

Summary
Interleukin-3 (IL-3) is capable of supporting the proliferation of a broad range of hematopoietic cell types, whereas granulocyte colony-stimulating factor (G-CSF) and macrophage CSF (M-CSF) represent critical cytokines in myeloid differentiation. When this was investigated in a pluripotent-stem-cell-based hematopoietic differentiation model, IL-3/G-CSF or IL-3/M-CSF exposure resulted in the continuous generation of myeloid cells from an intermediate myeloid-cell-forming complex containing CD34+ clonogenic progenitor cells for more than 2 months. Whereas IL-3/G-CSF directed differentiation toward CD45+CD11b+CD15+CD16+CD66b+ granulocytic cells of various differentiation stages up to a segmented morphology displaying the capacity of cytokine-directed migration, respiratory burst response, and neutrophil-extracellular-trap formation, exposure to IL-3/M-CSF resulted in CD45+CD11b+CD14+CD163+CD68+ monocyte/macrophage-type cells capable of phagocytosis and cytokine secretion. Hence, we show here that myeloid specification of human pluripotent stem cells by IL-3/G-CSF or IL-3/M-CSF allows for prolonged and large-scale production of myeloid cells, and thus is suited for cell-fate and disease-modeling studies as well as gene- and cell-therapy applications.
Graphical Abstract
Highlights
-
•
Myeloid specification of human PSCs by IL-3-/M-CSF, G-CSF, or GM-CSF
-
•
Large-scale and continuous generation of M2-MΦ or granulocytes by M-CSF or G-CSF
-
•
Functional iPSC-derived macrophages or granulocytes similar to in-vivo-derived cells
In this article, Moritz and colleagues show that IL-3 in combination with M-CSF/G-CSF or GM-CSF is able to instruct human pluripotent stem cells toward a myeloid lineage. The use of IL-3/G-CSF or IL-3/M-CSF allows for prolonged and large-scale production of human granulocytes or macrophages, respectively, that demonstrate similar functionalities compared with their in-vivo-derived counterparts.
Introduction
Hematopoietic in vitro differentiation of pluripotent stem cells (PSCs) such as embryonic stem cells (ESCs) and induced PSCs (iPSCs) holds great promise for disease modeling, drug testing, and the development of novel cell- and gene-therapy strategies. In the past, interest has been directed primarily toward reconstituting stem cells, a cell type that is difficult to generate from PSC sources. Recently, however, long-lived, mature myeloid cells have been described (Guilliams et al., 2013), and the organotropic transplantation of such cells may allow for new therapeutic scenarios (Happle et al., 2014; Suzuki et al., 2014).
During embryonic development, hematopoietic cells are generated by two distinct but partly overlapping programs termed primitive and definitive hematopoiesis. Both are orchestrated by a highly complex interaction of regulatory molecules, including transcription factors, cytokine-induced and intercellular signaling, and niche factors (Lancrin et al., 2009; Nostro et al., 2008; Sturgeon et al., 2014). Primitive hematopoietic development originates from distinct multipotent precursors known as hemangioblasts, which are able to generate both vascular and hematopoietic progeny via an intermediate, hemogenic endothelial stage (Lancrin et al., 2009). Subsequently, further hematopoietic specification and differentiation result in mature cells that are primarily of an erythroid and, to a lesser degree, myeloid lineage (Palis, 2014; Schulz et al., 2012). In a separate process originating in the dorsal aorta, definitive hematopoiesis allows for the generation of transplantable hematopoietic stem cells (HSCs) that are capable of repopulating the entire lympho-hematopoietic system long term. In this context, an important role for the cytokine interleukin-3 (IL-3) (Donahue et al., 1988; Robin et al., 2006; Wiles and Keller, 1991) as well as wnt signaling (Sturgeon et al., 2014) has been reported by a number of groups. Again, the fate of these repopulating HSCs, such as self-renewal, apoptosis, quiescence, and further differentiation and proliferation, is dependent on their exposure to other cells, matrix factors, or cytokines (Arai et al., 2004; Williams et al., 1991). For both programs, granulocyte-colony-stimulating factor (G-CSF) and monocyte-CSF (M-CSF) constitute the main driving forces for the generation and terminal differentiation of functional cells of a granulocytic or monocytic/macrophage lineage, respectively (Sengupta et al., 1988; Welte et al., 1985a, 1987).
G-CSF originally was identified by its capacity to promote the differentiation of human bone marrow progenitor cells toward neutrophils and is a critical component of this process (Welte et al., 1985b, 1987). However, the G-CSF receptor (CSF3R) is not exclusive to myeloid cells and has also been identified on HSCs, thus explaining the profound stem cell defects observed in congenital neutropenia patients suffering from defects in G-CSF signaling (Panopoulos and Watowich, 2008). In contrast, M-CSF, the crucial cytokine for generating mononuclear phagocytes or macrophages (MΦ) from HSC sources, appears to be primarily involved in terminal lineage differentiation (Yoshida et al., 1990). M-CSF was the first hematopoietic cytokine to be identified and cloned, and acts by activating its type III protein tyrosine kinase family receptor (c-fms) (Clark and Kamen, 1987; Sieff, 1987). Generating MΦ by M-CSF exposure, similarly to priming with IL-4/IL-10, results in alternatively activated M2-type Φ, in contrast to the classical pro-inflammatory M1Φ, which is differentiated from monocytes by GM-CSF or interferon-gamma (IFN−γ) exposure (Martinez et al., 2008; Sica and Mantovani, 2012).
To date, most protocols for hematopoietic differentiation of PSCs in vitro have utilized a multitude of cytokines or small molecules to mimic the modulation of signaling pathways at various stages of embryonic development (Choi et al., 2011; Kennedy et al., 2012; Sturgeon et al., 2014). However, many of the factors involved in this process remain ill defined. Therefore, this excessive priming may have unwanted effects on the differentiation or functionality of the desired target cells, hampering their use in disease modeling or cell- and gene-therapy applications. Thus, the development of simple but robust protocols for generating nonbiased and fully functional hematopoietic cells appears to be highly warranted.
Given the (1) emerging role of IL-3 in early primitive as well as definitive hematopoietic specification (Donahue et al., 1988; Robin et al., 2006; Yang et al., 1986), (2) the fundamental importance of G-CSF and M-CSF in terminal granulocyte and monocyte/macrophage differentiation (Clark and Kamen, 1987; Sengupta et al., 1988; Sieff, 1987; Welte et al., 1987), and (3) the synergy reported between IL-3 and late-acting factors such as G-CSF, M-CSF, and granulocyte-macrophage CSF (GM-CSF) (Donahue et al., 1988; Wiles and Keller, 1991), we sought to investigate the combined use of IL-3 with either M-CSF or G-CSF, employing embryoid body (EB)-based hematopoietic in vitro differentiation. For this purpose, we developed an innovative protocol that allows for the prolonged and large-scale production of functional granulocytes as well as monocytes/macrophages. Generation of immature myeloid cells was mediated by an intermediate myeloid-cell-forming complex (MCFC) containing CD34+ clonogenic progenitor cells, which upon continued G-CSF or M-CSF exposure generated terminally differentiated myeloid cells for ≥2 months. As generation of these cells was accomplished by exposure of PSCs to IL-3 and one additional cytokine only, this protocol can be expected to closely recapitulate many aspects of physiologic hematopoietic development. Thus, it may be particularly suitable for studying human hematopoietic development in vitro and generating mature functional cells for cell and gene therapy.
Results
Hematopoietic Specification of Human iPSCs
To investigate the effect of IL-3/G-CSF or IL-3/M-CSF on hematopoietic in vitro differentiation of PSCs, we established a four-step differentiation protocol (Figure 1A) utilizing human iPSCs (hiPSCs) previously generated from nonmobilized peripheral blood (PB)-derived CD34+ cells (hCD34iPSC11 and hCD34iPSC16) (Ackermann et al., 2014; Lachmann et al., 2014) or the human ESC line H9. To initiate differentiation, iPSCs were cultured on mouse feeder cells in the presence of basic fibroblast growth factor (bFGF), resulting in typical round-shaped colonies (Figure 1B, step 1). Induction of germ layer formation was induced within EBs for 5 days (d0–d5) by orbital shaking and reduction of bFGF concentrations by 4-fold. After 5 days, we obtained compact and spherical EB cells (Figure 1B, step 2), which showed a slight decrease in TRA1-60 expression (Figure 1C). Induction of mesoderm was confirmed by decreased expression levels of OCT4 (POU5F1) as well as increased expression of Brachyury (T) mRNA when compared with the original iPSCs (Figure 1D). From d0 onward, EBs were subjected to directed hematopoietic specification by using medium supplemented with IL-3 and either G-CSF or M-CSF. Culture of EBs under these conditions for 10–15 days resulted in the formation of endothelial-like stromal cells and MCFCs (Figure 1B, step 3). The stromal cells had endothelial structure (Figure S1A) and stained positive for CD31, CD309 (Flk1), and CD144 (VE-Cadherin), whereas cells isolated from MCFCs stained negative for CD31, CD309, and CD144 (Figure S1B). For the IL-3/M-CSF combination, hematopoietic specification within the MCFCs was demonstrated by a gradual reduction in expression of pluripotency (OCT4) and mesodermal (T) markers, whereas mRNA levels for the hematopoietic markers CD45 (PTPRC) and RUNX1 gradually increased during the differentiation process. Of note, the formation of endothelial-like stromal cells served as a reliable quality criterion for the subsequent success of hematopoietic differentiation (Figure S1).
Figure 1.
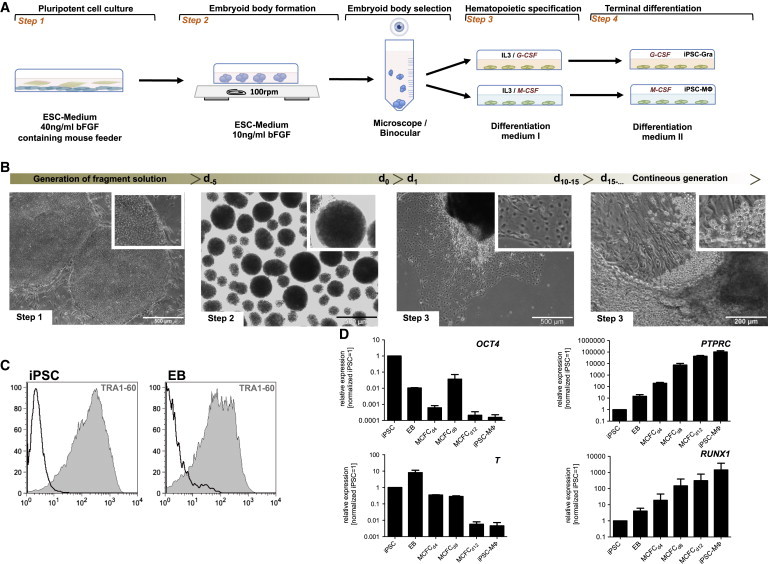
Four-Step Hematopoietic Differentiation Protocol for Human PSCs
(A) Schematic representation of the four-step hematopoietic differentiation protocol. Human PSCs were cultured on irradiated mouse feeder cells (step 1). Formation of EBs was induced by cultivating fragmented colonies in suspension plates on an orbital shaker (step 2). After 5 days, the largest EBs were manually selected under a binocular microscope and further cultured on adherent plates in differentiation medium I supplemented with either IL-3/G-SCF or IL-3/M-CSF for hematopoietic specification (step 3). From d10–d15 onward, granulocytes or monocyte/macrophages were generated by the MCFC. For terminal differentiation, cells were cultivated for another 7 days in differentiation medium II supplemented with G-CFS or M-CSF, leading to iPSC-MΦ or iPSC-Gra, respectively (step 4).
(B) Representative light microscopy images of steps 1–3 of hematopoietic differentiation (scale bars, 500 μM and 200 μM).
(C) Surface expression of Tra-1-60 (gray filled) in iPSCs and EBs determined by flow cytometry (black, respective isotype control).
(D) Expression of OCT4, Brachyury (T), PTPRC, and RUNX1 at different stages of differentiation (iPSCs, EBs [d0] in MCFCs at days 4, 8, and 12, and in terminally differentiated iPSC-MΦ) determined by quantitative RT-PCR analysis (expression normalized to undifferentiated iPSCs. GAPDH was used as a housekeeping control; n = 2 of independent experiments, mean ± SD). Data are shown for hCD34iPSC16.
See also Figures S1 and S2.
Further cultivation of the MCFCs in differentiation medium led to the continuous release of round-shaped suspension cells from the MCFCs from d12–d16 onward (Figure 1B, step 3). Remarkably, continuous shedding of cells from the MCFCs was observed for both the IL-3/G-CSF and IL-3/M-CSF combinations, for a period of 2–3 months. Furthermore, cells generated with IL-3/M-CSF showed high levels of PTPRC as well as RUNX1 mRNA from d12 onward, suggesting that these cells were of a hematopoietic lineage (Figure 1D). Of note, expression of PTPRC was also confirmed by flow cytometry, which demonstrated cells expressing CD45 as early as d8. In this context, cells appearing on d5 expressed CD34 only, whereas on d8 CD34+ cells became double positive for CD34 and CD45 (Figure S2A). Furthermore, when d8 CD34/CD45 double-positive cells were isolated from EBs and sorted by fluorescence-activated cell sorting (FACS) for additional evaluation of CD144 expression, only the CD144− fraction gave rise to myeloid colonies in semisolid medium (Figure S2B). Interestingly, almost no shedding of cells from MCFCs was observed for cultures grown in G-CSF alone (data not shown). As both M-CSF and G-CSF represent potent cytokines for the terminal differentiation of granulocytes and monocytes/macrophages, respectively, cells that shed from MCFCs were subsequently differentiated in the presence of a high concentration of G-CSF or M-CSF only for an additional 7 days (Figure 1A, step 4).
M-CSF Directs hiPSCs toward an M2-like Macrophage Phenotype
In vivo, monocytes that have differentiated from HSC sources can be polarized to either M1 or M2 macrophages (M1Φ and M2Φ, respectively), which play distinct roles within the immunoregulatory network. M1Φ (also known as classical or pro-inflammatory MΦ) are activated by IFN-γ, lipopolysaccharide (LPS), certain viruses, or GM-CSF, and promote TH1- or TH17-mediated immune responses, host defense, or antitumor immunity (Martinez et al., 2008; Sica and Mantovani, 2012). In contrast, M2Φ are activated by IL4, IL10, or M-CSF; regulate wound healing; and suppress T cell and host defense responses as well as antitumor immunity. Thus, we analyzed terminal differentiation along the monocyte/macrophage pathway in IL-3/M-CSF exposed cells and used PB monocytes polarized with either GM-CSF or M-CSF as controls for M1Φ or M2Φ, respectively.
Hematopoietic differentiation of iPSCs in the presence of IL-3/M-CSF resulted in the continuous production of cells by MCFCs for more than 2 months at a quantity of 5 × 105 to 1 × 106 cells/week/well (Figure 2A). Of note, myeloid cells could be harvested for up to 4–5 months before the cultures were exhausted. Cells in suspension were harvested twice a week and further differentiated in the presence of M-CSF only, resulting in adherent cells displaying long filopodia typical of M2Φ (Rey-Giraud et al., 2012; Figure 2B). The morphology of iPSC-derived macrophages (iPSC-MΦ) in culture resembled that of PB-derived M2Φ (PB-M2Φ) and was in contrast to the round shape of M1Φ generated from PB cells by GM-CSF exposure. Moreover, cytospins of iPSC-MΦ displayed a classical MΦ morphology indistinguishable from that of M1Φ or M2Φ control cells generated from the PB (Figure 2C). iPSC-MΦ, PB-M1Φ, and PB-M2Φ all displayed big organelles (vacuoles), a phenomenon frequently encountered in cell culture (also see Figure 3A). Flow-cytometric surface phenotyping of iPSC-MΦ revealed expression of classical MΦ markers such as CD45, CD14, CD163, and CD86 similar to those of M2Φ (Figure 2D), whereas surface markers associated with pluripotency (TRA1-60), HSCs (CD34), B cells (CD19), or granulocytes (CD66b) were absent (Figure 2E).
Figure 2.
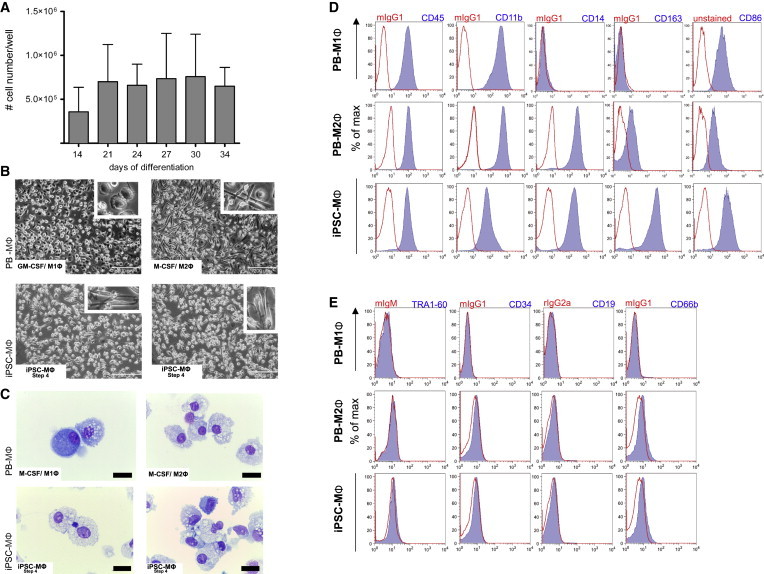
Phenotypical Characterization of iPSC-MΦ
(A) Quantities of monocyte/macrophages harvested from the supernatant at different days of differentiation (step 3). Quantities are given for one well of a six-well plate (n = 2 independent experiments, mean ± SD).
(B) Light microscopy of PB-derived macrophages (PB-MΦ) polarized with GM-CSF to M1Φ (top left), or with M-CSF to M2Φ (top right) and iPSC-MΦ (end of step 4, bottom panels; scale bar, 200 μM).
(C) Cytospins of PB-MΦ polarized to M1Φ (top left) or M2Φ (top right) and iPSC-MΦ (end of step 4, bottom panels; scale bar, 0.01 μM).
(D) Surface marker expression of typical MΦ maker (CD45, CD11b, CD14, CD163, and CD86; blue filled) on PB-M1Φ, PB-M2Φ, or iPSC-MΦ (end of step 4) analyzed by flow cytometry (red, respective isotype control).
(E) Surface marker expression of TRA-1-60, CD34, CD19, and CD66b (blue filled) on PB-M1Φ, PB-M2Φ or iPSC-MΦ (end of step 4) analyzed by flow cytometry (red, respective isotype control). Data are shown for hCD34iPSC16.
See also Figures S3–S5.
Figure 3.
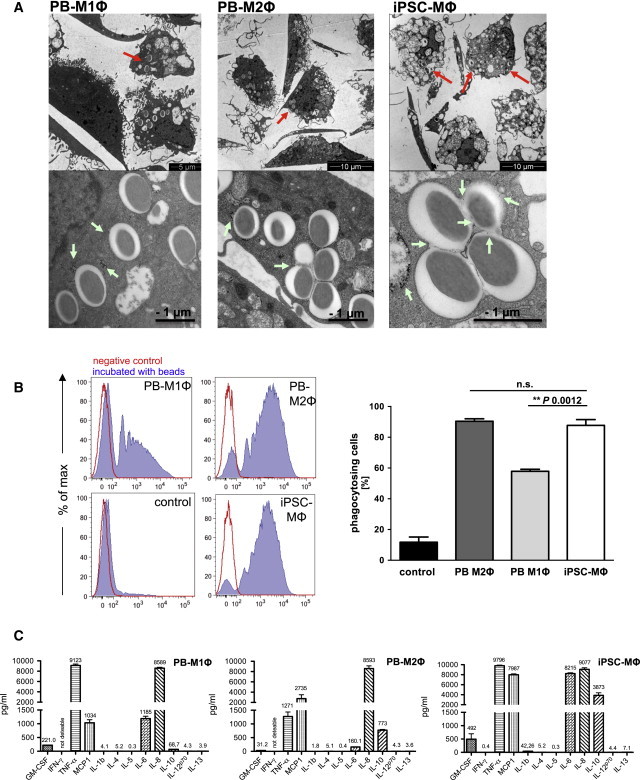
Functional Characterization of iPSC-MΦ
(A) Electron microscopy of latex-bead-containing phagosomes in M1Φ (top left), M2Φ (top center), and iPSC-MΦ (end of step 4, top right; scale bar, 5 or 10 μM), and higher magnifications of latex-bead-containing phagosomes (bottom row; scale bar, 1 μM). Red arrows indicate latex-bead-containing phagosomes. Green arrows indicate gold particles (∼5 nM) localized within a latex-bead-containing phagosome.
(B) Phagocytosis of fluorescein isothiocyanate (FITC)-labeled latex beads by control cells (undifferentiated iPSCs), PB-M1Φ, PB-M2Φ, or iPSC-MΦ (end of step 4) analyzed by flow cytometry. Left: diagrams of a representative experiment (blue filled, cells treated with 1 μm beads; red, untreated control. Right graph: percentage of phagocytosing cells (n = 3 independent experiments; mean ± SD of independent experiments; significance was calculated by one-way ANOVA with Tukey’s post hoc test).
(C) Cytokine production of PB-M1Φ, PB-M2Φ or iPSC (end of step 4) upon stimulation with LPS (n = 3 technical replicates, mean ± SD). Data are shown for hCD34iPSC16.
See also Figure S5.
A similar surface marker pattern was observed for macrophages derived from the human PSC line H9 (Figures S3A and S3B) or a second iPSC line (hCD34iPSC11) generated from human CD34+ cells (Figures S4A and S4B).
Of note, cells that recovered directly from the MCFC (end of step 3) and analyzed prior to terminal differentiation showed a clearly reduced CD14 and CD163 expression compared with iPSC-MΦ that differentiated for a further 7 days (step 4; Figures S5A and S5B). These findings highlight the need for terminal MΦ differentiation in the presence of high concentrations of M-CSF.
Functional Characterization of M-CSF-Primed iPSC-MΦ
In order to investigate the in vitro functionality of iPSC-MΦ, we analyzed cells for their ability to phagocytose latex beads or gold particles. As depicted in Figure 3A, electron microscopy revealed the ability of iPSC-MΦ to phagocytose latex beads (red arrow) and gold particles (green arrow) as efficiently as PB-M1Φ or PB-M2Φ (employed as controls). In addition, almost all phagosomes in iPSC-MΦ were co-localized with gold particles, indicating normal maturation by late endosome/lysosome fusion. Quantification of phagocytic efficiency by uptake of fluorescence-labeled latex beads revealed ∼90% positively labeled iPSC-MΦ or H9-derived macrophages, comparable to PB-M2Φ, whereas only ∼60% of PB-M1Φ were positive for latex beads (Figures 3B and S3C). Furthermore, we exposed iPSC-MΦ and cells freshly recovered from MCFC cultures to bacterial LPS to investigate the induction of pro-inflammatory cytokines and chemokines. Upon LPS stimulation, both iPSC-derived cell sources (fresh and terminally differentiated cells) produced IL-8 in similar quantities compared with the PB-M1Φ and PB-M2Φ controls, whereas release of IL1-β, IL-4, IL-5, IL-12p70, or IL-13 was hardly detectable (Figures 3C and S5C). Moreover, iPSC-MΦ and freshly MCFC-derived cells produced IL-6 and IL-10 at much higher levels than PB-M1Φ and PB-M2Φ, respectively. Of note, iPSC-MΦ and freshly MCFC-derived cells also secreted tumor necrosis factor alpha (TNF-α) and the chemokine monocyte chemotactic protein 1 (MCP-1), indicating a high capacity of these cells to induce inflammatory processes and monocyte attraction (Figures 3C and S5C), and a secretion pattern intermediate between M1Φ and M2Φ.
G-CSF Directs hiPSCs toward a Granulocyte Phenotype
Given the importance of G-CSF in directing multipotent HSCs toward a granulocyte lineage, we investigated the effect of G-CSF on the in vitro hematopoietic differentiation of hiPSCs by substituting M-CSF with G-CSF in our standard differentiation protocol. Similar to what was observed for the generation of macrophages, the IL-3/G-CSF combination resulted in the formation of MCFCs from settled EBs, which was observed within 10–15 days (Figure 4A). Again, the formation of endothelial-like stromal cells provided a quality criterion for the success of hematopoietic differentiation. After 13–16 days of IL-3/G-CSF exposure, shedding of round-shaped cells from the MCFCs was observed (Figure 4A). These cells could be harvested twice a week at a quantity of 4–5 × 106 cells/well/week for ≥7 weeks (Figure 4B). However, after 2–3 months, the number of cells harvested from the supernatant gradually decreased, indicating culture exhaustion. Further differentiation of these cells in high concentrations of G-CSF for 7 days yielded cells of granulocyte morphology (iPSC-Gra) closely resembling PB-Gra (Figure 4C). According to May-Giemsa-stained cytospins, iPSC-Gra reflected all stages of terminal granulocyte differentiation (average), including pro-myelocytes (4%), myelocytes (6%), meta-myelocytes (29%), n-banded neutrophils (22%), and segmented neutrophils (36%), as well as mature basophils (1%) and eosinophils (2%) (Figures 4D, 4E, and S6A). When analyzed by flow cytometry, iPSC-Gra were found to express the classical granulocyte surface markers CD45, CD11b, CD16, and CD66b, although in relation to PB-Gra, expression of CD11b, CD16, and CD66b was reduced (Figure 5A). This most likely reflects a failure of the in vitro differentiation process to completely recapitulate in vivo granulocyte maturation. This observation became even more prominent in cells that were harvested from the differentiation culture directly after they were shed from the MCFC (end of step 3) without further maturation in high-dose G-CSF (Figure S6B). In these cells, expression of CD16 and CD66b was even further reduced, although cells uniformly stained positive for CD45 and CD11b (Figure S6B). Similar to what was observed for iPSC-MΦ, iPSC-Gra and freshly MCFC-derived cells stained negative for the surface markers TRA1-60, CD34, and CD19, and the macrophage markers CD14 and CD163, further highlighting the granulocytic differentiation (Figures 5B and S6C).
Figure 4.
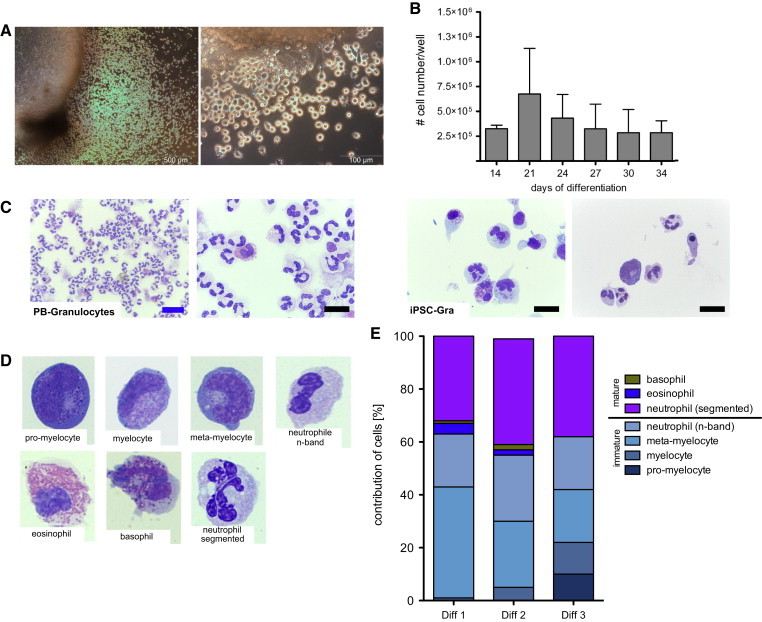
Phenotypical Characterization of iPSC-Gra
(A) Light microscopy of an MCFC generating iPSC-Gra (step 3; scale bar, 500 and 100 μM).
(B) Amount of granulocytes harvested from the supernatant at different days of differentiation (step 3; n = 2 independent experiments, mean ± SD). Quantities are given for one well of a six-well plate.
(C) Cytospins of PB-Gra and iPSC-Gra (end of step 4). Scale bars, 10 μM (black) and 20 μM (blue).
(D) Representative images for iPSC-Gra (end of step 4) resembling different maturation stages of granulopoiesis (magnification: 100×).
(E) Differential count of cytospins from iPSC-Gra. Data are shown for hCD34iPSC16.
See also Figure S7.
Figure 5.
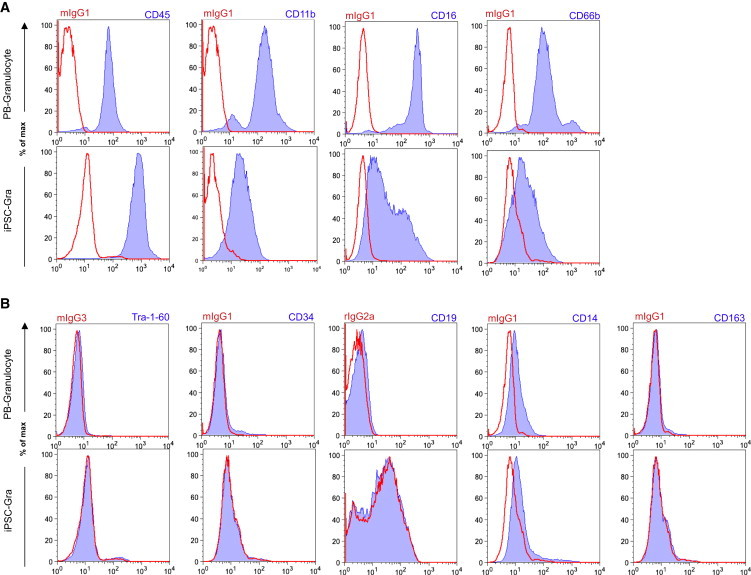
Surface Marker Profile of iPSC-Gra
(A) Surface marker expression of typical granulocytic maker (CD45, CD11b, CD16, and CD66b; blue filled) on PB-Gra or iPSC-Gra (end of step 4) analyzed by flow cytometry (red, respective isotype control).
(B) Surface marker expression of TRA-1-60, CD34, CD19, CD14, and CD163 (blue filled) on PB-Gra or iPSC-Gra (end of step 4) analyzed by flow cytometry (red, respective isotype control). Data are shown for hCD34iPSC16.
See also Figures S6 and S7.
We obtained similar results upon granulocytic differentiation with an iPSC line that was originally generated from human fibroblasts, employing a lentiviral reprogramming cassette. Subsequently, we excised the reprogramming cassette using Flp-recombinase and frt sites to obtain a transgene-free iPSC line (Kuehle et al., 2014). In the pluripotent state, this line demonstrated typical morphology and expressed typical pluripotency-associated surface markers, such as TRA1-60 and SSEA4 (A.S. and D.H., unpublished results; Figures S7A and S7B). Hematopoietic differentiation in our myeloid differentiation model utilizing IL-3/G-CSF resulted in cells with a classical granulocytic morphology, as shown on brightfield and cytospin images, as well as positive antigens for CD45 and CD66b (Figures S7C and S7D).
Functional Characterization of G-CSF-Primed iPSC-Gra
Given the classical morphology and cell-surface-marker phenotype of iPSC-Gra, we next assessed their functionality following a 7-day exposure to high-level G-CSF. First, we investigated the capacity of iPSC-Gra to generate hydrogen peroxide during the oxidative burst. We measured hydrogen peroxide activity indirectly by assessing the oxidation of nonfluorescent dihydrorhodamine 123 (DHR 123) to fluorescent rhodamine 123. As depicted in Figure 6A, only baseline rhodamine expression was detected in iPSC-Gra and PB-Gra (serving as positive controls). Upon activation of iPSC-Gra with phorbol myristate acetate (PMA), however, ∼90% of iPSC-Gra stained positive for rhodamine comparably to PB-Gra controls (Figure 6A). Similarly, upon activation with PMA, iPSC-Gra showed robust reactive oxygen species (ROS) induction, as measured by the conversion of dichlorofluorescein diacetate (DCFDA) to the cell-impermeable and fluorescent dye dichlorofluorescein (DCF) (Figure 6B). At all PMA concentrations investigated, iPSC-Gra induced significant levels of ROS within 2–6 hr, with close to plateau levels reached after 22–34 hr. Although this time course largely recapitulated the ROS response of PB-Gra control cells, the overall ROS levels achieved in iPSC-Gra were reduced by ∼50%.
Figure 6.
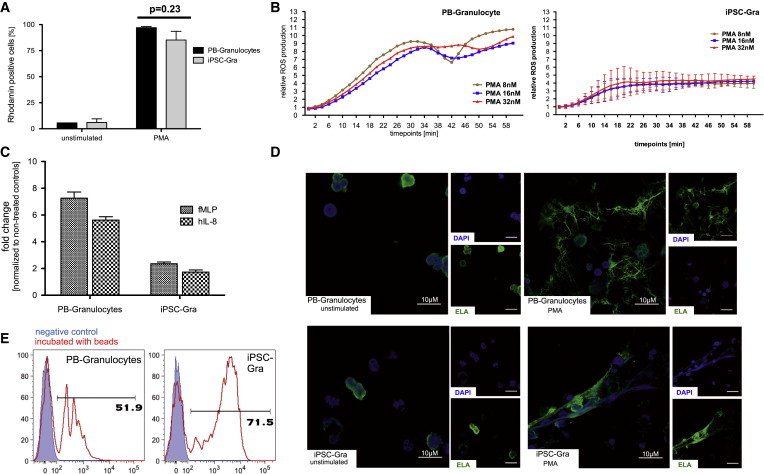
Functional Characterization of iPSC-Gra
(A) Dihydrorhodamine (DHR) assay measuring the production of reactive oxygen species (ROS) after activation with phorbol myristate acetate (PMA) in PB-Gra or iPSC-Gra by flow cytometry (n = 3 of independent experiments; mean ± SEM).
(B) Time dependence of ROS production in PB-Gra or iPSC-Gra after PMA stimulation measured by conversion of DCFDA to luminescent DCF (n = 1 for PB-Gra; n = 2 independent experiments, mean ± SD for iPSC-Gra).
(C) Migration potential of PB-Gra or iPSC-Gra toward an fMLP or hIL-8 gradient (n = 3 independent experiments; mean ± SD). Values are given as fold change over nonstimulated cells.
(D) Confocal fluorescence microscopy of NET formation by PB-Gra (top row) or iPSC-Gra (bottom row) after stimulation with PMA. Images represent nuclear-DAPI staining (blue) and ELA staining (green), as well as overlays (scale bar, 10 μM).
(E) Phagocytosis of FITC-labeled latex beads, PB-Gra, or iPSC-Gra analyzed by flow cytometry (a representative experiment is shown; blue filled, untreated control; red, cells treated with 1 μm beads). Data are shown for hCD34iPSC16.
iPSC-Gra were also able to actively migrate toward an IL-8 or N-formyl-methionyl-leucyl-phenylalanine (fMLP) gradient, and their migratory capacity was increased by ∼2-fold in comparison with their nonstimulated counterparts. Nevertheless, this property was clearly reduced in comparison with PB-Gra, which showed a 6- to 7-fold increase in mobility upon IL-8 or fMLP stimulation (Figure 6C). In addition, iPSC-Gra were able to form neutrophil extracellular traps (NETs) for extracellular pathogen killing. As illustrated in Figure 6D, neutrophil-elastase (ELA) expression in iPSC-Gra and PB-Gra co-localized with nuclear-DAPI staining under nonstimulated conditions, whereas upon stimulation with PMA, both cell types underwent characteristic changes in intracellular architecture and expelled their chromatin to form NETs, although this reaction was more pronounced in PB-Gra. Moreover, granulocytes represent professional phagocytes with the capacity to clear pathogens upon stimulation, as assessed by their ability to take up fluorescence-labeled beads. Also in this assay, iPSC-Gra demonstrated functional activity at least as good as that of PB-Gra (52% versus 72% uptake of latex beads, PB-Gra versus iPSC-Gra, respectively; Figure 6E).
Generating Macrophages and Granulocytes by Using GM-CSF
Finally, we investigated the effect of GM-CSF on hCD34iPSC16 in our myeloid differentiation protocol. Hematopoietic differentiation of iPSCs in the presence of IL-3/GM-CSF generated MCFCs with the same efficacy as observed with the M-CSF/IL-3 or G-CSF/IL-3 combination, and round-shaped cells were shed 10–15 days after cytokine application (Figure 7A). Thereafter, cells produced by the MCFCs were harvested continuously at a quantity of ∼4 × 105 cells/week/well. Of note, FACS analysis of freshly isolated cells revealed two populations expressing either CD45+, CD11b+, CD14+, and CD163+ (population 1; Figure 7B, top row) or CD66b−/dim, CD45+, and CD11b+ (population 2; Figure 7B, bottom row) surface markers. Differentiation of cells in 100 ng/ml GM-CSF for 7 more days provided both suspension and adherent cells (Figure 7C) with a granulocyte or macrophage morphology detected by cytospin (Figure 7D). On flow cytometry, this population expressed the surface markers CD66bdim, CD14, CD163, CD16, CD45, and CD11b, though to various extents (Figure 7E).
Figure 7.
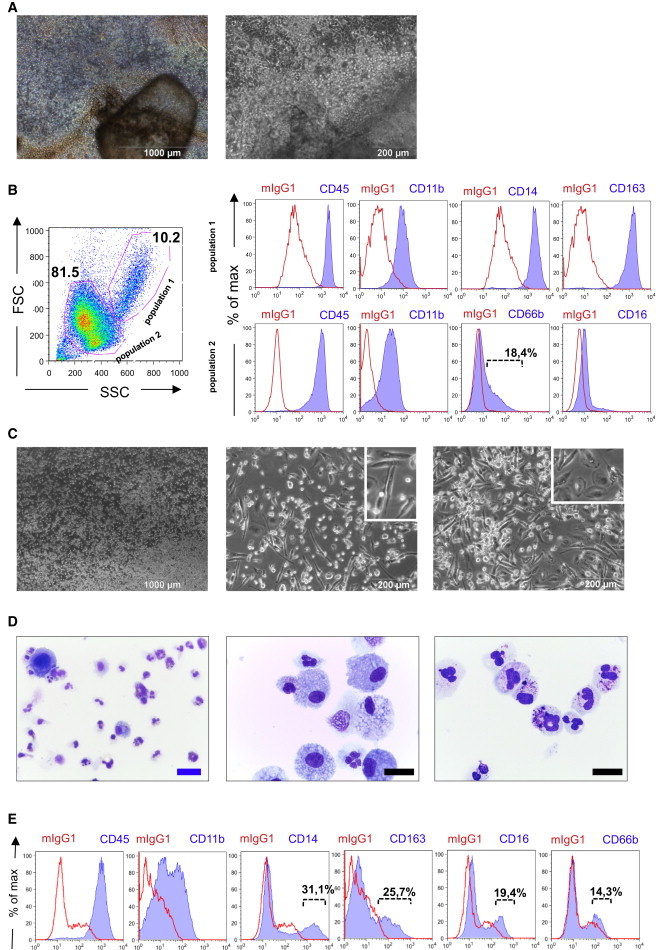
Generation of Granulocytes or Macrophages by GM-CSF
(A) Formation of an MCFC by addition of GM-CSF and IL-3 to differentiation medium I.
(B) Flow-cytometric analysis of cells freshly isolated from the MCFC (step 3). Cells were pre-gated according SSC/FSC gating for population 1 (top row) or population 2 (bottom row; red line represents isotype controls, blue line represents antigen).
(C–E) Brightfield (C; scale bars, 1,000 and 200 μM) and cytospin images (D; scale bars, 10 [black] and 20 μM [blue]), and flow-cytometric analysis (E) of cells harvested from the MCFC in the presence of GM-CSF/IL-3 and further differentiated in the presence of 100 ng/ml GM-CSF (end of step 4; red line represents isotype controls, blue line represents antigen). Data are shown for hCD34iPSC16.
Discussion
Hematopoietic differentiation of human PSCs represents a highly complex and finely tuned process in which the interaction of different cytokines, matrix factors, cell types, signaling pathways, and transcription factors results in specialized cells of the hematopoietic lineage (Clarke et al., 2013; Sumi et al., 2008). In this study, we provide experimental evidence that a combination of IL-3/G-CSF or IL-3/M-CSF in vitro is sufficient to drive differentiation of hiPSCs toward functional cells of granulocyte or monocyte/macrophage lineage. As a matter of fact, when only G-CSF was used, lower cell shedding from the MCFC was observed (data not shown). This implies that IL-3 is critical for the early part of this process. In the context of adult hematopoiesis, IL-3 has been demonstrated to support myelopoiesis, specifically when combined with other growth factors such as G-CSF or GM-CSF, and later work established its efficacy on the stem and early progenitor cell level (Donahue et al., 1988; Krumwieh et al., 1990). Similarly, a cooperative effect of IL-3 with cytokines such as IL-1, M-CSF, GM-CSF, and erythropoietin on both myeloid and erythroid in vitro differentiation of ESCs has been described (Wiles and Keller, 1991). More recently, IL-3 was also demonstrated to promote the development of HSCs in the yolk sac and the aorta-gonad-mesonephros (AGM) region of murine embryos, and a role of this cytokine in proliferation or survival of early HSCs was postulated (Robin et al., 2006). Given these results, our finding that IL-3 has a profound effect on the hematopoietic differentiation process comes as no surprise. Moreover, our own unpublished data suggest that IL-3 also cooperates with IL-4 in generating dendritic cells from hiPSCs, further supporting a pivotal role of IL-3 during hematopoietic in vitro differentiation of human PSC sources.
Clearly, in our hands, the formation of an endothelial-like stromal cell layer by the MCFC was predictive for successful hematopoietic differentiation later on. Although the requirement of endothelial support and even endothelial origin (hemogenic endothelium) for definitive hematopoiesis has been firmly established (Choi et al., 2012; Clarke et al., 2013; Kennedy et al., 2007; Sturgeon et al., 2014), it appears unlikely that the endothelial-like stromal cells that anchored the EBs to the plates in our cultures directly interact with the primitive cells within the MCFC that is responsible for the continuous output of hematopoietic cells. A much more likely explanation is that also within the MCFC, endothelial stromal cell support is required for efficient hematopoietic differentiation, and the endothelial-like stromal cell layer on the outside functions as an indicator of the efficacy of this interaction (Sandler et al., 2014).
Based on their well-documented activity profile, we employed M-CSF and G-CSF to direct terminal myeloid differentiation toward the monocyte/macrophage or granulocyte lineage in our studies. We found that the IL-3/M-CSF combination efficiently promoted the differentiation of iPSCs into iPSC-MΦ that resembled PB-MΦ in terms of morphology, surface phenotype, cytokine secretion potential, and phagocytic activity. Interestingly, iPSC-MΦ displayed surface marker expression and phagocytic properties similar to (alternatively) M2-polarized MΦ in the PB. In this respect, iPSC-MΦ strongly expressed CD14 and CD163, and showed a high capacity for latex bead phagocytosis similar to PB monocytes polarized to M2-Φ by M-CSF. However, with regard to a number of other characteristics, the cells that were recovered by our in vitro differentiation protocol did not strictly follow the dichotomy of M1 versus M2 polarization. Thus, CD86 (B7-2) was highly expressed on our iPSC-MΦ, a surface marker typically associated with pro-inflammatory M1Φ. However, this might be explained by the in vitro differentiation of our iPSCs using final differentiation medium (Rey-Giraud et al., 2012). Moreover, when we investigated secretion of pro- and anti-inflammatory cytokines and chemokines upon LPS stimulation, iPSC-MΦ showed secretion of TNF-α, MCP1, IL-6, IL-8, and IL-10 at levels indicating profound activation. Although M2Φ are known to be stimulated by LPS, classification of our iPSC-MΦ according to the M1/M2 scheme based on their chemokine release pattern appears problematic. LPS induces upregulation of its receptor, CD14 (Landmann et al., 1996; Meng and Lowell, 1997), in M1Φ and M2Φ, which then typically express TNF-α, IL1, and IL-6 (M1Φ), or TGF-β and IL-10 (M2Φ) (Cella et al., 1997; Hao et al., 2012; Lacey et al., 2012; Martinez et al., 2008). However, these patterns are by no means exclusive. Thus, for M2 polarization, various subtypes (M2a–c) with altered secretion patterns have been described, and furthermore, mixtures of M1 and M2 patterns may co-exist (Jones and Ricardo, 2013). Thus, the nonclassical secretion pattern of iPSC-MΦ is not surprising. Independently of their M1/M2 specification, however, iPSC-MΦ constitute remarkably active MΦ and a highly interesting source for potential cell-therapy approaches (e.g., for infections and malignant diseases). In this context, the high IL-8 levels that were induced in these cells are of considerable interest because this cytokine is a major chemotactic agent for recruiting granulocytes and other effector immune cells to the site of activity (Harada et al., 1994).
In a similar way, differentiation of iPSCs utilizing the IL-3/G-CSF combination resulted in granulocytes that mimicked the characteristic features of PB-Gra, such as surface marker expression, respiratory burst activity, formation of NETs, and migratory and phagocytotic capacity. Moreover, iPSC-Gra displayed all stages of terminal granulocyte maturation, ranging from the promyelocyte to the segmented-cell stage, a phenomenon that is most likely explained by the continuous shedding of less differentiated granulocyte lineage cells from the MCFC, which then further maturate in suspension culture. In line with this hypothesis, cells directly isolated from MCFC cultures showed a more immature surface phenotype with reduced CD16 (Fc gamma RIII), and CD66b (also known as CD67, NCA-95, or CGM6) expression, whereas expression of these markers increased upon exposure to G-CSF for 7 more days. In the human in vivo setting, expression of CD15 is associated with blast to pro-myelocyte differentiation, whereas CD66b and CD16 appear in pro-myelocytes to meta-myelocytes and persist to the segmented stage (Elghetany, 2002). However, iPSC-derived granulocytes also displayed certain functional deficiencies when compared with their PB counterparts. This was particularly obvious in their capacity to migrate toward IL-8 or fMLP gradients. Expression of the IL-8 receptor (CXCR2) on mature rather than immature granulocytes has been described (Nguyen-Jackson et al., 2010). Despite many similarities, exposing iPSC-Gra to G-CSF for 7–10 days might not be sufficient to drive granulopoiesis from iPSCs in vitro toward terminally differentiated cells. In this regard, either sorting cells for a more mature marker profile (CD15/CD16) or an additional cultivation of cells in G-CSF in combination with other differentiation stimuli, such as transferrin (Evans et al., 1986), may allow for the generation of more functional cells. Although the number of granulocytes reached 0.5 × 106/well/week in a laboratory scale, both more advanced scale-up techniques and GMP-grade final hematopoietic differentiation should be investigated before this protocol is extended to clinical applications. This is highly relevant given that granulocyte transfusion is performed with a minimal cell dose of 2–3 × 1010 (Lee et al., 2001).
In this work, we developed an innovative hematopoietic differentiation protocol that allows for the large-scale generation of functional, mature myeloid cells such as granulocytes or macrophages from single cultures over prolonged time periods. As an advantage in comparison with other protocols in the field, our protocol combines minimal cytokine support and hence a reduced complexity of the procedure with high and long-term cellular output. Even more importantly, our protocol allows one to generate different mature myeloid cell types by simply changing the cytokine support in the differentiation medium. Therefore, we expect it will also be suitable for producing other cell types, such as dendritic cells, platelets, and megakaryocytes. Clearly, a more precise characterization of the iPSC-derived myeloid cells by genome-wide expression analysis (which we have already initiated) is the next important step from a scientific perspective and is a prerequisite for potential clinical applications. Given the minimal addition of external cytokines, this protocol also appears to be particularly suited to gain further insights into hiPSC-based hematopoietic in vitro differentiation and its relation to the corresponding in vivo processes that take place in the embryo. Given the efficacy of the in vitro process, the protocol also appears to be useful for future cell-replacement or cell-based gene-therapy strategies based on terminally differentiated myeloid cells.
Experimental Procedures
Hematopoietic Differentiation of hiPSCs
We performed hematopoietic differentiation of hiPSCs using a modified version of the EB-based hematopoietic differentiation protocol described previously (Lachmann et al., 2014; van Wilgenburg et al., 2013). In brief, PSC colonies were disrupted to fragments using collagenase-V, and EB formation was induced by cultivation for 5 days in ESC medium supplemented with 10 ng/mL bFGF and 10 μM Rock inhibitor (Y-27632; Tocris) in six-well suspension plates on an orbital shaker (100 rpm). After manual transfer of EBs onto tissue culture six-well plates and cultivation in differentiation medium I (APEL medium; Stem Cell Technologies) supplemented with 1% penicillin-streptomycin (Life Technologies), 25 ng/ml human IL-3 (hIL-3) and 50 ng/ml human M-CSF (hM-CSF), human G-CSF (hG-CSF), or human GM-CSF (hGM-CSF; all from Peprotech), MCFCs were generated from the attached EBs within 7 days. From d10–d15 onward, monocytes/macrophages or granulocytes generated by the MCFCs were harvested twice a week from the supernatant and filtered through a 150-mM mesh. For further maturation, monocytes/macrophages or granulocytes were cultured in differentiation medium II (RPMI1640 medium supplemented with 10% fetal serum, 2 mM L-glutamine, 1% penicillin-streptomycin, and 100 ng/ml hM-CSF, hG-CSF, or hGM-CSF, respectively) for 7–10 days.
Phagocytosis Assay
For analysis of phagocytosis, cells were cultured overnight in 96-well plates at a concentration of 1 × 105 cells/ml in RPMI1640 medium supplemented with 10% fetal serum, 2 mM L-glutamine, 1% penicillin-streptomycin, and 100 ng/ml hM-CSF. The next day, carboxylate-modified red fluorescent latex beads with a mean diameter of 1 μm (L3030; Sigma-Aldrich) were added at a concentration of 1:200, and the cells were incubated with or without beads for 2 hr. After repeated washing, the cells were analyzed by flow cytometry.
Immunostaining of NETs
NET formation was analyzed using confocal laser-scanning microscopy. Neutrophils were suspended in RPMI and treated with either PMA (16 nM) or medium on coverslips and incubated for 4 hr at 37°C. Specimens were washed with warm PBS, fixed with 3% PBS-buffered formaldehyde, and permeabilized with 0.5% Triton X-100 in PBS. After blocking with 5% BSA, ELA was labeled using the monoclonal mouse anti-human ELA clone NP57 (Dako) primary antibody. Secondary antibody was conjugated with Alexa Fluor-488 (anti-mouse, catalog no. A11001; Invitrogen Life Technologies) and DAPI (catalog no. P36935; Sigma-Aldrich). Slides were analyzed using an Olympus FluoView confocal laser-scanning microscope in sequential scanning mode for two channels.
Author Contributions
N.L., M.A., and T.M., designed and performed research, analyzed data, and wrote the manuscript. E.F., S.L., C.G., C.H., O.K., S.B., M.P.K., S.J., and D.H. designed and performed research, and analyzed data. A.S., C.F., J.S., and G.H. designed research and analyzed data. D.L. and T.B. performed research.
Acknowledgments
The authors thank M. Ballmaier (Cell Sorting Facility, Hannover Medical School) for scientific support and Helena Lickei (Department of Respiratory Medicine, Hannover Medical School) for her excellent help with cytospin preparation and readout. This work was supported by grants from the Else Kröner-Fresenius Stiftung, the Deutsche Forschungsgemeinschaft (Cluster of Excellence REBIRTH, Exc 62/1, grants MO 886/6-1 and SFB738), the Bundesministerium für Bildung und Forschung (BMBF, PidNet), the German Center for Lung Research (DZL), and Hannover Medical School internal programs (Hochschulinterne Leistungsförderung [HiLF] and Young Academy).
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information
References
- Ackermann M., Lachmann N., Hartung S., Eggenschwiler R., Pfaff N., Happle C., Mucci A., Göhring G., Niemann H., Hansen G. Promoter and lineage independent anti-silencing activity of the A2 ubiquitous chromatin opening element for optimized human pluripotent stem cell-based gene therapy. Biomaterials. 2014;35:1531–1542. doi: 10.1016/j.biomaterials.2013.11.024. [DOI] [PubMed] [Google Scholar]
- Arai F., Hirao A., Ohmura M., Sato H., Matsuoka S., Takubo K., Ito K., Koh G.Y., Suda T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–161. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Cella M., Engering A., Pinet V., Pieters J., Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. 1997;388:782–787. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- Choi K.D., Vodyanik M., Slukvin I.I. Hematopoietic differentiation and production of mature myeloid cells from human pluripotent stem cells. Nat. Protoc. 2011;6:296–313. doi: 10.1038/nprot.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K.D., Vodyanik M.A., Togarrati P.P., Suknuntha K., Kumar A., Samarjeet F., Probasco M.D., Tian S., Stewart R., Thomson J.A., Slukvin I.I. Identification of the hemogenic endothelial progenitor and its direct precursor in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:553–567. doi: 10.1016/j.celrep.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S.C., Kamen R. The human hematopoietic colony-stimulating factors. Science. 1987;236:1229–1237. doi: 10.1126/science.3296190. [DOI] [PubMed] [Google Scholar]
- Clarke R.L., Yzaguirre A.D., Yashiro-Ohtani Y., Bondue A., Blanpain C., Pear W.S., Speck N.A., Keller G. The expression of Sox17 identifies and regulates haemogenic endothelium. Nat. Cell Biol. 2013;15:502–510. doi: 10.1038/ncb2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue R.E., Seehra J., Metzger M., Lefebvre D., Rock B., Carbone S., Nathan D.G., Garnick M., Sehgal P.K., Laston D. Human IL-3 and GM-CSF act synergistically in stimulating hematopoiesis in primates. Science. 1988;241:1820–1823. doi: 10.1126/science.3051378. [DOI] [PubMed] [Google Scholar]
- Elghetany M.T. Surface antigen changes during normal neutrophilic development: a critical review. Blood Cells Mol. Dis. 2002;28:260–274. doi: 10.1006/bcmd.2002.0513. [DOI] [PubMed] [Google Scholar]
- Evans W.H., Wilson S.M., Mage M.G. Transferrin induces maturation of neutrophil granulocyte precursors in vitro. Leuk. Res. 1986;10:429–436. doi: 10.1016/0145-2126(86)90073-1. [DOI] [PubMed] [Google Scholar]
- Guilliams M., De Kleer I., Henri S., Post S., Vanhoutte L., De Prijck S., Deswarte K., Malissen B., Hammad H., Lambrecht B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao N.B., Lü M.H., Fan Y.H., Cao Y.L., Zhang Z.R., Yang S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012;2012:948098. doi: 10.1155/2012/948098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happle C., Lachmann N., Skuljec J., Wetzke M., Ackermann M., Brennig S., Mucci A., Jirmo A.C., Groos S., Mirenska A. Pulmonary transplantation of macrophage progenitors as effective and long-lasting therapy for hereditary pulmonary alveolar proteinosis. Sci. Transl. Med. 2014;6:250ra113. doi: 10.1126/scitranslmed.3009750. [DOI] [PubMed] [Google Scholar]
- Harada A., Sekido N., Akahoshi T., Wada T., Mukaida N., Matsushima K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J. Leukoc. Biol. 1994;56:559–564. [PubMed] [Google Scholar]
- Jones C.V., Ricardo S.D. Macrophages and CSF-1: implications for development and beyond. Organogenesis. 2013;9:249–260. doi: 10.4161/org.25676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M., D’Souza S.L., Lynch-Kattman M., Schwantz S., Keller G. Development of the hemangioblast defines the onset of hematopoiesis in human ES cell differentiation cultures. Blood. 2007;109:2679–2687. doi: 10.1182/blood-2006-09-047704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M., Awong G., Sturgeon C.M., Ditadi A., LaMotte-Mohs R., Zúñiga-Pflücker J.C., Keller G. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:1722–1735. doi: 10.1016/j.celrep.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Krumwieh D., Weinmann E., Siebold B., Seiler F.R. Preclinical studies on synergistic effects of IL-1, IL-3, G-CSF and GM-CSF in cynomolgus monkeys. Int. J. Cell Cloning. 1990;8(Suppl 1):229–247. doi: 10.1002/stem.5530080722. discussion 247–228. [DOI] [PubMed] [Google Scholar]
- Kuehle J., Turan S., Cantz T., Hoffmann D., Suerth J.D., Maetzig T., Zychlinski D., Klein C., Steinemann D., Baum C. Modified lentiviral LTRs allow Flp recombinase-mediated cassette exchange and in vivo tracing of “factor-free” induced pluripotent stem cells. Mol. Ther. 2014;22:919–928. doi: 10.1038/mt.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey D.C., Achuthan A., Fleetwood A.J., Dinh H., Roiniotis J., Scholz G.M., Chang M.W., Beckman S.K., Cook A.D., Hamilton J.A. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J. Immunol. 2012;188:5752–5765. doi: 10.4049/jimmunol.1103426. [DOI] [PubMed] [Google Scholar]
- Lachmann N., Happle C., Ackermann M., Lüttge D., Wetzke M., Merkert S., Hetzel M., Kensah G., Jara-Avaca M., Mucci A. Gene correction of human induced pluripotent stem cells repairs the cellular phenotype in pulmonary alveolar proteinosis. Am. J. Respir. Crit. Care Med. 2014;189:167–182. doi: 10.1164/rccm.201306-1012OC. [DOI] [PubMed] [Google Scholar]
- Lancrin C., Sroczynska P., Stephenson C., Allen T., Kouskoff V., Lacaud G. The haemangioblast generates haematopoietic cells through a haemogenic endothelium stage. Nature. 2009;457:892–895. doi: 10.1038/nature07679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmann R., Knopf H.P., Link S., Sansano S., Schumann R., Zimmerli W. Human monocyte CD14 is upregulated by lipopolysaccharide. Infect. Immun. 1996;64:1762–1769. doi: 10.1128/iai.64.5.1762-1769.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.J., Chung I.J., Park M.R., Kook H., Hwang T.J., Ryang D.W., Kim H.J. Clinical efficacy of granulocyte transfusion therapy in patients with neutropenia-related infections. Leukemia. 2001;15:203–207. doi: 10.1038/sj.leu.2402007. [DOI] [PubMed] [Google Scholar]
- Martinez F.O., Sica A., Mantovani A., Locati M. Macrophage activation and polarization. Front. Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- Meng F., Lowell C.A. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J. Exp. Med. 1997;185:1661–1670. doi: 10.1084/jem.185.9.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Jackson H., Panopoulos A.D., Zhang H., Li H.S., Watowich S.S. STAT3 controls the neutrophil migratory response to CXCR2 ligands by direct activation of G-CSF-induced CXCR2 expression and via modulation of CXCR2 signal transduction. Blood. 2010;115:3354–3363. doi: 10.1182/blood-2009-08-240317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nostro M.C., Cheng X., Keller G.M., Gadue P. Wnt, activin, and BMP signaling regulate distinct stages in the developmental pathway from embryonic stem cells to blood. Cell Stem Cell. 2008;2:60–71. doi: 10.1016/j.stem.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palis J. Primitive and definitive erythropoiesis in mammals. Front. Physiol. 2014;5:3. doi: 10.3389/fphys.2014.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panopoulos A.D., Watowich S.S. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine. 2008;42:277–288. doi: 10.1016/j.cyto.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey-Giraud F., Hafner M., Ries C.H. In vitro generation of monocyte-derived macrophages under serum-free conditions improves their tumor promoting functions. PLoS ONE. 2012;7:e42656. doi: 10.1371/journal.pone.0042656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin C., Ottersbach K., Durand C., Peeters M., Vanes L., Tybulewicz V., Dzierzak E. An unexpected role for IL-3 in the embryonic development of hematopoietic stem cells. Dev. Cell. 2006;11:171–180. doi: 10.1016/j.devcel.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Sandler V.M., Lis R., Liu Y., Kedem A., James D., Elemento O., Butler J.M., Scandura J.M., Rafii S. Reprogramming human endothelial cells to haematopoietic cells requires vascular induction. Nature. 2014;511:312–318. doi: 10.1038/nature13547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz R., Marchenko N.D., Holembowski L., Fingerle-Rowson G., Pesic M., Zender L., Dobbelstein M., Moll U.M. Inhibiting the HSP90 chaperone destabilizes macrophage migration inhibitory factor and thereby inhibits breast tumor progression. J. Exp. Med. 2012;209:275–289. doi: 10.1084/jem.20111117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta A., Liu W.K., Yeung Y.G., Yeung D.C., Frackelton A.R., Jr., Stanley E.R. Identification and subcellular localization of proteins that are rapidly phosphorylated in tyrosine in response to colony-stimulating factor 1. Proc. Natl. Acad. Sci. USA. 1988;85:8062–8066. doi: 10.1073/pnas.85.21.8062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A., Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieff C.A. Hematopoietic growth factors. J. Clin. Invest. 1987;79:1549–1557. doi: 10.1172/JCI112988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgeon C.M., Ditadi A., Awong G., Kennedy M., Keller G. Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat. Biotechnol. 2014;32:554–561. doi: 10.1038/nbt.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi T., Tsuneyoshi N., Nakatsuji N., Suemori H. Defining early lineage specification of human embryonic stem cells by the orchestrated balance of canonical Wnt/beta-catenin, Activin/Nodal and BMP signaling. Development. 2008;135:2969–2979. doi: 10.1242/dev.021121. [DOI] [PubMed] [Google Scholar]
- Suzuki T., Arumugam P., Sakagami T., Lachmann N., Chalk C., Sallese A., Abe S., Trapnell C., Carey B., Moritz T. Pulmonary macrophage transplantation therapy. Nature. 2014;514:450–454. doi: 10.1038/nature13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wilgenburg B., Browne C., Vowles J., Cowley S.A. Efficient, long term production of monocyte-derived macrophages from human pluripotent stem cells under partly-defined and fully-defined conditions. PLoS ONE. 2013;8:e71098. doi: 10.1371/journal.pone.0071098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte K., Platzer E., Gabrilove J.L., Lu L., Levi E., Polivka A., Mertelsmann R., Moore M.A. Purification to apparent homogeneity and biochemical characterization of human pluripotent hematopoietic colony-stimulating factor. Haematol. Blood Transfus. 1985;29:398–401. doi: 10.1007/978-3-642-70385-0_82. [DOI] [PubMed] [Google Scholar]
- Welte K., Platzer E., Lu L., Gabrilove J.L., Levi E., Mertelsmann R., Moore M.A. Purification and biochemical characterization of human pluripotent hematopoietic colony-stimulating factor. Proc. Natl. Acad. Sci. USA. 1985;82:1526–1530. doi: 10.1073/pnas.82.5.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte K., Bonilla M.A., Gillio A.P., Boone T.C., Potter G.K., Gabrilove J.L., Moore M.A., O’Reilly R.J., Souza L.M. Recombinant human granulocyte colony-stimulating factor. Effects on hematopoiesis in normal and cyclophosphamide-treated primates. J. Exp. Med. 1987;165:941–948. doi: 10.1084/jem.165.4.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiles M.V., Keller G. Multiple hematopoietic lineages develop from embryonic stem (ES) cells in culture. Development. 1991;111:259–267. doi: 10.1242/dev.111.2.259. [DOI] [PubMed] [Google Scholar]
- Williams D.A., Rios M., Stephens C., Patel V.P. Fibronectin and VLA-4 in haematopoietic stem cell-microenvironment interactions. Nature. 1991;352:438–441. doi: 10.1038/352438a0. [DOI] [PubMed] [Google Scholar]
- Yang Y.C., Ciarletta A.B., Temple P.A., Chung M.P., Kovacic S., Witek-Giannotti J.S., Leary A.C., Kriz R., Donahue R.E., Wong G.G. Human IL-3 (multi-CSF): identification by expression cloning of a novel hematopoietic growth factor related to murine IL-3. Cell. 1986;47:3–10. doi: 10.1016/0092-8674(86)90360-0. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Hayashi S., Kunisada T., Ogawa M., Nishikawa S., Okamura H., Sudo T., Shultz L.D., Nishikawa S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.