

Abstract
The mechanisms by which neurons respond to inflammatory mediators such as interferons (IFNs) remain largely undefined. We previously showed that the activation and nuclear localization of the core IFN signaling molecule, Stat1, are muted and delayed in primary mouse hippocampal neurons treated with IFN gamma as compared to control mouse embryonic fibroblasts (MEF). Here, we show that the kinetics of Stat1 and Stat2 activation following type I IFN exposure are also unique in neurons, affecting gene expression and neuronal response. Specifically, despite lower basal expression of many IFN stimulated genes in neurons, basal expression of the type I IFN themselves is significantly higher in primary hippocampal neurons compared to MEF. Elevated homeostatic IFN in neurons is critical and sufficient for early control of viral infection. These data provide further evidence that neurons exploit unique signaling responses to IFNs, and define an important contribution of homeostatic IFN within the CNS. Such differences are likely critical for the ability of neurons to survive a viral challenge.
1. Introduction
The host immune response can efficiently resolve certain neurotropic infections, but unregulated or chronic immune responses in the central nervous system (CNS) can be pathogenic and often fatal. Immune dysregulation within the brain can result in encephalitis and meningitis, and contributes to many chronic neuroinflammatory diseases such as multiple sclerosis (1-6). Therefore, a balance must be achieved in which pathogen control or clearance is achieved with minimal neuropathology. This is particularly relevant for infections of CNS neurons, which are a chiefly non-renewable cell population. Well-defined immune mechanisms that clear viral infections in the periphery, including perforins and granzymes, are underutilized in the brain, perhaps protecting the neuronal population from immune-mediated cytolysis. Instead, cytokines, including the interferons (IFNs), are fundamental contributors to CNS virus clearance. Thus, an overarching goal of our studies is to elucidate the unique interactions of IFNs with neuronal targets, and to define how IFN signaling limits or clears neurotropic infections in the absence of CNS disease.
Viral reproduction in the CNS is a relatively rare, albeit serious, consequence of infection by a number of human viruses. While some viruses are well-known to be neurotropic (e.g., poliovirus, rabies virus, West Nile virus, and some herpesviruses), others that are primarily associated with peripheral infections, including influenza and measles, can also result in life-threatening CNS complications (7). For example, influenza has been associated with encephalitis, Reye's Syndrome, and acute necrotizing encephalopathy, particularly in children (8). Moreover, measles virus (MV) infection of CNS neurons is associated with invariably fatal diseases such as subacute sclerosing panencephalitis, which can occur months to years after primary virus exposure (9). While some of these viruses gain access to the brain parenchyma due to a weakened immune response (e.g., herpesviruses), most result in neuropathology via induction of the immune response (10).
An early and essential line of defense against viral infection is the induction of interferons (IFNs) that ultimately leads to antiviral gene expression (reviewed in 11). Briefly, in most mammalian cells, viruses are detected by pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) and retinoic acid-inducible gene-I (Rig-I). Recognition of viral nucleic acid in infected cells by these receptors leads to downstream signaling, including activation of transcription factors NF-kB and IFN-regulatory factors 3 and 7 (Irf3, Irf7). Migration of these transcription factors into the nucleus induces expression of genes encoding type I IFNs (IFNα, IFNβ). Once released from the cell, IFNs then bind to their receptor (Ifnar, composed of Ifnar1 and Ifnar2 subunits) on the surface of neighboring cells, leading to activation of Janus-activated kinase 1 (Jak1) and tyrosine kinase 2 (Tyk2). Signal transducers and activators of transcription 1 and 2 (Stat1, Stat2) are recruited to the activated receptor and are phosphorylated by Jak1 and Tyk2, resulting in a complex of Stat1, Stat2 and IFN regulatory factor 9 (Irf9). This ISGF3 complex then translocates to the nucleus and binds IFN-sensitive response elements (ISREs) in the promoters of IFN-stimulated genes (ISGs). Expression of ISGs leads to induction of an antiviral state (reviewed in 11).
While IFNs are generally recognized as indispensable for an effective immune response, cell type-specific properties may diversify how cells respond to the same extracellular ligand. This is governed in part by the variety, level and distribution of Stat molecules in a given cell type, as type I IFNs are able to activate all seven known Stat molecules. Thus, following engagement of the IFN receptor, distinct Stat homo- and heterodimer complexes may form, and interactions between such complexes and promoter elements determine what genes will be up-or down-regulated (reviewed in 12). For example, in immune cells, type I IFN aids in activation and Th1 skewing of naive T cells (13), promotes survival and cytolytic activity of CD8+ T cells, and ensures efficient antibody response by B cells (14-15). Non-immune cells have varied responses to IFNs as well. For example, in primary human hepatocytes, IFNα triggers an antiviral and antitumor state (16). Cardiac myocytes, which, like neurons, are a non-renewable cell population, express higher basal levels of IFNβ than cardiac fibroblasts, likely as a “pre-arming” mechanism to protect against viral infection (17). Within the CNS, distinct cellular responses to IFN could play a role in infection and immunity as well as neurodegenerative diseases and the response to injury or ischemia (18).
We utilize a mouse model of neuron-restricted measles virus (MV) infection, in which CNS neurons express a human MV vaccine strain receptor (CD46) under the transcriptional control of the neuron-specific enolase (NSE) promoter (NSE-CD46) (19). Using this model, we showed that adult mice clear MV infection from CNS neurons without neurological damage or neuronal loss in an IFNγ- and T cell-dependent manner (20). Primary hippocampal neurons can be explanted from these mice and grown in culture; while all neurons of NSE-CD46 transgenic mice are CD46-positive, and therefore permissive, the hippocampus of day e15-16 embryos is very neuron-rich, enabling virtually pure cultures. When we assessed IFNγ signaling in these hippocampal neurons, we found that the response to IFNγ treatment was distinct from that observed in control MEF. Specifically, neurons responded to IFNγ with delayed and attenuated kinetics of Stat1 expression and activation, resulting in a concomitantly delayed and reduced expression of classic IFNγ–dependent genes (18). These foundational studies indicate that IFNγ activates a critical antiviral program in neurons, but that Stat1 plays a subordinated role in this response.
Because Stat1 is also a key mediator in type I IFN signaling, we wished to characterize the response of CNS neurons to type I IFN in the context of viral infection. Utilizing NSE-CD46 mice and primary neurons explanted from these mice, we show here that hippocampal neurons respond to type I IFN with an unique ISG signature. Moreover, exogenous type I IFN enhances resistance to viral infection in primary fibroblasts, but not in primary neurons. We provide evidence that neurons express increased basal levels of type I IFNs compared to fibroblasts, which we hypothesize is critical for controlling early infection in the absence of exogenous IFN. This suggests that within the CNS, neurons respond uniquely to crucial cytokines produced soon after infection, and we hypothesize that this may be a protective mechanism to ensure survival during infection and the subsequent adaptive immune response.
2. Materials and Methods
2.1 Ethics statement
This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was reviewed and approved by the Fox Chase Cancer Center Institutional Animal Care and Use Committee (Office of Laboratory Animal Welfare assurance number A3285-01).
2.2 Cells, viruses, mice, and infections
Primary hippocampal neurons were prepared from embryonic (E15–16) mice, as described (18, 21-23). Neurons were plated on 15-mm glass coverslips or in 12-well plates coated with poly-L-lysine (Sigma-Aldrich) at a density of 2×105 cells/well, unless otherwise noted. Neuron cultures were quality-controlled, and were routinely >95% Map2-positive. Neurons were plated and incubated for 5 days (d) to allow for full differentiation prior to IFNβ treatment or infection. Primary mouse embryonic fibroblasts (MEF) were isolated from the same embryos and maintained in complete DMEM medium (DMEM supplemented with 10% fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 ng/ml streptomycin). All cells were maintained at 37°C, 5% CO2 in a humidified incubator.
MV-Edmonston (vaccine strain) was purchased from American Type Culture Collection and passaged and titered in Vero cells. Passages 2 or 3 of the MV stock were used for intracerebral (IC) injections and in vitro infection assays. LCMV Armstrong (LCMV-Arm; ATCC) was passaged in BHK-21 fibroblasts and plaque purified, and titers were determined on Vero fibroblasts.
Homozygous NSE-CD46+ transgenic mice (line 18; H-2b) (19) were maintained in the closed breeding colony of the Fox Chase Cancer Center. Homozygous NSE-CD46+ and haplotype-matched homozygous immune knockout (KO) mice were intercrossed for three or more generations to obtain NSE-CD46+ mice on the desired KO background. Ifnar-deficient mice (24) on 129S2/SvPas background were obtained from Luis Sigal (Fox Chase Cancer Center, Philadelphia, PA). Genotypes of all mice used in these experiments were confirmed by PCR analysis of tail biopsy DNA and/or flow cytometry on blood cells.
Isoflurane-anesthetized mice were infected with MV via IC inoculation (1×104 PFU in a volume of 30 μl, delivered along the midline using a 27g needle). Mice were monitored daily post-infection for signs of illness, including weight loss, ruffled fur, ataxia, and seizures. Moribund mice were euthanized in accordance with IACUC guidelines.
2.3 Infection and interferon treatment of primary cells
Five days post-plating, primary hippocampal neurons or MEF from NSE-CD46+ or CD46/Ifnar1 KO mice were infected with MV or LCMV (multiplicity of infection [MOI] = 1) for 1 h. Thereafter, the inoculum was removed and the cells were maintained in conditioned Neurobasal media. For cells treated with IFNβ, murine IFNβ (Millipore) was diluted in B27-free Neurobasal media, added to the cultures (100 U/ml final), and incubated for the indicated times prior to collection.
2.4 Immunoblots
Untreated and IFNβ-treated primary cells cultured on tissue culture plastic were lysed directly with protein solubilization buffer (106 mM Tris HCl, 141 mM Tris Base, 0.51 mM EDTA, 2% SDS). Where indicated, protein isolated from 2×105 cells per sample were separated on a NuPAGE 7% Tris-Acetate gel or 10% Bis-Tris gel (Invitrogen), and transferred (semi-dry) to PVDF-FL (Millipore). Within an experiment, corresponding samples from neurons and MEF were run on the same gel, to allow direct comparison. The blots were blocked for one hour in Odyssey Blocking Buffer (Licor). The blots were subsequently incubated in primary antibody solution: anti-Stat1 (1:500), anti-phospho-specific Stat1 (pY701; 1:500), both from BD Biosciences Pharmingen; anti-Stat2 (1:600), from Santa Cruz Biotechnology Inc., Santa Cruz, CA; anti-phospho-specific Stat2 (pY689; 1:500), from Millipore; and anti-glyceraldehyde-3-phosphate dehydrogenase (Gapdh; 1:1000; Millipore). All antibodies were diluted in Odyssey Blocking Buffer and incubated overnight at 4°C. After three washes in TBS-T (10 min each), the blots were incubated in secondary antibody (Licor) solution: goat anti-rabbit IRDye 680RD for anti-Stat2 (1:20,000) and anti-Stat2p689 (1:20,000), goat anti-mouse IRDye 680RD for anti-Stat1 (1:10,000) and anti-Stat1pY701 (1:20,000) and anti-Gapdh (1:20,000); all were diluted in Odyssey Blocking Buffer for 1 h at room temperature. The blots were washed as described above, then imaged on the Odyssey CLx infrared imaging system (Licor).
2.5 Reverse transcriptase quantitative real-time PCR (qRT-PCR)
RNA was purified from whole cell lysates using the RNeasy Mini kit (Qiagen, Valencia, CA). RNA was quantified using a Nanodrop spectrophotometer. RNA was reverse-transcribed using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Gene-specific primers in combination with Universal Probe Library probes and Universal Master mix (Roche) were run on a Mastercycler Realplex2 system (Eppendorf). Cycling conditions were 50°C, 2 min; 95°C, 10 min; followed by 40 (2-step) cycles (95°C, 15 sec; 60°C, 60 sec). Relative quantification to the control (cyclophilin B) was done using the comparative Ct method. The values plotted are the average from 3 PCR reactions.
3. Results
3.1 Expression and activation of Stat1 and Stat2 differ between neurons and fibroblasts following type I IFN exposure
We previously identified unique attributes of the neuronal signaling response to the type II IFN, IFNγ, as compared to control fibroblasts (18). To determine whether similar differences exist in neurons following type I IFN exposure, primary mouse hippocampal neurons were treated with IFNβ, and their response compared with primary mouse embryonic fibroblasts (MEF) treated in a similar manner (25). The dose of 100U/mL IFNβ was chosen to be consistent with (or below) the majority of published work on type I IFNs. We first assessed the activation of Stat1 and Stat2 following IFNβ exposure. As previously shown, primary neurons have a lower homeostatic expression of Stat1 than control fibroblasts, which we originally hypothesized was the basis for the muted and delayed transcriptional response following IFNγ engagement (18). Despite lower basal levels of Stat1 in neurons, however, both neurons and MEFs showed rapid induction of activated Stat1 (Fig. 1A). Though MEFs had a more robust pStat1 signal at early times post-IFN exposure, phosphorylation of Stat1 was prolonged in neurons. Similarly, basal Stat2 levels were also lower in neurons, but again, the early kinetics of Stat2 phosphorylation were approximately equal between the two cell populations, with neuronal Stat2 phosphorylation sustained through 24 hours post-exposure (Fig. 1B). Thus, despite similar induction profiles between the two primary cell populations, the activation of canonical signaling molecules is sustained in neurons following IFNβ addition.
Figure 1. Kinetics of Stat1/2 phosphorylation in response to IFNβ are distinct in neurons and MEF.
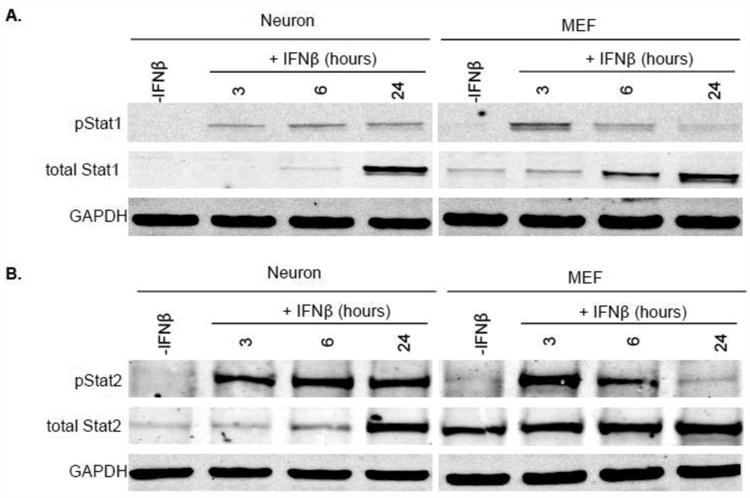
Primary hippocampal neurons and MEF were treated with 100U/mL recombinant IFNβ or vehicle control. Protein lysates were collected 3, 6, and 24 h thereafter and immunoblotted for total Stat1, pStat1 (Tyr-701), and Gapdh (A) or total Stat2, pStat2 (Tyr-690), and Gapdh (B). Western blots shown in the Figure are representative of three, independent experiments with identical outcomes.
3.2 Neurons and MEF induce distinct patterns of ISGs in response to IFNβ
We hypothesized that prolonged activation of Stat molecules in neurons would result in a sustained transcriptional response following type I IFN exposure. We thus evaluated the neuronal and MEF gene expression profiles to IFNβ by evaluating a limited number of canonical ISGs for analysis by qRT-PCR over a 24 hour period. We chose representative ISGs from two categories: i) those that encode transcription factors, or proteins that complex with transcription factors (Stat1, Stat2, Irf7, and Irf9); and ii) those that encode proteins that directly participate in the antiviral response (Oas1a and Isg15)(26).
The pattern and magnitude of gene induction differed significantly between neurons and MEF following IFNβ exposure, even at timepoints when the pStat1 and pStat2 expression levels were similar between cell types. Antiviral genes Oas1a and Isg15 were highly induced in MEF, whereas neuronal expression of these genes was negligible (Fig. 2Ai-ii). In sharp contrast, neuronal mRNA synthesis of Stat1 and Irf7 was higher than in MEF, while no significant differences between cell populations were evident in Stat2 and Irf9 expression (Fig. 2B). These results, albeit using a limited number of type I IFN target genes, indicate that neurons and MEF mount distinct responses to IFNβ, likely diversifying the cell type-specific response to infection at the level of gene expression.
Figure 2. Neurons and MEF respond with different patterns of ISG induction in response to IFNβ.
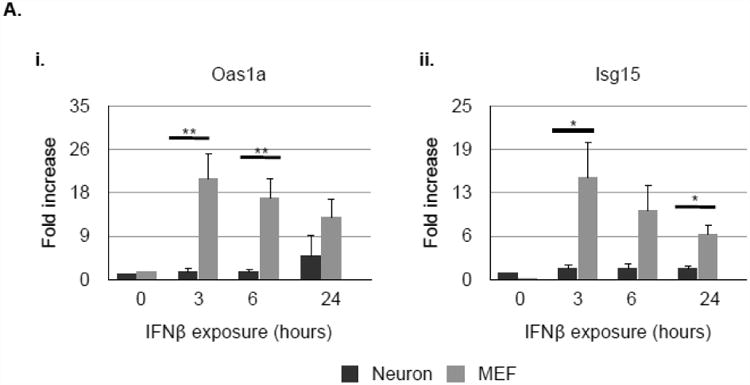
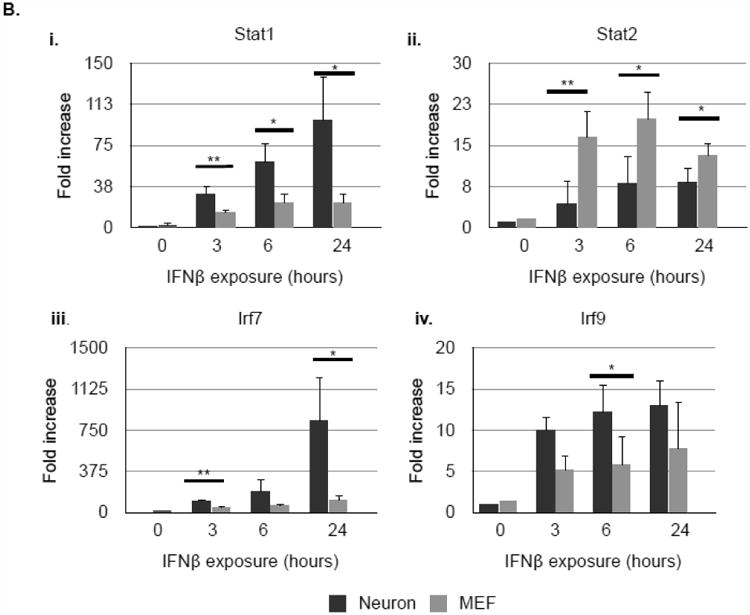
Primary hippocampal neurons and MEF were treated with 100U/mL recombinant IFNβ. RNA lysates were collected 3, 6, and 24 h thereafter and analyzed for expression of Oas1A (Ai.), Isg15 (Aii.), Stat1 (Bi.), Stat2 (Bii.), Irf7 (Biii.), and Irf9 (Biv.) by qRT-PCR. Relative gene expression level represents pooling of data from three independent experiments after normalization within a given experiment. *p<0.05, **p<0.01.
We next sought to define the underlying cause of the distinct gene expression profiles between these cell types. We hypothesized that expression of some genes may not be significantly elevated following IFN exposure if basal expression was already high. To determine whether this was the case, we compared basal expression levels of these genes in neurons and MEF (Fig. 3). Raw data (expressed as differences in dCt values) are shown in Fig. 3A, and fold-differences are represented in Fig. 3B (Fig. 3Bi: genes with higher basal levels in MEF; Fig. 3Bii: genes with higher basal levels in neurons). Notably, both Stat1 and Irf7, which were more highly induced in neurons following IFNβ exposure, were expressed at significantly higher levels in MEF under basal conditions. Interestingly, there was no difference in basal levels of Irf3, which together with Irf7 plays a role in regulating expression of type I IFNs. Also of interest were the basal expression levels of both Oas1a and Isg15. While MEF, but not neurons, highly up-regulate Oas1a in response to IFNβ, there were no significant differences in the basal expression levels of this gene in either cell type. In contrast, Isg15, which is also disproportionately induced in MEF following IFNβ exposure, was expressed at significantly higher levels in untreated neurons as compared to MEF. Importantly, expression of the Ifnar1 and Ifnar 2 subunits at the RNA level do not differ significantly between the two cell types (data not shown). Thus, basal (or homeostatic) cell-type specific gene expression profiles appear to be a variable in determining the magnitude of induction of these genes following exposure to type I IFN.
Figure 3. Basal levels of select ISGs differ between neurons and MEF.
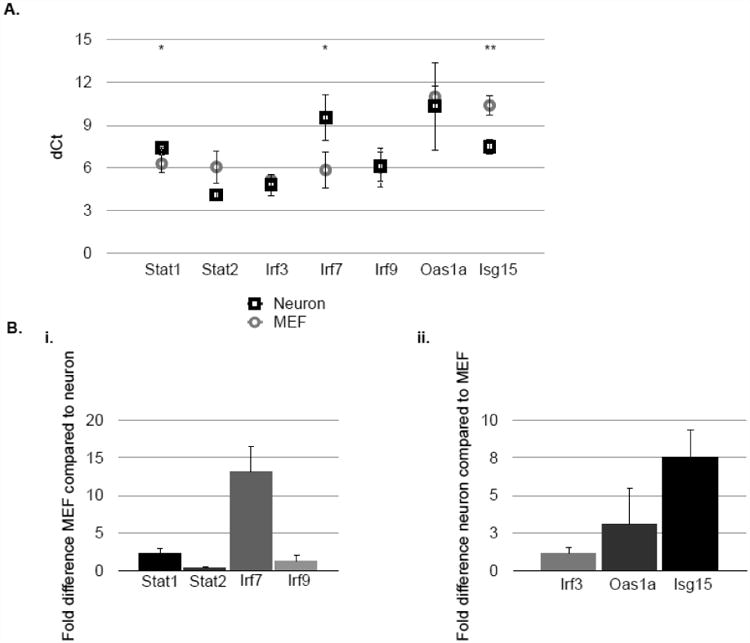
RNA lysates were collected from untreated primary hippocampal neurons or MEF and analyzed for expression of Stat1, Stat2, Irf7, Irf9, Oas1a, and Isg15 by qRT-PCR. A: dCt value following normalization to ppib (cyclophilin B). (Recall that each increase in dCt value represents two-fold less RNA in the sample). B: fold-difference in gene expression comparing MEF to neuron (i) or neuron to MEF (ii). Relative gene expression levels represents pooling of data from independent experiments after normalization within a given experiment. Results are representative of 3 independent experiments. *p<0.05, **p<0.01.
3.3 Basal expression of type I interferons is higher in neurons compared to MEF
Differences in homeostatic expression of type I IFNs themselves may drive the unique signatures of basal ISG expression. Expression of the majority (7/12 genes) of type I IFNs examined at an RNA level was significantly higher in neurons than MEF under unstimulated conditions (Fig. 4A-B). Notably, type I IFN types known to be important during viral infection were among the most disproportionately expressed in neurons. To exclude the possibility that elevated type I IFN expression was an artifact of primary neuron cultures, the expression of type I IFN was quantified in freshly isolated embryonic mouse hippocampi, which at day E15-16, are comprised of mostly neurons (22). The expression of type I IFNs in hippocampi were comparable to in vitro cultured neurons from the same litter of embryos (data not shown).
Figure 4. Expression of type I IFNs under basal conditions is higher in neurons than in MEF.
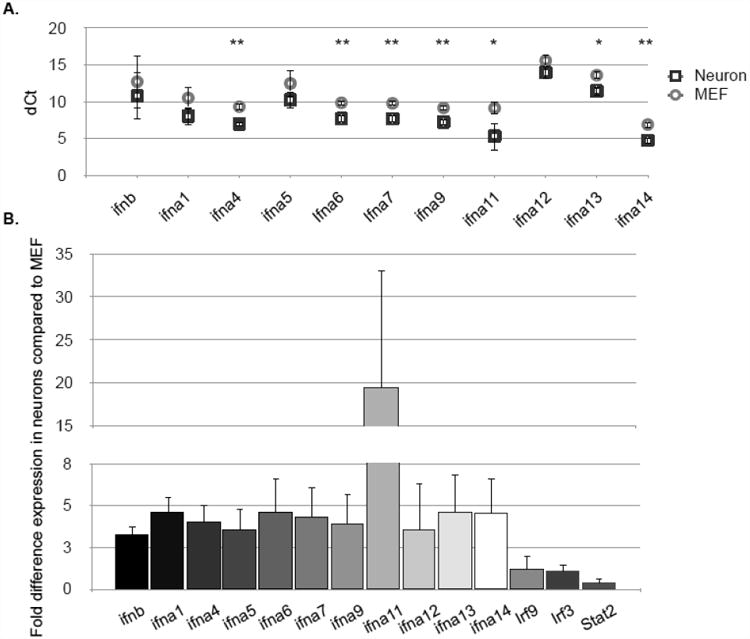
RNA from untreated primary hippocampal neurons or MEF was collected and expression of IFNβ, IFNα1, IFNα4-9, and IFNα11-14 was detected by qRT-PCR and expressed as dCt following normalization to ppib (cyclophilin B) (A) and fold difference in neuronal expression compared to MEF (B). *p<0.05, **p<0.01. A representative experiment of four is shown.
3.4 Type I IFN restricts viral replication in MEF but not primary neurons in vitro
To determine the effect of elevated basal type I IFN expression in neurons on virus infection, primary hippocampal neurons and MEF were infected with LCMV (MOI=1) with or without addition of recombinant IFNβ (100U/mL). Addition of recombinant IFNβ did not appreciably decrease LCMV levels in neurons over a 48 h time period (Fig. 5A, inset). In sharp contrast, IFNβ had potent antiviral activity against LCMV infection of MEF (Fig. 5A). To determine whether this was a virus-specific effect, primary hippocampal neurons were infected with MV (MOI=1) with or without the addition of IFNβ. Again, addition of type I IFN did not have an effect on viral load at 24 or 28 h following infection (Fig. 5B). This further supports the hypothesis that the neuronal response to IFNβ is distinct from the canonical response observed in MEF (25), and may primarily favor induce genes that are not directly involved in viral clearance.
Figure 5. IFNβ restricts viral replication in MEF but not primary neurons.
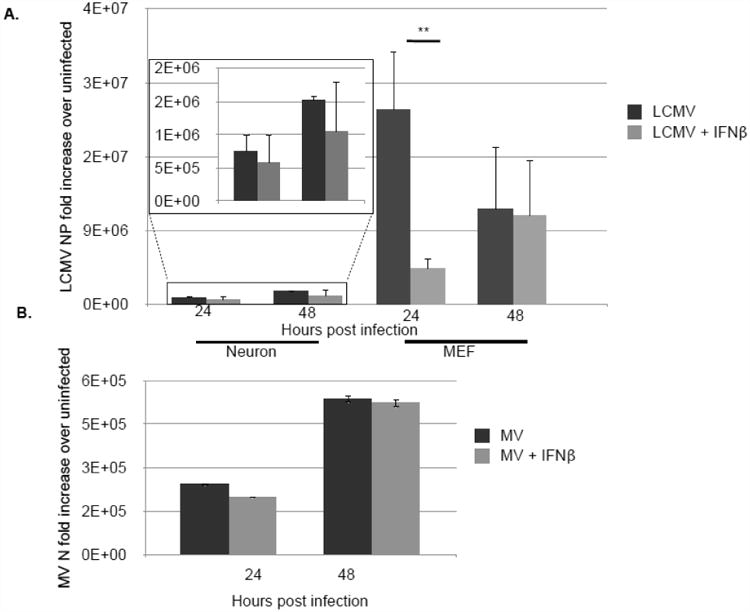
Primary hippocampal neurons or MEF were treated with 100U/mL recombinant IFNβ or vehicle control and infected with LCMV (MOI=1) (A) or MV-Edmonston (MOI=1) (B). RNA was collected at 24 and 48 h post-infection and viral replication was detected by qRT-PCR. Relative gene expression level represents pooling of data from independent experiments after normalization within a given experiment. Results are representative of 3 independent experiments. **p<0.01.
3.5 Type I IFN expression is required for early control of MV in vitro and in vivo but is dispensable for survival
The surprising observation that exogenous IFNβ did not have an effect on viral load in neuronal cultures prompted us to confirm that type I IFN was actually required for neuronal control of viral infection. To do so, CD46 and CD46/Ifnar KO primary hippocampal neurons were infected with MV (MOI=1), but in this experiment, no exogenous IFN was added. Ablation of homeostatic type I IFN signaling in primary neurons renders them significantly more vulnerable to viral infection, as evidenced by significantly elevated levels of viral RNA in CD46/Ifnar KO neurons (Fig. 6A).
Figure 6. Type I IFNs play a role in restriction of early viral replication in vitro and in vivo but are dispensable for survival following MV infection.
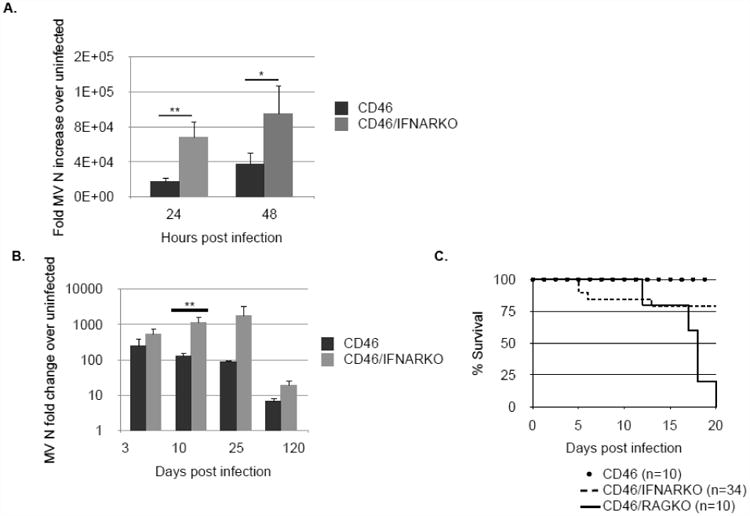
A: CD46 and CD46/Ifnar KO primary hippocampal neurons were infected with MV (MOI=1). RNA was collected at 24 and 48 h post-infection and viral replication was detected by qRT-PCR. B: MV-infected animals were sacrificed at various timepoints and brains collected for RNA analysis. Relative gene expression level represents pooling of data from independent experiments after normalization within a given experiment. Results are representative of 3 independent experiments. *p<0.05, **p<0.01. C: CD46, CD46/Rag KO, and CD46/Ifnar KO were infected intracranially with 104 PFU MV-Edmonston and monitored daily for morbidity and mortality.
We previously showed (20) that T cells and IFNγ are required for clearance of MV from the CNS. To define the contribution of type I IFNs in the control of MV infection in adult mice, we initially evaluated the survival of MV-permissive Ifnar1-deficient mice following MV challenge. In these mice, only CNS neurons are permissive, allowing us to determine how the presence or absence of IFN signaling influenced viral pathogenesis. Surprisingly, greater than 75 percent of MV-challenged CD46/Ifnar KO survived infection, in contrast to CD46/Rag-2 KO mice, which all showed significant weight loss coincident with the appearance of signs of CNS disease, including ataxia, ruffled appearance, and seizures (Fig. 6C). While viral titers from CD46/Ifnar KO mice were substantially higher than control, immunocompetent CD46 mice at early times post-infection (days 3-10), CD46/Ifnar KO mice eventually cleared the infection with similar kinetics to control mice (Fig. 6B), likely owing to the presence of the protective adaptive immune response which appears within the CNS by 7-10 days post-infection. (Of note, CD46/Ifnar KO mice succumb to a peripheral infection with wild type vesicular stomatitis virus (VSV) much earlier than immunocompetent mice, as previously published (data not shown)(27)). These data suggest that, in vivo, there is a virus-type dependence on type I IFNs, and that the outcome of a neuronal infection is influenced both by the basal IFN expression in neurons as well as the challenge virus.
4. Discussion
We can draw three conclusions from our data. First, the kinetics of Stat1 and Stat2 activation in response to type I IFN differ in neurons and fibroblasts, and the resultant differences in signaling have a significant impact on the resulting pattern of gene expression. We attribute this to inherent variations in basal (homeostatic) expression of a number of these genes. Second, we provide evidence that despite lower basal expression of select ISGs in neurons, basal expression of type I IFNs themselves is significantly higher in neurons compared to fibroblasts. The elevated homeostatic expression of neuronal IFN is critical and sufficient for early control of viral infection. Finally, we demonstrate a requirement for IFN signaling in early control of viral infection in vivo, while confirming that survival during viral infection in the absence of type I IFN signaling is virus- and cell type-specific.
These data contribute to a growing literature which suggests that responsiveness to a particular cytokine is regulated in a cell type-specific manner (12, 17-18, 28-29). Both type I and type II IFNs activate multiple Stat proteins, depending on which are bioavailable within a given cell (28). We have previously shown that neurons respond uniquely to IFNγ, with markedly delayed, attenuated, and prolonged phosphorylation of Stat1 (18). It was surprising to find that, in contrast, neurons and fibroblasts respond equally rapidly to IFNβ. There is evidence to suggest that expression levels of various Stats influence which are actually activated by type I IFN (26). It is possible that, due to reduced basal expression of both Stat1 and Stat2 in neurons, other Stat molecules or other IFN-induced pathways are more highly activated, thus skewing the cellular response to a soluble ligand.
It is also interesting to note that basal expression, as well as patterns of induction of IFN-stimulated genes, differs significantly between primary hippocampal neurons and fibroblasts. While differences in gene induction between the two cell types can likely be attributed to the variation in basal expression of these genes, it will be of interest to determine the mechanisms underlying selective gene profiles such as Isg15, which are more highly and consistently expressed in neurons. We have not determined whether the higher levels of IFN expression in neurons are functioning in autocrine loops or whether mRNA or protein is stored for rapid synthesis and release in the event of infection or injury (30-33). However, given that, of the two primary transcription factors that drive expression of type I IFN, Irf7 expression is lower in neurons and Irf3 expression is comparable between the two cell types, it is reasonable to speculate that increased type I IFN mRNA in neurons results from increased stability or neuronal storage of the RNA rather than higher levels of transcription. Cardiac myocytes, which are similar to neurons in that they are a non-renewable cell population, express increased levels of IFNβ and several components of the Jak-Stat signaling pathway (29), suggesting that vulnerable cell populations may utilize higher basal IFN expression as a mechanism to protect against viral infection. Combined with data presented here, a compelling case can be made that vulnerable cell populations such as neurons may be “pre-armed” to respond quickly and specifically under conditions of viral challenge.
We hypothesize that altered profiles of ISG and IFN expression are one mechanism by which neurons protect themselves from the potentially deleterious immune response triggered by infection. It is well established that IFNs (as well as a variety of other cytokines) have the potential to be neurotoxic (34-40). However, exposure of various cell types to type I IFN renders cells less responsive to future exposure to the same cytokine (25). It is conceivable, then, that neurons produce sufficient quantities of type I IFN to desensitize them from future exposure to IFN, and possibly other cytokines, produced by resident glial cells and incoming immune cells. This would render them less responsive to, and therefore protected from, the incoming immune response to infection.
Type I IFNs are necessary for early control of many viral infections (41-47). Interestingly, while the absence of type I IFN signaling leads to increased viral load in MV-infected NSE-CD46 mice, its absence has a minimal impact on survival. Using a similar mouse model, it was shown that survival of Ifnar-deficient mice following MV infection was dependent on viral dose (48). Morever, unrelated viruses that strongly induce a type I IFN response have been shown to be controlled by different profiles of ISGs specifically in the CNS (49). It is possible, therefore, that survival in the absence of type I IFN signaling may be influenced in part by the kinetics of viral replication and spread, as well as the timing of the incoming adaptive immune response. Presumably, in our model, incoming IFNγ-producing T cells are able to efficiently control infection in mice lacing type I IFNs. It will be of interest to determine whether the ∼20% of Ifnar-deficient mice that succumb following infection die due to unrestricted infection, or whether neurons lacking type I IFN signaling are rendered hypersensitive to other cytokines, resulting in immune-mediated neuronal damage and loss. Some interferons are more effective against particular viruses, in part because distinct type I interferons engage the receptor with different affinity. For these studies, we chose IFN-beta because it is generally used as a “representative” IFN in the literature, though future efforts will look at differential type I IFN expression following infection, and outcomes following neuronal exposure.
The differences observed between neurons and fibroblasts may afford some protection to generally non-renewable neurons: for example, Oas1 and Pkr tend to be associated with cell suicide, and thus their robust induction in fibroblasts may help to eliminate potential viral factories, whereas a similar response in neurons may result in catastrophic neuronal loss. In related work from our lab, we have shown that IFNγ addition in neurons does not trigger the usual antiviral transcriptional program, but rather selectively induces pro-survival pathways and genes (O'Donnell, et al, unpublished observations). Efforts are underway to explore how the altered cellular responses to these critical immune mediators may shape viral clearance and influence host survival.
Our data provide further evidence for complex, cell type-specific responses to extracellular cytokines, and suggest that vulnerable cell populations (for example, those that cannot be repopulated), can be pre-armed to contend with infections and the potentially damaging immune responses that such infections induce. These data could be especially interesting if interpreted in light of current treatment for multiple sclerosis. The primary treatment still used is recombinant IFNβ, though the mechanism of action remains incompletely understood (reviewed in 50, 51). While research geared toward understanding this mechanism has focused primarily on the effect of IFN on immune cells, it will be interesting to determine how IFNβ treatment affects the cellular response to incoming immune cell populations. An overarching conclusion of this study, and similar studies like it, would be that the contributions of cytokines in infection and other diseases must be considered in a disease- and cell type-specific manner if they are to be completely understood and therapeutically exploited.
Highlights.
Stat1 and Stat2 activation in response to type I IFN differs in neurons and fibroblasts.
Despite lower basal expression of select ISGs in neurons, basal expression of type I IFNs is higher in neurons.
Mouse survival during infection in the absence of type I IFN signaling is virus- and cell type-specific.
Acknowledgments
The authors would like to thank Siddharth Balachandran, Christine Matullo, Kevin O'Regan, Andreas Solomos, Kristen Henkins, and Karen Lancaster for their contributions to this study and to this manuscript.
This work was supported by the following sources: RO1 NS40500 (GFR), F31 NS076202-01A1 (SEC), the F. M. Kirby Foundation (GFR), and P30CA006927 from the National Cancer Institute.
The project described was supported by Grant Number F31NS076202 from the National Institute Of Neurological Disorders And Stroke. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders And Stroke or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kundig TM, Hengartner H, Zinkernagel RM. T cell-dependent IFN-gamma exerts an antiviral effect in the central nervous system but not in peripheral solid organs. J Immunol. 1993;150(6):2316–21. [PubMed] [Google Scholar]
- 2.Binder GK, Griffin DE. Interferon-gamma-mediated site-specific clearance of alphavirus from CNS neurons. Science. 2001;293(5528):303–6. doi: 10.1126/science.1059742. [DOI] [PubMed] [Google Scholar]
- 3.Dorries R. The role of T-cell-mediated mechanisms in virus infections of the nervous system. Curr Top Microbiol Immunol. 2001;253:219–45. doi: 10.1007/978-3-662-10356-2_11. [DOI] [PubMed] [Google Scholar]
- 4.Griffin DE, Metcalf T. Clearance of virus infection from the CNS. Curr Opin Virol. 2011;1(3):216–21. doi: 10.1016/j.coviro.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith JA, et al. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2012;87(1):10–20. doi: 10.1016/j.brainresbull.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilms H, et al. Inflammation in Parkinson's diseases and other neurodegenerative diseases: cause and therapeutic implications. Curr Pharm Des. 2007;13(18):1925–8. doi: 10.2174/138161207780858429. [DOI] [PubMed] [Google Scholar]
- 7.Johnson RT. Viral Infections of the Nervous System. Philadelphia: Lippincott-Raven; 1998. [Google Scholar]
- 8.Studahl M. Influenza virus and CNS manifestations. J Clin Virol. 2003;28(3):225–32. doi: 10.1016/s1386-6532(03)00119-7. [DOI] [PubMed] [Google Scholar]
- 9.Dubois-Dalcq M, Coblentz JM, Pleet AB. Subacute sclerosing panencephalitis. Unusual nuclear inclusions and lengthy clinical course. Arch Neurol. 1974;31(6):355–63. doi: 10.1001/archneur.1974.00490420021001. [DOI] [PubMed] [Google Scholar]
- 10.James SH, Kimberlin DW, Whitley RJ. Antiviral therapy for herpesvirus central nervous system infections: neonatal herpes simplex virus infection, herpes simplex encephalitis, and congenital cytomegalovirus infection. Antiviral Res. 2009;83(3):207–13. doi: 10.1016/j.antiviral.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stark GR, et al. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–64. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 12.von Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25(3):361–72. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 13.Hibbert L, et al. IL-27 and IFN-alpha signal via Stat1 and Stat3 and induce T-Bet and IL-12Rbeta2 in naive T cells. J Interferon Cytokine Res. 2003;23(9):513–22. doi: 10.1089/10799900360708632. [DOI] [PubMed] [Google Scholar]
- 14.Nishikomori R, et al. Activated Stat4 has an essential role in Th1 differentiation and proliferation that is independent of its role in the maintenance of IL-12R beta 2 chain expression and signaling. J Immunol. 2002;169(8):4388–98. doi: 10.4049/jimmunol.169.8.4388. [DOI] [PubMed] [Google Scholar]
- 15.Curtsinger JM, et al. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. 2005;174(8):4465–9. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 16.Radaeva S, et al. Interferon-alpha activates multiple Stat signals and down-regulates c-Met in primary human hepatocytes. Gastroenterology. 2002;122(4):1020–34. doi: 10.1053/gast.2002.32388. [DOI] [PubMed] [Google Scholar]
- 17.Zurney J, Howard KE, Sherry B. Basal expression levels of IFNar and Jak-Stat components are determinants of cell-type-specific differences in cardiac antiviral responses. J Virol. 2007;81(24):13668–80. doi: 10.1128/JVI.01172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rose RW, et al. Altered levels of Stat1 and Stat3 influence the neuronal response to interferon gamma. J Neuroimmunol. 2007;192(1-2):145–56. doi: 10.1016/j.jneuroim.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rall GF, et al. A transgenic mouse model for measles virus infection of the brain. Proc Natl Acad Sci U S A. 1997;94(9):4659–63. doi: 10.1073/pnas.94.9.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patterson CE, et al. Immune-mediated protection from measles virus-induced central nervous system disease is noncytolytic and gamma interferon dependent. J Virol. 2002;76(9):4497–506. doi: 10.1128/JVI.76.9.4497-4506.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rall GF, Mucke L, Oldstone MB. Consequences of cytotoxic T lymphocyte interaction with major histocompatibility complex class I-expressing neurons in vivo. J Exp Med. 1995;182(5):1201–12. doi: 10.1084/jem.182.5.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banker G, Goslin K. Culturing Nerve Cells. Cambridge: MIT Press; 1991. [Google Scholar]
- 23.Pasick JM, Kalicharran K, Dales S. Distribution and trafficking of JHM coronavirus structural proteins and virions in primary neurons and the OBL-21 neuronal cell line. J Virol. 1994;68(5):2915–28. doi: 10.1128/jvi.68.5.2915-2928.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller U, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264(5167):1918–21. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 25.Sakamoto S, et al. Cells previously desensitized to type 1 interferons display different mechanisms of activation of stat-dependent gene expression from naive cells. J Biol Chem. 2004;279(5):3245–53. doi: 10.1074/jbc.M309631200. [DOI] [PubMed] [Google Scholar]
- 26.Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol. 2011;1(6):519–25. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Detje CN, et al. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J Immunol. 2009;182(4):2297–304. doi: 10.4049/jimmunol.0800596. [DOI] [PubMed] [Google Scholar]
- 28.Ramana CV, et al. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23(2):96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 29.Qing Y, Stark GR. Alternative activation of Stat1 and Stat3 in response to interferon-gamma. J Biol Chem. 2004;279(40):41679–85. doi: 10.1074/jbc.M406413200. [DOI] [PubMed] [Google Scholar]
- 30.Campbell DS, Holt CE. Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron. 2001;32(6):1013–26. doi: 10.1016/s0896-6273(01)00551-7. [DOI] [PubMed] [Google Scholar]
- 31.Kleiman R, Banker G, Steward O. Differential subcellular localization of particular mRNAs in hippocampal neurons in culture. Neuron. 1990;5(6):821–30. doi: 10.1016/0896-6273(90)90341-c. [DOI] [PubMed] [Google Scholar]
- 32.Poon MM, et al. Identification of process-localized mRNAs from cultured rodent hippocampal neurons. J Neurosci. 2006;26(51):13390–9. doi: 10.1523/JNEUROSCI.3432-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eberwine J, et al. Analysis of subcellularly localized mRNAs using in situ hybridization, mRNA amplification, and expression profiling. Neurochem Res. 2002;27(10):1065–77. doi: 10.1023/a:1020956805307. [DOI] [PubMed] [Google Scholar]
- 34.Fritz-French C, Tyor W. Interferon-alpha (IFNalpha) neurotoxicity. Cytokine Growth Factor Rev. 2012;23(1-2):7–14. doi: 10.1016/j.cytogfr.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 35.Sas AR, et al. Interferon-alpha causes neuronal dysfunction in encephalitis. J Neurosci. 2009;29(12):3948–55. doi: 10.1523/JNEUROSCI.5595-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hashioka S, et al. Stat3 inhibitors attenuate interferon-gamma-induced neurotoxicity and inflammatory molecule production by human astrocytes. Neurobiol Dis. 2011;41(2):299–307. doi: 10.1016/j.nbd.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 37.Patten SB. Psychiatric side effects of interferon treatment. Curr Drug Saf. 2006;1(2):143–50. doi: 10.2174/157488606776930562. [DOI] [PubMed] [Google Scholar]
- 38.Mizuno T, et al. Interferon-gamma directly induces neurotoxicity through a neuron specific, calcium-permeable complex of IFN-gamma receptor and AMPA GluR1 receptor. FASEB J. 2008;22(6):1797–806. doi: 10.1096/fj.07-099499. [DOI] [PubMed] [Google Scholar]
- 39.Mehla R, et al. Programming of neurotoxic cofactor CXCL-10 in HIV-1-associated dementia: abrogation of CXCL-10-induced neuro-glial toxicity in vitro by PKC activator. J Neuroinflammation. 2012;9(1):239. doi: 10.1186/1742-2094-9-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Surace MJ, Block ML. Targeting microglia-mediated neurotoxicity: the potential of NOX2 inhibitors. Cell Mol Life Sci. 2012;69(14):2409–27. doi: 10.1007/s00018-012-1015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodgers BC, Mims CA. Role of macrophage activation and interferon in the resistance of alveolar macrophages from infected mice to influenza virus. Infect Immun. 1982;36(3):1154–9. doi: 10.1128/iai.36.3.1154-1159.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soike KF, Kramer MJ, Gerone PJ. In vivo antiviral activity of recombinant type alpha interferon A in monkeys with infections due to simian varicella virus. J Infect Dis. 1983;147(5):933–8. doi: 10.1093/infdis/147.5.933. [DOI] [PubMed] [Google Scholar]
- 43.Carr DJ, et al. RNA-dependent protein kinase is required for alpha-1 interferon transgene-induced resistance to genital herpes simplex virus type 2. J Virol. 2005;79(14):9341–5. doi: 10.1128/JVI.79.14.9341-9345.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ida-Hosonuma M, et al. The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J Virol. 2005;79(7):4460–9. doi: 10.1128/JVI.79.7.4460-4469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samuel MA, Diamond MS. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol. 2005;79(21):13350–61. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horisberger MA, Haller O, Arnheiter H. Interferon-dependent genetic resistance to influenza virus in mice: virus replication in macrophages is inhibited at an early step. J Gen Virol. 1980;50(1):205–10. doi: 10.1099/0022-1317-50-1-205. [DOI] [PubMed] [Google Scholar]
- 47.Brzozka K, Finke S, Conzelmann KK. Identification of the rabies virus alpha/beta interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J Virol. 2005;79(12):7673–81. doi: 10.1128/JVI.79.12.7673-7681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mrkic B, et al. Measles virus spread and pathogenesis in genetically modified mice. J Virol. 1998;72(9):7420–7. doi: 10.1128/jvi.72.9.7420-7427.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kalinke U, Prinz M. Endogenous, or therapeutically induced, type I interferon responses differentially modulate Th1/Th17-mediated autoimmunity in the CNS. Immunol Cell Biol. 2012;90(5):505–9. doi: 10.1038/icb.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fensterl V, et al. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog. 2012;8(5):e1002712. doi: 10.1371/journal.ppat.1002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kieseier BC. The mechanism of action of interferon-beta in relapsing multiple sclerosis. CNS Drugs. 2011;25(6):491–502. doi: 10.2165/11591110-000000000-00000. [DOI] [PubMed] [Google Scholar]