

Abstract
The haloacid dehalogenase (HAD) superfamily is one of the largest enzyme families, consisting mainly of phosphatases. Although intracellular phosphate plays important roles in many cellular activities, the biological functions of HAD enzymes are largely unknown. Pho13 is 1 of 16 putative HAD enzymes in Saccharomyces cerevisiae. Pho13 has not been studied extensively, but previous studies have identified PHO13 to be a deletion target for the generation of industrially attractive phenotypes, namely, efficient xylose fermentation and high tolerance to fermentation inhibitors. In order to understand the molecular mechanisms underlying the improved xylose-fermenting phenotype produced by deletion of PHO13 (pho13Δ), we investigated the response of S. cerevisiae to pho13Δ at the transcriptomic level when cells were grown on glucose or xylose. Transcriptome sequencing analysis revealed that pho13Δ resulted in upregulation of the pentose phosphate (PP) pathway and NADPH-producing enzymes when cells were grown on glucose or xylose. We also found that the transcriptional changes induced by pho13Δ required the transcription factor Stb5, which is activated specifically under NADPH-limiting conditions. Thus, pho13Δ resulted in the upregulation of the PP pathway and NADPH-producing enzymes as a part of an oxidative stress response mediated by activation of Stb5. Because the PP pathway is the primary pathway for xylose, its upregulation by pho13Δ might explain the improved xylose metabolism. These findings will be useful for understanding the biological function of S. cerevisiae Pho13 and the HAD superfamily enzymes and for developing S. cerevisiae strains with industrially attractive phenotypes.
INTRODUCTION
Phosphate, one of the most abundant intracellular molecules, is carried in ATP and transferred to many other metabolites and proteins (1). Phosphate starvation in medium results in cell cycle arrest at the G1 stage (2). Moreover, the intracellular inorganic phosphate pool mediates the proper initiation and homeostasis of metabolic flux in the glycolytic pathway (3). In signal transduction pathways, protein phosphorylation plays a key role by regulating protein localization, protein interactions, and enzyme activity (4).
The haloacid dehalogenase (HAD) superfamily is one of the largest enzyme families, consisting of ∼80,000 sequences that encode proteins whose functions are largely unknown (5). The family name originated from the first enzyme that was structurally characterized (6), but 70% of the family members are phosphatases (http://sfld.rbvi.ucsf.edu/django/superfamily/). The active site of HAD enzymes consists of highly conserved core domains and a versatile cap domain (7, 8). The presence and location of the cap domain allow diverse substrate specificities, which form the basis for the classification of the enzymes into several subfamilies (7).
Saccharomyces cerevisiae Pho13 is structurally classified as a member of HAD subfamily IIA (9), which contains ∼3,000 sequences, including eukaryotic 2-phosphoglycolate phosphatase (PGP or PGLP) and pyridoxal phosphatase. Although two bacterial enzymes, AraL from Bacillus subtilis (a Gram-positive bacterium) and NagD from Escherichia coli (a Gram-negative bacterium), are classified into this subfamily as well, their biochemical properties differ from those of eukaryotic enzymes (9, 10). Moreover, the physiological importance of this group of enzymes is not yet understood.
Although little is known about the biochemical and physiological functions of S. cerevisiae Pho13, deletion of the PHO13 gene (pho13Δ) results in significant and industrially relevant phenotypic changes in engineered strains capable of fermenting xylose (11–16). Introduction of a heterologous metabolic pathway consisting of either xylose reductase/xylitol dehydrogenase/xylulokinase [X(R/X)D(H/X)K] or xylose isomerase/xylulokinase [X(I/X)K] allows S. cerevisiae to metabolize xylose (17). However, the rates of xylose metabolism in the engineered strains are lower than those of glucose metabolism. In one study, random transposon mutagenesis of an S. cerevisiae strain overexpressing X(R/X)D(H/X)K generated a mutant strain with an insertional mutation in PHO13 that grew much better on xylose (11). In another independent study, adaptive evolution of an S. cerevisiae strain overexpressing X(R/X)D(H/X)K on xylose led to a spontaneous single nucleotide polymorphism (SNP) in PHO13, which increased xylose consumption 5-fold (12). In both cases, the loss of Pho13 function (pho13Δ) was shown to be responsible for the improvement in xylose metabolism (12, 14). Moreover, pho13Δ in S. cerevisiae strains overexpressing X(R/X)D(H/X)K improved tolerance to common fermentation inhibitors (weak acids and sugar degradation products) (13, 15). In addition to its effects in strains overexpressing X(R/X)D(H/X)K, pho13Δ in a strain overexpressing X(I/X)K improved the growth rate on xylose 8-fold (16). Thus, different research groups have confirmed the positive effect of pho13Δ on xylose metabolism in both xylose pathways and in various strain backgrounds. As such, there is a need to determine the biological function of the Pho13 enzyme and the mechanism underlying the effect of pho13Δ on xylose metabolism.
This study focused on the Pho13 enzyme. In transcriptome sequencing (RNA-seq) analysis, we found that pho13Δ induced transcriptional changes when cells were grown on xylose or glucose. We propose that pho13Δ triggers transcriptional reprogramming, which directly improves xylose metabolism in engineered S. cerevisiae strains capable of fermenting xylose.
MATERIALS AND METHODS
Strain construction.
To construct the pho13Δ and stb5Δ mutants, PCR-based gene deletion (18) was performed using the plasmids and primers listed in Tables 1 and 2. Specifically, using a pUG6 plasmid as the template, the kanMX marker gene with PHO13-specific overhanging sequences (the nucleotide sequence in lowercase letters in Table 2) was amplified using primers SOO615 and SOO428. The PCR product was purified and transformed into yeast strains using the lithium acetate/single-stranded carrier DNA–polyethylene glycol method (19). The pho13Δ::kanMX deletion mutant was selected on YPD (10 g/liter yeast extract, 20 g/liter peptone, 20 g/liter glucose) agar medium containing 300 μg/ml G418. To construct the stb5Δ::hghMX mutant, a pUG75 plasmid, the SOO411 and SOO412 primers, and YPD agar medium containing 300 μg/ml hygromycin B were used instead. Several colonies were confirmed through colony PCR using primers that amplified each end of the integration site: SOO575/SOO148 and SOO576/SOO149 for pho13Δ::kanMX and SOO431/SOO148 and SOO455/SOO149 for stb5Δ::hghMX.
TABLE 1.
Plasmids and strains used in this study
| Plasmid or strain | Description | Reference or source |
|---|---|---|
| Plasmids | ||
| pUG6 | A template plasmid containing a kanMX marker for PCR-based gene knockout | 30 |
| pUG75 | A template plasmid containing an hphMX marker for PCR-based gene knockout | 18 |
| p41N-Cas9 | A single-copy plasmid containing Cas9 and a natMX marker | This study |
| p42H-GND1.1 | A multicopy plasmid containing a gRNA and an hphMX marker | This study |
| p41K-TEF1P-STB5 | A single-copy plasmid containing STB5 under the control of the TEF1 promoter and a kanMX marker | This study |
| Strains | ||
| BY4742 | A laboratory wild-type strain of S. cerevisiae | 31 |
| BY4742 pho13Δ | BY4742 pho13::kanMX | 32 |
| D452-2 | A laboratory wild-type strain of S. cerevisiae | 33 |
| D452-2 pho13Δ | D452-2 pho13::kanMX | This study |
| SR7 | D452-2 with a xylose pathway (S. stipitis XYL1, XYL2, and XYL3) | 12 |
| SR7 pho13Δ | SR7 pho13::kanMX | 12 |
| SR7 stb5Δ | SR7 stb5::hphMX | This study |
| SR7 pho13Δ stb5Δ | SR7 pho13::kanMX stb5::hphMX | This study |
| SR7 pho13Δ Cas9 | SR7 pho13Δ pRS41N-Cas9 | This study |
| SR7 pho13Δ GND1.1 | SR7 pho13Δ with a mutation in the upstream GND1 | This study |
| SR7 TEF1P-STB5 | SR7 p41K-TEF1P-STB5 | This study |
TABLE 2.
Primers used in this study
| Name | Sequencea | Descriptionb |
|---|---|---|
| SOO615 | 5′-tatcaagctcgagccaaatcacaaaaaaagccttatagcttgccctgacaaagaatatacaactcgggaaaCAGCTGAAGCTTCGTACGC | pho13::loxP-F |
| SOO428 | 5′-aaaacaacaaacctgaatatttttccttttcaaaaagtaattctacccctagattttgcattgctcctGCATAGGCCACTAGTGGATCTG | pho13::loxP-R |
| SOO411 | 5′-cgtaacaagaggtataatatccgagcgtacagggctaaaaaattaatacaaaggtgtaaaagaaggacatgCAGCTGAAGCTTCGTACGC | stb5::loxP-F |
| SOO412 | 5′-ttaaacggcagcaacaagccgctgccgtatagtatgacgacatgacaaaactcggtgaacatatgtcaGCATAGGCCACTAGTGGATCTG | stb5::loxP-R |
| SOO575 | 5′-GCCGGATCCACTGTGATACTAACGGGCAACTAC | PHO13_883-up-F |
| SOO576 | 5′-GCCGTCGACGAATTGGTCAACACTCTGAGCG | PHO13_384-down-R |
| SOO431 | 5′-GCGCTTAGCACTGTTGAATC | STB5_527-up-F |
| SOO455 | 5′-AAGTTCCGCCTCTTGGAGAC | STB5_434-down-R |
| SOO148 | 5′-GGATGTATGGGCTAAATG | B-M (paired with up) |
| SOO149 | 5′-CCTCGACATCATCTGCCC | C-M (paired with down) |
| SOO613 | 5′-GCCTTCTACGTTTCCATCCA | RT-qPCR_ACT1-F |
| SOO614 | 5′-GGCCAAATCGATTCTCAAAA | RT-qPCR_ACT1-R |
| SOO355 | 5′-TGCTCGTTATGCGTCTTATCC | RT-qPCR_ZWF1-F |
| SOO356 | 5′-CTGCACCTGTGGCAATTATTC | RT-qPCR_ZWF1-R |
| SOO429 | 5′-GCGGACGAATCACTATCTTCTC | RT-qPCR_SOL3-F |
| SOO430 | 5′-GCTCTCTTGAAGGCACCATAA | RT-qPCR_SOL3-R |
| SOO417 | 5′-CTGCTACTTATGGCTGGAAACT | RT-qPCR_GND1-F |
| SOO418 | 5′-GGTTCTTCTCTGTAGGCCTTTG | RT-qPCR_GND1-R |
| SOO365 | 5′-CCTCAAGACTCCACAACTAACC | RT-qPCR_TAL1-F |
| SOO366 | 5′-ACCATGCTTCTTACCGTATTCC | RT-qPCR_TAL1-R |
| SOO567 | 5′-TTGTACCTATTGTGGCTGTCGGTGTTACATTACGGTGCCTCTGTGTTACGCGGTGCTTCG | GND1.1_Donor-F |
| SOO568 | 5′-CCACCAAGGCCAAAGCGCAAGGCCGCGTGGACCACCAGGACGAAGCACCGCGTAACACAG | GND1.1_Donor-R |
Lowercase nucleotides indicate PHO13-specific overhanging sequences; the underlined nucleotide in SOO568 represents an SNP.
F, forward; R, reverse.
Strain construction using CRISPR-Cas.
The following method was modified from the first paper describing S. cerevisiae genome engineering with a clustered regularly interspaced short palindromic repeat (CRISPR)-Cas system (20), as follows. (i) p41N-Cas9 was constructed by transferring the Cas9 cassette from the p414-TEF1p-Cas9-CYC1t plasmid (Addgene) to p41N, a single-copy plasmid containing a natMX marker.
(ii) Guide RNA (gRNA) with 20 bp of a GND1.1-specific sequence was designed as described in Table 3. The GND1.1-specific sequence was the only modification to the gRNA described previously (20). It was chosen from the sequence immediately 5′ of the NGG protospacer-associated motif (PAM) sequence, which was located in the Stb5-binding sequence (CGGTGTTA, where the PAM sequence is underlined). The gRNA was synthesized by IDT (Coralville, IA, USA) as gBlocks gene fragments with 5′ phosphorylated ends. The gBlocks gene fragments were then ligated with a p42H vector that had been linearized and dephosphorylated by treatment with EcoRV and rSAP. The resulting plasmid, p42H-GND1.1, was treated with BamHI and SalI to confirm the presence of the insert.
TABLE 3.
gRNA (gBlocks) sequences (388 bp)
| Parts | Sequence | Length (bp) |
|---|---|---|
| SNR52 promoter | TCTTTGAAAAGATAATGTATGATTATGCTTTCACTCATATTTATACAGAAACTTGATGTTTTCTTTCGAGTATATACAAGGTGATTACATGTACGTTTGAAGTACAACTCTAGATTTTGTAGTGCCCTCTTGGGCTAGCGGTAAAGGTGCGCATTTTTTCACACCCTACAATGTTCTGTTCAAAAGATTTTGGTCAAACGCTGTAGAAGTGAAAGTTGGTGCGCATGTTTCGGCGTTCGAAACTTCTCCGCAGTGAAAGATAAATGATC | 269 |
| GND1.1 gRNA (target specific) | GGTGTTACATTACGGTGCCT | 20 |
| Structural crRNA | GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGGTGC | 79 |
| SUP4 terminator | TTTTTTTGTTTTTTATGTCT | 20 |
(iii) Donor DNA was PCR amplified using the primers SOO567/SOO568, which generated double-stranded DNA of 100 bp that covered the GND1.1-specific sequence area and contained a mutation in the Stb5-binding sequence (CTGTGTTA, where the mutation is underlined). Because the mutation site overlapped the PAM sequence, there was no need to introduce an additional mutation in the PAM sequence to prevent gRNA binding.
(iv) The target strain (SR7 pho13Δ) was first transformed with p41N-Cas9 and plated on a YPD agar plate containing 120 μg/ml nourseothricin (clonNAT). The resulting strain was transformed with the p42H-GND1.1 plasmid and donor DNA and plated on a YPD agar plate containing 120 μg/ml nourseothricin and 300 μg/ml hygromycin B. The colonies were restreaked on the same type of agar plates, and the mutation was confirmed by sequencing 1 kb upstream of GND1.
Culture conditions.
All strains were aerobically precultured in 5 ml of YPD at 30°C for 24 h. For the growth profile and RNA extraction, precultured cells were inoculated into 50 ml of YP (yeast extract-peptone) medium containing either 40 g/liter glucose or 40 g/liter xylose at an initial cell density of 2 × 106 cells. For sugar fermentation, precultured cells were inoculated into 50 ml of YP medium containing both 40 g/liter glucose and 40 g/liter xylose at an initial cell density of 2 × 107 cells. The culture was incubated in a 250-ml Erlenmeyer flask at 30°C and 100 rpm. RNA was extracted when the absorbance at 600 nm (A600) reached 1, as shown in Fig. 1. For testing of cell growth on xylose, a Bioscreen C plate reader system was used (Growth Curves USA, Piscataway, NJ) as previously described (12).
FIG 1.
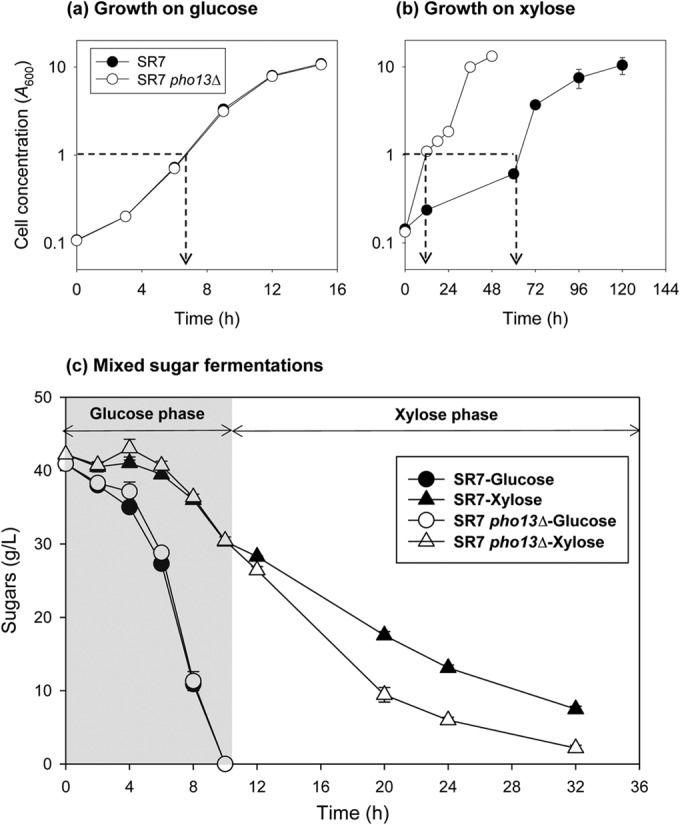
pho13Δ significantly improves the xylose metabolism of the mutant, while there is no visible effect when the cells are metabolizing glucose (overlapping symbols). Cell growth was measured during growth on a single carbon source, glucose (a) or xylose (b). (c) Sugar consumption was measured during growth on a mixture of glucose and xylose, and the results from the two strains were combined. When cells reached the mid-exponential phase (arrows in panels a and b), samples were collected for RNA extraction.
RNA extraction.
For RNA-seq and reverse transcription (RT)-quantitative PCR (qPCR), RNA was extracted from the fresh cultures described above. First, 1 × 107 cells (equivalent to an A600 of 0.5) were harvested and resuspended in a buffer containing 1 M sorbitol, 0.1 M EDTA, 0.1% 2-mercaptoethanol, and 50 U Zymolyase (pH 7.4). The solution was incubated at 30°C and 100 rpm for 30 min to generate spheroplasts. RNA was immediately extracted using a Qiagen RNeasy minikit. The RNA yield was 10 to 40 μg.
RNA-seq.
The concentration and quality of the RNA samples were evaluated on a Bioanalyzer RNA chip. Samples with high-quality total RNA were used to construct a bar-coded library with poly(A)+ RNA. Sequencing was performed on an Illumina HiSeq 2000 system at the W. M. Keck Center for Comparative and Functional Genomics at the University of Illinois at Urbana-Champaign. The detailed procedure for Illumina sequencing has been described previously (12). The sequencing results were analyzed using the CLC Genomic Workbench (version 6.5) program as follows. First, ∼100-bp sequencing reads (fastq files) were trimmed and mapped to the S. cerevisiae S288C reference genome sequence modified with a xylose pathway. All mapped reads at exons were counted, and the numbers were converted to the total number of exon reads per kilobase of exon length per million mapped reads (RPKM). The numbers of RPKM from three biological replicates of the pho13Δ strain were compared to those of the wild-type strain, and t tests were applied. Finally, genes with significantly different expression levels (>2-fold, P < 0.05) were identified (see Data Set S1 in the supplemental material).
RT-qPCR of pentose phosphate (PP) pathway genes.
For quantitative PCR with RNA samples (RT-qPCR), cDNA was generated from 1 μg of RNA using iScript reverse transcription supermix (Bio-Rad, Hercules, CA, USA). Then, cDNA solution (1 μl) was used directly for qPCR with prepared primer solutions and SYBR green I master mix (Roche Applied Science, Indianapolis, IN, USA). qPCRs were performed in a 96-well plate on a LightCycler 480 apparatus (Roche). All primers (Table 2) were designed using the IDT PrimerQuest program.
The qPCR results were calculated by the comparative threshold cycle (CT) method (the ΔΔCT method) (21). Specifically, the transcript abundance (the change in the CT [ΔCT] ± s [where s is the standard deviation of the ΔCT value]) of a target gene (ZWF1, SOL3, GND1, and TAL1) relative to that of a reference gene (ACT1) was calculated from three biological replicates. The difference in the ΔCT value (ΔΔCT ± s [where s is the standard deviation of the ΔCT value of a pho13Δ mutant]) for a pho13Δ mutant compared to that for a wild-type strain was then converted into a fold change [2(−ΔΔCT)] with an error range [2(−ΔΔCT + s) − 2(−ΔΔCT − s)].
RESULTS
pho13Δ enhances xylose metabolism but does not affect glucose metabolism.
To characterize the phenotypic changes induced by pho13Δ, we compared wild-type and pho13Δ mutant strains of xylose-fermenting S. cerevisiae SR7 grown under glucose (Fig. 1a), xylose (Fig. 1b), and the mixed sugar (Fig. 1c) conditions. The SR7 strain, created from the wild-type S. cerevisiae D452-2 strain, contains a heterologous xylose metabolic pathway consisting of the Scheffersomyces stipitis XYL1, XYL2, and XYL3 genes (12). When the strains were grown on glucose, no phenotypic differences were observed (Fig. 1a). However, when the strains were grown on xylose, the SR7 pho13Δ strain grew significantly faster than the SR7 strain (Fig. 1b). Specifically, the SR7 pho13Δ strain grew from a cell density (A600) of 0.1 to one of 1 in 12 h, while the SR7 strain took four times longer. When fermenting a mixture of glucose and xylose (Fig. 1c), the growth differences between the SR7 and SR7 pho13Δ strains were not as obvious as the growth differences on xylose only. However, during a xylose-consuming phase followed by a glucose-consuming phase, the xylose consumption rate of the SR7 pho13Δ strain was faster than that of the SR7 strain, which confirmed that the metabolic benefits induced by pho13Δ were specific to xylose metabolism and consistent for the single sugar and the mixed sugar fermentations (Fig. 1c).
pho13Δ induces the PP pathway and NADPH gene expression regardless of the type of sugar substrate.
To elucidate the mechanisms underlying the phenotypic changes induced by pho13Δ, the global transcript profiles of SR7 and SR7 pho13Δ grown in glucose or xylose were analyzed. Three biological replicates of RNA samples were subjected to RNA-seq. The genes differentially expressed (DE; >2-fold) in the pho13Δ strain compared to their expression in the wild-type strain when the strains were grown in the same sugar were identified with a P value of <0.05 (see Data Set S1 in the supplemental material). Although the deletion phenotype was observed only in cells grown on xylose (Fig. 1), transcriptional changes induced by pho13Δ were observed with both glucose and xylose: 12 DE genes were detected for growth on glucose, and 277 DE genes were detected for growth on xylose (Fig. 2). When those DE genes were presented on the basis of the direction of changes, we found that 9 genes were upregulated under both conditions (Table 4). Most genes were directly or indirectly involved in NADPH regeneration: PP pathway genes (GND1, SOL3, and TAL1), NADPH-specific oxidoreductase genes (GCY1 and GOR1), and NADH kinase genes (YEF1). These results suggest that the loss of Pho13 induces the transcriptional upregulation of genes involved in the PP pathway and redox balance.
FIG 2.
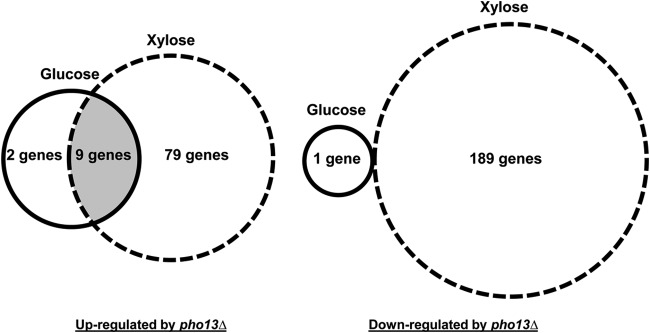
Number of genes upregulated and downregulated (>2-fold, P < 0.05) by pho13Δ during exponential growth on glucose and xylose.
TABLE 4.
Nine genes upregulateda by the pho13Δ strain on both glucose and xylose
| Gene name | Fold change in expressionb |
Function | Pathway | |
|---|---|---|---|---|
| Glucose | Xylose | |||
| SOL3 | 6.2 | 8.8 | 6-Phosphogluconolactonase | PP pathway |
| GND1 | 6.8 | 15.0 | 6-Phosphogluconate dehydrogenase | PP pathway |
| TAL1 | 3.5 | 4.2 | Transaldolase | PP pathway |
| GCY1 | 5.1 | 7.8 | Glycerol dehydrogenase | NADPH regeneration |
| GOR1 | 4.1 | 5.3 | Glyoxylate reductase | NADPH regeneration |
| YEF1 | 2.6 | 13.8 | ATP-NADH kinase | NADPH regeneration |
| YDR248C | 4.3 | 4.9 | Putative gluconokinase | |
| YHR182C-A | 6.0 | 16.6 | Transposable element gene | |
| YLR152C | 4.4 | 6.0 | Uncharacterized | |
Upregulated >2-fold (P < 0.05). None of the genes was downregulated.
Relative number of reads per kilobase of exon length per million reads (RPKM) of the pho13Δ mutant compared to that of the wild-type strain.
pho13Δ upregulates fermentative pathways under xylose conditions.
In terms of the numbers of genes affected and the degree of changes, pho13Δ had greater effects during xylose metabolism than during glucose metabolism. We reasoned that the pho13Δ-induced changes in the transcriptional profile in cells grown on xylose might be related to the phenotypic improvement in xylose metabolism (Fig. 1). To confirm this hypothesis, we investigated the transcriptional changes of genes in central metabolic pathways (the PP pathway, glycolysis, and the tricarboxylic acid [TCA] cycle) (Fig. 3). When cells were grown on xylose, most genes involved in the PP pathway and fermentative metabolism (glycolysis and ethanol production) were upregulated by pho13Δ (black-line arrows in Fig. 3). On the other hand, some genes involved in respiratory metabolism (TCA cycle and ethanol utilization) were downregulated (dotted-line arrows in Fig. 3). The RKI1 gene, coding for ribose-5-phosphate (ribose-5P) isomerase, was the only gene from the PP pathway that was downregulated by pho13Δ. Because ribose-5P is a precursor for the histidine, purine, and pyrimidine biosynthetic pathways, the RKI1 gene was considered a component of anabolic metabolism. In summary, the pho13Δ mutant induced transcriptomic changes that were favorable for xylose fermentation.
FIG 3.

Transcriptomic changes in central metabolic pathways induced by pho13Δ during growth on different carbon sources. The fold change in expression in the pho13Δ mutant relative to that in the wild-type strain is presented. Glyceraldehyde-3P, glyceraldehyde-3-phosphate; Fructose-6P, fructose-6-phosphate; Acetyl-CoA, acetyl coenzyme A.
The pho13Δ-induced upregulation of the PP pathway genes is not specific to the strain background.
To confirm the RNA-seq results, RT-qPCR was performed to assess SOL3, GND1, and TAL1 gene expression using RNA samples from strains SR7 and SR7 pho13Δ grown on glucose. As a negative control, ZWF1 (encoding glucose-6-phosphate dehydrogenase) was also tested because its transcript abundance was not affected by pho13Δ when cells were grown on glucose. The transcript abundance of the three genes was 4- to 8-fold higher in the SR7 pho13Δ strain than in the SR7 strain (Fig. 4), in agreement with the RNA-seq results. To determine if the transcriptional changes induced by pho13Δ were associated with the heterologous xylose pathway in SR7 or were dependent on the strain background (S. cerevisiae D452-2), RT-qPCR for the same gene set was performed to assess transcripts in the D452-2 pho13Δ and BY4742 pho13Δ strains, with the respective wild-type strains being used as controls. In both pho13Δ strains, the SOL3, GND1, and TAL1 genes were upregulated, while ZWF1 expression remained the same. These results suggest that the transcriptional upregulation induced by the loss of Pho13 did not depend either on the xylose pathway or on the strain background.
FIG 4.

RT-qPCR confirmation of transcriptional changes induced by pho13Δ in three different strain backgrounds. The fold change in expression in the pho13Δ mutant relative to that in the corresponding wild-type strain is presented. Three genes (SOL3, GND1, and TAL1) upregulated by pho13Δ were tested along with one gene that was not affected (ZWF1); all genes are in the pentose phosphate pathway. A fold change of 1 (dashed line) indicates no change in the level of transcription.
Transcription factor Stb5 is upregulated by pho13Δ.
The RNA-seq and RT-qPCR results indicated that pho13Δ resulted in global transcriptional changes. Because Pho13 does not have a DNA binding domain, we hypothesized that at least one transcription factor was involved in the pho13Δ-induced transcriptional changes. However, we did not find any transcription factors that were differentially expressed (>2-fold, P < 0.05) by pho13Δ in our RNA-seq results (see Data Set S1 in the supplemental material). To detect minor changes in the expression of transcription factors, the RNA-seq data were reprocessed using a P value of <0.05. With this criterion, 30 transcription factors were differentially expressed because of pho13Δ in cells grown on glucose or xylose (Fig. 5). Among the six transcription factors that were differentially expressed in the pho13Δ strain on glucose, STB5 was the only gene that was regulated in the same direction (upregulation) under both conditions (1.3-fold increase on glucose and 1.5-fold increase on xylose). YAP5 was upregulated during growth on glucose but downregulated during growth on xylose. STB5 is a zinc finger protein that upregulates PP pathway and NADPH-producing genes under oxidative stress and NADPH limitation (22, 23). The results suggest that Stb5 mediates the transcriptional changes induced by pho13Δ.
FIG 5.
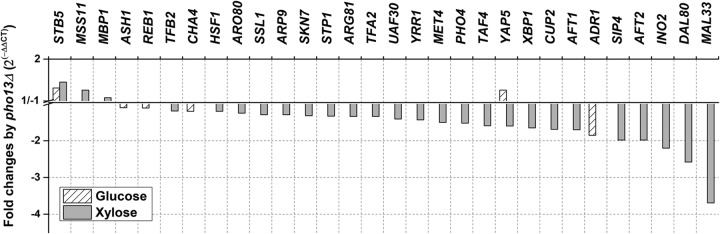
Fold change in the expression of transcription factors induced by pho13Δ during growth on different carbon sources (P < 0.05).
Stb5 is required for the transcriptional changes induced by pho13Δ.
To determine if Stb5 plays a role in the transcriptional changes induced by pho13Δ, the transcript abundance of the gene that was the most strongly upregulated by pho13Δ, GND1, was compared in strains lacking PHO13 or STB5 (Fig. 6). The relative expression of GND1 was significantly higher in the pho13Δ mutant than in the wild-type strain, as found earlier. However, in the stb5Δ mutant, the level of GND1 was only 10% of that in the wild-type strain, which supported the conclusion that the Stb5 transcription factor is required for PP pathway activation. Deletion of PHO13 in the stb5Δ mutant (resulting in a pho13Δ stb5Δ mutant) did not upregulate GND1. These results suggest that Stb5 is necessary for the transcriptional upregulation induced by pho13Δ.
FIG 6.

Upregulation of GND1 by pho13Δ requires the transcription factor Stb5 and the Stb5-binding site of GND1. RT-qPCR was used to measure the expression of GND1 in strains with pho13Δ or stb5Δ. The pho13Δ GND1.1 mutant had an SNP (CTGTGTTA, where the SNP is underlined) in the Stb5-binding sequence (CGGTGTTA) upstream of the GND1 coding region.
To confirm that Stb5 was responsible for the transcriptional changes induced by pho13Δ, we created a mutant in which Stb5 could not mediate the transcriptional regulation of GND1. A mutation was introduced in the Stb5-binding sequence (CGGTGTTA), located 262 bp upstream from the GND1 coding region. Chromatin immunoprecipitation with microarray technology analysis has demonstrated that DNA oligonucleotides with an SNP in the binding sequence, such as CTGTGTTA (where the SNP is underlined), do not exhibit any detectable binding to Stb5 (22). Using a CRISPR-Cas system (20), we introduced an SNP (CTGTGTTA, where the SNP is underlined) into the Stb5-binding sequence of GND1 in vivo, to generate GND1.1. When GND1.1 was introduced into the pho13Δ strain, GND1 upregulation was not detected, and the expression was the same as that in the wild-type strain (Fig. 6). The result strongly supports the conclusion that Stb5 mediates the transcriptional changes induced in the pho13Δ mutant.
STB5 overexpression did not lead to the transcriptional activation of the pho13Δ-response genes.
Because the pho13Δ mutation resulted in the upregulation of STB5, we tested if the overexpression of STB5 alone could activate the pho13Δ-response genes, such as GND1. When the STB5 gene was overexpressed under the control of the strong and constitutive TEF1 promoter, the mutant had a lower level of expression of GND1 than the control strain (Fig. 7a). Moreover, the STB5-overexpressing mutant was not able to grow on xylose under a condition where the SR7 strain grew significantly (Fig. 7b). These results suggest that the upregulation of STB5 is not sufficient to explain transcriptional changes induced by pho13Δ.
FIG 7.

A strain overexpressing STB5 did not upregulate GND1 (a) and did not grow on xylose (b). RT-qPCR was used to measure the expression of GND1 in the control strain (SR7 41K), the pho13 deletion mutant (SR7 pho13Δ), and the STB5-overexpressing mutant (SR7 41K-TEF1P-STB5). The cell growth of the three strains was measured in YP medium containing 40 g/liter xylose for 80 h using a Bioscreen C plate reader system.
DISCUSSION
The Pho13 enzyme in S. cerevisiae has received little attention because it is not directly related to any of the metabolic pathways in native strains and because its deletion does not produce a significant phenotype under standard culture conditions with glucose. On the other hand, three independent studies have found that pho13Δ improves xylose metabolism in strains engineered with a heterologous xylose-consuming pathway (11–13). Although the phenotypic improvement was significant, the mechanism was difficult to explain because xylose metabolism itself is not clearly understood in the engineered strains. Nevertheless, one study proposed that the xylulose-5-phosphate phosphatase activity of Pho13 might create a futile cycle when coupled with strong xylulokinase activity (12). Xylulokinase is a key enzyme connecting the heterologous xylose-consuming pathway to the native PP pathway (17).
However, the results of the present study suggest that Pho13 has other biochemical properties that are not dependent on xylose or the presence of the xylose pathway. Moreover, a previous study found that Pho13 had protein phosphatase activity against phosphoseryl proteins (24), suggesting that Pho13 might be involved in protein signaling. Meanwhile, a spontaneous mutation in Pho13 improved the protein's specificity; the mutant form of Pho13 (G208D) was able to dephosphorylate a toxic intermediate in the purine biosynthesis pathway, 5-amino-4-imidazole N-succinocarboxamide ribonucleotide-5-phosphate (SAICAR), to generate the nontoxic riboside (25).
One hypothesis regarding the function of Pho13 is that S. cerevisiae Pho13 has a broad range of substrates and little specificity for the detoxification of sugar phosphates that accumulate to high levels. When 23 HAD-like phosphatases from E. coli were tested against 80 phosphorylated substrates, most enzymes exhibited substrate promiscuity with very wide and overlapping substrate ranges (26). Specifically, E. coli NagD, an S. cerevisiae Pho13 ortholog, had high specificities for both nucleotide monophosphates (UMP and GMP) and sugar phosphates (ribose-5-phosphate and glucose-1-phosphate) (10, 26). Although NagD is expressed from the N-acetylglucosamine (GlcNAc) metabolic operon, it has no effect on metabolism (27). Because nonspecific HAD enzymes have broad and low substrate specificities and do not participate in specific metabolic pathways, it was previously proposed that nonspecific HAD enzymes detoxify sugar phosphates that accidentally accumulate at metabolic bottlenecks (9).
On the other hand, some eukaryotic orthologs of S. cerevisiae Pho13 have unique biological functions and specific substrates. Phosphoglycolate phosphatase 1 (Pgp1), a Pho13 ortholog in the plant Arabidopsis thaliana, contributes to the photorespiration pathway in chloroplasts (28). When the gene coding for Pgp1 was disrupted, the mutant was unable to grow in normal air (28). Interestingly, disruption of a cytosolic homolog of Pgp1, Pgp2, produced no phenotype, despite the fact that both enzymes have the same biochemical properties (28). Meanwhile, an S. cerevisiae Pho13 ortholog in humans was found to have pyridoxal phosphatase activity in the vitamin B6 metabolic pathways (29).
Currently, it is not clear whether S. cerevisiae Pho13 is closely related to any bacterial or eukaryotic orthologs. However, other yeast orthologs, which have not been characterized, could provide more information about the enzymes in this subfamily. Specifically, investigation of native xylose-fermenting yeast, such as S. stipitis and Neurospora crassa, might be a good strategy for determining if the pho13Δ-induced signaling mechanism is conserved in yeast.
In this study, we identified a global transcriptional response induced by deletion of PHO13 (pho13Δ). In the pho13Δ mutant, an oxidative stress response was constitutively activated via the transcription factor Stb5. The stress response involved upregulation of PP pathway and NADPH-producing genes. The connection between Pho13 and Stb5 might be important for understanding the biological function of S. cerevisiae Pho13. Given that Stb5 is activated under NADPH-limiting conditions (22, 23), Pho13 could also be involved in cellular redox maintenance.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from the Energy Biosciences Institute to Soo Rin Kim and Yong-Su Jin.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03474-14.
REFERENCES
- 1.Ljungdahl PO, Daignan-Fornier B. 2012. Regulation of amino acid, nucleotide, and phosphate metabolism in Saccharomyces cerevisiae. Genetics 190:885–929. doi: 10.1534/genetics.111.133306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saldanha AJ, Brauer MJ, Botstein D. 2004. Nutritional homeostasis in batch and steady-state culture of yeast. Mol Biol Cell 15:4089–4104. doi: 10.1091/mbc.E04-04-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Heerden JH, Wortel MT, Bruggeman FJ, Heijnen JJ, Bollen YJM, Planqué R, Hulshof J, O'Toole TG, Wahl SA, Teusink B. 2014. Lost in transition: start-up of glycolysis yields subpopulations of nongrowing cells. Science 343:1245114. doi: 10.1126/science.1245114. [DOI] [PubMed] [Google Scholar]
- 4.Hunter T. 1995. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 5.Akiva E, Brown S, Almonacid DE, Barber AE, Custer AF, Hicks MA, Huang CC, Lauck F, Mashiyama ST, Meng EC, Mischel D, Morris JH, Ojha S, Schnoes AM, Stryke D, Yunes JM, Ferrin TE, Holliday GL, Babbitt PC. 2014. The structure-function linkage database. Nucleic Acids Res 42:D521–D530. doi: 10.1093/nar/gkt1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Motosugi K, Esaki N, Soda K. 1982. Purification and properties of a new enzyme, dl-2-haloacid dehalogenase, from Pseudomonas sp. J Bacteriol 150:522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lahiri SD, Zhang G, Dai J, Dunaway-Mariano D, Allen KN. 2004. Analysis of the substrate specificity loop of the HAD superfamily cap domain. Biochemistry 43:2812–2820. doi: 10.1021/bi0356810. [DOI] [PubMed] [Google Scholar]
- 8.Koonin EV, Tatusov RL. 1994. Computer analysis of bacterial haloacid dehalogenases defines a large superfamily of hydrolases with diverse specificity: application of an iterative approach to database search. J Mol Biol 244:125–132. doi: 10.1006/jmbi.1994.1711. [DOI] [PubMed] [Google Scholar]
- 9.Godinho LM, de Sá-Nogueira I. 2011. Characterization and regulation of a bacterial sugar phosphatase of the haloalkanoate dehalogenase superfamily, AraL, from Bacillus subtilis. FEBS J 278:2511–2524. doi: 10.1111/j.1742-4658.2011.08177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tremblay LW, Dunaway-Mariano D, Allen KN. 2006. Structure and activity analyses of Escherichia coli K-12 NagD provide insight into the evolution of biochemical function in the haloalkanoic acid dehalogenase superfamily. Biochemistry 45:1183–1193. doi: 10.1021/bi051842j. [DOI] [PubMed] [Google Scholar]
- 11.Ni H, Laplaza JM, Jeffries TW. 2007. Transposon mutagenesis to improve the growth of recombinant Saccharomyces cerevisiae on d-xylose. Appl Environ Microbiol 73:2061–2066. doi: 10.1128/AEM.02564-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim SR, Skerker JM, Kang W, Lesmana A, Wei N, Arkin AP, Jin Y-S. 2013. Rational and evolutionary engineering approaches uncover a small set of genetic changes efficient for rapid xylose fermentation in Saccharomyces cerevisiae. PLoS One 8:e57048. doi: 10.1371/journal.pone.0057048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujitomi K, Sanda T, Hasunuma T, Kondo A. 2012. Deletion of the PHO13 gene in Saccharomyces cerevisiae improves ethanol production from lignocellulosic hydrolysate in the presence of acetic and formic acids, and furfural. Bioresour Technol 111:161–166. doi: 10.1016/j.biortech.2012.01.161. [DOI] [PubMed] [Google Scholar]
- 14.Van Vleet JH, Jeffries TW, Olsson L. 2008. Deleting the para-nitrophenyl phosphatase (pNPPase), PHO13, in recombinant Saccharomyces cerevisiae improves growth and ethanol production on d-xylose. Metab Eng 10:360–369. doi: 10.1016/j.ymben.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Li Y-C, Gou Z-X, Liu Z-S, Tang Y-Q, Akamatsu T, Kida K. 2014. Synergistic effects of TAL1 over-expression and PHO13 deletion on the weak acid inhibition of xylose fermentation by industrial Saccharomyces cerevisiae strain. Biotechnol Lett 36:2011–2021. doi: 10.1007/s10529-014-1581-7. [DOI] [PubMed] [Google Scholar]
- 16.Lee S-M, Jellison T, Alper H. 2014. Systematic and evolutionary engineering of a xylose isomerase-based pathway in Saccharomyces cerevisiae for efficient conversion yields. Biotechnol Biofuels 7:122. doi: 10.1186/s13068-014-0122-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim SR, Park Y-C, Jin Y-S, Seo J-H. 2013. Strain engineering of Saccharomyces cerevisiae for enhanced xylose metabolism. Biotechnol Adv 31:851–861. doi: 10.1016/j.biotechadv.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Hegemann J, Heick S. 2011. Delete and repeat: a comprehensive toolkit for sequential gene knockout in the budding yeast Saccharomyces cerevisiae, p 189–206. In Williams JA. (ed), Strain engineering, vol 765 Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 19.Gietz RD, Schiestl RH. 2007. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- 20.DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM. 2013. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41:4336–4343. doi: 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Larochelle M, Drouin S, Robert F, Turcotte B. 2006. Oxidative stress-activated zinc cluster protein Stb5 has dual activator/repressor functions required for pentose phosphate pathway regulation and NADPH production. Mol Cell Biol 26:6690–6701. doi: 10.1128/MCB.02450-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hector RE, Bowman MJ, Skory CD, Cotta MA. 2009. The Saccharomyces cerevisiae YMR315W gene encodes an NADP(H)-specific oxidoreductase regulated by the transcription factor Stb5p in response to NADPH limitation. New Biotechnol 26:171–180. doi: 10.1016/j.nbt.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 24.Tuleva B, Vasileva-Tonkova E, Galabova D. 1998. A specific alkaline phosphatase from Saccharomyces cerevisiae with protein phosphatase activity. FEMS Microbiol Lett 161:139–144. doi: 10.1111/j.1574-6968.1998.tb12940.x. [DOI] [PubMed] [Google Scholar]
- 25.Hürlimann HC, Laloo B, Simon-Kayser B, Saint-Marc C, Coulpier F, Lemoine S, Daignan-Fornier B, Pinson B. 2011. Physiological and toxic effects of purine intermediate 5-amino-4-imidazolecarboxamide ribonucleotide (AICAR) in yeast. J Biol Chem 286:30994–31002. doi: 10.1074/jbc.M111.262659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuznetsova E, Proudfoot M, Gonzalez CF, Brown G, Omelchenko MV, Borozan I, Carmel L, Wolf YI, Mori H, Savchenko AV, Arrowsmith CH, Koonin EV, Edwards AM, Yakunin AF. 2006. Genome-wide analysis of substrate specificities of the Escherichia coli haloacid dehalogenase-like phosphatase family. J Biol Chem 281:36149–36161. doi: 10.1074/jbc.M605449200. [DOI] [PubMed] [Google Scholar]
- 27.Peri KG, Goldie H, Waygood EB. 1990. Cloning and characterization of the N-acetylglucosamine operon of Escherichia coli. Biochem Cell Biol 68:123–137. doi: 10.1139/o90-017. [DOI] [PubMed] [Google Scholar]
- 28.Schwarte S, Bauwe H. 2007. Identification of the photorespiratory 2-phosphoglycolate phosphatase, PGLP1, in Arabidopsis. Plant Physiol 144:1580–1586. doi: 10.1104/pp.107.099192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jang YM, Kim DW, Kang T-C, Won MH, Baek N-I, Moon BJ, Choi SY, Kwon O-S. 2003. Human pyridoxal phosphatase: molecular cloning, functional expression, and tissue distribution. J Biol Chem 278:50040–50046. doi: 10.1074/jbc.M309619200. [DOI] [PubMed] [Google Scholar]
- 30.Güldener U, Heck S, Fiedler T, Beinhauer J, Hegemann JH. 1996. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res 24:2519–2524. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker Brachmann C, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14:115–132. doi:. [DOI] [PubMed] [Google Scholar]
- 32.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian K-D, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, et al. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 33.Hosaka K, Nikawa J-i, Kodaki T, Yamashita S. 1992. A dominant mutation that alters the regulation of INO1 expression in Saccharomyces cerevisiae. J Biochem 111:352–358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.