. G. Jørgensen and J.-W. Veening, unpublished data). This deletion is similar to a deletion that was described previously by Ramos-Montañez et al. (29).
(iv) MK147 (PhlpA-hlpA_gfp).
In strain MK147, sfgfp(Bs)was inserted downstream of hlpA on the same transcriptional unit. The sequence upstream of sfgfp(Bs) (including the ribosomal binding site) is identical to the upstream sequence of hlpA. The hlpA gene and its upstream region were amplified from genomic DNA of S. pneumoniae D39 using primers hlpA-up-F and hlpA-R-SphI. Furthermore, sfgfp(Bs), cat, and the region downstream of hlpA were amplified from genomic DNA of strain JWV500 using primers gfp(dsm)_F_rbshlpA_SphI and hlpA-down-R. The two fragments were digested with SphI, ligated, and transformed into S. pneumoniae D39. Transformants were selected on Columbia blood agar with chloramphenicol. Correct transformants were verified by PCR and sequencing.
Microscopy of pneumococcal cells. (i) Epifluorescence microscopy.
Epifluorescence microscopy of pneumococcal cells was performed as described previously (28). Briefly, S. pneumoniae was grown to exponential phase (OD600 of 0.15) and spotted onto agarose slides. Microscopy was performed using a DV Elite microscope (Applied Precision, USA) with Solid-State Illumination (Applied Precision) using a scientific complementary metal-oxide-semiconductor (sCMOS) camera with a 100× oil immersion objective. To visualize red fluorescence, an mCherry filter set with 562- to 588-nm excitation and 602- to 648-nm emission wavelengths was used with a quad polychroic mirror (mCherry, 580 to 630 nm). To visualize GFP fluorescence, GFP/fluorescein isothiocyanate (FITC) filters with excitation at 461 to 489 nm and emission at 501 to 559 nm was used with a quad polychroic mirror (GFP, 480 to 540 nm). For comparison of GFP signals between strains, images were acquired using Softworx (Applied Precision) with an exposure time of 0.2 s with 50% excitation light. Quantification of fluorescence signals was done using ImageJ (http://rsb.info.nih.gov/ij/). Relative standard deviation was defined as σp/<p>, where <p> is the mean fluorescence and σp is the standard deviation, and was used as a measure of cell-to-cell variability (30).
(ii) Time-lapse fluorescence microscopy.
Time-lapse fluorescence microscopy of S. pneumoniae was performed with a DV Elite microscope (Applied Precision) as described previously (28, 31). Images were modified for publication using Softworx and ImageJ. Where appropriate, images were deconvolved using Softworx.
(iii) FRAP.
S. pneumoniae cells were grown to an OD600 of 0.2 and immobilized on 1.5% (wt/vol) agarose pads. A fluorescence recovery after photobleaching (FRAP) experiment was performed using a DV Elite microscope (Applied Precision) with a 100× objective equipped with an sCMOS camera and a X4 laser module. The cells were imaged three times before photobleaching and then bleached at 488 nm (50 mW) for 5 ms with 10% laser power. Cells were imaged every second after bleaching with epifluorescence microscopy. Images were analyzed using ImageJ.
Western blotting.
Cells were grown in 4-ml cultures and harvested by centrifugation at 8,500 × g for 5 min when the OD600 reached 0.2. Cells were lysed by resuspending the pellet in 100 μl of SEDS lysis buffer (32) containing 0.02% (wt/vol) SDS, 15 mM EDTA, 0.01% (wt/vol) deoxycholate, and 150 mM NaCl, and the cell suspension was incubated at 37°C for 5 min. Proteins from whole-cell extract were separated by SDS-PAGE (12% [wt/vol] polyacrylamide). One gel was stained with Coomassie to verify that similar protein quantities were loaded for each sample. Proteins were then blotted onto a polyvinylidene fluoride (PVDF) membrane, and GFP proteins and GFP fusion proteins were detected using polyclonal anti-GFP from rabbit (Invitrogen, The Netherlands) as the primary antibody and horseradish peroxidase (HRP)-conjugated anti-rabbit IgG antibody (GE Healthcare, The Netherlands) as the secondary antibody. Protein bands were quantified using ImageLab software (Bio-Rad, USA).
Studies of S. pneumoniae in mouse lung tissue. (i) Ethics statement.
All animal experiments were done at the University of Leicester and were conducted in strict accordance with guidelines of the Home Office of the United Kingdom. The University of Leicester Ethical Committee and the Home Office of the United Kingdom approved the protocol. All mice were scored for signs of disease using the method described by Morton and Griffiths (33). Any mouse that became severely lethargic was culled, in accordance with the Home Office License.
(ii) Mice.
Female MF1 mice were purchased from Charles River Laboratories (United Kingdom) and were acclimatized for 1 week prior to use. Mice used for infection experiments were between 9 and 10 weeks of age.
(iii) Infection.
For intranasal infection, animals were lightly anesthetized with a mixture of O2 and 2.5% (vol/vol) isoflurane (Abbott Laboratories, Maidenhead, United Kingdom) and infected intranasally with an inoculum of 1 × 106 CFU in 50 μl of phosphate-buffered saline (PBS) (34) unless stated otherwise. Mice were regularly monitored for clinical signs of disease (33) and were culled at predetermined time points or if they became severely lethargic. Blood was taken after 24 h from the tail vein for CFU counts.
(iv) Preparation of lung tissue sections.
At necropsy, whole lungs were completely immersed in 22-oxacalcitriol (OCT) embedding matrix (CellPath, Powys, United Kingdom) to slowly freeze (21). Tissue sections (15 μm) were cut using a microtome blade (Bright, Huntingdon, United Kingdom). Unstained sections were permanently preserved with a drop of DPX mounting resin (BDH, Poole, United Kingdom) and a coverslip.
(v) Imaging of lung tissue sections.
Microscopy of lung tissue sections was performed using a DV Elite microscope (Applied Precision) with an sCMOS camera using Solid-State Illumination (Applied Precision) through a 100× oil immersion objective (phase contrast; 1.30 numerical aperture [NA]). Phase-contrast images and GFP fluorescence images (0.5-s exposure time with 100% excitation light) were acquired as z-stacks (28 slices with 0.2 μm between each slice) using Softworx (Applied Precision). Images were modified for publication using Softworx and ImageJ (http://rsb.info.nih.gov/ij/).
(vi) Statistical analysis.
GraphPad Prism, version 5.0, software was used to analyze the data. A log rank (Mantel-Cox) test and a Gehan-Breslow-Wilcoxon test were used to analyze the survival data. An unpaired t test was used to analyze the CFU counts from blood. Results were considered significant at P values of <0.05.
Studies of interactions between S. pneumoniae and human epithelial cells. (i) Cell culture.
The human type II lung epithelial cell line A549 (ATCC CCL-185) was routinely cultured in Dulbecco's modified Eagle medium–nutrient mixture F-12 with GlutaMAX (Life Technologies, The Netherlands) supplemented with 10% (vol/vol) fetal bovine serum (FBS; VWR, The Netherlands) and maintained at 37°C in a humidified 5% (vol/vol) CO2 atmosphere.
(ii) Coincubation of S. pneumoniae and the cell line for fluorescence imaging.
A coincubation experiment was performed as described by Mlacha et al. (35) with some modifications. A549 cells were plated on eight-chamber microscopy slides (μ-slide; Ibidi, Germany), and the monolayer confluence was confirmed by phase-contrast microscopy. Prior to coincubation, the layer was rinsed twice with phosphate-buffered saline (PBS). S. pneumoniae strains were grown in C+Y medium (pH 6.8) until mid-logarithmic phase (OD600 of ∼0.2) and then centrifuged and resuspended in RPMI 1640 medium without phenol red (Life Technologies, The Netherlands) but supplemented with 1% (vol/vol) PBS. Prior to mixing unencapsulated and encapsulated strains (1:1), suspensions of S. pneumoniae (JWV500, MK128, MK119, and MK127) were adjusted to the multiplicities of infection (MOI) of 10 (i.e., 10 bacteria for every A549 cell) and then added onto the A549 monolayer. In order to optimize cell interaction, the slides were centrifuged (at 1,400 × g for 5 min) and then incubated at 37°C in 5% (vol/vol) CO2 for 2 h. To remove nonadhering bacteria, the supernatants were aspirated, and subsequently the slides were washed twice with RPMI 1640 medium supplemented with 1% (vol/vol) FBS.
(iii) Fluorescence imaging.
During the process of imaging, the slides were incubated at 37°C under a humidified 5% (vol/vol) CO2 atmosphere. Imaging was performed on a DV Elite microscope (Applied Precision) with an sCMOS camera using Solid-State Illumination (Applied Precision) through a 60× oil immersion objective (bright field; 1.42 NA; working distance [WD], 0.15 mm). The images were generated by first focusing on the monolayer of A549 using bright-field microscopy and then imaging on the FITC channel (excitation, 475 nm; emission, 523 nm) and Alexa 594 channel (excitation, 575 nm; emission, 625 nm). In order to quantify fluorescence intensity for adherent bacteria, microscopy stacks were split into the three channels: bright field, GFP, and RFP. Next, background signals were removed from the GFP and RFP channels by adjusting their thresholds in ImageJ. Arbitrary values (RFP, minimum 200; GFP, minimum 250) were chosen to remove background fluorescence while retaining signals from bacterial cells. This redefinition of threshold converted the channels into binary images. In each channel, the amount of signal was calculated by multiplying the mean signal value by the area. For each image, the ratio of the RFP to GFP channel was calculated by dividing the RFP signal by GFP signal. Images were modified for publication using softWoRx, version 6.1 (Applied Precision), and ImageJ.
(iv) Coincubation of S. pneumoniae and A549 for CFU counts.
Coincubation of S. pneumoniae mutant strains with the lung epithelial cell line for CFU counting was performed similarly to the coincubation protocol for fluorescence imaging, except that A549 cells were plated on a 24-well plate. After 2 h of incubation, the supernatant was removed, and the cell layer was washed twice with PBS to remove nonadherent pneumococci. Trypsin-EDTA solution was added, and samples were incubated at 37°C for 5 min to dislodge the epithelial layer along with adherent S. pneumoniae. The suspension was centrifuged (at 1,400 × g for 5 min) and resuspended in C+Y medium, diluted, and plated in 2% (vol/vol) blood Columbia agar. After incubation at 37°C overnight, colonies were counted manually. The data shown in Fig. 5B are a collection from three different experiments performed on different days. Statistical analysis was performed using an unpaired two-tailed t test (GraphPad Prism 6).
FIG 5.

Unencapsulated S. pneumoniae cells adhere more efficiently to lung epithelial cells than to encapsulated bacteria. Adhesion of S. pneumoniae reporters on a confluent monolayer of A549, a type II lung epithelial cell line, was imaged using wide-field epifluorescence microscopy. Imaging was performed at 2 h postcoincubation. (A) Schematic overview of the coincubation system, which involves an equimolar mixture of encapsulated and unencapsulated S. pneumoniae strains expressing different fluorescent proteins, on an epithelial A549 monolayer. (B) Classical adherence assay: enumeration of pneumococcal CFU that adhere to A549 2 h after coincubation. The unencapsulated reporter strain, MK128, showed a significantly (P < 0.01) higher propensity to adhere to the lung cell line (7.92 × 106 CFU/well) than to the encapsulated JWV500 reporter strain (average, 6.54 × 104 CFU/well). The adhesion assays were performed with three biological replicates. Panels C to L show the adherence of mixtures of encapsulated and unencapsulated S. pneumoniae reporter strains to human A549 cells, as follows: bright-field micrographs of A549 monolayers (C and H), GFP micrographs (D and I), RFP micrographs (E and J), merged images (F and K), enlarged images of the boxed regions in panels F and K, respectively (G and L). Arrowheads point to encapsulated pneumococci adhering to A549 cells.
Studies of interactions between S. pneumoniae and neutrophils. (i) Growth and imaging conditions.
On the day of the experiment, S. pneumoniae strains JWV500, MK119, MK127, and MK128 were inoculated from a plate and grown in C+Y medium until the OD600 was 0.3. Bacteria were washed and resuspended in RPMI 1640 medium containing 25 mM HEPES, l-glutamine (Lonza Biowhittaker, Basel, Switerland), and 0.05% human serum albumin (HSA; Sanquin, Amsterdam, The Netherlands) (RPMI-HSA). Strain JWV500 was mixed 1:1 with strain MK128, and strain MK119 was mixed 1:1 with strain MK127. Bacteria were diluted in RPMI-HSA medium to a final concentration of 5 × 106 bacteria/ml. Wells of an eight-well Lab-Tek II chambered cover glass (Thermo Scientific, Rochester, USA) were loaded with 200 μl of RPMI-HSA medium containing the mixed S. pneumoniae strains. Subsequently, 5 × 105 freshly isolated neutrophils (36) were added (MOI of 10) to the well, and imaging was started immediately while the neutrophils were settling at the cover glass bottom of the well. Microscopic image acquisition was performed using a Leica TSC SP5 inverted microscope equipped with an HCX PL APO 40× 0.85 objective (Leica Microsystems, The Netherlands). The microscope was encased in a dark-environment chamber that was maintained at 37°C. The cells and bacteria were monitored for GFP (GFP ET filter cube), RFP (N21 filter cube), and bright field every 10 s. To create a time-lapse movie of the interaction between the neutrophils and the bacteria, the separate channels were combined and rendered as a time-lapse movie using Leica LAS AF software. Informed written consent was obtained from all donors and was provided in accordance with the Declaration of Helsinki. Approval was obtained from the medical ethics committee of the University Medical Center Utrecht (Utrecht, The Netherlands).
(ii) Flow cytometry.
S. pneumoniae strains JWV500 and MK127 were grown as described above, and 5 × 106 bacteria were mixed with 5 × 105 freshly isolated neutrophils (MOI of 10) in the presence of 10% normal human pooled serum (15 donors) in a final volume of 100 μl of RPMI-HSA medium. Phagocytosis was initiated at 37°C with shaking for 15 min and subsequently measured by flow cytometry (FACSCalibur; Becton Dickinson, San Jose, CA). Neutrophils were gated based on their forward and side scatter profiles, and the percentage of neutrophils positive for GFP was determined. Under these conditions both ingested bacteria as well as bacteria bound to the neutrophil surface are measured.
RESULTS
Generating brightly fluorescent strains of S. pneumoniae.
Imaging of interactions between live pneumococci and host cells is a problem due to lack of sufficiently bright fluorescent strains. Recently, we have benchmarked a set of fluorescent proteins for the use as promoter fusions in S. pneumoniae (27; K. Beilharz, M. Kjos, R. van Raaphorst, and J.-W. Veening, submitted for publication). The brightest variants were a B. subtilis codon-optimized superfolder GFP, sfGFP(Bs), and an S. pneumoniae codon-optimized far-red fluorescent protein, mKate2 (27; Beilharz et al., submitted). While these reporters generated relatively good fluorescent signals when present as single copies stably integrated in the chromosome and driven by strong promoters, they were still not bright enough to be used in complex host-pathogen experiments in which high levels of experimental autofluorescence are present (data not shown). In general, untagged GFP is difficult to image because the fluorescence signal spreads through the entire cytoplasm by fast diffusion during the image acquisition time and is overwhelmed by cellular autofluorescence (37, 38). To overcome this problem, we fused sfGFP(Bs) and mKate2 (here called gfp and rfp, respectively) to the 3′ end of hlpA (SPD_0997), encoding the only known nucleoid binding protein in Streptococcus (39), and stably integrated the fusions by double crossover in the pneumococcal chromosome at the hlpA locus (Fig. 1A). Besides potentially limiting diffusion of the fluorescent proteins by localizing them to the nucleoid (see below), we have shown previously by high-throughput sequencing of RNA transcripts (RNA-Seq) that hlpA is highly transcribed (40).
The fusion constructs were integrated into the pathogenic encapsulated S. pneumoniae D39 genetic background (41), resulting in strains JWV500 (HlpA-GFP) and MK119 (HlpA-RFP) (Fig. 1A). Note that in strain MK119, the fusion gene is integrated as a second hlpA copy downstream of the native hlpA gene. As shown in Fig. 1B, the reporters displayed very bright, nucleoid localized fluorescence with an average maximum fluorescence 70-fold higher (for JWV500) or 35-fold higher (for MK119) than the strongest GFP and RFP reporters, respectively, from our other studies (27; Beilharz et al., submitted). In fact, we have recently shown that HlpA-RFP can be used as an accurate marker for the nucleoid (28, 40). Importantly, the fluorescent signal remained high during growth and division, as shown by time-lapse microscopy (Fig. 1B), demonstrating that the HlpA fusions are expressed and active at all stages of the S. pneumoniae cell cycle. HlpA is an essential gene in S. pneumoniae D39 (M. Kjos and J.-W. Veening, unpublished data), but no defects in cell morphology (Fig. 1B) or growth (Fig. 1C) were observed in these genetically labeled fluorescent strains, suggesting that the fusion and the chloramphenicol marker had no detrimental effect on hlpA or on downstream genes (the closest downstream gene is located >400 bp away from the construct). Finally, since the HlpA fusions are active in all growth phases, the level of fluorescence can also be used as a proxy for growth in these strains when a microtiter plate reader with appropriate filters is used (data not shown).
HlpA-GFP is efficiently translated and shows low cellular diffusion.
To understand the underlying reason for the bright fluorescence of the HlpA fusions, we first checked if high transcription from the hlpA promoter is responsible. To test this, a construct was made where gfp was integrated downstream of hlpA on the same transcriptional unit and containing the same ribosomal binding site as hlpA, resulting in strain MK147 (Fig. 2A). Quantification of the fluorescence signals from strains JWV500 and MK147 showed that the translational fusion was approximately 100-fold brighter than the promoter fusion strain (Fig. 2B). This clearly demonstrated that the protein fusion is essential for the high fluorescence signal and that merely high levels of transcription cannot explain its brightness. Immunoblotting using anti-GFP antibodies further demonstrated that the HlpA-GFP protein level is significantly higher than that of GFP alone when they are expressed from the same promoter and the same ribosomal binding site (approximately 25-fold higher) (Fig. 2C). Thus, the HlpA-GFP fusion provides high fluorescence signals, probably due to efficient translation, although it is possible that the transcript or fusion protein stability is also affected. Since HlpA is a nucleoid binding protein, the fluorescent signal from the protein fusion is concentrated on the nucleoids and not distributed across the whole cytoplasm (as in, for example, MK147), and this may also contribute to increasing the strength of the fluorescent signal. Indeed, fluorescence recovery after photobleaching (FRAP) experiments suggest that diffusion of HlpA-GFP is slower and less than that of cytoplasmic GFP (Fig. 2D; see also Fig. S1 in the supplemental material). Together, these results show that a C-terminal GFP fusion with HlpA is highly efficiently translated and localized to the nucleoid, leading to bright fluorescent signals.
FIG 2.

Benchmarking of the HlpA-GFP strain. (A) Fluorescence microscopy of S. pneumoniae expressing the protein fusion hlpA-gfp (JWV500), the transcriptional fusion hlpA_gfp (MK147), and strain P92 carrying the multicopy GFPmut3-expressing plasmid pGFP1 (21). (B) Quantitative comparison of maximum fluorescence intensities in the different strains. The inset shows the same plot on a different y scale. The relative standard deviation, σp/<p>, is shown as a measure of cell-to-cell variability. More than 500 cells were measured for each strain. (C) Immunoblotting of whole-cell extracts to compare GFP protein levels between strains. The relative protein level was determined from the intensities of the bands in the blot. The relative GFP intensity was determined based on the fluorescence values plotted in panel B. α, anti. (D) Fluorescence recovery after photobleaching experiment of strains expressing a cytoplasmic GFP (P92) and the HlpA-GFP fusion (JWV500). Fluorescence recovery curves are shown to the left, and selected images acquired from the experiment are shown to the right. The time of bleaching is indicated with a dashed line in the plot. Plots and images of more cells are shown in Fig. S1 in the supplemental material.
Benchmarking cells expressing HlpA-GFP.
To our knowledge, the only known published examples of GFP-labeled S. pneumoniae used for imaging host interactions are strains constitutively expressing GFP from a multicopy plasmid (plasmids named pGFP1 [21] and pMV158GFP [22, 42]). The pGFP1-carrying strain was used to image S. pneumoniae in mouse bronco-epithelial cells (21). We compared the fluorescent intensity of an S. pneumoniae strain carrying the pGFP1 plasmid (strain P92) with the HlpA-GFP fusion strain JWV500. Comparisons of fluorescent intensities showed that strain JWV500 displayed 25-fold stronger fluorescence than P92 (Fig. 2A and B). Immunoblotting showed that the protein level was approximately 10-fold lower in P92, despite the fact that GFP in this strain was expressed from a multicopy plasmid (Fig. 2C). It should be noted that the GFP variant used in pGFP, GFPmut3 (43), is probably less intrinsically bright than the one in JWV500, sfGFP(Bs) (27). It is also worth noting that the heterogeneity in fluorescence signals between individual cells is larger in the pGFP-carrying strain than in those expressing GFP from the chromosome (Fig. 2B) (with a relative standard deviation, σp/<p>, higher for P92 than for JWV500 and MK147). This may be caused by differences in the copy numbers of plasmids inside the cells; however, other factors, such as poor GFP folding, may also contribute to such heterogeneity (30). Taken together, the fluorescent and genetic properties of HlpA-GFP-expressing S. pneumoniae are superior to the system previously described.
GFP-expressing S. pneumoniae cells are fully virulent and can be localized in mouse lung tissue.
Given the favorable in vitro properties of the constructs described above, we wanted to check whether these strains were virulent and to test whether they were also suitable for imaging in an in vivo animal model. Mice were challenged intranasally with PBS (mock treatment), the unlabeled parental D39 strain, or the HlpA-GFP strain, and disease signs and survival time were followed (Fig. 3). All mice showed clear signs of illness (starry coat and hunched appearance), and there was no significant difference in pathogenesis between unlabeled and labeled bacteria (Fig. 3; see also Fig. S2 in the supplemental material). To limit the number of sacrificed mice, only two mice were infected with the HlpA-RFP strain, and both developed disease similar to that caused by the parental unlabeled strain (data not shown). Lung tissue sections taken from mice infected for 48 h were investigated by three-dimensional (3D) wide-field microscopy, and highly fluorescent pneumococci, with strong homogenous signals, were readily identified within bronchial epithelial cells when the HlpA-GFP strain was used (Fig. 4). We were not able to detect red fluorescent pneumococci when HlpA-RFP was used. Possibly, the histology fixation procedure abolishes the red fluorescence signal, or the HlpA-RFP is unstable under these conditions since bacteria isolated from infected mice displayed strong red fluorescence when recultured in vitro (data not shown). Optimization of the fixation and embedding protocol may therefore help to overcome this problem. It is also possible that the low level of O2 in the tissue does not allow proper folding and maturation of RFP. In any case, these experiments demonstrate that HlpA-GFP is an excellent reporter that can be used for in vivo tracing of pneumococcal disease pathogenesis.
FIG 3.
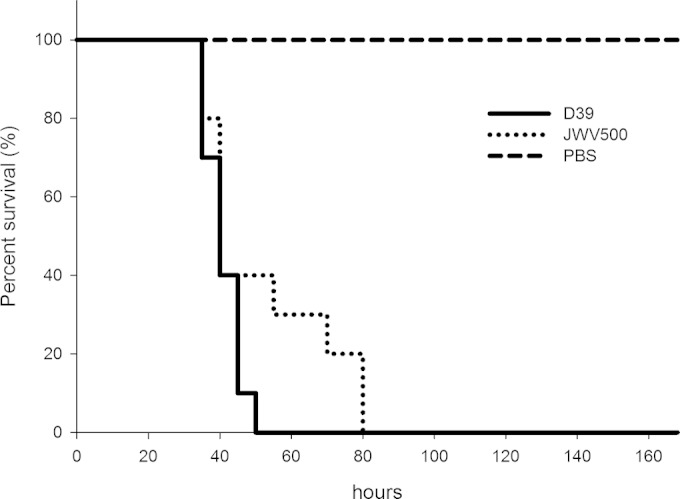
Percent survival of mice intranasally infected with 2.0 × 106 CFU of S. pneumoniae D39 (wild type) or 1.7 × 106 CFU of S. pneumoniae JWV500 (hlpA-gfp) suspended in 50 μl of PBS. For each of the bacterial strains, 10 mice were infected. PBS (50 μl) without bacteria was used as a negative control (n = 5). The experiment was ended after 168 h. There were no significant differences in survival rates between the wild-type and JWV500 strains (P > 0.05 with a log rank test and Gehan-Breslow-Wilcoxon test).
FIG 4.

Imaging of S. pneumoniae JWV500 (hlpA-gfp) in mouse lung tissue. Images of the mouse lung tissue were acquired as z-stacks (28 slices, with 0.2-μm distance; total depth of 5.6 μm) using wide-field epifluorescence microscopy. One overlay between phase-contrast (red) and GFP (green) of the middle stack image is shown on top, with a side volume view of all 28 images from the stack on the right. The fluorescent S. pneumoniae cells are clearly distinguishable within the tissue. GFP images of four depths from the z-stack are shown in the bottom panel. The white and yellow arrowheads point to cells visible only in certain depths within the mouse lung tissue. Scale bar, 5 μm.
Unencapsulated bacteria adhere more efficiently than encapsulated bacteria: imaging the interactions between S. pneumoniae and human epithelial cells.
The exopolysaccharide capsule is a major virulence factor in S. pneumoniae and is important at several stages of infection (8, 44–46). We wanted to test whether our reporters could be used to directly image the impact of the capsule on different stages of the infection process. First, to study how the capsule affects cell adhesion in a human infection model, we cultured A549 (ATCC CCL-185) type II human lung carcinoma epithelial cells and imaged the adhesion of S. pneumoniae by a dual-color experiment (Fig. 5A). To do so, we introduced a capsule mutation in both the HlpA-GFP and HlpA-RFP reporter strains. The capsule mutation introduced was Δcps2E (here designated Δcps) which has been used in other studies (29, 47) and has been shown to produce capsule-deficient cells. Next, we performed competition of bacterial adherence between HlpA-GFP (strain JWV500) and a Δcps strain expressing HlpA-RFP (Δcps/HlpA-RFP; MK128) or between HlpA-RFP (MK119) and a Δcps/HlpA-GFP (MK127) strain and added them to a confluent A549 monolayer (Fig. 5A) at an MOI (multiplicity of infection) of 10 (10 bacteria to 1 human cell). Traditional plating assays showed an increased propensity of unencapsulated reporter strains to adhere to A549 cells (Fig. 5B). Live-cell imaging showed that already after 2 h, only capsule mutants adhere efficiently to the human epithelial cells (Fig. 5C to J). By quantifying the fluorescence intensity of adhered bacteria, we found that the ratio between RFP and GFP signals of the Δcps/HlpA-RFP and HlpA-GFP strains (Fig. 5D and E) was 230 (average ratio of three analyzed images), which closely corresponds to the result of traditional CFU counting (Fig. 5B) that showed a 2-log increase of mutant capsule adherence compared to encapsulated strain. Furthermore, a similar ratio of fluorescence intensity between GFP and RFP (360; average of two analyzed images) was found for the Δcps/HlpA-GFP and HlpA-RFP strains (Fig. 4I and J). This single experiment provides direct evidence in live cells that the capsule limits pneumococcal adherence to the host cells and is fully in line with reports in the literature (8, 44–46).
The capsule protects S. pneumoniae against phagocytosis: imaging of pneumococcal interactions with human neutrophils.
While the above-mentioned experiments clearly show that unencapsulated strains adhere more efficiently to human cells, it was previously shown using elegant biochemical and immunological experiments that the capsule protects the pneumococci from recognition of the human immune system (3, 4, 38). To examine this conundrum in more detail, we tested whether our reporter strains could be used to directly visualize encapsulated and unencapsulated pneumococci in the presence of human neutrophils in a dual-color experiment. We mixed the encapsulated and unencapsulated fluorescently labeled pneumococci as described above with human neutrophils in the presence of 10% human serum and performed time-lapse microscopy. As shown in Fig. 6A and Movie S1 in the supplemental material, human neutrophils specifically moved to and phagocytosed unencapsulated pneumococci but not encapsulated cells. Our reporter strains are also bright enough to be used in flow cytometry. Based on sorting of neutrophils after phagocytosis, we could show that the capsule mutant was more efficiently phagocytosed than encapsulated cells (Fig. 6B). These results clearly demonstrate the protective effect of the capsule on phagocytosis by human neutrophils. Furthermore, this is further proof that the described fluorescent markers for both GFP and RFP are bright enough to be visualized inside eukaryotic cells.
FIG 6.

Interaction of S. pneumoniae with human neutrophils. Phagocytosis of encapsulated (A to D) and unencapsulated (ΔcpsE::kan strain) (E to H) S. pneumoniae strains by human neutrophils was imaged using wide-field epifluorescence microscopy. Both encapsulated and unencapsulated strains were labeled with either GFP or RFP and mixed in a 1:1 ratio before neutrophils were added and phagocytosis was allowed to occur. Neutrophils efficiently phagocytose the ΔcpsE strains, whereas wild-type bacteria with capsule are not taken up. Scale bar, 50 μm. The bright GFP signals from S. pneumoniae also allow sorting of phagocytosing neutrophils from nonphagocytosing neutrophils by flow cytometry (I and J); the ΔcpsE strain is phagocytosed much more efficiently than the encapsulated strain.
DISCUSSION
Bacterial strains appropriate for fluorescent live-cell imaging in vivo should meet certain criteria to function optimally for this purpose. Most important, the fluorescence signal should be sufficiently bright to distinguish the bacterial cells from the background fluorescence. The fluorescence signal should also be stable, preferably not fade during the course of the experiment, and show low cell-to-cell variability. Furthermore, the labeled strains should be genetically similar to the original strain to faithfully reflect the pathogenesis cycle. By stably integrating genes expressing fusions of the brightest available GFP [sfGFP(Bs)] (27) or RFP (mKate2) (Beilharz et al., submitted) in S. pneumoniae to the nucleoid binding protein HlpA, we generated intensely fluorescent strains which were 25-fold brighter than the best known GFP-expressing strain used hitherto for in vivo imaging (21). The bright fluorescence is caused by high and stable levels of the fusion protein HlpA-GFP compared to nonfused GFP, as well as by limited diffusion due to binding of HlpA-GFP to the nucleoid (Fig. 2). Also important, HlpA is highly conserved at the DNA-sequence level and present in all S. pneumoniae strains sequenced so far (data not shown); thus, the reporters can straightforwardly be transferred to different medically relevant transformable pneumococcal strains. The principle presented here of fusing fluorescent proteins to nucleoid binding proteins to obtain highly fluorescent bacterial cells suitable for in vivo and in vitro studies should be generally applicable and be of particular use for other bacterial species when obtaining bright enough cells for live cell imaging is a challenge.
Our novel, brightly fluorescent strains were fully virulent in a mouse model, and pneumococci labeled with HlpA-GFP could readily be detected in infected mouse lung tissue (Fig. 3 and 4; see also Fig. S2 in the supplemental material). Notably, the signal-to-noise ratio of HlpA-GFP cells within infected mouse tissue is high enough that even regular wide-field epifluorescence can be used for imaging, without the need for expensive laser-based confocal fluorescence microscopy. Use of the HlpA-GFP-labeled strain will thus simplify the detection of S. pneumoniae during mouse infection experiments.
We used our fluorescently labeled pneumococci to directly image different stages of pneumococcal infection using live cells. To study adhesion to the epithelial layer, which is important for the initial stages of infection, we used A549 (ATCC CCL-185) type II human lung carcinoma epithelial cells. A549 has been used extensively to elucidate bacterial adherence and invasion to the host (48), and the pneumococcal coincubation model has been successfully used to identify essential host-pathogen virulence factors, including PsaA (49), PavA (50), and PspC (51). We could confirm and demonstrate that the exopolysaccharide capsule inhibits adhesion of S. pneumoniae to human epithelial cells and thus also the infection process (Fig. 5). This result is in line with a study of Hammerschmidt et al. (52), where fixed samples of pneumococci and the A549 cell line were used for electron microscopy to show that pneumococci adhering to the human host have reduced levels of capsule. Possibly, the capsule covers crucial pneumococcal adhesion-mediating surface proteins that are otherwise exposed on the bacterial cell surface (45, 52), and this might also partly explain the success of colonization of nontypeable strains within human populations (9).
While the capsule is disadvantageous for adhesion to host cells, it is highly advantageous and necessary for pneumococcal evasion of the host immune system. For example, the capsule protects pneumococci against mucus-mediated clearing in the very early stages of infection since unencapsulated bacteria agglutinate within mucus (44). Using dual-color strains, we were able to image in real time the protective function of the capsule against phagocytosis by human neutrophils (Fig. 6). This protection may be mediated by sterically hindering the neutrophils or by blocking of complement binding proteins (4, 7, 45, 53).
By the use of relatively straightforward imaging approaches, this work, for the first time, uses live-cell imaging to demonstrate the opposing attributes of the pneumococcal capsule at different stages of infection. These results confirm that regulation of capsule production is critical for colonization of S. pneumoniae within the human host. The presence of such regulation is reflected by the observation of phase variation of pneumococci (8, 54), and studies suggest that little capsule is produced when pneumococci are in contact with the epithelial cell layer (52). However, the mode of regulation remains to be further unraveled, and a number of genes and environmental factors seem to be important (45, 54–57). The imaging tools used in this work will pave the way for new types of experiments which will help further our knowledge on the pathogenesis of the pneumococcus.
Supplementary Material
ACKNOWLEDGMENTS
M.K. was supported by a long-term fellowship from the European Biochemical Societies. Work in the lab of J.-W.V. is supported by the EMBO Young Investigator Programme, a VIDI fellowship from the Netherlands Organization for Scientific Research, Earth and Life Sciences (864.12.001), and a European Research Council starting grant (337399-PneumoCell).
We thank W. J. Quax and R. Setroikromo from the Department of Pharmaceutical Biology, Groningen Research Institute of Pharmacy, University of Groningen, the Netherlands, for their kind gift of human cell line A549.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02221-14.
REFERENCES
- 1.O'Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, Lee E, Mulholland K, Levine OS, Cherian T. 2009. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374:893–902. doi: 10.1016/S0140-6736(09)61204-6. [DOI] [PubMed] [Google Scholar]
- 2.Griffith F. 1928. The significance of pneumococcal types. J Hyg (Lond) 27:113–159. doi: 10.1017/S0022172400031879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hyams C, Camberlein E, Cohen JM, Bax K, Brown JS. 2010. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect Immun 78:704–715. doi: 10.1128/IAI.00881-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abeyta M, Hardy GG, Yother J. 2003. Genetic alteration of capsule type but not PspA type affects accessibility of surface-bound complement and surface antigens of Streptococcus pneumoniae. Infect Immun 71:218–225. doi: 10.1128/IAI.71.1.218-225.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim JO, Romero-Steiner S, Sørensen UB, Blom J, Carvalho M, Barnard S, Carlone G, Weiser JN. 1999. Relationship between cell surface carbohydrates and intrastrain variation on opsonophagocytosis of Streptococcus pneumoniae. Infect Immun 67:2327–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinberger DM, Trzciński K, Lu Y-J, Bogaert D, Brandes A, Galagan J, Anderson PW, Malley R, Lipsitch M. 2009. Pneumococcal capsular polysaccharide structure predicts serotype prevalence. PLoS Pathog 5:e1000476. doi: 10.1371/journal.ppat.1000476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henriques-Normark B, Tuomanen EI. 2013. The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harb Perspect Med 3:a010215. doi: 10.1101/cshperspect.a010215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiser JN, Austrian R, Sreenivasan PK, Masure HR. 1994. Phase variation in pneumococcal opacity: relationship between colonial morphology and nasopharyngeal colonization. Infect Immun 62:2582–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chewapreecha C, Harris SR, Croucher NJ, Turner C, Marttinen P, Cheng L, Pessia A, Aanensen DM, Mather AE, Page AJ, Salter SJ, Harris D, Nosten F, Goldblatt D, Corander J, Parkhill J, Turner P, Bentley SD. 2014. Dense genomic sampling identifies highways of pneumococcal recombination. Nat Genet 46:305–309. doi: 10.1038/ng.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z, Clarke TB, Weiser JN. 2009. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J Clin Invest 119:1899–1909. doi: 10.1172/JCI36731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez CJ, Shivshankar P, Stol K, Trakhtenbroit S, Sullam PM, Sauer K, Hermans PWM, Orihuela CJ. 2010. The pneumococcal serine-rich repeat protein is an intra-species bacterial adhesin that promotes bacterial aggregation in vivo and in biofilms. PLoS Pathog 6:e1001044. doi: 10.1371/journal.ppat.1001044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parker D, Soong G, Planet P, Brower J, Ratner AJ, Prince A. 2009. The NanA neuraminidase of Streptococcus pneumoniae is involved in biofilm formation. Infect Immun 77:3722–3730. doi: 10.1128/IAI.00228-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elhaik-Goldman S, Kafka D, Yossef R, Hadad U, Elkabets M, Vallon-Eberhard A, Hulihel L, Jung S, Ghadially H, Braiman A, Apte RN, Mandelboim O, Dagan R, Mizrachi-Nebenzahl Y, Porgador A. 2011. The natural cytotoxicity receptor 1 contribution to early clearance of Streptococcus pneumoniae and to natural killer-macrophage cross talk. PLoS One 6:e23472. doi: 10.1371/journal.pone.0023472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srivastava A, Casey H, Johnson N, Levy O, Malley R. 2007. Recombinant bactericidal/permeability-increasing protein rBPI21 protects against pneumococcal disease. Infect Immun 75:342–349. doi: 10.1128/IAI.01089-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colino J, Shen Y, Snapper CM. 2002. Dendritic cells pulsed with intact Streptococcus pneumoniae elicit both protein- and polysaccharide-specific immunoglobulin isotype responses in vivo through distinct mechanisms. J Exp Med 195:1–14. doi: 10.1084/jem.20011432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mook-Kanamori BB, Rouse MS, Kang C-I, van de Beek D, Steckelberg JM, Patel R. 2009. Daptomycin in experimental murine pneumococcal meningitis. BMC Infect Dis 9:50. doi: 10.1186/1471-2334-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Francis KP, Yu J, Bellinger-Kawahara C, Joh D, Hawkinson MJ, Xiao G, Purchio TF, Caparon MG, Lipsitch M, Contag PR. 2001. Visualizing pneumococcal infections in the lungs of live mice using bioluminescent Streptococcus pneumoniae transformed with a novel Gram-positive lux transposon. Infect Immun 69:3350–3358. doi: 10.1128/IAI.69.5.3350-3358.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orihuela CJ, Gao G, Francis KP, Yu J, Tuomanen EI. 2004. Tissue-specific contributions of pneumococcal virulence factors to pathogenesis. J Infect Dis 190:1661–1669. doi: 10.1086/424596. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Barke RA, Charboneau R, Schwendener R, Roy S. 2008. Morphine induces defects in early response of alveolar macrophages to Streptococcus pneumoniae by modulating TLR9-NF-κB signaling. J Immunol 180:3594–3600. doi: 10.4049/jimmunol.180.5.3594. [DOI] [PubMed] [Google Scholar]
- 20.Johnson AW, Sidman JD, Lin J. 2013. Bioluminescent imaging of pneumococcal otitis media in chinchillas. Ann Otol Rhinol Laryngol 122:344–352. doi: 10.1177/000348941312200510. [DOI] [PubMed] [Google Scholar]
- 21.Kadioglu A, Sharpe JA, Lazou I, Svanborg C, Ockleford C, Mitchell TJ, Andrew PW. 2001. Use of green fluorescent protein in visualisation of pneumococcal invasion of broncho-epithelial cells in vivo. FEMS Microbiol Lett 194:105–110. doi: 10.1111/j.1574-6968.2001.tb09454.x. [DOI] [PubMed] [Google Scholar]
- 22.Ribes S, Ebert S, Regen T, Agarwal A, Tauber SC, Czesnik D, Spreer A, Bunkowski S, Eiffert H, Hanisch U-K, Hammerschmidt S, Nau R. 2010. Toll-like receptor stimulation enhances phagocytosis and intracellular killing of nonencapsulated and encapsulated Streptococcus pneumoniae by murine microglia. Infect Immun 78:865–871. doi: 10.1128/IAI.01110-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin B, Garcia P, Castanié M-P, Claverys J-P. 1995. The recA gene of Streptococcus pneumoniae is part of a competence-induced operon and controls lysogenic induction. Mol Microbiol 15:367–379. doi: 10.1111/j.1365-2958.1995.tb02250.x. [DOI] [PubMed] [Google Scholar]
- 24.Sorg RA, Kuipers OP, Veening J-W. Gene expression platform for synthetic biology in the human pathogen Streptococcus pneumoniae. ACS Synth Biol, in press. doi: 10.1021/sb500229s. [DOI] [PubMed] [Google Scholar]
- 25.Arai R, Ueda H, Kitayama A, Kamiya N, Nagamune T. 2001. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng 14:529–532. doi: 10.1093/protein/14.8.529. [DOI] [PubMed] [Google Scholar]
- 26.Yuzenkova Y, Gamba P, Herber M, Attaiech L, Shafeeq S, Kuipers OP, Klumpp S, Zenkin N, Veening J-W. 2014. Control of transcription elongation by GreA determines rate of gene expression in Streptococcus pneumoniae. Nucleic Acids Res 42:10987–10999. doi: 10.1093/nar/gku790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Overkamp W, Beilharz K, Detert Oude Weme R, Solopova A, Karsens H, Kovács ÁT, Kok J, Kuipers OP, Veening J-W. 2013. Benchmarking various green fluorescent protein variants in Bacillus subtilis, Streptococcus pneumoniae, and Lactococcus lactis for live cell imaging. Appl Environ Microbiol 79:6481–6490. doi: 10.1128/AEM.02033-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kjos M, Veening J-W. 2014. Tracking of chromosome dynamics in live Streptococcus pneumoniae reveals that transcription promotes chromosome segregation. Mol Microbiol 91:1088–1105. doi: 10.1111/mmi.12517. [DOI] [PubMed] [Google Scholar]
- 29.Ramos-Montañez S, Kazmierczak KM, Hentchel KL, Winkler ME. 2010. Instability of ackA (acetate kinase) mutations and their effects on acetyl phosphate and ATP amounts in Streptococcus pneumoniae D39. J Bacteriol 192:6390–6400. doi: 10.1128/JB.00995-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jørgensen MG, van Raaphorst R, Veening J-W. 2013. Noise and stochasticity in gene expression: a pathogenic fate determinant, p 157–176. In Harwood C, Wipat A (ed), Microbial synthetic biology Academic Press, Oxford, United Kingdom. [Google Scholar]
- 31.De Jong IG, Beilharz K, Kuipers OP, Veening J-W. 2011. Live cell imaging of Bacillus subtilis and Streptococcus pneumoniae using automated time-lapse microscopy. J Vis Exp 53:3145. doi: 10.3791/3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shoemaker NB, Guild WR. 1972. Kinetics of integration of transforming DNA in pneumococcus. Proc Natl Acad Sci U S A 69:3331–3335. doi: 10.1073/pnas.69.11.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morton DB, Griffiths PH. 1985. Guidelines on the recognition of pain, distress and discomfort in experimental animals and an hypothesis for assessment. Vet Rec 116:431–436. doi: 10.1136/vr.116.16.431. [DOI] [PubMed] [Google Scholar]
- 34.Kadioglu A, Gingles NA, Grattan K, Kerr A, Mitchell TJ, Andrew PW. 2000. Host cellular immune response to pneumococcal lung infection in mice. Infect Immun 68:492–501. doi: 10.1128/IAI.68.2.492-501.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mlacha SZK, Romero-Steiner S, Hotopp JCD, Kumar N, Ishmael N, Riley DR, Farooq U, Creasy TH, Tallon LJ, Liu X, Goldsmith CS, Sampson J, Carlone GM, Hollingshead SK, Scott JAG, Tettelin H. 2013. Phenotypic, genomic, and transcriptional characterization of Streptococcus pneumoniae interacting with human pharyngeal cells. BMC Genomics 14:383. doi: 10.1186/1471-2164-14-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Surewaard BGJ, van Strijp JAG, Nijland R. 2013. Studying interactions of Staphylococcus aureus with neutrophils by flow cytometry and time lapse microscopy. J Vis Exp 77:e50788. doi: 10.3791/50788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu J, Xiao J, Ren X, Lao K, Xie XS. 2006. Probing gene expression in live cells, one protein molecule at a time. Science 311:1600–1603. doi: 10.1126/science.1119623. [DOI] [PubMed] [Google Scholar]
- 38.Davis RW, Timlin JA, Kaiser JN, Sinclair MB, Jones HDT, Lane TW. 2010. Accurate detection of low levels of fluorescence emission in autofluorescent background: Francisella-infected macrophage cells. Microsc Microanal 16:478–487. doi: 10.1017/S1431927610000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu D, Yumoto H, Murakami K, Hirota K, Ono T, Nagamune H, Kayama S, Matsuo T, Miyake Y. 2008. The essentiality and involvement of Streptococcus intermedius histone-like DNA-binding protein in bacterial viability and normal growth. Mol Microbiol 68:1268–1282. doi: 10.1111/j.1365-2958.2008.06232.x. [DOI] [PubMed] [Google Scholar]
- 40.Slager J, Kjos M, Attaiech L, Veening J-W. 2014. Antibiotic-induced replication stress triggers bacterial competence by increasing gene dosage near the origin. Cell 157:395–406. doi: 10.1016/j.cell.2014.01.068. [DOI] [PubMed] [Google Scholar]
- 41.Avery OT, Macleod CM, McCarty M. 1944. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: induction of transformation by a deoxyribonucleic acid fraction isolated from pneumococcus type III. J Exp Med 79:137–158. doi: 10.1084/jem.79.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nieto C, Espinosa M. 2003. Construction of the mobilizable plasmid pMV158GFP, a derivative of pMV158 that carries the gene encoding the green fluorescent protein. Plasmid 49:281–285. doi: 10.1016/S0147-619X(03)00020-9. [DOI] [PubMed] [Google Scholar]
- 43.Cormack BP, Valdivia RH, Falkow S. 1996. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 44.Nelson AL, Roche AM, Gould JM, Chim K, Ratner AJ, Weiser JN. 2007. Capsule enhances pneumococcal colonization by limiting mucus-mediated clearance. Infect Immun 75:83–90. doi: 10.1128/IAI.01475-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kadioglu A, Weiser JN, Paton JC, Andrew PW. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol 6:288–301. doi: 10.1038/nrmicro1871. [DOI] [PubMed] [Google Scholar]
- 46.Weiser JN. 2010. The pneumococcus: why a commensal misbehaves. J Mol Med 88:97–102. doi: 10.1007/s00109-009-0557-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cartee RT, Forsee WT, Bender MH, Ambrose KD, Yother J. 2005. CpsE from type 2 Streptococcus pneumoniae catalyzes the reversible addition of glucose-1-phosphate to a polyprenyl phosphate acceptor, initiating type 2 capsule repeat unit formation. J Bacteriol 187:7425–7433. doi: 10.1128/JB.187.21.7425-7433.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Talbot UM, Paton AW, Paton JC. 1996. Uptake of Streptococcus pneumoniae by respiratory epithelial cells. Infect Immun 64:3772–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berry AM, Paton JC. 1996. Sequence heterogeneity of PsaA, a 37-kilodalton putative adhesin essential for virulence of Streptococcus pneumoniae. Infect Immun 64:5255–5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pracht D, Elm C, Gerber J, Bergmann S, Rohde M, Seiler M, Kim KS, Jenkinson HF, Nau R, Hammerschmidt S. 2005. PavA of Streptococcus pneumoniae modulates adherence, invasion, and meningeal inflammation. Infect Immun 73:2680–2689. doi: 10.1128/IAI.73.5.2680-2689.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hammerschmidt S, Agarwal V, Kunert A, Haelbich S, Skerka C, Zipfel PF. 2007. The host immune regulator factor H interacts via two contact sites with the PspC protein of Streptococcus pneumoniae and mediates adhesion to host epithelial cells. J Immunol 178:5848–5858. doi: 10.4049/jimmunol.178.9.5848. [DOI] [PubMed] [Google Scholar]
- 52.Hammerschmidt S, Wolff S, Hocke A, Rosseau S, Müller E, Rohde M. 2005. Illustration of pneumococcal polysaccharide capsule during adherence and invasion of epithelial cells. Infect Immun 73:4653–4667. doi: 10.1128/IAI.73.8.4653-4667.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S, Henriques-Normark B. 2007. Capsule and d-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol 9:1162–1171. doi: 10.1111/j.1462-5822.2006.00857.x. [DOI] [PubMed] [Google Scholar]
- 54.Manso AS, Chai MH, Atack JM, Furi L, De Ste Croix M, Haigh R, Trappetti C, Ogunniyi AD, Shewell LK, Boitano M, Clark TA, Korlach J, Blades M, Mirkes E, Gorban AN, Paton JC, Jennings MP, Oggioni MR. 2014. A random six-phase switch regulates pneumococcal virulence via global epigenetic changes. Nat Commun 5:5055. doi: 10.1038/ncomms6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shainheit MG, Mulé M, Camilli A. 2014. The core promoter of the capsule operon of Streptococcus pneumoniae is necessary for colonization and invasive disease. Infect Immun 82:694–705. doi: 10.1128/IAI.01289-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Geno KA, Hauser JR, Gupta K, Yother J. 2014. Streptococcus pneumoniae phosphotyrosine phosphatase CpsB and alterations in capsule production resulting from changes in oxygen availability. J Bacteriol 196:1992–2003. doi: 10.1128/JB.01545-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yother J. 2011. Capsules of Streptococcus pneumoniae and other bacteria: paradigms for polysaccharide biosynthesis and regulation. Annu Rev Microbiol 65:563–581. doi: 10.1146/annurev.micro.62.081307.162944. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.