

Abstract
Small molecule drugs are the foundation of modern medical practice yet their use is limited by the onset of unexpected and severe adverse events (AEs). Regulatory agencies rely on post-marketing surveillance to monitor safety once drugs are approved for clinical use. Despite advances in pharmacovigilance methods that address issues of confounding bias, clinical data of AEs are inherently noisy. Systems pharmacology– the integration of systems biology and chemical genomics – can illuminate drug mechanisms of action. We hypothesize that these data can improve drug safety surveillance by highlighting drugs with a mechanistic connection to the target phenotype (enriching true positives) and filtering those that do not (depleting false positives). We present an algorithm, the modular assembly of drug safety subnetworks (MADSS), to combine systems pharmacology and pharmacovigilance data and significantly improve drug safety monitoring for four clinically relevant adverse drug reactions.
Introduction
Small molecule drugs are essential in modern medical practice. However, all drugs have the potential to cause severe side effects and even the most efficacious drugs can turn out to be dangerous (e.g. Vioxx, Avandia).1,2 Indeed, one of the primary reasons drugs fail during clinical trials is that they are found to cause adverse events (AEs).3 While clinical trials aim to address drug safety issues, their inherent limitations (including number of patients, duration of study, and homogeneity of the study population) lead to new AEs often being discovered only after a drug has been approved.4,5 The FDA relies on pharmacovigilance methods to monitor drug safety in the post-marketing phase. These methods primarily rely on spontaneous reporting systems (SRSs), such as the FDA Adverse Event Reporting System (FAERS), that collect voluntary submissions from healthcare providers and patients as well as mandatory submissions from pharmaceutical companies. However, because these data are passive collections of events their use is limited in cases where reporting lags behind safety events. Interest has shifted to Medicare claims data (e.g. Observational Medical Outcomes Partnership) and the electronic health records (e.g. FDA’s Mini-Sentinel) where adverse drug events may potentially be detected in near real time.
Multiple quantitative signal detection algorithms have been developed to mine observational health data for adverse drug events.6,7 These methods are primarily based on disproportionality analysis, wherein a ratio of the observed occurrence of a drug-AE combination to the expected occurrence for other drugs is calculated to quantify the combination’s “unexpectedness”.8 In spite of the utility of these methods, they suffer from known limitations due to both sampling variance (e.g. under- or over-reporting of events depending on how established the drug-event relationship is) and reporting biases (such as reporting disease symptoms as adverse events).8,9 Pharmacovigilance methods, such as the multi-item gamma Poisson shrinker (MGPS) currently used by the FDA, correct for sampling variance by estimating confidence intervals for the disproportionality statistics to dampen unsubstantiated drug-event signals.10,11 High-dimensional propensity scoring techniques5 and self-controlled case series12 have been developed to address issues of reporting biases. Both of these methods work by defining a well-matched set of controls. Despite these advances, however, pharmacovigilance methods continue to suffer from both high false positive and false negative rates.7,8,10
These persistent limitations suggest that biological data regarding a drug’s targeted proteins and pathways may represent a complementary avenue for predicting drug safety. In addition, it has become increasingly apparent that the traditional pharmacological paradigm of “one drug one target” has broken down,13 with off-target, unknown interactions leading to unintended consequences. It is imperative, therefore, to investigate drug effects in a more holistic context.14
Systems pharmacology (also referred to as chemical systems biology) is an emerging field integrating physiological, biochemical, genomic, and chemical data to analyze drug actions and side effects in the context of the molecular interactions in the cell (the “interactome”).15 For example, chemical data (e.g. a drug’s chemical structure) and biological data (e.g. a drug’s protein targets) were recently integrated to explore common mechanisms of adverse events.16 To do so the authors looked for common chemical substructures or protein features across drugs or their targets for a subset of drugs known to cause a given side effect. A typical approach in systems pharmacology is to convert these data to a “network” consisting of nodes and edges. Nodes represent biological entities, such as proteins or small molecules, and edges represent relationships between these entities, such as protein-protein interactions or drug-target binding affinities. This representation enables the application of graph theory – a mature sub-domain of mathematics – to systems pharmacology data.14 Graph theoretic approaches were used to discover that proteins commonly form highly intra-connected sub-networks called modules according to shared biological function.14 These modules have been used to identify pathways that mediate the therapeutic and adverse effects of drugs17,18 and to predict previously unknown AEs.19 While systems pharmacology data has been used in specific cases to predict drug side effects, it has not yet been established that integrating these approaches would improve pharmacovigilance.
We hypothesized that incorporating systems pharmacology data into drug safety surveillance would improve pharmacovigilance by reducing the rate of false positives while simultaneously enriching for true positives. Here we present a method called the Modular Assembly of Drug Safety Subnetworks (MADSS). For a given adverse event, MADSS integrates multiple chemical and biological data sources into a common network and identifies a module, which we refer to as an “AE neighborhood,” representing a putative AE mechanistic pathway. The AE-module and the network are then used to evaluate each drug for its potential relationship to the AE. Our hypothesis is that drugs targeting proteins in this “AE neighborhood” are more likely to cause the AE. A recent “medication-wide association study” (MWAS) performed a multivariate analysis to generate pharmacovigilance statistics for four clinically important adverse events - upper gastrointestinal bleeding, acute liver failure, acute myocardial infarction, and acute kidney failure - but was unable to eliminate many false positives and false negatives.20 We validate MADSS by showing that the combination of systems pharmacology models and MWAS statistics leads to significant improvements in safety predictions for all four AEs individually and combined compared to MWAS alone. We evaluate multiple network analysis parameters to demonstrate the flexibility of the method in evaluating either an individual or combination of AEs. Finally, using the chemical and biological data integrated in MADSS we investigate the potential mechanisms of drug AEs and show that multiple drug classes can act through shared functional clusters to elicit AEs.
Results
We used the Modular Assembly of Drug Safety Subnetworks (MADSS) algorithm (Figure 1) to integrate human protein-protein interaction (PPI) data from the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING)21 with genetic data for four adverse events: gastrointestinal bleeding (GI), acute liver failure (LF), acute myocardial infarction (MI), and acute kidney failure (KF). The PPI network from STRING contains 13,926 proteins (nodes) and 217,823 interactions (edges) derived from physical interaction experiments, co-expression data, literature co-mentions, and molecular pathway databases. We curated a set of proteins with primary data linking them to each of the four conditions irrespective of drug involvement (8, 58, 320, and 41 genes for GI, LF, MI, and KF, respectively; see Materials and Methods). We pared down these protein sets to ensure high interconnectivity (Materials and Methods), resulting in 8, 15, 10, and 35 proteins in our “AE seed sets” for GI, LF, MI, and KF, respectively (Supplementary Table S1).
Figure 1.

Overview of Modular Assembly of Drug Safety Subnetworks (MADSS). Orange boxes indicate data sources used in this analysis. Gray boxes indicate additional data sources not used in this study but supported by the method. Beginning with a human protein-protein interaction network (interactome) built from such data as experimental evidence, metabolic pathway databases, text mining, and interactions predicted from co-expression data, we isolate all medium-confidence interactions and above. Seed proteins with demonstrated genetic links to the adverse event (AE) are subsequently annotated. We next apply four adapted network analysis functions to score all proteins in the interactome on their connectivity to the seed set. Proteins with high scores embody an AE neighborhood (gray dotted circle); drugs targeting proteins in this subnetwork are predicted to elicit AEs. We assign positive and negative control drugs to their highest-scoring target. We then combine the four AE neighborhoods (one for each pairwise network function) by training a random forest classifier to generate a subnetwork (SubNet) model (red dotted circle). We integrate MWAS and systems pharmacology (SubNet) models using a logistic regression classifier to predict drug safety.
Our hypothesis is that drugs targeting proteins with high connectivity to the seed sets will be more likely to cause the adverse event. We scored all 13,926 proteins for their connectivity to each AE seed set using four functions (Supplementary Figure S1; see Materials and Methods). We then used a reference standard containing a total of 149 positive and negative control drugs for each of the four adverse events (GI, LF, MI, and KF) created by combining literature review and natural language processing of product labels.20 Of 149 total drugs, there were 77, 95, 79, and 53 controls for GI, LF, MI, and KF, respectively.
We assigned each drug the score of its most highly connected target protein that was itself not a seed protein (Supplementary Figure S2). This results in a dataset with each drug being represented by four scores of connectivity functions. We used these connectivity scores to train two machine learning algorithms, logistic regression and random forests. This results in four drug safety subnetwork (SubNet) models (one for each AE). In addition, we also grouped all AEs together to build a global model of adverse effects (Materials and Methods). We found that the random forest algorithm significantly outperformed logistic regression at linking drugs to their known side effects for each of the four events (Supplementary Figure S3).
Improving drug safety predictions using systems pharmacology
We investigated the additive contribution of systems pharmacology models (SubNet) to pharmacovigilance statistics (MWAS) in predicting drug safety. We found that, individually, both MWAS (β=0.79 ±0.18, P = 1.05e−5) and SubNet (β=4.34 ±0.58, P = 7.42e−14) were significant predictors of adverse events. In addition, we found the combined model outperformed the univariate models (χ2= 75.9, P< 1 ×10−15). The Area Under the Receiver Operating Characteristic Curve (AUROC) is the probability of successfully differentiating two randomly chosen drugs, one that causes the AE and one that does not. For the combined model we found an AUROC of 0.85 compared to 0.81 and 0.69 for SubNet-alone and MWAS-alone, respectively (Figure 2A). In addition to outperforming overall, the combined model also outperformed for each adverse event individually (Figure 2B) with improvements in AUROC of 6.2% (P = 0.10), 33.9% (P = 0.047), 15.3% (P = 0.01), and 27.3% (P = 0.007) for GI, LF, MI, and KF, respectively. We found these results are reliable across different performance measures (Figure 2C). Finally, we observed improvements in both sensitivity and specificity in MWAS+SubNet compared to MWAS and SubNet alone for each AE (Table 1, Supplementary Table S4). At a false positive rate of 20%, sensitivity (i.e. recall) improves from 42%, for MWAS alone, to 70% when drug safety statistics are combined with systems pharmacology data.
Figure 2.
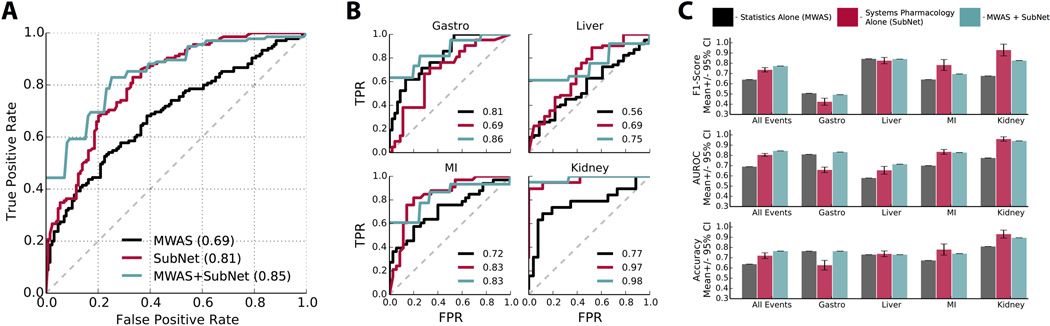
Systems pharmacology data significantly improve drug safety predictions. (A) Receiver operating characteristic (ROC) curve showing performance of pharmacovigilance statistics (MWAS) alone, systems pharmacology (SubNet) alone, and MWAS+SubNet for four adverse events (AEs) combined. The true positive rate, or sensitivity, is plotted against the false positive rate, or 1-specificity. Area under the ROC curve (AUROC) is indicated in parentheses; an AUROC of 0.50 is equivalent to random classification and 1 represents perfect classification. MWAS+SubNet performs significantly better than MWAS alone. (B) ROC curves demonstrating performance for individual AEs: gastrointestinal bleeding (Gastro), acute liver failure (Liver), acute myocardial infarction (MI), and acute kidney failure (Kidney). AUROCs for MWAS alone (black), SubNet alone (red), and MWAS+SubNet (green) are indicated. (C) Quantification of classifier performance using the commonly applied metrics of F1 score (measuring classifier precision and recall), AUROC, and accuracy.
Table 1.
Comparison of sensitivity (true positive rate, TPR) and specificity (true negative rate, TNR) for drugs receiving high MWAS+SubNet (Both) scores across all four adverse events (AEs). GI: gastrointestinal bleeding; LF: acute liver failure; MI: acute myocardial infarction; KF: acute kidney failure.
| Drug | AE Caused |
MWAS Sensitivity |
SubNet Sensitivity |
Both Sensitivity |
MWAS Specificity |
SubNet Specificity |
Both Specificity |
|---|---|---|---|---|---|---|---|
| Diflunisal | GI | 62% | 62% | 100% | 87% | 71% | 100% |
| Ibuprofen | GI | 52% | 57% | 100% | 91% | 71% | 100% |
| Flurbiprofen | GI | 10% | 33% | 100% | 100% | 89% | 100% |
| Indomethacin | GI | 24% | 29% | 50% | 98% | 89% | 100% |
| Oxaprozin | GI | 19% | 19% | 50% | 100% | 89% | 100% |
| Lamotrigine | LF | 87% | 87% | 100% | 13% | 48% | 100% |
| Nevirapine | LF | 39% | 90% | 100% | 78% | 39% | 100% |
| Ofloxacin | LF | 66% | 79% | 83% | 39% | 52% | 100% |
| Stavudine | LF | 58% | 66% | 83% | 52% | 61% | 100% |
| Acetazolamide | LF | 52% | 68% | 71% | 52% | 61% | 100% |
| Desipramine | MI | 70% | 85% | 100% | 60% | 74% | 100% |
| Darbepoetinalfa | MI | 49% | 73% | 100% | 80% | 86% | 100% |
| Estradiol | MI | 67% | 52% | 75% | 60% | 89% | 100% |
| Frovatriptan | MI | 42% | 64% | 75% | 89% | 86% | 100% |
| Imipramine | MI | 64% | 58% | 67% | 71% | 89% | 100% |
| Captopril | KF | 84% | 84% | 100% | 35% | 100% | 100% |
| Cyclosporine | KF | 63% | 90% | 100% | 92% | 100% | 100% |
| Lisinopril | KF | 47% | 79% | 100% | 92% | 100% | 100% |
| Etodolac | KF | 37% | 32% | 100% | 92% | 100% | 100% |
| Hydrochlorothiazide | KF | 5% | 95% | 100% | 100% | 89% | 100% |
Evaluating choice of model parameters
MADSS has many parameters (network connectivity function, validated versus predicted drug targets, PPI confidence, PPI relationship, PPI data source, and drug target type) (Figure 1). We evaluated the effects of each of these parameters on model performance for each AE while holding all other parameters constant (Supplementary Material, Supplementary Figure S4). We found the best performance when using all PPIs of “medium” confidence and above and all known drug targets (see Supplementary Material).
Exploring mechanisms of adverse events
To interrogate potential mechanisms of adverse events, we investigated the interactions between high-scoring drug targets and seed proteins. While some drug targets were direct neighbors of seeds, others were linked to seed proteins through intermediates. We calculated the shortest paths from high-scoring drug targets to all seeds and mapped the AE neighborhood by clustering drugs, drug targets, and intermediate proteins into functional groups (Figure 3, Supplementary Figures S5, S6, S7). The AE neighborhood for MI is enriched for drug targets involved in G-protein signaling coupled to cyclic nucleotide second messengers (P = 3.4E−7) (HTR1A, HTR1B, ADRB2, CHRM2, and DRD2; see Figure 3). Intermediate proteins connecting targets to MI seeds are involved in cAMP biosynthesis (P = 1.9E−17) and inflammatory responses (P = 7.2E−17). Seeds integrating the greatest number of shortest paths were the beta-1 adrenergic receptor (ADRB1) and interleukin 6 (IL6) (61 and 52 shortest paths (sp), respectively).
Figure 3.

Network flow representation of acute myocardial infarction AE neighborhood. Red triangles represent drug classes. Blue nodes with red borders are high-scoring drug targets; red nodes are seed proteins. Blue nodes in the center represent intermediates linking drug targets to seeds. Intermediate node size and edge thickness are representative of the number of shortest paths traveling through them. The AE neighborhood for MI constructed using MADSS is enriched for genes involved in cAMP biosynthesis and inflammatory response.
We found enrichment for cytochrome P450 enzymes for both GI and LF high-scoring targets (P = 2.4E−7 and P = 5.5E−4, respectively) (Supplementary Figures S5 and S6). Intermediates in the AE neighborhood for GI were enriched for proteins involved in arachidonic acid metabolism (P = 4.3E−37), and the seeds integrating the majority of shortest paths were COX-1 and COX-2 (PTGS1 (97 sp) and PTGS2 (108 sp), respectively). In the AE neighborhood for LF, we observed enrichment of intermediates involved in cellular calcium ion homeostasis (P = 2.8E−27), with the seeds liver activation regulated chemokine (CCL20, 115 sp) and corticotropin (POMC, 124 sp) integrating most of the shortest paths. Lastly, high-scoring drug targets within the AE neighborhood for KF were enriched for proteins involved in blood pressure regulation (P = 1.3E−5) (Supplementary Figure S7). Intermediates consisted mainly of organic ion transporters (P = 1.4E−3), with renin (REN, 6 sp), albumin (ALB, 4 sp), kinogen-1 (KNG1, 4 sp), and hepatocyte growth factor (HGF, 4 sp) acting as the most highly integrative seeds.
Discussion
Drug safety surveillance resources (e.g. spontaneous adverse event reports and electronic health records) suffer from issues of confounding bias, noise, and missing data. These challenges limit the usefulness of pharmacovigilance algorithms because they lead to inadvertently flagging false signals and hiding true ones. We reasoned that incorporation of an approach never exposed to these biases –systems pharmacology models of the molecular connections between drugs and AEs – could help alleviate these shortcomings. Furthermore, with ever-growing resources of interaction data on the horizon,22 a mechanistically driven method that scales to large and diverse data sets has the potential to shape the drug safety landscape. We demonstrate this potential by successfully combining our systems pharmacology approach (MADSS) with traditional pharmacovigilance statistics to significantly improve the prediction of four serious adverse drug events.
High-scoring targets are biologically relevant mediators of adverse events
Many of the high-scoring drug targets in our AE neighborhoods received support in the literature for their involvement in mediating adverse events. For example, decreases in prostacyclin synthesis due to COX-2 (PTGS2) inhibition is well-known to increase risk of myocardial infarction.23 Serotonin has also been implicated in coronary artery disease,24 a major cause of myocardial infarction,25 and serotonin 5-HT1B receptors (HTR1B) mediate coronary vasoconstriction.26 HTR1A and HTR1B ranked 245 and 269 (top 2%), respectively in the AE neighborhood for MI constructed using mean first passage time (MFPT); activation of another serotonin receptor (HTR2A, rank 379) is known to increase synthesis of interleukin-6 (IL6, one of the MI seeds) in vascular smooth muscle, contributing to pro-inflammatory pathways.27
Other high-scoring targets such as RXRA suggest less-characterized potential mechanisms of adverse events. Etodolac, a non-steroidal anti-inflammatory drug (NSAID), binds to the retinoid X receptor-α (RXRα), leading to its degradation.28 Complexes of peroxisome proliferator-activated receptors (PPARs) and RXRα are thought to play anti-inflammatory and anti-atherogenic roles in coronary artery disease.29 Additionally, decreased expression of RXRαhas been associated with more pronounced carotid atherosclerotic disease progression;30 carotid atherosclerosis has been shown to be predictive of future MI.31
AE neighborhood intermediates are important transducers of drug action
Pathways enriched in our AE neighborhoods are also supported in the literature. For MI, inflammatory pathways are a key element of coronary artery disease progression.32 The role of cAMP biosynthesis by adenylyl cyclase downstream of beta adrenergic receptor activation during heart failure is also well-established.33 In the AE neighborhood for GI, genes related to arachidonic acid metabolism have long been recognized to be involved in NSAID-induced gastric bleeding.34 Additionally, perturbations to calcium ion homeostasis have been implicated as a mechanism of drug-induced liver injury.35
Limitations of method
There are several existing limitations to the method described. First, the method is dependent on the existence of a seed set of proteins that are, ideally, causally linked to the adverse event. Except for a few instances, such genes are largely unavailable for adverse events. To address this issue in this study we used genes linked to the 4 conditions (i.e., gastrointestinal bleeding, acute liver failure, acute myocardial infarction, and acute kidney failure) irrespective of drug association. In addition, we currently assume a single mechanism of action for each AE. Off-target screening and quantitative structure-activity relationship (QSAR) models would complement our approach in cases when drug target information is lacking or absent.
The results of our model evaluation showed that optimization across multiple AEs occasionally involved making sacrifices to the quality of an individual AE’s predictions. For example, we observed better performance for MI alone when only protein-protein interactions related to expression were used. Additionally, the performance for GI was lower than for the other three AEs; allowing the use of seeds for drug scoring led to an increase in performance for GI with no comparable improvement for the other AEs, suggesting that more drugs in the GI reference standard (such as non-steroidal anti-inflammatory agents) act mechanistically through the seeds than through alternative targets (Supplementary Figure S2). Future applications of MADSS can benefit from the flexibility of optimizing across multiple or individual AEs.
We were unable to completely eliminate incidence of false positives using MADSS, although we observed improvements in both sensitivity and specificity compared to MWAS for each AE (Table 1, Supplementary Table S4). In addition, the performance of our approach may suffer when there is little known about a drug, as is the case when a drug is “first-in-class.” We evaluated the performance under this situation (Supplementary Material) and found that MADSS was still able to eliminate many of the false positives found using traditional drug safety statistics (Supplementary Table S5).
In addition to predicting single drug adverse events, systems pharmacology is also poised to help identify drug-drug interactions (DDIs).36–38 Future iterations of MADSS could combine network-based predictions of DDIs with statistical predictions.5
In conclusion, we present a new method leveraging protein-protein interaction network analysis to improve pharmacovigilance. The method is by design modular, allowing for the incorporation of diverse data sets and optimization for the desired adverse event(s). While we elected to use the self-controlled case series statistics used in the MWAS study in combination with SubNet, an individual or regulatory agency using MADSS can easily substitute any desired pharmacovigilance statistic. In addition, seed sets for new adverse events can be generated with minimal input, and new drugs can quickly be evaluated using predicted targets. Regulatory agencies can flag signals enriched using our method for follow-up study. We demonstrate that combining systems pharmacology models with pharmacovigilance leads to significant and meaningful improvements in predicting drug safety.
Materials and Methods
Modular Assembly of Drug Safety Subnetworks (MADSS)
An outline of MADSS (Modular Assembly of Drug Safety Subnetworks) can be found in Figure 1. We applied MADSS to four pathological conditions, which may be drug-induced adverse events (AEs) –upper gastrointestinal bleeding (GI), acute liver failure (LF), acute myocardial infarction (MI), and acute kidney failure (KF).
Pruning network
We first pruned an initial protein-protein interaction (PPI) network representative of a composite human cell to eliminate low-confidence interactions. We used STRING 9.1 (Search Tool for the Retrieval of Interacting Genes/Proteins), a meta-database of PPIs including data from BioGRID, MINT, KEGG, HPRD, and Reactome, as well as co-occurrence and natural language processing text mining.21 Beginning with all PPIs in humans, we pruned the network to only utilize PPIs with a confidence score ≥700 (out of a maximum score of 1000; see Supplementary Material). We refer to this pruned network as the interactome.
Assigning adverse event (AE) seeds
We identified a small set of proteins with established relationships to each of these four conditions and annotated these proteins as AE seeds. In general, the molecular etiology of adverse events is not well understood. Therefore, we curated initial lists of seed proteins by manually mining GeneCards39 and PubMed for gene or protein names and aliases co-occurring with these four conditions irrespective of drug involvement (Supplementary Table S2). We then refined these lists to a core set of interconnected proteins which we call the “AE seed set” (see Supplementary Material).
Building AE neighborhoods
Multiple metrics exist for characterizing the connectivity between two nodes in a given network. These include mean first passage time (which measures the proximity of two nodes); betweenness centrality (the fraction of shortest paths containing a node of interest); shared neighbors (the fraction of shared adjacent nodes between two nodes of interest); and inverse shortest path (the smallest number of edges connecting two nodes). We adapted each of these functions to score proteins in the interactome on the basis of their connectivity to the seed proteins. Proteins receiving high connectivity scores are on average more connected to the seeds than to the rest of the network and thus constitute a subnetwork of the global interactome which we call an AE neighborhood. See Supplementary Material for equations and descriptions of each of the four connectivity functions.
Connecting drugs to their targets
We downloaded DrugBank 3.0, a manually annotated database connecting 1691 approved drugs to 2074 protein targets.40 Only those drugs in the reference standard which could be mapped to DrugBank IDs (143 of 149) were used in this analysis. For a given adverse event and reference standard drug, we compared the connectivity scores of each protein target and assigned the drug to its highest scoring non-seed target. The result of this is a data matrix for each adverse event where each row represents a drug and each column represents one of the four connectivity functions. The value at a given row and column is the connectivity score for the given drug’s highest scoring target (Supplementary Table S3).
Fitting subnetwork model (SubNet) and predicting drug safety
For each adverse event, we trained a random forest (RF) classifier using the drug scores for the four network functions as input parameters (or “features”). The classifier uses patterns within these features to predict whether a given drug will or will not cause an AE. We utilized out-of-bag (OOB) scores to get an unbiased estimate of classifier performance (for a primer on machine learning approaches, see Supplementary Material). To account for the stochastic nature of random forests we used the mean and standard deviation of 20 replicates to characterize performance. We refer to these systems pharmacology subnetwork models as SubNet. SubNet defines a network link between a drug, target proteins, AE neighborhood proteins, and AE.
To determine the combined performance of SubNet models and pharmacovigilance statistics (MWAS), we trained a logistic regression classifier on the predictions (OOB estimates) from the final SubNet models and the MWAS drug scores as features. We characterized the generalization error using mean and standard deviation 10-fold cross-validation (MWAS+SubNet). We used an analysis of variance (ANOVA) to determine the additive contribution of the systems pharmacology models (SubNet) with the statistical pharmacovigilance methods (MWAS).
Evaluating the subnetwork (SubNet) models
We performed two sets of evaluations: (i) evaluating the overall performance combining all four adverse events and (ii) evaluating the performance for each of the four adverse events independently. In either case we generated receiver operating characteristic (ROC) curves for MWAS alone, SubNet alone, and then for MWAS and SubNet combined. In addition we also calculated the mean and 95% confidence intervals for area under ROC curve (AUROC), F1 score (a measure combining both precision and recall), and accuracy. We evaluated “first-in-class” performance using drugs in the reference standard which were alone in their Anatomical Therapeutic Chemical (ATC) level 2 classification category (Supplementary Table S5). All evaluations were conducted using estimates derived from OOB predictions or cross-validation.
Identifying shared mechanisms of adverse events
For each adverse event, we calculated shortest paths through our derived systems pharmacology AE neighborhoods to each of the seeds from the drug targets receiving high mean first passage time connectivity scores. We manually grouped intermediate proteins (those between drug targets and AE seed proteins) by their connectivity profiles in this network and performed enrichment analysis to assign functional labels. We consolidated edges from multiple targets to the same intermediate or seed. We weighted edges between a seed and the adverse event by the number of shortest paths that were integrated by that particular seed. All intermediate proteins involved in shortest paths from targets to seeds were used to generate the representation in Figure 3.
Implementation
All scripts were written in Python 2.7.5. Network analysis was performed using custom scripts and NetworkX.41 Machine learning was performed using Scikit-learn.42 Logistic regression and ANOVA were performed using glm in R 3.0.3. AUROC comparison (DeLong’s test) was performed using pROC in R 3.0.3.43 Enrichment analysis was performed using DAVID Functional Annotation.44 Network representations were created using Cytoscape 3.0.2 and 3.1.0.45
Supplementary Material
Study Highlights.
• What is the current knowledge on the topic?
State-of-the-art pharmacovigilance algorithms suffer from high false positive and false negative rates. Systems pharmacology and network analysis offer complementary approaches for integrating biological data towards mechanistic insight of drug action.
• What question did this study address?
Will the combination of systems pharmacology models and pharmacovigilance statistics improve drug safety surveillance?
• What this study adds to our knowledge?
Through the modular assembly of drug safety subnetworks, we demonstrate that systems pharmacology models can be successfully combined with current pharmacovigilance statistics across a range of etiologically diverse adverse events to improve predictive power.
• How this might change clinical pharmacology and therapeutics?
Post-marketing surveillance strategies should incorporate systems pharmacology models to enrich for potentially dangerous candidate drugs for follow-up study, ultimately reducing the health and economic impact of adverse events.
Acknowledgements
TL is supported by the National Heart, Lung, and Blood Institute (T32HL120826). SV and GH are supported by the National Library of Medicine (R01 LM006910). MJK is supported by NIH grants GM93456 and MH099712. TL and NPT are supported by the National Institute of General Medical Sciences (R01GM107145) and a PhRMA Foundation Research Starter Award.
Footnotes
All supplementary materials can also be found at http://tatonettilab.org/sm/madss.
Author Contributions
TL and NPT designed the research. TL, MN, MJK, and SV performed the research. TL and NPT analyzed the data. GH and NPT contributed new reagents/ analytical tools. TL and NPT wrote the manuscript.
The authors declare that they have no conflict of interest.
References
- 1.Graham DJ, et al. Risk of acute myocardial infarction and sudden cardiac death in patients treated with cyclo-oxygenase 2 selective and non-selective non-steroidal anti-inflammatory drugs: nested case-control study. Lancet. 2005;365:475–481. doi: 10.1016/S0140-6736(05)17864-7. [DOI] [PubMed] [Google Scholar]
- 2.Nissen SE, Nissen SE, Wolski K, Wolski K. Effect of Rosiglitazone on the Risk of Myocardial Infarction and Death from Cardiovascular Causes. N. Engl. J. Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 3.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug. Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 4.Berlin JA, et al. Adverse event detection in drug development: recommendations and obligations beyond phase 3. Am. J. Public Health. 2008;98:1366–1371. doi: 10.2105/AJPH.2007.124537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tatonetti NP, Ye PP, Daneshjou R, Altman RB. Data-Driven Prediction of Drug Effects and Interactions. Science Translational Medicine. 2012;4:125ra31–125ra31. doi: 10.1126/scitranslmed.3003377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harpaz R, et al. Novel Data-Mining Methodologies for Adverse Drug Event Discovery and Analysis. Clin. Pharmacol. Ther. 2012;91:1010–1021. doi: 10.1038/clpt.2012.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryan PB, et al. Empirical assessment of methods for risk identification in healthcare data: results from the experiments of the Observational Medical Outcomes Partnership. Statist. Med. 2012;31:4401–4415. doi: 10.1002/sim.5620. [DOI] [PubMed] [Google Scholar]
- 8.Bate A, Evans SJW. Quantitative signal detection using spontaneous ADR reporting. Pharmacoepidem. Drug Safe. 2009;18:427–436. doi: 10.1002/pds.1742. [DOI] [PubMed] [Google Scholar]
- 9.Stephenson WP, Hauben M. Data mining for signals in spontaneous reporting databases: proceed with caution. Pharmacoepidem. Drug Safe. 2007;16:359–365. doi: 10.1002/pds.1323. [DOI] [PubMed] [Google Scholar]
- 10.DuMouchel W. Bayesian data mining in large frequency tables, with an application to the FDA spontaneous reporting system. The American Statistician. 1999;53:177–190. [Google Scholar]
- 11.Szarfman A, Machado SG, O’Neill RT. Use of screening algorithms and computer systems to efficiently signal higher-than-expected combinations of drugs and events in the US FDA’s spontaneous reports database. Drug Saf. 2002;25:381–392. doi: 10.2165/00002018-200225060-00001. [DOI] [PubMed] [Google Scholar]
- 12.Simpson SE. A Positive Event Dependence Model for Self-Controlled Case Series with Applications in Postmarketing Surveillance. BIOM. 2012;69:128–136. doi: 10.1111/j.1541-0420.2012.01795.x. [DOI] [PubMed] [Google Scholar]
- 13.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 14.Jacunski A, Tatonetti NP. Connecting the Dots: Applications of Network Medicine in Pharmacology and Disease. Clin. Pharmacol. Ther. 2013 doi: 10.1038/clpt.2013.168. [DOI] [PubMed] [Google Scholar]
- 15.Berger SI, Iyengar R. Network analyses in systems pharmacology. Bioinformatics. 2009;25:2466–2472. doi: 10.1093/bioinformatics/btp465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duran-Frigola M, Aloy P. Analysis of Chemical and Biological Features Yields Mechanistic Insights into Drug Side Effects. Chemistry & Biology. 2013;20:594–603. doi: 10.1016/j.chembiol.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 17.Berger SI, Ma'ayan A, Iyengar R. Systems Pharmacology of Arrhythmias. Science Signaling. 2010;3:ra30–ra30. doi: 10.1126/scisignal.2000723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gottlieb A, Altman RB. Integrating systems biology sources illuminates drug action. Clin. Pharmacol. Ther. 2014 doi: 10.1038/clpt.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cami A, Arnold A, Manzi S, Reis B. Predicting Adverse Drug Events Using Pharmacological Network Models. Science Translational Medicine. 2011;3:114ra127–114ra127. doi: 10.1126/scitranslmed.3002774. [DOI] [PubMed] [Google Scholar]
- 20.Ryan PB, Madigan D, Stang PE, Schuemie MJ, Hripcsak G. Medication-Wide Association Studies. CPT: Pharmacomet. Syst. Pharmacol. 2013;2:e76. doi: 10.1038/psp.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franceschini A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Research. 2012;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Law V, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Research. 2013;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krotz F, et al. Selective COX-2 inhibitors and risk of myocardial infarction. J. Vasc. Res. 2005;42:312–324. doi: 10.1159/000086459. [DOI] [PubMed] [Google Scholar]
- 24.Vikenes K, Farstad M, Nordrehaug JE. Serotonin Is Associated with Coronary Artery Disease and Cardiac Events. Circulation. 1999;100:483–489. doi: 10.1161/01.cir.100.5.483. [DOI] [PubMed] [Google Scholar]
- 25.Nabel EG, Nabel EG, Braunwald E, Braunwald E. A Tale of Coronary Artery Disease and Myocardial Infarction. N. Engl. J. Med. 2012;366:54–63. doi: 10.1056/NEJMra1112570. [DOI] [PubMed] [Google Scholar]
- 26.Nilsson T, et al. Characterisation of 5-HT receptors in human coronary arteries by molecular and pharmacological techniques. Eur. J. Pharmacol. 1999;372:49–56. doi: 10.1016/s0014-2999(99)00114-4. [DOI] [PubMed] [Google Scholar]
- 27.Ito T, Ikeda U, Shimpo M, Yamamoto K, Shimada K. Serotonin Increases Interleukin-6 Synthesis in Human Vascular Smooth Muscle Cells. Circulation. 2000;102:2522–2527. doi: 10.1161/01.cir.102.20.2522. [DOI] [PubMed] [Google Scholar]
- 28.Kolluri SK, et al. The R-enantiomer of the nonsteroidal antiinflammatory drug etodolac binds retinoid X receptor and induces tumor-selective apoptosis. Proceedings of the National Academy of Sciences. 2005;102:2525–2530. doi: 10.1073/pnas.0409721102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Libby P. Current Concepts of the Pathogenesis of the Acute Coronary Syndromes. Circulation. 2001;104:365–372. doi: 10.1161/01.cir.104.3.365. [DOI] [PubMed] [Google Scholar]
- 30.Giaginis C, et al. Correlation of Peroxisome Proliferator-Activated Receptor-gamma (PPAR-gamma) and Retinoid X Receptor-alpha (RXR-alpha) expression with clinical risk factors in patients with advanced carotid atherosclerosis. Med. Sci. Monit. 2011;17:CR381–CR391. doi: 10.12659/MSM.881849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hald EM, et al. Carotid Atherosclerosis Predicts Future Myocardial Infarction But Not Venous Thromboembolism: The Tromso Study. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;34:226–230. doi: 10.1161/ATVBAHA.113.302162. [DOI] [PubMed] [Google Scholar]
- 32.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 33.Marks AR. Calcium cycling proteins and heart failure: mechanisms and therapeutics. J. Clin. Invest. 2013;123:46–52. doi: 10.1172/JCI62834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langenbach R, et al. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 35.Sturgill MG, Lambert GH. Xenobiotic-induced hepatotoxicity: mechanisms of liver injury and methods of monitoring hepatic function. Clin. Chem. 1997;43:1512–1526. [PubMed] [Google Scholar]
- 36.Zhao X-M, et al. Prediction of Drug Combinations by Integrating Molecular and Pharmacological Data. PLoS Comput. Biol. 2011;7:e1002323. doi: 10.1371/journal.pcbi.1002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y-Y, et al. Exploring drug combinations in genetic interaction network. BMC Bioinformatics. 2012;13:S7. doi: 10.1186/1471-2105-13-S7-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng F, Zhao Z. Machine learning-based prediction of drug-drug interactions by integrating drug phenotypic, therapeutic, chemical, and genomic properties. Journal of the American Medical Informatics Association. 2014 doi: 10.1136/amiajnl-2013-002512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stelzer G, et al. In-silico human genomics with GeneCards. Hum. Genomics. 2011;5:709–717. doi: 10.1186/1479-7364-5-6-709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knox C, et al. DrugBank 3.0: a comprehensive resource for ‘Omics’ research on drugs. Nucleic Acids Research. 2010;39:D1035–D1041. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hagberg A, Swart P, S Chult D. Exploring network structure, dynamics, and function using NetworkX. 2008 [Google Scholar]
- 42.Pedregosa F, et al. Scikit-learn: Machine learning in Python. The Journal of Machine Learning Research. 2011;12:2825–2830. [Google Scholar]
- 43.Robin X, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 45.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.