

Abstract
Sodium (Na) homeostasis is crucial for life, and the Na+ level ([Na+]) of body fluids is strictly maintained at a range of 135–145 mM. However, the existence of a [Na+] sensor in the brain has long been controversial until Nax was identified as the molecular entity of the sensor. This review provides an overview of the [Na+]-sensing mechanism in the brain for the regulation of salt intake by summarizing a series of our studies on Nax. Nax is a Na channel expressed in the circumventricular organs (CVOs) in the brain. Among the CVOs, the subfornical organ (SFO) is the principal site for the control of salt intake behavior, where Nax populates the cellular processes of astrocytes and ependymal cells enveloping neurons. A local expression of endothelin-3 in the SFO modulates the [Na+] sensitivity for Nax activation, and thereby Nax is likely to be activated in the physiological [Na+] range. Nax stably interacts with Na+/K+-ATPase whereby Na+ influx via Nax is coupled with activation of Na+/K+-ATPase associated with the consumption of ATP. The consequent activation of anaerobic glucose metabolism of Nax-positive glial cells upregulates the cellular release of lactate, and this lactate functions as a gliotransmitter to activate GABAergic neurons in the SFO. The GABAergic neurons presumably regulate hypothetic neurons involved in the control of salt intake behavior. Recently, a patient with essential hypernatremia caused by autoimmunity to Nax was found. In this case, the hypernatremia was considered to be induced by the complement-mediated cell death in the CVOs, where Nax specifically populates.
Keywords: Salt homeostasis, Na+ sensing, Nax channel, Sensory circumventricular organs, Subfornical organ
Introduction
Terrestrial animals are exposed to considerable risks of dehydration and salt deficiency, and their life depends on the maintenance of water and salt in the body fluids [3, 4]. Changes in cell volume caused by severe hypertonicity or hypotonicity in body fluids can lead to irreversible damage to organs including nervous systems [5, 7, 62]. To escape from such risks, mammals have a set of homeostatic mechanisms that work together to maintain body fluid osmolality at approximately 300 mOsm/kg mainly through the intake or excretion of water and salt [37, 55]. When animals are dehydrated, both sodium ion concentration ([Na+]) and osmolality in body fluids increase because Na+ is the major cationic component of extracellular fluids and the main determinant of body fluid osmolality. Na+ homeostasis is thus inseparably linked with body fluid control, and [Na+] in body fluids needs to be continuously monitored to maintain its physiological range (135–145 mM for mammals) [4, 58].
Bengt Andersson hypothesized the existence of a putative [Na+] sensor within the brain nearly four decades ago that is distinct from osmosensors [3, 22]. Since then, a series of experimental evidences has suggested that the [Na+] increase in body fluids is detected in the brain [17, 48]. In addition, experimental ablation studies suggested that the anterior wall of the third ventricle is involved in [Na+] sensing [4, 17, 39]. However, the precise location of the [Na+]-sensing cells and the molecular entity of the [Na+]-specific sensor were not identified for a long time, though it has been a major interest of physiologists.
The most likely candidate sites for [Na+] sensing were postulated to be the circumventricular organs (CVOs) of the brain [16, 17, 57]. The CVOs, midline structures found in the brain of all vertebrates, are so named because of their proximity to the ventricles of the brain [49]. Their specialized common features are extensive vascularization, no blood-brain barrier (BBB), and atypical ependymal cells being exposed to cerebrospinal fluid (CSF). Among the CVOs, only three loci, the subfornical organ (SFO), organum vasculosum of the lamina terminalis (OVLT), and area postrema (AP), harbor neuronal cell bodies that have efferent neural connections to many other areas of the brain. Therefore, these three CVOs are termed sensory CVOs [38] (Fig. 1a). Because of the lack of a BBB, their component cells are exposed to the chemical environment of the systemic circulation, unlike other neural cells in the central nervous system (CNS). The SFO and OVLT situated on the anterior wall of the third ventricle are supposed to be involved in the sensing of [Na+] and osmolality .
Fig. 1.
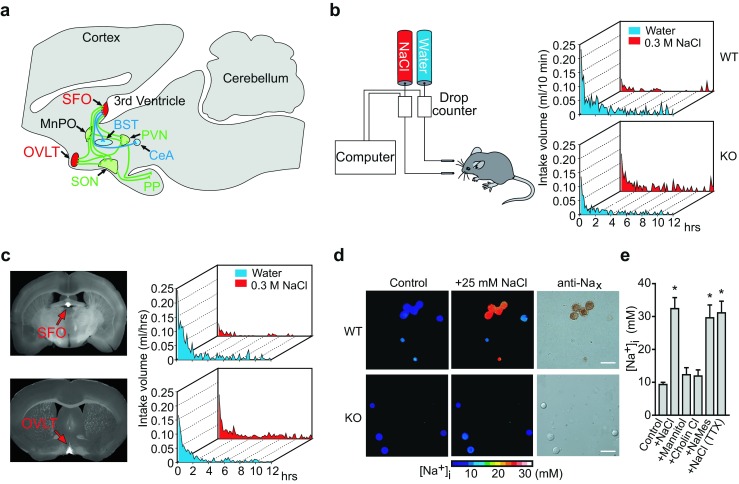
SFO is the primary locus of [Na+] sensing by Nax channel for the control of salt intake behavior. a Neural connections for body fluid control in the brain. Nax-positive sensory circumventricular organs in the midsagittal section are schematically represented in red. Of note, Nax is not expressed in the MnPO. Green lines indicate the neural connections involved in regulating the release of vasopressin; blue lines indicate putative neural connections involved in the control of water or salt intake. SFO subfornical organ, MnPO median preoptic area, OVLT organum vasculosum of the lamina terminalis, BST bed nucleus of the stria terminalis, SON supraoptic nucleus, PVN paraventricular nucleus, CeA central nucleus of the amygdala, PP posterior pituitary. b Averaged time course of water and saline (0.3 M NaCl) intake in wild-type (WT) and Na x-KO (KO) mice during the dark phase immediately after 48-h dehydration. Each point shows the averaged quantity per 10-min period of ten mice. c Coronal sections of mouse brains obtained from Na x-KO mice showing the infected loci by the expression of EGFP (left column). Time course of water and saline (0.3 M NaCl) intake by the infected mice after 48-h dehydration (right column). Behavioral data are the average of six mice that were successfully infected at a specific site in the brain by vectors encoding Na x and egfp. All of the Na x-KO mice that were conferred with the Na aversion behavior showed a common infection in the SFO (upper). On the other hand, transduction of the Nax gene into the OVLT could not rescue the abnormal salt intake behavior of the Na x-KO mice (lower). d Pseudocolor images showing the [Na+]i of the cells in the control ([Na+]o = 145 mM) and high-Na+ ([Na+]o = 170 mM) solutions (left and middle columns) and immunocytochemical images using anti-Nax antibody (right column). Scale bars, 50 μm. e The [Na+]i response to various stimulations. The response was dependent on [Na+]o, but not on extracellular [Cl−]o or osmotic pressure. Instead of 25 mM NaCl, 50 mM mannitol, 25 mM choline chloride (Cholin Cl), or 25 mM sodium methanesulfonate (NaMes) was added to the control solution. The response was not affected by 1 μM tetrodotoxin (TTX). *P < 0.001 by one-tailed Mann-Whitney tests. Reproduced with permission from [55] (a), [33] (b, c), and [34] (d, e)
The cell type of [Na+]-sensing cells has also been a concern whether they are neurons or glial cells. It has been long considered that neurons transmit neural information and glial cells provide nourishment and physical scaffold for neurons. However, it is now widely accepted that glial cells can actively modulate the information processing by neurons via several mechanisms [28, 63].
The primary locus of [Na+] sensing and the molecular entity of the [Na+] sensor
We have long been studying the functional roles of an atypical Na channel Nax, which was initially classified as a subfamily of voltage-gated Na channels and called Nav2 [51, 53, 54]. Nax was originally cloned by several independent groups from rat astrocytes [23], the human heart [24], a mouse atrial tumor cell line [21], and the rat dorsal root ganglia (DRG) [2]. The primary structure of Nax markedly differed from that of the other Nav channel members, including the key regions for voltage sensing and inactivation [25, 51]. Because previous attempts at the functional expression of Nax in heterologous systems had failed, we generated Na x-gene-knockout (Na x-KO) mice by inserting the lacZ gene in frame to examine the distribution and physiological roles of this channel [51, 65]. Nax was revealed to be expressed in some limited loci in the brain, including the SFO and OVLT [65]. Nax expression in these loci was confirmed by immunohistochemistry [67].
As the SFO and OVLT were the potential loci for [Na+] sensing, we examined the salt intake behaviors of the Na x-KO mice using the two-bottle test, providing them with both distilled water and a 0.3 M NaCl solution to drink [33, 65]. As long as wild-type (WT) and Na x-KO mice were fully satiated with water, they showed no marked preference for either [33]. However, when they were dehydrated, WT mice showed extensive water intake and aversion to saline, while Na x-KO mice did not show such an aversion to the saline (Fig. 1b) [33].
Because Na x-KO mice have a normal tasting ability, including that for salt, the behavioral defects in the Na x-KO mice were supposed to be attributable to some internal sensing mechanisms for [Na+] in body fluids [65]. Consistent with this view, infusion of a hypertonic Na+ solution into the cerebral ventricle did not induce aversion to salt in Na x-KO mice, in contrast to wild-type animals [33]. Importantly, the aversion to salt was not induced by the infusion of a hyperosmotic mannitol solution with physiological [Na+] in either genotype of mice, suggesting that Nax is involved in the [Na+]-specific sensing mechanism in the brain [33].
After water deprivation, Na x-KO mice showed marked neuronal activation in the SFO and OVLT compared with WT mice, as estimated by Fos immunoreactivity [65]. The behavioral phenotype of Na x-KO mice was completely recovered by a site-directed transfer of the Na x gene with an adenoviral vector into the SFO (Fig. 1c) [33]. These data clearly indicate that the SFO is the primary locus of [Na+] sensing in the brain for the control of salt intake behavior and that Nax plays a critical role in the sensing mechanism.
Molecular properties of Nax in vitro
We speculated that Nax may open in response to changes in extracellular [Na+] ([Na+]o) and function as the [Na+] sensor in the brain. We verified this possibility by imaging analysis of changes in the intracellular [Na+] ([Na+]i) when the [Na+]o was raised stepwise from the lower amount [34]. When a series of Na+ solutions higher than the physiological level were applied to Nax-positive cells isolated from the SFO, persistent Na+ influx appeared (Fig. 1d) [34]. The threshold value of Nax for [Na+]o was ∼150 mM ([19]; see also Fig. 2a). These [Na+]-sensitive cells were insensitive to the rise in osmolality or [Cl−]o (Fig. 1e) [34]. As expected, no SFO cells derived from Na x-KO mice showed such responses, and transfection of Nax cDNA conferred [Na+]o sensitivity on the cells from Na x-KO mice [53]. Notably, the Nax-immunoreactive cells in these loci were revealed to be glial fibrillary acidic protein (GFAP)-positive glial cells (astrocytes and ependymal cells) [67]. These data indicate that Nax is a Na channel sensitive to an increase in the [Na+]o and that glial cells are sensing cells.
Fig. 2.

Endothelin-3 (ET-3) signaling shifted the [Na+]o dependency of Nax activation to the lower side. a Relationships between the current density and [Na+]o in the presence or absence of 1 nM ET-3; n = 6 for each. b In situ hybridization for detection of ET-3 expression in the SFO of WT mice. Brains were obtained from mice provided freely with food and water (0 h) or from those provided only with food during the indicated period (12, 24, 36, and 48 h). Sections on the same slide are shown. c Activation cascades of Nax by ETBR signaling. The pathway indicated by dotted lines was suggested not to work for the Nax activation. Reproduced with permission from [35]
[Na+] dependency of Nax in vivo
As aforementioned, [Na+] is strictly controlled at 135–145 mM in the blood and CSF of mammals, including humans [58]. In order to maintain the physiological level strictly, the active range of sensitivity of brain [Na+] sensor(s) should be within this range. However, the apparent threshold value of Nax activation was ∼150 mM in vitro, as described above [34]. Therefore, it was presumed that the threshold value of Nax for [Na+]o must be modulated in vivo by some unknown mechanism.
Endothelin receptor B (ETBR) is predominantly expressed in glial cells in the brain [36] and extremely highly expressed in the SFO [31]. Furthermore, ET peptides and their receptors are intimately involved in the physiological control of systemic blood pressure and Na homeostasis [43]. It was thus tempting to speculate that ET is involved in signaling mechanisms mediated by sensor molecules such as Nax in the SFO.
By our in situ hybridization, ET-3, but not ET-1 or ET-2 messenger RNA (mRNA), was detected in some cells inside the SFO [35]. Moreover, ET-converting enzymes (Ece1 and Ece2), proteases responsible for the conversion of inactive ET precursors (big endothelin) to bioactive mature forms [41], were also expressed in the SFO in a similar manner [35]. This situation suggested the presence of autocrine or paracrine signaling mechanisms for ET in the SFO (see below).
Enhancement mechanism of Nax sensitivity in vivo by ET-3
We examined the effects of ET-3 on the [Na+]o dependency of Nax by using the patch clamp method [35]. [Na+]o-sensitive inward currents were observed when the “high Na+ solution” ([Na+]o = 170 mM) was applied to Nax-positive SFO cells derived from WT mice [35]. The dose response curve of the [Na+]o-dependent response reached a maximum at ∼170 mM without ET-3; C 1/2 was 161 mM (Fig. 2a; [ET-3] = 0 nM). When 1 nM ET-3 was applied, the response curve of [Na+]o dependency shifted to the lower side; the C 1/2 value shifted to 133 mM (Fig. 2a; [ET-3] = 1 nM). This condition may reflect a physiological situation in vivo.
During dehydration, ET-3 mRNA levels in the SFO increased in a time-dependent manner (Fig. 2b; approximately ten fold at 36 h compared to 0 h). On the other hand, the expression of ETBR was not regulated by dehydration [35]. The activation of Nax via ETBR was revealed to be mediated by a PKC pathway that activates ERK1/2 downstream [35] (Fig. 2c). The phosphorylation of ERK1/2 was consistently and markedly enhanced in the SFO tissues of dehydrated mice [35].
Cells in the SFO, which lacks a BBB, may be exposed to circulating hormones, including ETs [61]. However, ET-3 levels in the plasma and CSF remained low even after 1–2 days of dehydration (ranging from 13 to 32 pM) [35]. Because ET-3 at 50 pM did not affect Nax gating, ET-3 locally produced in the SFO probably stimulates Nax-positive glial cells through ETBR in an autocrine or paracrine fashion.
We further demonstrated that a specific blocker of ETBR attenuated the salt-aversive behavior in WT mice induced by dehydration [35]. Even in slightly dehydrated animals, induction of the expression of ET-3 in the SFO may lead to a stimulation of ETBR signaling and a significant enhancement of the sensitivity of Nax to [Na+]o, which may help animals respond to dehydration robustly.
Direct interaction between Nax channels and α subunits of Na+/K+-ATPase
Our analyses using electron microscopy revealed that Nax channels are specifically expressed in perineuronal processes of astrocytes and ependymal cells enveloping particular neural populations, including GABAergic neurons in the SFO (Fig. 3a). These Nax-positive glial cells were sensitive to an increase in the [Na+]o [67], indicating that glial cells, not neurons, are the primary site of [Na+] sensing (Fig. 3b). These findings imply that there exists a signal transfer from Nax-positive glial cells to SFO neurons because neuronal activity in the SFO is involved in the control of body fluid homeostasis. However, there was no clue as to the signaling substance or mechanism at that time.
Fig. 3.
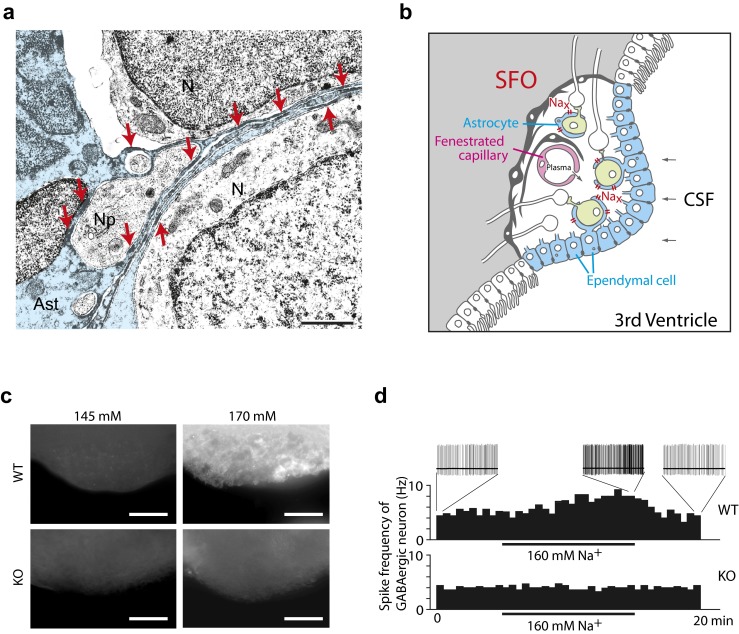
Nax channels control lactate signaling from glial cells to neurons for [Na+] sensing in the SFO. a Immunoelectron microscopy of the SFO using anti-Nax antibody. Neurons (N) and their processes (Np) are enveloped with the immunopositive thin processes of an astrocyte (Ast; blue). Red arrows point to immunopositive signals. Neurons and their processes, including synapses, are surrounded by immunopositive thin processes of astrocytes. Scale bars, 1 μm. b Schematic drawing of Nax-positive ependymal cells and astrocytes in the SFO. The SFO is characterized by the presence of neuronal cell bodies and extensive networks of fenestrated capillaries that allow components of the plasma to leak into the intercellular space. The SFO has contact with the CSF through a single layer of Nax-positive ependymal cells. c Imaging analyses of the uptake of glucose in the SFO using a fluorescent glucose derivative. The SFO tissues isolated from wild-type (WT) and Na x-KO (KO) mice were incubated with the fluorescent glucose analog in 145 mM (left column) or 170 mM (right column) Na+ solution. Scale bars, 50 μm. d Control of spike frequency of GABAergic neurons in the SFO by Na+. The SFO tissues from WT and Na x-KO mice were treated with the high-Na+ solution. Nax is indispensable for [Na+]-dependent potentiation of the GABAergic firing in the SFO. Reproduced with permission from [67] (a), [52] (b), and [60] (c, d)
We therefore started with a screening for molecules interacting with the cytoplasmic domains of Nax to better understand the physiological processes involving Nax in glial cells. This screening revealed that Nax channels stably interact with α1 and α2 subunits of Na+/K+-ATPase via its carboxyl-terminus region [60]. Subsequent detailed analyses revealed a close physical and functional coupling between Nax and Na+/K+-ATPase: Binding of Nax to Na+/K+-ATPase is requisite to [Na+]i-dependent activation of Na+/K+-ATPase, and [Na+]o-dependent activation of Nax leads to stimulation of Na+/K+-ATPase activity [60].
Na+-dependent metabolic enhancement of the Nax-positive glial cells
Activation of Na+/K+-ATPase potentially stimulates anaerobic metabolism of glucose in glial cells and produces lactate as the end product. To examine whether the Nax channel is indeed involved in the energy control system in the Nax-positive glial cells in vivo, we performed an imaging analysis of the uptake of glucose in the SFO using a fluorescent glucose derivative. [Na+]o-sensitive glucose uptake was obviously detected selectively in the WT but not in the Na x-KO tissues: After incubation with a hypertonic Na+ solution, an intensively labeled mesh-like structure became apparent in the SFO obtained from WT mice [60] (Fig. 3c). This result suggested that fine glial processes in the SFO actively took up the fluorescent derivative of glucose. The enhancement of glucose uptake was completely abolished by an Na+/K+-ATPase inhibitor, ouabain, indicating that the activity of Na+/K+-ATPase plays an essential role in the glucose demand induced by the elevation of the [Na+]o [60]. Consistently, lactate release from the SFO tissue of WT, but not of Na x-KO mice, was upregulated by incubation with hypertonic Na+ solution [60].
Lactate signaling from glial cells to neurons
We next examined the possibility that lactate mediates the signal transfer from glial cells to neurons to control activity. In the SFO, GABAergic neurons are one of the major neuronal types surrounded by Nax-positive glial processes [67]. In electrophysiological experiments using tissue slices, we found that the GABAergic neurons in the SFO are spontaneously firing, and the firing frequency in the slices of WT mice gradually increased on application of hypertonic Na+ (Fig. 3d, WT). In contrast, the activity of GABAergic neurons in the SFO of Na x-KO mice was not potentiated by hypertonic Na+ (Fig. 3d, KO). GABAergic neurons of both genotypes were increased when lactate was directly added at 1 mM to the perfusate [60]. An inhibitor of monocarboxylate transporters (MCTs), which transport lactate across the membrane, inhibited the Na+-dependent potentiation of the GABAergic firing [60]. Subsequent analyses revealed that the underlying mechanism of the activation was depolarization of GABAergic neurons, due, in part, to the inactivation of the ATP-sensitive K channel (Kir6.2/KATP channel): the KATP channel closes in response to an increase of intracellular ATP level as a result of lactate metabolism in neurons [60]. These data clearly indicate that lactate released from glial cells serves as an energy substrate to upregulate the firing activity of the GABAergic neurons. This lactate signaling appears to play a crucial role in the control of neuronal activities involved in the Na intake behavior in the brain.
Summary of the [Na+]-sensing mechanism in the SFO for the control of salt intake behavior
Based on our findings, a schematic overview of the cellular mechanisms for [Na+] sensing and [Na+]-dependent regulation of neural activities in the SFO is presented in Fig. 4. The sensory CVOs, including the SFO, are characterized by the extensive networks of fenestrated capillaries which allow ingredients of plasma to be released to the intercellular space. Their ventricular side is partitioned by an ependymal cell layer facing the third ventricle. Nax channels populate perineural processes of astrocytes and ependymal cells in the SFO. Even under hydrated (normal) conditions, ET-3 level expressed in the SFO could modulate the [Na+]o dependency of Nax and make Nax sensitive to an increase in [Na+]o in the physiological range. When animals are dehydrated, [Na+] in plasma and CSF significantly increases above the usual level. Under such conditions, the [Na+]o exceeds the threshold of Nax, Nax channels open, and the [Na+]i is increased. This leads to activation of Na+/K+-ATPase in these cells. Activated Na+/K+-ATPase consumes ATP higher than the usual level to pump out Na+. To fuel Na+/K+-ATPase with ATP, the glial cells enhance glucose uptake to stimulate anaerobic glycolysis. Lactate, the end product of the anaerobic glycolysis, is released from the glial cells and supplied to neurons, including GABAergic neurons, through the processes enveloping them. Lactate stimulates the activity of the GABAergic neurons through production of ATP, which presumably leads to the regulation of hypothetic neurons involved in the control of salt intake behavior. In dehydrated KO mice, the [Na+]-dependent stimulation of glycolysis is impaired and the activity of the GABAergic neurons is not promoted.
Fig. 4.

Overview of the [Na+]-sensing mechanism and Nax-dependent regulation of neuronal activity in the SFO. Reproduced with permission from [55]
Essential hypernatremia caused by autoimmunity to Nax
“Essential hypernatremia” is clinically characterized by chronic elevation of plasma [Na+] with an inappropriate lack of thirst and upward resetting of the osmotic set point for vasopressin release, thereby resulting in persistent hypernatremia with a euvolemic state [6, 18, 68]. In most cases of essential hypernatremia, structural abnormalities are commonly detected in the hypothalamic-pituitary area, as a result of trauma, tumors, or inflammation. However, several cases of essential hypernatremia without demonstrable hypothalamic structural lesions have been reported, although the precise mechanism(s) has(have) not yet been elucidated [8, 14, 15, 20, 29, 59].
We examined a patient with essential hypernatremia associated with abnormal reductions in water intake and vasopressin release without demonstrable hypothalamic structural lesions and found that she developed autoantibodies to Nax [32, 54]. Passive transfer of the immunoglobulin (Ig) fraction of the patient’s serum in WT mice reproduced her symptoms. This was revealed to be induced by complement-mediated cell death in the CVOs, where Nax is specifically expressed [32]. The SFO and OVLT have projections to the SON and PVN, which are responsible for regulating the production/release of vasopressin (Fig. 1a) [46, 64]. Histological damage to the SFO and OVLT may be the reason for the dysregulation in vasopressin production/release. This defect in the regulation of vasopressin appears to have caused serious symptoms in the patient.
Since sensory CVOs lack a BBB, antibodies easily leak from blood vessels into these loci [11]. This indicates that cell surface proteins, including channels and transporters in the sensory CVOs, may be easy targets of autoantibodies. Neurons or glial cells in the sensory CVOs are known to express receptors for multiple circulating peptides, including angiotensin II and natriuretic peptides [49]. These receptors may also be potential targets for autoantibodies. We very recently identified more patients with essential hypernatremia whose sera contained autoantibodies that were specifically reactive to the SFO (in preparation). Thus, examination of autoantibodies reactive to the sensory CVOs represents an option for clinical tests of patients with chronic body-fluid disorders.
Nax expression in the other loci in the brain and peripheral tissues
Beside the SFO and OVLT, clusters of lacZ expression in the Na x-KO mice were also observed in some specific loci in the brain: the medial preoptic area, the anterior and dorsomedial part of the hypothalamic area, dorsomedial part of the interpeduncular nucleus, medial part of the median raphe (so-called rhabdoid nucleus), mesencephalic nucleus of the fifth cranial nerve, medial habenular nucleus, median eminence, and neurohypophysis [65]. In addition, relatively weak lacZ expression was detected in the cerebral cortex in layer IV of the lateral area (from the most anterior portion to the end of the posterior portion of the cortex) and the basolateral amygdala [65]. This expression was confirmed by immunohistochemistry using our antibody (our unpublished data).
Grob et al. reported that neurons in the median preoptic nucleus (MnPO) in rats responded to a change in the extracellular sodium concentration [27]. MnPO is a midline structure situated on the anterior wall of the third ventricle between the SFO and OVLT and receives neural connections from these two loci [49]. They also showed that the MnPO in rats was positive for Nax expression by in situ hybridization. As aforementioned, we detected X-gal staining in the medial preoptic area but not in the MnPO in Na x-KO mice [65]. Recently, the same group reported that this was attributed to the species difference by showing that Nax expression in the MnPO is observed in rats but not in mice [50]. However, as far as we examined with our antibody, of which specificity was confirmed by immunostaining using Na x-KO mice [34], we could not detect any signals in rat MnPO immunohistochemically (our unpublished data). Because MnPO has a BBB, it may not primarily participate in the [Na+] sensing in the brain, though it should be an important locus for integration of the neural information for body fluid homeostasis.
In the peripheral nervous systems, Nax expression was detected in the dorsal root ganglia and non-myelinating Schwann cells [65, 66]. In previous papers by other groups, relatively high levels of Na x mRNA were detected outside the nervous system, particularly in the lung, heart, and perinatal uterine smooth muscle by Northern blot analysis [2, 23], and in the kidney by reverse transcription-polymerase chain reaction (RT-PCR) [2]. We examined the localization of Nax throughout the visceral organs at the cellular level [66]: In the lung especially of neonates, robust Nax signals were observed in the alveolar type II cells, which actively absorb sodium and water to aid gas exchange through the alveolar surface [47]. The myometrium of the pregnant uterus was significantly positive for Nax expression [65]. In visceral organs including lung, heart, intestine, bladder, kidney and tongue, a subset of Schwann cells within the peripheral nerve trunks and ganglia were highly positive for Nax. Further studies revealed that these Nax-positive cells were non-myelinating Schwann cells of sympathetic and/or parasympathetic nerve fibers surrounding blood vessels [66]. Recently, Lara et al. reported that Nax is expressed in the thick ascending limb and collecting duct cells in rat kidney [44]. However, as far as we examined lacZ expression in the Na x-KO mice and Nax expression in wild-type mice by immunohistochemistry, we could not verify the expression in the kidney (our unpublished data).
Future directions
In this review, we summarized a series of our studies on the [Na+]-sensing mechanism in the brain for the control of salt intake behavior. Although some candidates for osmosensors have been postulated, the mechanism for osmosensing is still controversial [42, 55], in contrast to the [Na+] sensing. A [Na+]-sensing mechanism in the brain is also considered to be involved in the control of blood pressure. Excess dietary salt is one of the decisive factors for the rise of blood pressure [40]. Slightly elevated plasma [Na+] with apparent normovolemia is often observed in hypertensive humans [10]. Increases in [Na+] in CSF by central infusion of Na+-rich artificial CSF cause sympathetic hyperactivity and hypertension [45]. Therefore, some sensing mechanism for [Na+] in CSF is considered to be a decisive factor in salt-sensitive hypertension, though the precise mechanism has not been elucidated [10, 56]. It is noteworthy that lesions of the anteroventral third ventricle region (AV3V), which encompasses the SFO and OVLT, prevent the development or reverse hypertension in several hypertensive rats including Dahl salt-sensitive and DOCA-salt rats [9, 12, 13, 19, 26, 30]. In addition, it was reported that the ventral lamina terminalis mediates enhanced cardiovascular responses of rostral ventrolateral medulla neurons during increased dietary salt [1]. It would be important to examine whether the [Na+] sensing by Nax in the SFO and/or OVLT is involved in the salt-sensitive control of blood pressure.
Acknowledgments
We thank Ms. Akiko Kodama for her secretarial assistance.
References
- 1.Adams JM, Bardgett ME, Stocker SD. Ventral lamina terminalis mediates enhanced cardiovascular responses of rostral ventrolateral medulla neurons during increased dietary salt. Hypertension. 2009;54:308–314. doi: 10.1161/HYPERTENSIONAHA.108.127803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akopian AN, Souslova V, Sivilotti L, Wood JN. Structure and distribution of a broadly expressed atypical sodium channel. FEBS Lett. 1997;400:183–187. doi: 10.1016/S0014-5793(96)01389-0. [DOI] [PubMed] [Google Scholar]
- 3.Andersson B. Thirst—and brain control of water balance. Am Sci. 1971;59:408–415. [PubMed] [Google Scholar]
- 4.Andersson B. Regulation of water intake. Physiol Rev. 1978;58:582–603. doi: 10.1152/physrev.1978.58.3.582. [DOI] [PubMed] [Google Scholar]
- 5.Arieff AI. Management of hyponatraemia. BMJ. 1993;307:305–308. doi: 10.1136/bmj.307.6899.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avioli LV, Earley LE, Kashima HK. Chronic and sustained hypernatremia, absence of thirst, diabetes insipidus, and adrenocorticotrophin insufficiency resulting from widespread destruction of the hypothalamus. Ann Intern Med. 1962;56:131–140. doi: 10.7326/0003-4819-56-1-131. [DOI] [PubMed] [Google Scholar]
- 7.Ayus JC, Armstrong DL, Arieff AI. Effects of hypernatraemia in the central nervous system and its therapy in rats and rabbits. J Physiol. 1996;492:243–255. doi: 10.1113/jphysiol.1996.sp021305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bappal B, Sheikh HA, Radhakrishan A, Mula-Abed WA. Adipsic hypernatremia with a reset osmostat. Saudi Med J. 2006;27:727–729. [PubMed] [Google Scholar]
- 9.Berecek KH, Barron KW, Webb RL, Brody MJ. Vasopressin-central nervous system interactions in the development of DOCA hypertension. Hypertension. 1982;4:131–137. [PubMed] [Google Scholar]
- 10.Blaustein MP, Leenen FH, Chen L, Golovina VA, Hamlyn JM, Pallone TL, Van Huysse JW, Zhang J, Wier WG. How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol. 2012;302:H1031–H1049. doi: 10.1152/ajpheart.00899.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Broadwell RD, Sofroniew MV. Serum proteins bypass the blood-brain fluid barriers for extracellular entry to the central nervous system. Exp Neurol. 1993;120:245–263. doi: 10.1006/exnr.1993.1059. [DOI] [PubMed] [Google Scholar]
- 12.Buggy J, Fink GD, Johnson AK, Brody MJ. Prevention of the development of renal hypertension by anteroventral third ventricular tissue lesions. Circ Res. 1977;40:I110–I117. [PubMed] [Google Scholar]
- 13.Buggy J, Fink GD, Haywood JR, Johnson AK, Brody MJ. Interruption of the maintenance phase of established hypertension by ablation of the anteroventral third ventricle (AV3V) in rats. Clin Exp Hypertens. 1978;1:337–353. doi: 10.3109/10641967809068612. [DOI] [PubMed] [Google Scholar]
- 14.Cauble MS, Mack-Shipman L, Schaefer GB, Balakrishnan S, Larsen JL. Idiopathic hypothalamic dysfunction with precocious puberty and adipsic hypernatremia first presenting in adolescence. J Pediatr Endocrinol Metab. 2001;14:1163–1167. doi: 10.1515/jpem-2001-0816. [DOI] [PubMed] [Google Scholar]
- 15.Conley SB, Brocklebank JT, Taylor IT, Robson AM. Recurrent hypernatremia; a proposed mechanism in a patient with absence of thirst and abnormal excretion of water. J Pediatr. 1976;89:898–903. doi: 10.1016/S0022-3476(76)80593-8. [DOI] [PubMed] [Google Scholar]
- 16.Cox PS, Denton DA, Mouw DR, Tarjan E. Natriuresis induced by localized perfusion within the third cerebral ventricle of sheep. Am J Physiol Regul Integr Comp Physiol. 1987;252:R1–R6. doi: 10.1152/ajpregu.1987.252.1.R1. [DOI] [PubMed] [Google Scholar]
- 17.Denton DA, McKinley MJ, Weisinger RS. Hypothalamic integration of body fluid regulation. Proc Natl Acad Sci U S A. 1996;93:7397–7404. doi: 10.1073/pnas.93.14.7397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeRubertis FR, Michelis MF, Davis BB. “Essential” hypernatremia. Report of three cases and review of the literature. Arch Intern Med. 1974;134:889–895. doi: 10.1001/archinte.1974.00320230099021. [DOI] [PubMed] [Google Scholar]
- 19.de Wardener HE. The hypothalamus and hypertension. Physiol Rev. 2001;81:1599–1658. doi: 10.1152/physrev.2001.81.4.1599. [DOI] [PubMed] [Google Scholar]
- 20.duRivage SK, Winter RJ, Brouillette RT, Hunt CE, Noah Z. Idiopathic hypothalamic dysfunction and impaired control of breathing. Pediatrics. 1985;75:896–898. [PubMed] [Google Scholar]
- 21.Felipe A, Knittle TJ, Doyle KL, Tamkun MM. Primary structure and differential expression during development and pregnancy of a novel voltage-gated sodium channel in the mouse. J Biol Chem. 1994;269:30125–30131. [PubMed] [Google Scholar]
- 22.Fitzsimons JT. Bengt Andersson’s pioneering demonstration of the hypothalamic “drinking area” and the subsequent osmoreceptor/sodium receptor controversy. Acta Physiol Scand Suppl. 1989;583:15–25. [PubMed] [Google Scholar]
- 23.Gautron S, Dos Santos G, Pinto-Henrique D, Koulakoff A, Gros F, Berwald-Netter Y. The glial voltage-gated sodium channel: cell- and tissue-specific mRNA expression. Proc Natl Acad Sci U S A. 1992;89:7272–7276. doi: 10.1073/pnas.89.15.7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.George ALJ, Knittle TJ, Tamkun MM. Molecular cloning of an atypical voltage-gated sodium channel expressed in human heart and uterus: evidence for a distinct gene family. Proc Natl Acad Sci U S A. 1992;89:4893–4897. doi: 10.1073/pnas.89.11.4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/S0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 26.Goto A, Ganguli M, Tobian L, Johnson MA, Iwai J. Effect of an anteroventral third ventricle lesion on NaCl hypertension in Dahl salt-sensitive rats. Am J Physiol Heart Circ Physiol. 1982;243:H614–H1618. doi: 10.1152/ajpheart.1982.243.4.H614. [DOI] [PubMed] [Google Scholar]
- 27.Grob M, Drolet G, Mouginot D. Specific Na+ sensors are functionally expressed in a neuronal population of the median preoptic nucleus of the rat. J Neurosci. 2004;24:3974–3984. doi: 10.1523/JNEUROSCI.3720-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamilton NB, Attwell D. Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci. 2010;11:227–238. doi: 10.1038/nrn2803. [DOI] [PubMed] [Google Scholar]
- 29.Hayek A, Peake GT. Hypothalamic adipsia without demonstrable structural lesion. Pediatrics. 1982;70:275–278. [PubMed] [Google Scholar]
- 30.Haywood JR, Fink GD, Buggy J, Boutelle S, Johnson AK, Brody MJ. Prevention of two-kidney, one-clip renal hypertension in rat by ablation of AV3V tissue. Am J Physiol Heart Circ Physiol. 1983;245:H683–H689. doi: 10.1152/ajpheart.1983.245.4.H683. [DOI] [PubMed] [Google Scholar]
- 31.Hindmarch C, Fry M, Yao ST, Smith PM, Murphy D, Ferguson AV. Microarray analysis of the transcriptome of the subfornical organ in the rat: regulation by fluid and food deprivation. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1914–R1920. doi: 10.1152/ajpregu.90560.2008. [DOI] [PubMed] [Google Scholar]
- 32.Hiyama TY, Matsuda S, Fujikawa A, Matsumoto M, Watanabe E, Kajiwara H, Niimura F, Noda M. Autoimmunity to the sodium-level sensor in the brain causes essential hypernatremia. Neuron. 2010;66:508–522. doi: 10.1016/j.neuron.2010.04.017. [DOI] [PubMed] [Google Scholar]
- 33.Hiyama TY, Watanabe E, Okado H, Noda M. The subfornical organ is the primary locus of sodium-level sensing by Nax sodium channels for the control of salt-intake behavior. J Neurosci. 2004;24:9276–9281. doi: 10.1523/JNEUROSCI.2795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hiyama TY, Watanabe E, Ono K, Inenaga K, Tamkun MM, Yoshida S, Noda M. Nax channel involved in CNS sodium-level sensing. Nat Neurosci. 2002;5:511–512. doi: 10.1038/nn0602-856. [DOI] [PubMed] [Google Scholar]
- 35.Hiyama TY, Yoshida M, Matsumoto M, Suzuki R, Matsuda T, Watanabe E, Noda M. Endothelin-3 expression in the subfornical organ enhances the sensitivity of Nax, the brain sodium-level sensor, to suppress salt intake. Cell Metab. 2013;17:507–519. doi: 10.1016/j.cmet.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 36.Hori S, Komatsu Y, Shigemoto R, Mizuno N, Nakanishi S. Distinct tissue distribution and cellular localization of two messenger ribonucleic acids encoding different subtypes of rat endothelin receptors. Endocrinology. 1992;130:1885–1895. doi: 10.1210/endo.130.4.1312429. [DOI] [PubMed] [Google Scholar]
- 37.Hussy N, Deleuze C, Desarménien MG, Moos FC. Osmotic regulation of neuronal activity: a new role for taurine and glial cells in a hypothalamic neuroendocrine structure. Prog Neurobiol. 2000;62:113–134. doi: 10.1016/S0301-0082(99)00071-4. [DOI] [PubMed] [Google Scholar]
- 38.Johnson AK, Gross PM. Sensory circumventricular organs and brain homeostatic pathways. FASEB J. 1993;7:678–686. doi: 10.1096/fasebj.7.8.8500693. [DOI] [PubMed] [Google Scholar]
- 39.Johnson RF, Beltz TG, Thunhorst RL, Johnson AK. Investigations on the physiological controls of water and saline intake in C57BL/6 mice. Am J Physiol Regul Integr Comp Physiol. 2003;285:R394–R403. doi: 10.1152/ajpregu.00130.2003. [DOI] [PubMed] [Google Scholar]
- 40.Kaplan NM. Kaplan’s clinical hypertension. 9. Philadelphia: Lippincott, Williams & Wilkins; 2006. [Google Scholar]
- 41.Kedzierski RM, Yanagisawa M. Endothelin system: the double-edged sword in health and disease. Annu Rev Pharmacol Toxicol. 2001;41:851–876. doi: 10.1146/annurev.pharmtox.41.1.851. [DOI] [PubMed] [Google Scholar]
- 42.Kinsman B, Cowles J, Lay J, Simmonds SS, Browning KN, Stocker SD. Osmoregulatory thirst in mice lacking the transient receptor potential vanilloid type 1 (TRPV1) and/or type 4 (TRPV4) receptor. Am J Physiol Regul Integr Comp Physiol. 2014;307:R1092–R1100. doi: 10.1152/ajpregu.00102.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev. 2011;91:1–77. doi: 10.1152/physrev.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lara LS, Satou R, Bourgeois CR, Gonzalez AA, Zsombok A, Prieto MC, Navar LG. The sodium-activated sodium channel is expressed in the rat kidney thick ascending limb and collecting duct cells and is upregulated during high salt intake. Am J Physiol Renal Physiol. 2012;303:F105–F109. doi: 10.1152/ajprenal.00490.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leenen FH. The central role of the brain aldosterone—“ouabain” pathway in salt-sensitive hypertension. Biochim Biophys Acta. 2010;1802:1132–1139. doi: 10.1016/j.bbadis.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 46.Mangiapane ML, Thrasher TN, Keil LC, Simpson JB, Ganong WF. Role for the subfornical organ in vasopressin release. Brain Res Bull. 1984;13:43–47. doi: 10.1016/0361-9230(84)90006-6. [DOI] [PubMed] [Google Scholar]
- 47.Matalon S, O’Brodovich H. Sodium channels in alveolar epithelial cells: molecular characterization, biophysical properties, and physiological significance. Annu Rev Physiol. 1999;61:627–661. doi: 10.1146/annurev.physiol.61.1.627. [DOI] [PubMed] [Google Scholar]
- 48.McKinley MJ, Denton DA, Weisinger RS. Sensors for antidiuresis and thirst—osmoreceptors or CSF sodium detectors? Brain Res. 1978;141:89–103. doi: 10.1016/0006-8993(78)90619-4. [DOI] [PubMed] [Google Scholar]
- 49.McKinley MJ, McAllen RM, Davern P, Giles ME, Penschow J, Sunn N, Uschakov A, Oldfield BJ. The sensory circumventricular organs of the mammalian brain. Adv Anat Embryol Cell Biol. 2003;172:1–122. doi: 10.1007/978-3-642-55532-9_1. [DOI] [PubMed] [Google Scholar]
- 50.Nehmé B, Henry M, Mouginot D, Drolet G. The expression pattern of the Na+ sensor, Nax in the hydromineral homeostatic network: a comparative study between the rat and mouse. Front Neuroanat. 2012;6:26. doi: 10.3389/fnana.2012.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Noda M. The subfornical organ, a specialized sodium channel, and the sensing of sodium levels in the brain. Neuroscientist. 2006;12:80–91. doi: 10.1177/1073858405279683. [DOI] [PubMed] [Google Scholar]
- 52.Noda M. Hydromineral neuroendocrinology: mechanism of sensing sodium levels in the mammalian brain. Exp Physiol. 2007;92:513–522. doi: 10.1113/expphysiol.2006.035659. [DOI] [PubMed] [Google Scholar]
- 53.Noda M, Hiyama TY. Sodium-level-sensitive sodium channel and salt-intake behavior. Chem Senses. 2005;30(suppl 1):i44–i45. doi: 10.1093/chemse/bjh105. [DOI] [PubMed] [Google Scholar]
- 54.Noda M, Hiyama TY. The Nax channel: what it is and what it does. Neuroscientist. 2014 [Google Scholar]
- 55.Noda M, Sakuta H. Central regulation of body-fluid homeostasis. Trends Neurosci. 2013;36:661–673. doi: 10.1016/j.tins.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 56.Orlov SN, Mongin AA. Salt-sensing mechanisms in blood pressure regulation and hypertension. Am J Physiol Heart Circ Physiol. 2007;293:H2039–H2053. doi: 10.1152/ajpheart.00325.2007. [DOI] [PubMed] [Google Scholar]
- 57.Park R, Denton DA, McKinley MJ, Pennington G, Weisinger RS. Intracerebroventricular saccharide infusions inhibit thirst induced by systemic hypertonicity. Brain Res. 1989;493:123–128. doi: 10.1016/0006-8993(89)91006-8. [DOI] [PubMed] [Google Scholar]
- 58.Peruzzo M, Milani GP, Garzoni L, Longoni L, Simonetti GD, Bettinelli A, Fossali EF, Bianchetti MG. Body fluids and salt metabolism—part II. Ital J Pediatr. 2010;36:78–85. doi: 10.1186/1824-7288-36-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schaad U, Vassella F, Zuppinger K, Oetliker O. Hypodipsia-hypernatremia syndrome. Helv Paediatr Acta. 1979;34:63–76. [PubMed] [Google Scholar]
- 60.Shimizu H, Watanabe E, Hiyama TY, Nagakura A, Fujikawa A, Okado H, Yanagawa Y, Obata K, Noda M. Glial Nax channels control lactate signaling to neurons for brain [Na+] sensing. Neuron. 2007;54:59–72. doi: 10.1016/j.neuron.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 61.Smith PM, Ferguson AV. Circulating signals as critical regulators of autonomic state—central roles for the subfornical organ. Am J Physiol Regul Integr Comp Physiol. 2010;299:R405–R415. doi: 10.1152/ajpregu.00103.2010. [DOI] [PubMed] [Google Scholar]
- 62.Stiefel D, Petzold A. H2O coma. Neurocrit Care. 2007;6:67–71. doi: 10.1385/NCC:6:1:67. [DOI] [PubMed] [Google Scholar]
- 63.Tasker JG, Oliet SH, Bains JS, Brown CH, Stern JE. Glial regulation of neuronal function: from synapse to systems physiology. J Neuroendocrinol. 2012;24:566–576. doi: 10.1111/j.1365-2826.2011.02259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thrasher TN, Keil LC, Ramsay DJ. Lesions of the organum vasculosum of the lamina terminalis (OVLT) attenuate osmotically-induced drinking and vasopressin secretion in the dog. Endocrinology. 1982;110:1837–1839. doi: 10.1210/endo-110-5-1837. [DOI] [PubMed] [Google Scholar]
- 65.Watanabe E, Fujikawa A, Matsunaga H, Yasoshima Y, Sako N, Yamamoto T, Saegusa C, Noda M. Nav2/NaG channel is involved in control of salt-intake behavior in the CNS. J Neurosci. 2000;20:7743–7777. doi: 10.1523/JNEUROSCI.20-20-07743.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Watanabe E, Hiyama TY, Kodama R, Noda M. Nax sodium channel is expressed in non-myelinating Schwann cells and alveolar type II cells in mice. Neurosci Lett. 2002;330:109–113. doi: 10.1016/S0304-3940(02)00708-5. [DOI] [PubMed] [Google Scholar]
- 67.Watanabe E, Hiyama TY, Shimizu H, Kodama R, Hayashi N, Miyata S, Yanagawa Y, Obata K, Noda M. Sodium-level-sensitive sodium channel Nax is expressed in glial laminate processes in the sensory circumventricular organs. Am J Physiol Regul Integr Comp Physiol. 2006;290:R568–R576. doi: 10.1152/ajpregu.00618.2005. [DOI] [PubMed] [Google Scholar]
- 68.Welt LG. Hypo- and hypernatremia. Ann Intern Med. 1962;56:161–164. doi: 10.7326/0003-4819-56-1-161. [DOI] [PubMed] [Google Scholar]