

Abstract
Naturally occurring guanidine derivatives frequently display medicinally useful properties. Among them, the higher order pyrrole-imidazole alkaloids, the dragmacidins, the crambescidins/batzelladines, and the saxitoxins/tetradotoxins have stimulated the development of many new synthetic methods over the past decades. We provide here an overview of the syntheses of these cyclic guanidine-containing natural products.
Keywords: Natural Products, Alkaloids, Guanidine, Total Synthesis
1. Introduction
Guanidine plays an important role in medicinal chemistry, coordination chemistry, organocatalysis, and biochemistry.1-4 The ability of this highly polar and basic functional group in establishing strong ionic/hydrogen bonding5 and cation-π stacking6,7 interactions with various functional groups has made it particularly useful in the design of small-molecule drugs, metal ligands, and organocatalysts. Additionally, many naturally occurring polycyclic guanidine derivatives possess useful and potent biological properties.8-13 However, the mode of action of many naturally occurring guanidine derivatives is poorly understood because of the synthetic challenges associated with the preparation of their structural derivatives for in-depth biological studies.14-17 Some examples are shown in Fig. 1.
Figure 1.
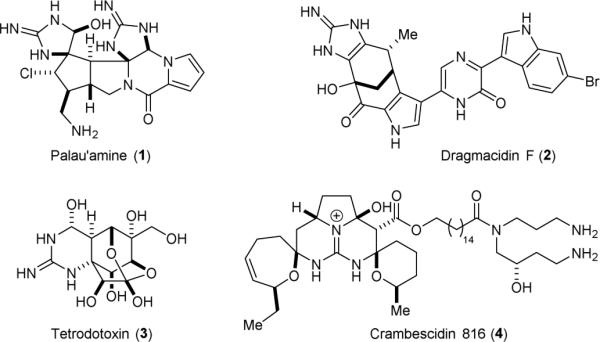
Structures of four representative polycyclic guanidine-containing natural products.
Alkaloids 1-4 each represents a class of natural products that are important to the field of total synthesis. Palau’amine (1) is a highly nitrogenated, noncrystalline, redox labile, and pH sensitive alkaloid.18 Its unusual physical and chemical properties preclude the use of many traditional tools for its synthesis. Dragmacidin F (2) is another polycyclic alkaloid with exceptionally high nitrogen content.19 Its drug-like skeleton encompasses four different heterocycles, including a cyclic guanidine-embedding aminoimidazole ring. This unique molecular architecture provides a valuable platform for the development of new chemical reactions. Tetrodotoxin (3) is a potent neurotoxin well known for its ability to block the sodium ion channels.20 The compact and densely functionalized polycyclic skeleton of 3 makes its synthesis a daunting task. Crambescidin 816 (4) is a selective calcium ion channel inhibitor that is more potent than the FDA-approved hypertension drug nifedipine.21 Its guanidine group is enclosed in a pentacyclic moiety. Several review articles exist highlighting the isolation, structural determination, biogenesis, biological functions, and total syntheses of these alkaloids.22-46 We provide herein an overview of the total syntheses of these four classes of cyclic guanidine-containing natural products.
2. The higher order pyrrole-imidazole alkaloids
The pyrrole-imidazole alkaloids, also known as the oroidin family of alkaloids, are a large group of natural products isolated from a variety of marine sponges.22,29,32,33 Many of these molecules possess anticancer, antimicrobial, antiviral, or immunosuppressive properties. Structurally, oroidin and its debrominated congeners (5) serve as the building blocks of this family of alkaloids in a way similar to isoprene in the world of terpenoids. Oxidation, cyclization, and oligomerization of 5 provide a diverse set of new molecules.47-53 Among the monomeric family members, hymenialdisines (6),54-61 cyclooroidin (7),52,62-64 agelastatins (8),53,65-82 and dibromophakellin/dibromophakellstatin (9)48,83-90 have been the most popular synthetic targets (Fig. 2).22,23,26,29,32
Figure 2.
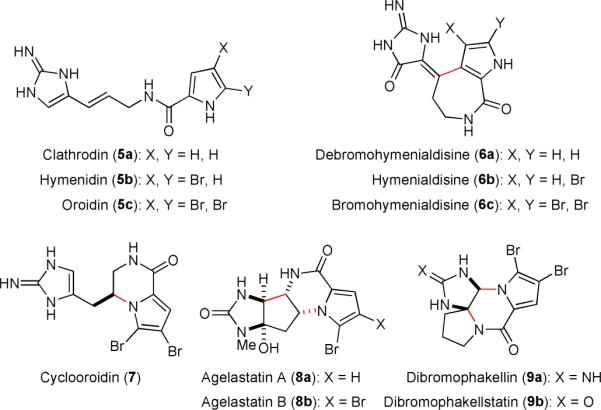
Structures of the basic units of the pyrrole-imidazole alkaloids and some monomeric members.
Several polycyclic pyrrole–imidazole alkaloids deriving from the dimerization of 5 have been found in nature (Fig. 3). With 15–30% nitrogen and 8–42% halogen by weight, these molecules are among the most synthetically challenging natural products. Since the discovery of the first oroidin dimer sceptrin (10a) in 1981, many research groups have devoted to studying the synthesis of these structurally unique alkaloids. Constitutionally, sceptrins (10)91-97 are the [2+2] dimers; ageliferins/nagelamides (11)92,98-100 are the [4+2] dimers; and palau’amines/konbu'acidins (1),18,101-103 stylloguanidines/carteramines (12),104-107 axinellamines (13),108,109 massadines (14),110-112 and donnazoles (15)51 are the [3+2] dimers of 5. Stylissadines (16),113 the only tetrameric member known to date, are the dimers of massadine (14a) and 2-epi-massadine, which is yet to be identified as a natural product itself. The existence of nagelamides E–G (11d–11f), the C10-epimers of ageliferins (11a–11c), and the 2-epi-massadine fragment in 16 indicate that the biogenic dimerization of 5 is not a concerted cycloaddition reaction. Instead, a single-electron oxidation is involved as indicated by Molinski and Romo's metabiosynthetic studies.49,50
Figure 3.

Structures of the dimeric and tetrameric pyrrole-imidazole alkaloids.
The structural determination of palau’amine (1) is quite an interesting story. Kinnel and Scheuer reported in 1993 the isolation of palau’amine, and proposed its structure to be 1’ with a cis-H11/H12 configuration (Fig. 4).18 Although the H11/H12 coupling constant of palau’amine was unusually large for a cis configuration and the NOE/ROESY data could not be reconciled for the C20 aminal stereocenter with 1’,101 they considered that the trans-H11/H12 configured ring system would be too strained to exist. Styloguanidines (12a-c)104 and konbu’acidins (1c,d)102 are believed to have the same stereochemistry as palau’amine. It was not until 2007 that Köck, Quinn, and Matsunaga elucidated the correct structure of palau’amines, konbu’acidins, and styloguanidines.103,105,106,114. Surprisingly, palau’amine embeds a trans-fused 5,5-bicyclic ring system. Notably, although transbicyclo[3.3.0]octane is 6.4 kcal/mol more strained than its cisisomer,115 incorporation of a heteroatom (a nitrogen atom for palau’amine) into the ring system can help relieve the torsional strain.116 We found that ΔΔG(gas)298K (DFT, B3LYP/6-31G*) for the protonated “palau’amines” 1(H+)2 and 1’(H+)2 is only 3.7 kcal/mol although Köck reported that “cis-styloguanidine” is 6.5 kcal/mol more stable than 12a (DFT, B3LYP/6-31G*).25 Interestingly, 1 is not the only natural product bearing a trans-fused 5,5-bicyclic ring system. Epoxydictymene (17) isolated from the brown algae Dictyota dichotoma also possesses a heteroatom-containing trans-fused 5,5 ring system.117 The structure of 17 was determined by X-ray analysis of its dihydroxylated derivative and confirmed by total synthesis by Schreiber118,119 and Paquette120 in 1994 and 1997, respectively.
Figure 4.
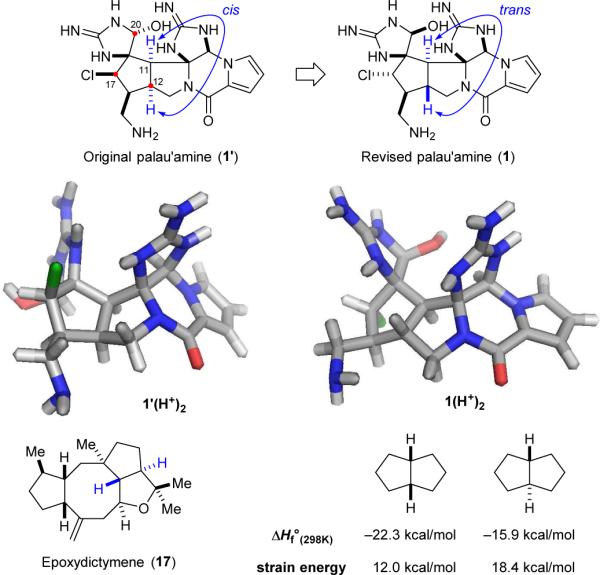
Structures of the originally proposed and revised palau’amine (1’ and 1) and epoxydictymene (17), the equilibrium geometry of 1’(H+)2 and 1(H+)2, and the strain energies of cis- and trans-bicyclo[3.3.0]octane.
2.1. Synthetic contributions from the Overman group
The Overman group reported the use of an azomethine imine cycloaddition reaction to establish the core structure of cis-“palau’amine” (1’) in 1997.121-123 This approach allowed for an efficient synthesis of a highly functionalized cis-palau’amine analog 23 in 2007 (Fig. 5).124 The NMR data of 23, in particular, the H11/H12 coupling constant, are significantly different from those of the natural palau’amine, supporting the then proposed structural revision of palau’amine. The synthesis started with a ring-closing metathesis of 17 to deliver dihydropyrrole 18. Heating an ethanolic solution of 18 with thiosemicarbazide (19) initiated a tandem reaction sequence comprising a condensation of 18 and 19, an intramolecular [3+2] cycloaddition reaction of the resulting 20, and a cyclization of the cycloaddition product 21 to give 22. The intramolecular cycloaddition of 20 was highly stereoselective, giving the 3-azabicyclo[3.3.0]octane molecular framework and the two nitrogen-containing quaternary centers with complete stereochemical control. Cleavage of the N–N linkage of 22 followed by installation of the second iminohydantoin group furnished 23.
Figure 5.
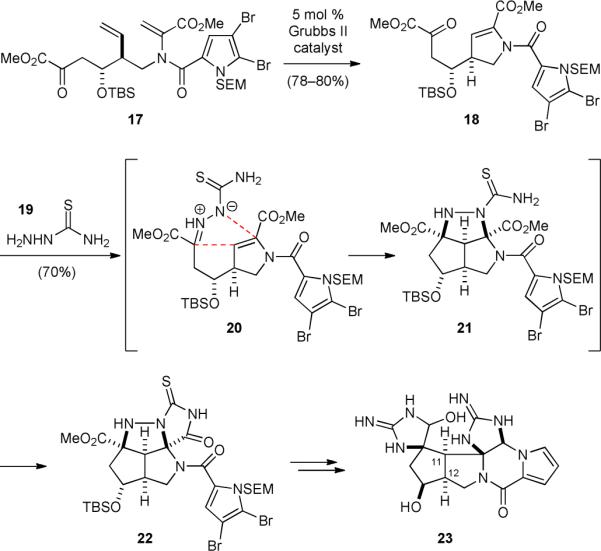
Overman's synthetic approach to “cis-palau’amine” (1’).
2.2. Synthetic contributions from the Carreira group
The first asymmetric synthesis of the core skeleton of the [3+2]-type pyrrole-imidazole dimers was reported by the Carreira group in 2000.125,126 They also successfully constructed a fully functionalized massadine synthetic intermediate in 2011.127,128 Specifically, they prepared ketone 24 from allyldimethylsilyl cyclopentadiene and dimethyl fumarate by a Diels–Alder reaction followed by Tamao oxidation (Fig. 6). Subjecting 24 to an Ugi four-component coupling reaction gave 25. The amide group of 25 was then cleaved through reduction of the nitro group with tin(II) chloride and treatment of the resulting aniline with isoamyl nitrite to give a diazo intermediate that reacted with the amide group to yield an acylbenzotriazole. Subsequent reduction of the acylbenzotriazole group was accompanied with a lactam formation to yield 26. After the N-protecting group switch and amide reduction, the olefin group of 27 was cleaved by ozonolysis to reveal the cyclopentyl core skeleton of massadine. Cleavage of the ozonide in the presence of methanol using the method developed by Schreiber129 gave the terminally differentiated product 28. Lindgren oxidation followed by Barton decarboxylation then yielded 29. Finally, installation of the two guanidine groups provided the fully functionalized massadine core structure 30.
Figure 6.
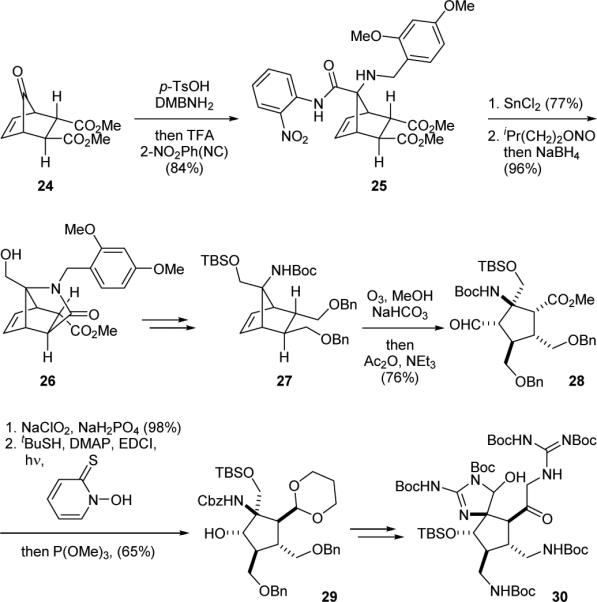
Carreira's synthetic approach to massadine (14a).
2.3. Synthetic contributions from the Austin group
The Austin group used a [2+2+1] cycloaddition approach to construct the cyclopentyl core of palau’amine in 2000.130,131 The intramolecular Pauson–Khand cyclization of enyne 31 gave 32 and 33 as a 4:1 mixture of diastereomers (Fig. 7). Subsequent reduction of the enone group and cleavage of the N–O linkage by single-electron reduction revealed the core skeleton of 1’. Interestingly, without prior saturation of the enone group, reduction of 32 directly with samarium(II) iodide would lead to the cleavage of the enone γ-C–O bond instead of the N–O bond.
Figure 7.
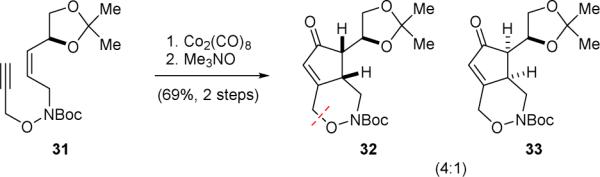
Austin's synthetic approach to “cis-palau’amine” (1’).
2.4. Synthetic contributions from the Romo group
The Romo group has made significant contributions to understanding of the special reactivity of the pyrrole-imidazole family of alkaloids.132-136 They have also provided valuable insights into the biogenesis of both the monomeric53 and the dimeric132 pyrrole-imidazole alkaloids with biomimetic syntheses. Through collaboration with the Molinski group, they further provided evidence that the biogenic dimerization of 5 is promoted by single-electron oxidation.49,50,137
It was generally believed that the [3+2]-type pyrrole-imidazole dimers were derived from the [4+2]-type dimers through an oxidative skeletal rearrangement. For example, the biosynthesis of “palau’amine” (1’) might involve a [4+2] cycloaddition reaction of 3-amino-1-(2-aminoimidazolyl)prop-1-ene (34) and 11,12-dehydrophakellin (35) (Fig. 8) as proposed by Kinnel and Scheuer in 1998.101 A chloroperoxidase-mediated oxidative ring contraction of the resulting 36 would then yield 1’. While attempts to thermally induce the homodimerization of oroidin (5c) failed to generate dibromoageliferin (11c),52 the Romo group showed in 2001 that the ageliferin skeleton could be obtained from a Diels–Alder reaction of a vinylimidazolinone derivative and an electron-deficient dienophile.132 They also demonstrated that the regioselectivity of this [4+2] cycloaddition reaction could be controlled by tuning the electronic properties of the imidazolinone group. For example, heating a solution of 37 bearing a tosylvinyl (Tsv) protecting group with enone 38 gave 39 exclusively (Fig. 9).133 However, hydrogenation of the electron-deficient Tsv group of 38 to an electron-rich tosylethyl (Tse) group before reacting with 39 led to the formation of a 2.5:1 mixture of regioisomers.
Figure 8.
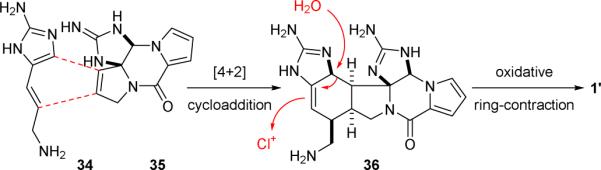
The Kinnel–Scheuer biosynthetic hypothesis.
Figure 9.
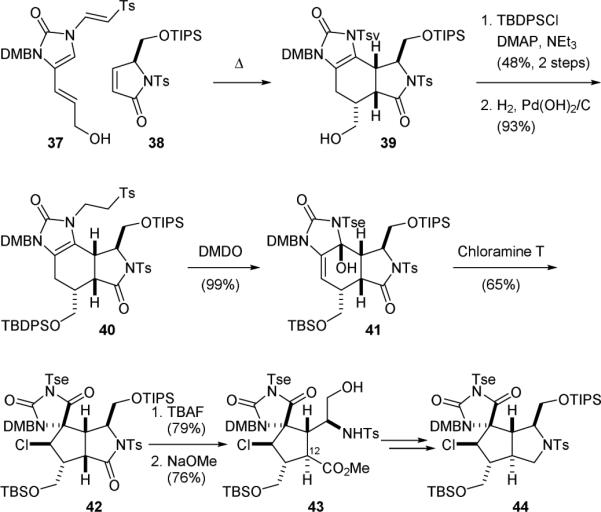
Romo's synthetic approach to palau’amine (1).
To induce the Scheuer oxidative skeletal rearrangement, the hydroxyl group of 39 was protected and the Tsv group was saturated to give the Tse-protected 40. Oxidation of 40 with dimethyldioxirane (DMDO) provided allylic alcohol 41. Subsequent treatment of 41 with chloramine T promoted the Scheuer-type ring contraction reaction to afford 42. Notably, a fine control of the reaction conditions is crucial to the success of this rearrangement reaction. Oxidation of 40 with meta-chloroperoxybenzoic acid (mCPBA) would destroy the ageliferin skeleton. Oxidation of 41 with N-chlorosuccinimide (NCS), or replacing the electron-rich Tse group with the electron-deficient Tsv or Ts group would also lead to aromatization of the six-membered ageliferin core skeleton.
Since the relative stereochemistry of palau’amine was revised in 2007,103,105,106,114 the Romo group has redirected their focus to establishing the trans-fused 5,5-bicyclic molecular framework of 1.136 Removal of the triisopropylsilyl (TIPS) protecting group of 42 followed by cleavage of the lactam group and C12-epimerization provided 43. Subsequent protection of the hydroxyl group, reduction of the ester group, and cyclization under Mitsunobu conditions gave pyrrolidine 44. The coupling constants and observed NOE signals of 44 are consistent with those reported for palau’amine (1), thus providing the first direct experimental support for the trans-H11/H12 configuration for palau’amine.
2.5. Synthetic contributions from the Lovely group
The Lovely group also reported the use of a Diels–Alder approach to construct the core skeleton of ageliferin in 2001.138-140 They found that vinylimidazole could serve as either a diene or a dienophile in the Diels–Alder reactions. Heating a solution of 45 gave the Diels–Alder adduct 46 as the major product (Fig. 10). Interestingly, the inverse electron-demand Diels–Alder reaction product 47 was also obtained. Cleavage of the N–O linkage of 46 by single-electron reduction yielded 48. Subsequent treatment of 48 with the Davis reagent or DMDO induced the Scheuer-type oxidative ring-contraction reaction to afford the palau’amine core 49.
Figure 10.
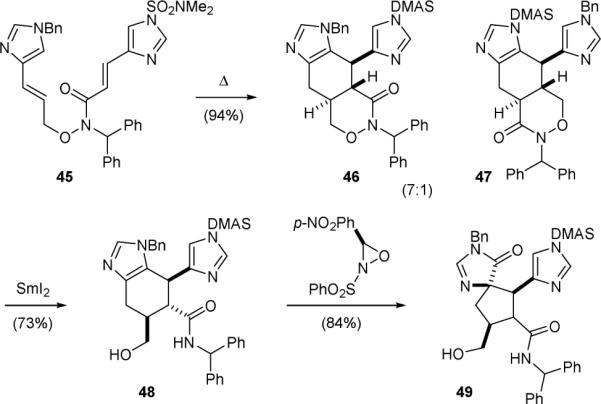
Lovely's synthetic approach to palau’amine (1).
2.6. Synthetic contributions from the Ohta group
The Ohta group has used the Diels-Alder approach to accomplish the biomimetic synthesis of 12,12’-dimethylageliferin in 2002.141,142 Thermolysis of 50 promoted a homodimerization to give a 50:1 mixture of 51 and 52, favoring the exo-Diels-Alder reaction product (endo/exo relative to the ester group) (Fig. 11). Computational studies confirmed that there is a favorable HOMO/LUMO interaction for the homodimerization of 50. The ester groups of 51 were then reduced and the thiophenol groups were cleaved by nickel boride reduction to give 53 after protection of the hydroxyl groups. Lithiation of 53 followed by reaction with trisyl azide yielded 54. The azido groups were reduced and protected as imines to provide 55. Finally, installation of the pyrrole groups and deprotection furnished 12,12’-dimethylageliferin (56). This work is the first synthesis of a fully functionalized dimer pyrrole-imidazole alkaloid.
Figure 11.
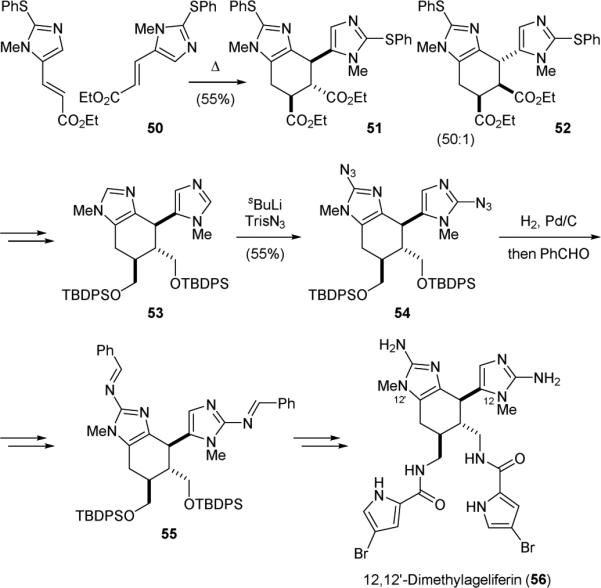
Ohta's synthesis of 12,12’-dimethylageliferin (56).
2.7. Synthetic contributions from the Baran group
The Baran group has completed the total syntheses of all cyclic pyrrole-imidazole dimers. They first achieved the synthesis of sceptrin (10a) and ageliferin (11a) in 2004.143-147 They then accomplished the syntheses of axinellamines (13) and massadines (14) in 2008.148-150 Subsequently, they completed the total synthesis of the legendary natural product palau’amine (1) in 2010.151 In 2011, the Baran group further reported the asymmetric synthesis of 1, 13, and 14 using an enantioselective Diels-Alder reaction to set up the absolute stereochemistry of these alkaloids.152 They also disclosed a practical approach that enables the gram-scale synthesis of 13 in 2011.153
Whereas the [2+2] photocycloaddition would be the most straightforward synthetic strategy for assembling sceptrin (10a), both the Faulkner group and the Baran group found that photolysis of hymenidin (5b) under various conditions failed to deliver the C2 symmetric dimer 10a.91,143 To date, the only successful photo-dimerization of a vinylimidazole derivative was reported by the D'Auria group in 1998.154 Photolysis of allyl urocanate (57) in the presence of the photosensitizer benzophenone gave the head-to-head dimer 58 (Fig. 12). Computational studies suggest that both the HOMO/LSOMO and the HSOMO/LUMO interactions play a role in this reaction. Intriguingly, low stereoselectivity was observed for the photo-dimerization of methyl and ethyl urocanates.
Figure 12.
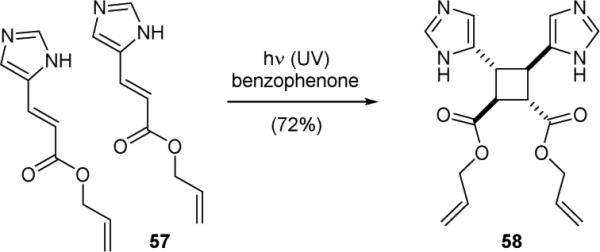
D'Auria's synthetic studies of sceptrin (10a).
Because of the difficulty associated with the photodimerization of 5b, the Baran group used an alternative photocycloaddition approach to construct the cyclobutane ring of 10a. Photolysis of 59 led to a facile [2+2] cycloaddition reaction to give 60 (Fig. 13). An acid-promoted 3-oxaquadricyclane rearrangement reaction143 then occurred to give 62 via cation 61. Subsequent installation of the pyrrole group afforded 63. Chlorination of 63 followed by amination and hydrolysis of the formyl groups gave a bis-α-aminoketone. Subsequent reaction with cyanamide then provided sceptrin (10a). This approach allowed for a gram-scale synthesis of 10a with an impressive 24% overall yield.
Figure 13.
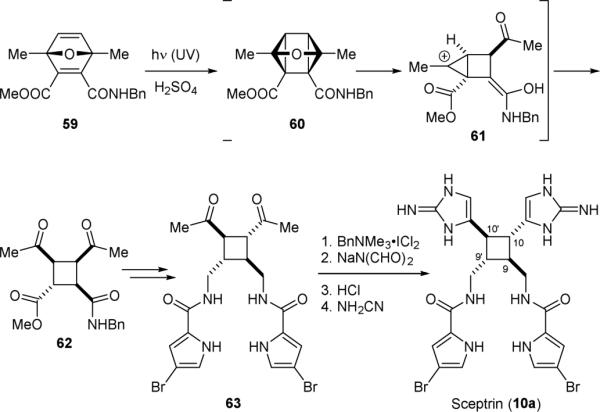
Baran's synthesis of sceptrin (10a).
Based on the observation that sceptrin (10a) and ageliferin (11a) were frequently isolated together in sponges, the Baran group proposed that 11a is derived from 10a through a [1,3]-sigmatropic rearrangement reaction.155,156 Supportive of this hypothesis is the observation that thermolysis of 10a•2HCl in water gave 11a•2HCl, together with a small amount of nagelamide E (11d), the C10 diastereomer of 10a (Fig. 14).144 Computational studies by the Houk group suggest that this rearrangement reaction proceeds through a dicationic diradical intermediate.146
Figure 14.
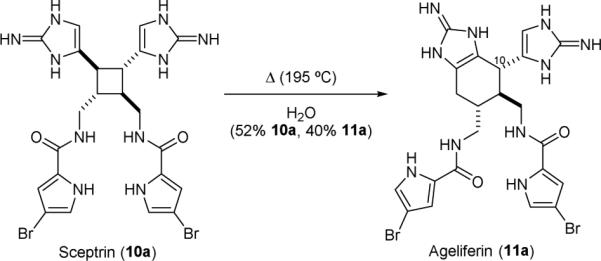
Baran's synthesis of ageliferin (11a).
The Baran group has also accomplished the syntheses of all [3+2]-type pyrrole-imidazole dimers. They first completed the synthesis of axinellamines (13), using an intramolecular aldol reaction to establish its central cyclopentyl core skeleton.148,149 Specifically, ozonolysis of 64 obtained from a Diels-Alder reaction provided diketone 65 (Fig. 15). Bromination of 65 followed by treatment of the resulting bis-α-bromoketone with dry silica gel induced an intramolecular aldol reaction to yield 66. Subsequent installation of the two cyclic guanidine groups and the chlorine atom gave 67. Oxidation of the aminoimidazole group of 67 with DMDO gave diol 68, which cyclized in acid to afford 69. Subsequent oxidation of 69 with silver(II) picolinate occurred selectively at the α-position of the cyclic guanidine group to provide 70. Finally, installation of the pyrrole groups furnished 13.
Figure 15.
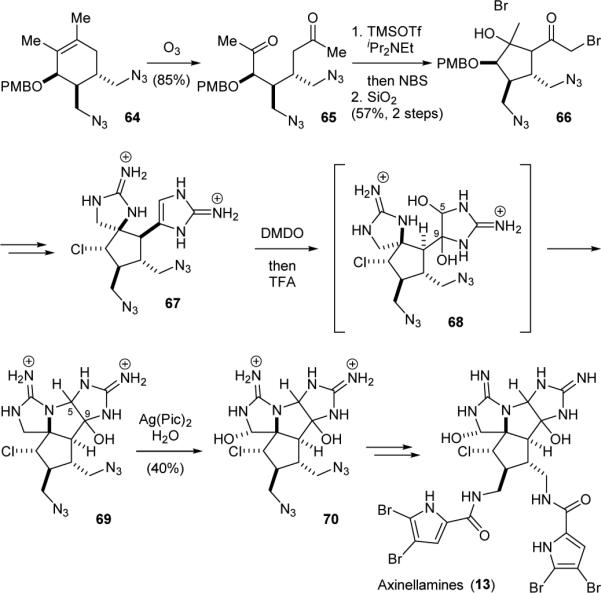
Baran's synthesis of axinellamines (13).
After completing the synthesis of axinellamines (13), the Baran group developed a modified route to enable the synthesis of massadines (14).150 Reaction of α-chloroketone 71, an advanced axinellamine synthetic intermediate, with diformylamide followed by removal of the guanidine protecting groups gave 72 (Fig. 16). Oxidation of 72 with silver(II) picolinate and hydrolysis of the formyl groups afforded α-aminoketone 73. The aminoimidazole group was then introduced by reacting 73 with cyanamide to provide 74. Partial hydrolysis of the chloride yielded 76. Consistent with Romo's previous observation in the study of palau’amine synthesis,135 this reaction proceeded through the intermediacy of aziridinium ion 75 leading to the retention of the C14 configuration. Oxidation of 74/76 with DMDO followed by an acid-promoted cyclization gave a mixture of 77 and 78, favoring the undesired C3,C7-configurations. Finally, installation of the pyrrole groups completed the synthesis of massadines (14). It is believed that 3,7-epi-massadines are also natural products.
Figure 16.
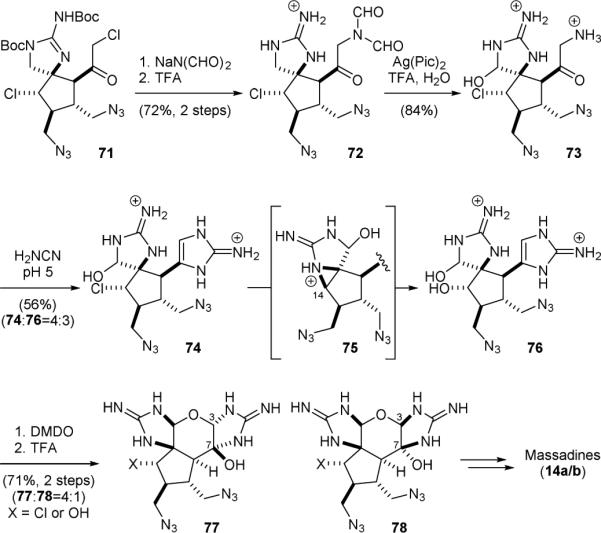
Baran's synthesis of massadines (14).
The Baran group accomplished the synthesis of legendary alkaloid palau’amine (1) in 2010, using an intriguing macrocyclization approach to construct its “phakellin” subunit.151 The strategy of constructing highly strained natural products via macrocyclization was introduced by Kishi for the mitomycin synthesis in 1971.157-160 To synthesize 1, the aminoimidazole group of the massadine synthetic intermediate 74 was brominated to give 79 (Fig. 17). The pyrrole group was then installed through a more nucleophilic acylpyrrole surrogate to give 80. Treatment of 80 with acid promoted the formation of pyrrole and the removal of the tert-butyl ester group to yield 81. Reduction of the azido groups followed by an intramolecular amide coupling gave “macro-palau’amine” (82). Heating 82 in acid induced a smooth transannular cyclization and established the highly strained trans-fused 5,5 bicyclic ring system, furnishing the long-sought target palau’amine (1). In addition to minimizing the use of protecting groups, this elegant synthesis also exploits the concept of redox economy. The asymmetric syntheses of axinellamines (13), massadines (14), and palau’amine (1) were accomplished in 2011 by the development of a catalytic asymmetric Diels-Alder reaction to generate optically active 64.152
Figure 17.
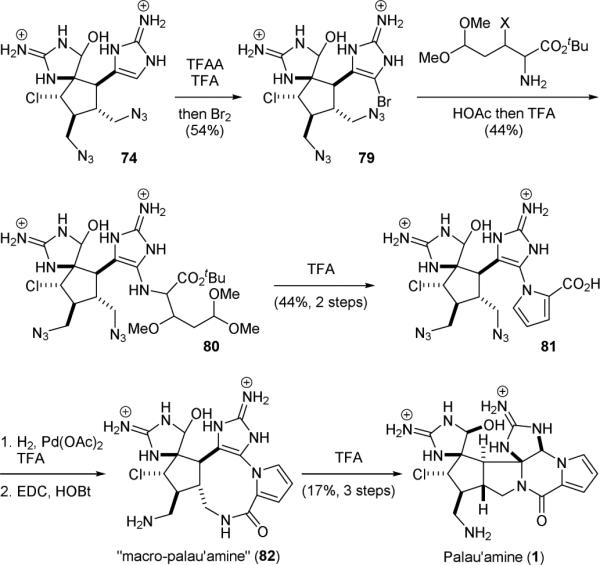
Baran's synthesis of palau’amine (1).
In 2011, the Baran group reported a new synthetic route for achieving the gram-scale synthesis of axinellamines (13).153 The central cyclopentane core was assembled by an intermolecular Pauson–Khand reaction of 83 and 84 (Fig. 18). Subsequent reduction of the resulting enone 85 followed by chlorination gave 86. Next, an interesting In(0)/Zn(0)-promoted Barbier-type reaction was used to deliver the amino alcohol side-chain for constructing the aminoimidazole group. Introduction of the nitrogen functionalities to 87 then yielded 88, which was subjected to a chloroamidation reaction in the presence of a catalytic amount of trifluoromethanesulfonamide (TfNH2) to afford spiro-guanidine 89. Finally, installation of the aminoimidazole group gave 67, an advanced axinellamine synthetic intermediate in their previous approach. Only seven purifications were used for the entire synthesis and over 1 g of 13 was prepared in one run.
Figure 18.
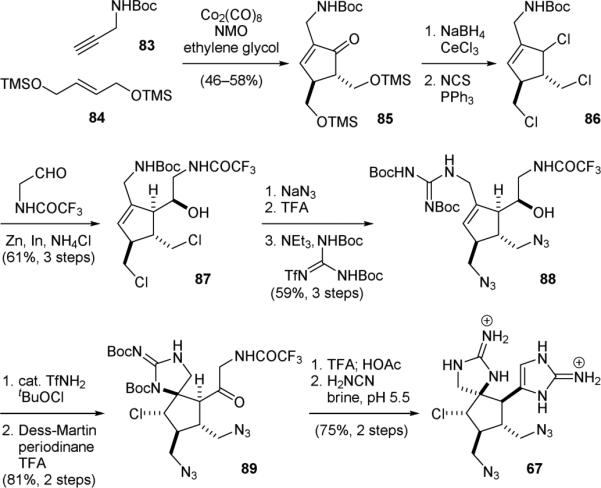
Baran's new synthesis of axinellamines (13).
2.8. Synthetic contributions from the Birman group
The Birman group also accomplished the synthesis of sceptrin (10a) in 2004.161 Both Baran and Birman recognized the symmetry of sceptrin and constructed its core skeleton using a [2+2] photocycloaddition reaction with a non-biogenic disconnection. Photolysis of 90 and 91 in the presence of benzophenone induced the [2+2] cycloaddition reaction to give 92 (Fig. 19). Methanolysis of the succinic anhydride group followed by the introduction of the nitrogen atoms for attaching the pyrrole groups provided 93. Saponification of 94 was accompanied with C10-epimerization to give the desired all-trans configuration of 10a. Subsequent Arndt-Eistert homologation followed by bromination afforded bis-α-bromoketone 94. The aminoimidazole groups were then constructed by reacting 94 with Boc-guanidine to afford 95. Finally, introduction of the bromopyrrole or dibromopyrrole groups concluded the syntheses of sceptrin (10a) and dibromosceptrin (10c).
Figure 19.
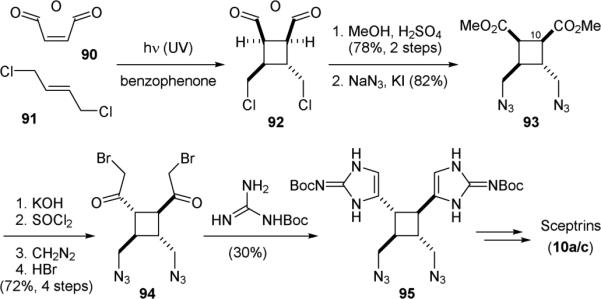
Birman's synthesis of sceptrin (10a).
2.9. Synthetic contributions from the Harran group
The Harran group reported a unique approach for the synthesis of palau’amine (1) in 2005.162 A refined route allowing for the construction of the entire axinellamine skeleton was later published in 2012.163 Using a retro-Scheuer-type rearrangement, they have also achieved the synthesis of ageliferin (11a).164 To construct the axinellamine skeleton, they first oxidized the titanium enolate of 96 to initiate a homocoupling reaction (Fig. 20). Hydrogenation of the resulting C2-symmetric 97 followed by bromination yielded 98. Deprotonation of 98 at the C9 and C14 positions led to the cleavage of the N–N linkages. Cyclization of the resulting amide anions with the imines gave 99 as a mixture of diastereomers. Deprotonation of the two major ismoers with a strong amine base regenerated imines and induced the imineenamine isomerization. Addition of the C10-enamine to the C14-imine followed by hydrolysis of the oxadiazine group and removal of the protecting group of the pyrroles gave 100 as a mixture of C14- and E/Z-isomers. Reduction of the enone group using Myers’ lithium amidotrihydroborate (LAB)165 gave 101. Interestingly, the natural C10-epimer was obtained as the minor product. Oxidation of the correct isomer 101 with the Davis reagent gave the axinellamine skeleton. Finally, reduction of the iminohydantoin group proceeded with debromination to provide 13-dechloro-6’,6”-didebromo-axinellamines (102).
Figure 20.

Harran's synthetic approach to axinellamine (13).
The Harran group found that 101 readily equilibrated upon thermolysis to give a mixture of C10- and C14-diastereomers, with 101a and 101b being the major diastereomers (Fig. 21).164 The cleavage of the C10–C14 linkage in this reaction is reminiscent of the sceptrin-ageliferin rearrangement reaction reported by Baran.146 Reduction of the major product 101a gave 103, which underwent an acid-promoted ring-expansion reaction to yield ageliferin (11a).
Figure 21.
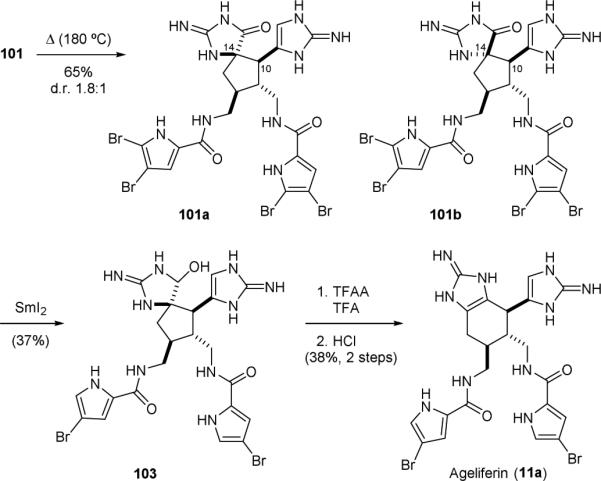
Harran's synthesis of ageliferin (11a).
2.10. Synthetic contributions from the Gleason group
The Gleason group reported an efficient synthesis of the skeleton of the originally proposed palau’amine (1’) in 2008.166,167 They also used a Diels-Alder/oxidative cleavage strategy to establish the key cyclopentyl ring of 1’. Although similar to the approach used by Carreira, the Gleason's approach allows for a more direct introduction of the requisite functionalities. They found that reaction of diene 104 and chloromethyleneoxazolone 105 gave a 1:1 ratio of 106 and 107 (Fig. 22). Diene 104 is rather stable toward 1,5-hydrogen shift and has a half-life time of 37 h at 23 °C. Subsequent Rubottom oxidation of 106/107 and methanolysis of the oxazolone group gave 108 and 109. Oxidative cleavage of the α-hydroxy ketone group of 109 yielded 110 bearing the “cis-palau’amine” stereochemistry.
Figure 22.
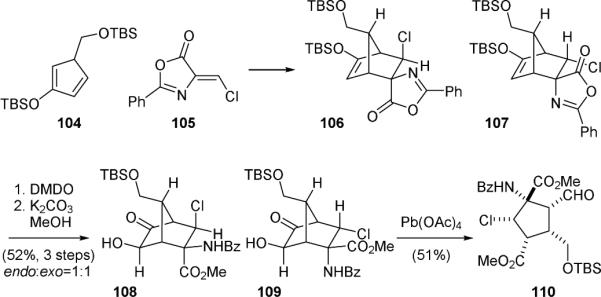
Gleason's synthetic approach to “cis-palau’amine” (1’).
2.11. Synthetic contributions from the Gin group
The Gin group also reported a creative and flexible approach for the syntheses of both the original and revised core structures of palau’amine (1’/1) in 2008.168 Dihydroquinone 111 obtained from the Diels-Alder reaction of cyclopentadiene and benzoquinone was oxidized to afford epoxide 112 (Fig. 23). Treating 112 with sodium ethoxide induced a Favoskii-type rearrangement to give 113. Alkylation of 113 then provided 114, which underwent a spontaneous [3,3]-sigmatropic rearrangement to give an equilibrium mixture of 114 and 115 in a ratio of 2.6:1. The subsequent Meerwein-Verley-Pondorf reduction proceeded under Curtin-Hammett kinetic control through 115 to yield 116 after chlorination. Curtis rearrangement of 116 then provided 117. Reaction of 117 with hydroxylamine followed by treatment of the resulting oxime with thionyl chloride promoted the Beckmann rearrangement to give 118. The amide group was next protected and the olefin group was cleaved by ozonolysis. Reductive workup gave a diol that was treated with acid to induce the lactone formation. The alcohol group was protected in situ to give 119 bearing the correct relative stereochemistry of 1. Elaboration of 114/115 through 114 could also be achieved by treating this mixture with benzeneselenol to protect the enone group of 114. Further elaboration through a reaction sequence similar to that used for preparing 119 provided the “all-cis” diastereomer of 119 bearing the relative stereochemistry of 1’.
Figure 23.
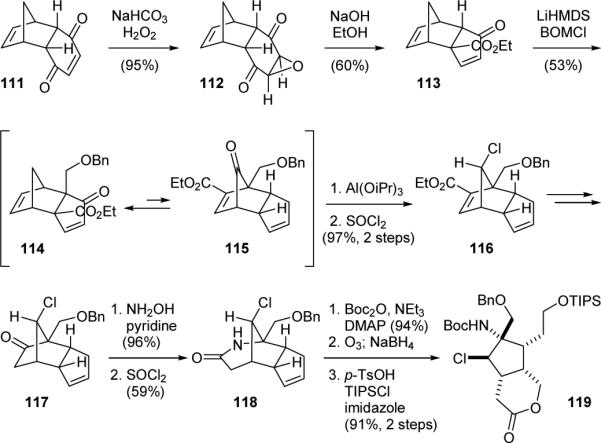
Gin's synthetic approach to palau’amine (1).
2.12. Synthetic contributions from the Namba–Nishigawa group
The Namba and Nishigawa groups disclosed an interesting cyclization approach for the construction the core skeleton of palau’amine in 2009.169,170 In short, treating tosyl hydrazide 120 with a catalytic amount of Hg(II) triflate gave 121 (Fig. 24). Subsequent oxidation of the hydroxyl group to an enone followed by introduction of the two side-chains yielded 122.
Figure 24.
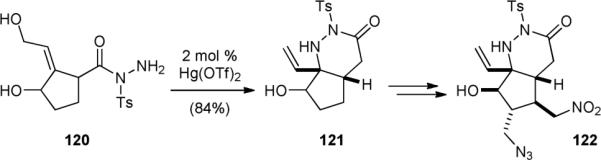
Namba and Nishigawa's synthetic approach to palau’amine (1).
2.13. Synthetic contributions from the Williams group
The Williams group reported the use of an asymmetric [3+2] dipolar cycloaddition approach to synthesize the core skeleton of palau’amine (1) in 2010.171 Condensation of oxazinone 123 with formaldehyde gave azomethine ylide 124 that underwent an intramolecular [3+2] cycloaddition reaction to give 125 (Fig. 25). Reductive removal of the chiral auxiliary group and protection of the primary hydroxyl and secondary amino groups afforded 126. Sequential oxidation of 126 then yielded enone 127. Finally, Morita-Baylis-Hillman hydroxymethylation of 127 followed by lactam cleavage, Michael addition, and alcohol reduction provided the palau’amine core skeleton 128.
Figure 25.
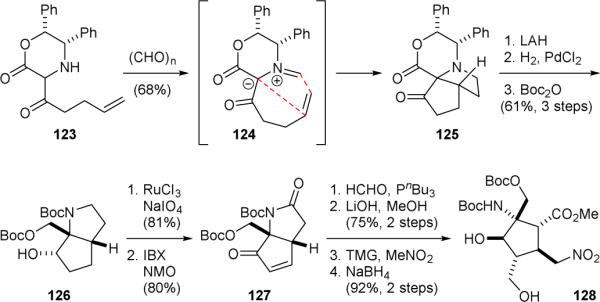
William's synthetic approach to palau’amine (1).
2.14. Synthetic contributions from the Feldman group
The Feldman group also used a Scheuer-type ring-contraction approach to synthesize the core skeleton of palau’amine (1) in 2010.172,173 They first constructed the phakellin subunit using a Pummerer-type oxidation reaction. Oxidation of 129 with the Stang's reagent gave 130 (Fig. 26). Oxymercuration of 130 followed by oxidation provided 131 along with the regioisomers of the β-diketone. Finally, diazotransfer of this mixture of isomers and photolysis induced a Wolff rearrangement to give 132.
Figure 26.
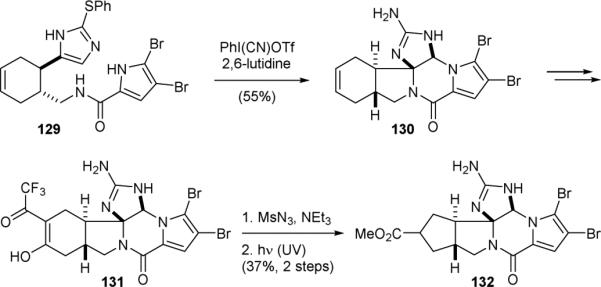
Feldman's synthetic approach to palau’amine (1).
2.15. Synthetic contributions from our group
Our lab is interested in studying the biomimetic syntheses of the pyrrole-imidazole family of alkaloids.33,174-180 We have developed a palladium-catalyzed C–H functionalization reaction to enable an efficient synthesis of dibromophakellstatin in 2007.175 We have also accomplished the asymmetric synthesis of ageliferins (11) in 2011, using a tandem radical cyclization approach as the key step to construct its cyclohexenyl core.177,178 Recently, we have further achieved the asymmetric synthesis of sceptrin (10a) and massadine (14a), and corrected the absolute stereochemistries of sceptrins (10) and ageliferins (11).179
We believed that the biogenic dimerization of 5 proceeds through a radical cyclization mechanism, and sought to provide support for the single-electron transfer (SET)181-185 biosynthetic hypothesis put forth by Molinski and Romo.49,50 Our biomimetic synthesis of ageliferins (11) involves the use of a SET-oxidation for setting up its cyclohexenyl core skeleton.174,177,178 To improve the efficiency of the dimerization process, we tethered the two monomeric units with an ester linker. We also replaced one olefin with an enol, a highly electron-rich olefin, to enable the selective SET-oxidation. The synthesis started with an aldol reaction of 133 and 134 followed by an alcohol oxidation to provide β-ketoester 135 (Fig. 27). The oxidative radical cyclization of 135 proceeded smoothly to yield 136. Subsequent decarboxylation revealed the ageliferin core skeleton and gave 137. Installation of the N7/N7’ nitrogen atoms at the C8/C8’ positions was challenging for steric reasons but could be achieved by sequential mesylation, iodination, and azidation to afford 138. Installation of the bromopyrrole groups yielded 139, and subsequent introduction of the second aminoimidazole group completed the synthesis of ageliferin (11a). The iminophosphorane of 139 served as a good protecting group of aminoimidazole. Because the N7 and N7’ groups of 138 were differentially protected, pyrrole groups with different bromine patterns could be introduced selectively to give bromoageliferin (11b) and dibromoageliferin (11c).
Figure 27.
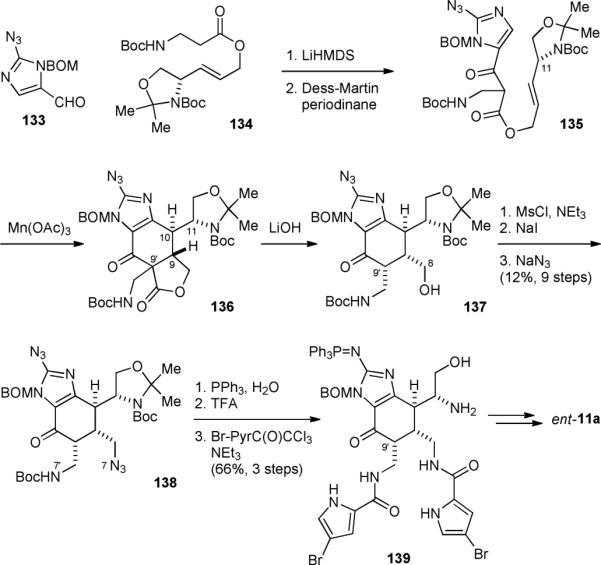
Our synthesis of ageliferin (11a).
Surprisingly, while we targeted ageliferin in its natural enantiomeric form, ent-ageliferin was obtained instead.177 In our synthesis, L-serine was used as the source of chirality to establish the absolute stereochemistry of 11a. Repeating the synthesis with D-serine gave nat-ageliferins.178 MMFF calculations of the equilibrium geometry of 135 confirmed that the conformation of 135 was controlled by the A1,3 strain created by the C11 stereocenter and oxidation of 135 should give 136.176 NMR analyses also suggested that 136 was the correct reaction product. However, this study could not provide direct evidence for the misassignment of the absolute stereochemistry of ageliferins because the C11 stereogenic center was transformed into a sp2-hybridized carbon in ageliferins. Recently, we carried out the Scheuer-type oxidation on 138 and obtained three crystal structures confirming the oxidative cyclization product of 135 to be 136.179 Therefore, the originally assigned absolute stereochemistry of ageliferins (11) must be incorrect. Because the absolute stereochemistry of 11 was determined based on that of sceptrins (10),145 the originally assigned absolute stereochemistry of 10 should also be reconsidered.
To provide independent evidence for the misassignment of the original absolute stereochemistry of 10, we developed a SET-mediated [2+2] cycloaddition approach for the asymmetric synthesis of 10a.179 l-Glutamic acid was used as the source of chirality for establishing the absolute stereochemistry of 141 (Fig. 28). Coupling of 140 and 141 via electrophilic selenation afforded 142 after oxidative elimination of the phenylselenyl group and reduction of the azido group. Irradiation of a solution of 142 in the presence of a photoredox catalyst186-195 promoted a reversible SET reaction and induced the [2+2] cycloaddition196-206 to give 143. Transthioketalization of 143 revealed the cyclobutane core skeleton and gave 144. Subsequent C9 epimerization and installation of the side-chain functional groups furnished 10a. Consistent with our ageliferin synthesis, we obtained ent-sceptrin while targeting sceptrin in its originally assigned natural enantiomeric form.
Figure 28.
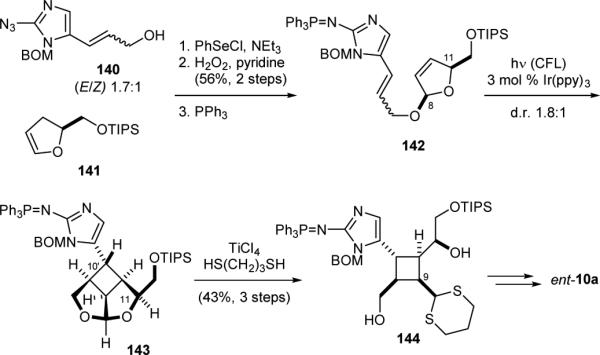
Our synthesis of sceptrin (10a).
The absolute stereochemistry of sceptrin (10a) was previously deduced from two X-ray crystallographic studies. A crystal structure of natural sceptrin was obtained by Faulkner and Clardy in 1981.91 The Hamilton test favored ent-10a slightly (R-factors: 0.090 vs. 0.094) but not conclusively. Although Baran's synthetic study supported ent-10a to be the natural enantiomer, the synthetic intermediate used to co-crystallize with a chiral amine had only 75% ee.145 Recently, the Baran group reexamined the absolute stereochemistry of natural sceptrin by anomalous X-ray scattering. With a Flack factor of –0.002(5) for 10a, they confirmed that the original absolute stereochemistry of sceptrin was misassigned.179
For the synthesis of massadine (14a), we sought to follow the Scheuer hypothesis and verify whether the [3+2] dimers could be derived from the [4+2] dimers through an oxidative ring-contraction reaction.179 We first reduced the carbonyl group of 145, a protected 10’-oxo-dibromoageliferin obtained from our dibromoageliferin synthesis (Fig. 29).177,178 The resulting 146 was oxidized in the presence of titanium(IV) isopropoxide to afford iminohydantoin 147. The hydroxyl group of 146 was used to control the stereochemistry of the Scheuer-type oxidative ring-contraction reaction to give the desired spiro-configuration. Reduction of 147 then gave hemiaminal 148. Subsequent removal of the protecting groups yielded “pre-massadine” (149). Oxidation of 149 with N-bromosuccinimide (NBS) in methanol gave a diastereomeric mixture of massadine-methanol adducts, which slowly converted into massadine (14a) and 3,7-epimassadine150 upon treatment with hydrochloric acid. Surprisingly, oxidation and deprotection of 145 gave nat-14a while deoxygenation and deprotection of 145 gave ent-11c. Consequently, massadine (14a) and dibromoageliferin (11c) must be antipodal and 14a cannot be produced from oxidation of 11c or its derivatives in nature. Although enantiomeric biosynthesis that produces both enantiomers of a natural product has been reported,207 the biogenic dimerization of 5 giving antipodal pyrrole-imidazole dimers is the first example of enantiodivergent biosynthesis that produces opposite enantiomers of natural products as congeners.
Figure 29.

Our synthesis of massadine (14a).
3. The dragmacidin alkaloids
Dragmacidins (2 and 150–152) are a group of indole alkaloids that possess a central pyrazine/pyrazinone ring (Fig. 30).19,208-212 Among them, dragmacidin D–F (2, 151, and 152) that contain an aminoimidazole or a cyclic guanidine unit in a highly complex molecular framework are particularly challenging synthetic targets. Biosynthetically, 2 and 152 are likely derived from 151.34 Notable biological activities of these marine sponge metabolites include selective inhibition of serine-threonine protein phosphatases 1 (PP1) over the 2A isoform (PP2A),212 brain nitric oxide synthase (bNOS) over the inducible isoform (iNOS),1 and HIV-1 over HSV virus.19
Figure 30.
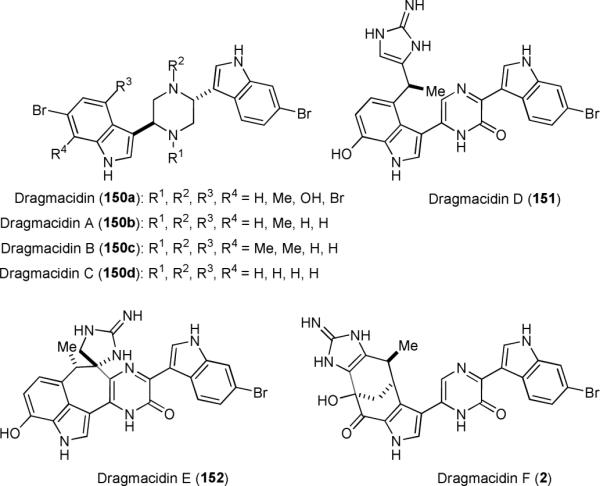
Structures of the dragmacidin alkaloids.
3.1. Synthetic contributions from the Stoltz group
The first synthesis of dragmacidin D (151) was completed by the Stoltz group in 2002, using a sequential Suzuki coupling approach.213 A selective Suzuki coupling of bromoiodopyrazine 153 and boronic acid 154 gave heterobiaryl 155 (Fig. 31). Subsequent Suzuki coupling of dibromide 155 and pinacolborane 156 proceeded selectively to yield 157. Removal of the alcohol protecting group, oxidation, and Henry reaction provided α-nitroketone 158. Reduction of 158 followed by cleavage of the benzyl and methyl ethers afforded α-aminoketone 159. Condensation of 159 with cyanamide concluded the synthesis of dragmacidin D (151). Impressively, this synthesis takes only 17 linear steps.
Figure 31.
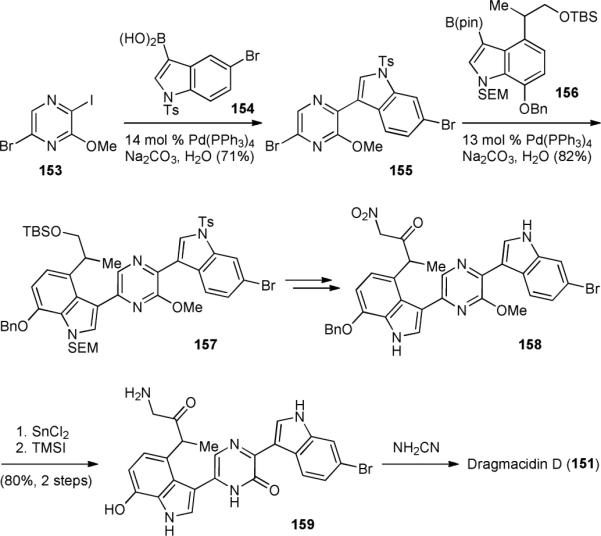
Stoltz's synthesis of dragmacidin D (151).
The Stoltz group also accomplished the asymmetric synthesis of dragmacidin F (2) and established its absolute stereochemistry in 2004.214-216 Starting from (–)-quinic acid (160), lactonization and selective alcohol protection gave 161 (Fig. 32). Oxidation of 161 followed by Wittig olefination and palladium-catalyzed hydrogenation provided acid 162. Subsequent installation of the pyrrole group afforded 163, which was subjected to a palladium-catalyzed oxidative Heck carbocyclization to yield 164. Hydrogenation of 164 followed by alcohol protection, and borylation gave 165. Coupling of pinacolborane 165 with bromoindole 155 proceeded smoothly to give 166. To install the aminoimidzole group, 166 was first converted into tosyloxime 167 and then treated with base to induce a Neber rearrangement. The resulting azirine 168 was hydrolyzed to give α-aminoketone 169. Deprotection of 169 followed by condensation with cyanamide furnished ent-dragmacidin F (2). A slightly modified method has also been developed to provide ent-162 from 160, allowing for the asymmetric synthesis of 2.217
Figure 32.

Stoltz's synthesis of dragmacidin F (2).
3.2. Synthetic contributions from the Yamaguchi–Itami group
The Yamaguchi–Itami group reported a highly efficient synthesis of dragmacidin D (151) in 2011.218 They used a series of C–H functionalization reactions to assemble its molecular skeleton. First, coupling of 4-iodoindole 170 with thiophene 171, a 2-butanone surrogate, through thiophene C–H functionalization gave 172 (Fig. 33). Next, removal of the silyloxy group, desulfation, debenzylation, and protection of the indole functional groups provided 173. Subsequently, the pyrazinone group was introduced through another C–H functionalzation reaction. The C–H/C–H coupling of 173 and pyrazine N-oxide 174 afforded 175, which was treated with trifluoroacetic anhydride to induce a rearrangement reaction to yield pyrazone 176. An acid-promoted oxidative C–H/C–H coupling of 176 and 177 then afforded 178. Finally, installation of the aminoimidazole group completed the synthesis of 151.
Figure 33.
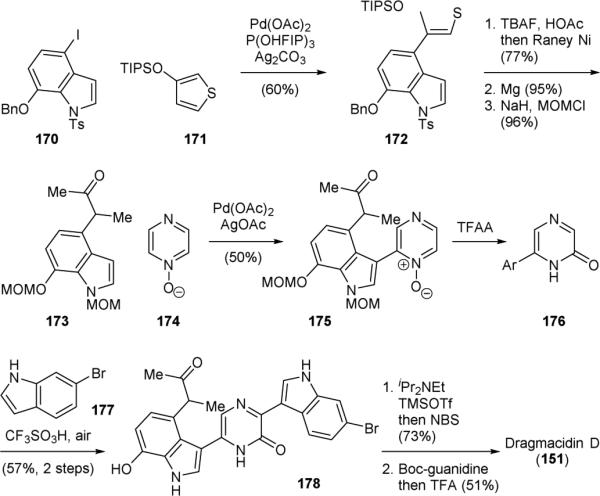
Yamaguchi and Itami's synthesis of dragmacidin D (151).
3.3. Synthetic contributions from the Feldman group
The Feldman group completed the synthesis of dragmacidin E (152) in 2011, using a Witkop cyclization reaction to establish its C4″-C6‴ linkage.219-222 Briefly, photolysis of the tryptophan derivative 179 afforded 180 (Fig. 34). Reduction of the enone group of 180 induced an intramolecular Dieckmann cyclization to provide 181. Subsequent decarbonylative cleavage of the lactam linkage and C5‴-aminocyanation gave 182 as a 5:1 mixture of C5‴-diastereomers. The imidazolidinone group was then constructed and the C-6 position was functionalized through indolic oxidation to yield 183. After reduction of the azido group and coupling with 184, the resulting 185 was subjected to a cyclodehydration reaction to generate the complete skeleton of 152. The dihydropyrazinone group was then oxidized to give 186. Finally, conversion of the urea group into a guanidine group and deprotection furnished 152.
Figure 34.

Feldman's synthesis of dragmacidin E (152).
3.4. Synthetic contributions from the Oikawa group
The Oikawa group also disclosed an approach for constructing the dragmacidin D skeleton in 2008.223,224 Similar to Feldman's strategy, a late-stage pyrazinone construction was planned. A Suzuki coupling of vinyl bromide 187 with thioimidazole group 188 was used to provide 189 (Fig. 35). After converting the amino ester group of 189 into a differentiated diamine, amine 190 was coupled with 184 to give 191, an advanced synthetic intermediate of dragmacidin D (151).
Figure 35.
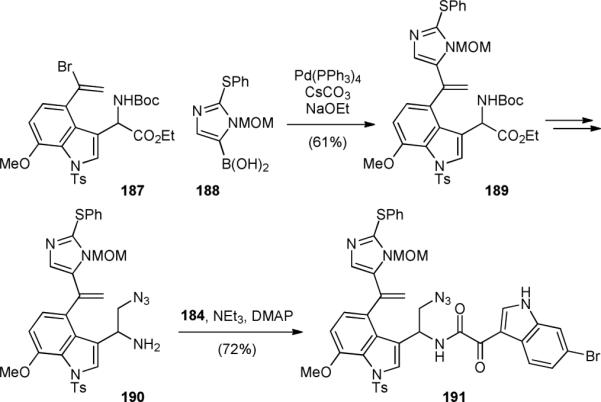
Oikawa's synthetic approach of dragmacidin D (151).
3.5. Synthetic contributions from the Jiang, Funk, and Jia groups
The Jiang, Funk, and Jia groups have each developed a unique synthetic approach to construct the pyrazinone core skeleton of dragmacidins.225-228 The Jiang group reported the use of a sequential cross-coupling strategy for the synthesis of the core skeleton of dragmacidin D (151) in 2002.225,226 The Funk group successfully constructed the molecular framework of dragmacidin E (152) in 2006,227 using an electrocyclization reaction as the key step . The Jia group demonstrated the utility of the intramolecular Friedel–Crafts reaction in preparing the indole skeleton of dragmacidin E (152) in 2014.228
4. The saxitoxin/tetrodotoxin alkaloids
Saxitoxins/gonyautoxins (STXs/GTXs, 192),229-236 zetekitoxins (ZTXs, 193),237-239 tetrodotoxins (TTXs, 3),20,240-248 and chiriquitoxin (CHTX, 194)249,250 are highly toxic cyclic guanidine-containing alkaloids that block the voltage-gated ion channels (Fig. 36).36-43 Among them, saxitoxin (STX, 192a) and tetrodotoxin (TTX, 3) are arguably the most infamous small-molecule toxins. STX (192) is responsible for the paralytic shellfish poisoning (PSP) and TTX (3) is the principle toxic component of puffer fish. Several different approaches have been developed for achieving the de novo synthesis of STXs/GTXs (192) and TTXs/CHTX (3/194). However, there is no report of the synthesis of ZTXs (193). The structure of ZTX C (193b) has also not been fully characterized. While the biosynthetic pathways of these exotic natural products are poorly understood, it is known that these neurotoxins are produced by symbiotic bacteria as they can be found in various animal species. For example, STX (192a) is also present in some puffer fish251 and TTX (3) can be found in certain frogs and newts.228,249,252,253
Figure 36.

Structures of saxitoxins/gonyautoxins (STX/GTX, 192), zetekitoxins (ZTX, 193), tetrodotoxins (3), and chiriquitoxin (194).
4.1. Synthetic contributions from the Kishi group
The first de novo synthesis of tetrodotoxin (3) was accomplished by the Kishi group in 1972.254-257 In this seminal synthesis, a regioselective Diels-Alder reaction was first used to prepare 197 from butadiene (195) and quinone 196 after mesylation (Fig. 37). A Beckmann rearrangement was then used to introduce the nitrogen atom at the 8a-position. The subsequent dihydroquinone reduction proceeded with high regioselectivity to give 198 because of the steric effects of the axial acetamide group. Next, epoxidation of 198 in the presence of camphorsulfonic acid gave 199, which was transformed into 200 through a series of redox and protection operations. Baeyer-Villiger oxidation of 200 gave 201 exclusively. Treating the resulting lactone 201 with potassium acetate in acetic acid led to the cleavage of the lactone ring and the formation of the oxonium ion 202. Trap of the oxonium ion with acetate and intramolecular attack of the epoxide of 202 by the carboxylate group yielded 203. After installation of the guanidine group, selective removal of one of the acetyl protecting groups of 204 followed by oxidative cleavage of the dihydrofuran ring and global deprotection concluded the synthesis of 3.
Figure 37.

Kishi's synthesis of tetrodotoxin (3).
The first synthesis of saxitoxin (192a) was accomplished also by the Kishi group in 1977.258-262 This classic synthesis started with condensation of benzyloxyacetaldehyde, vinylogous carbamate 205, and silicon tetraisothiocyanate to give 206 (Fig. 38). The ester group of 206 was then converted into a hydrazide, which was then treated with nitrosyl chloride to give an acyl azide. The subsequent Curtis-type rearrangement and trap of the resulting isocyanate with ammonia yielded 207. After transthioketalization, an acid-promoted cyclization established the saxitoxin skeleton and provided 208. The thiourea group of 208 was then converted into guanidine by treating with Meerwein salt followed by heating in melted ammonium propionate. Removal of the protecting groups of the resulting 209 afforded 210. Finally, installation of the carbamate group completed the synthesis of saxitoxin (192a). The asymmetric synthesis of ent-192c was also accomplished in 1992 by condensing a 1,3-dithiane analog of 205 with (R)-glyceraldehyde to prepared ent-dithia-206 after diol cleavage.262
Figure 38.
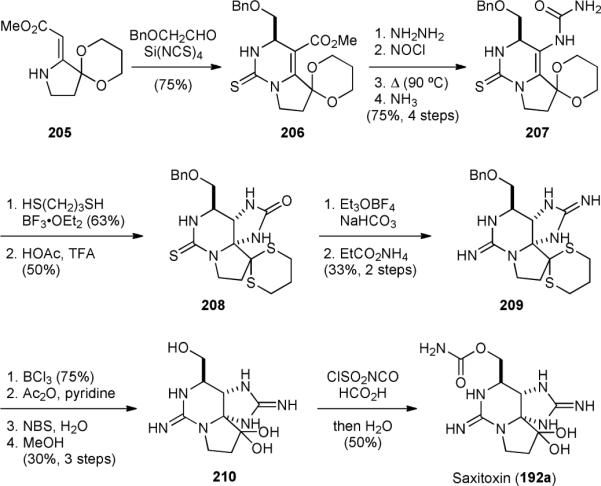
Kishi's synthesis of saxitoxin (192a).
4.2. Synthetic contributions from the Jacobi group
The Jacobi group reported the development of a practical synthesis of saxitoxin (192a) in 1984.263-265 The first step of this elegant synthesis is the activation of imidazolinone 211 by the formation of acyl imidazolinone 212 (Fig. 39). The highly electrophilic compound 212 was then reacted with benzylhydrazine to give hydrazide 213. Condensation of 213 with methyl glyoxylate hemiacetal provided azomethine imine 214, which underwent a 1,3-dipolar cycloaddition reaction under kinetic control to afford pyrazolidine 215. Subsequently, epimerization of the C6 stereocenter followed by reduction of the ester group yielded 216. After hydrazide reduction, deprotection of the resulting hydrazide group, and introduction of the thiocarbamate group, the N–N linkage was cleaved to induce a cyclization reaction to provide thiourea 217. Acetylation of the hydroxyl group followed by conversion of the urea and thiourea groups into cyclic guanidine groups afforded 218, a late-stage intermediate of Kishi's synthesis. This route is remarkably efficient and requires no chromatographic purification. Impressively, more than 0.5 g of 217 was prepared in a single run by this route. This intramolecular azomethine imine cycloaddition strategy was later adapted by the Overman group to construct the core skeleton of cis-palau’amine (1’) (Fig. 5).
Figure 39.
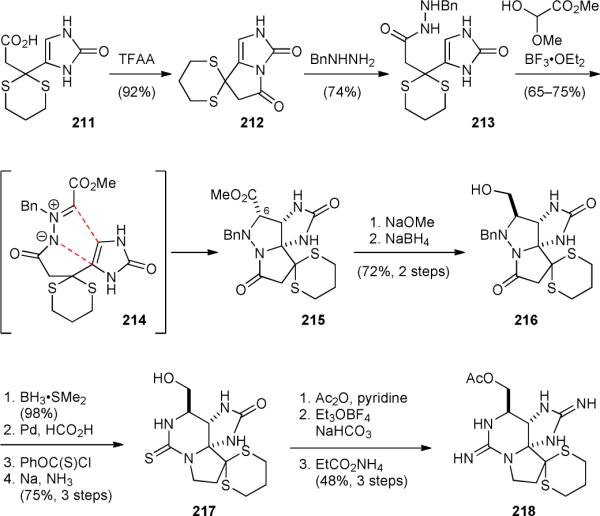
Jacobi's synthesis of saxitoxin (192a).
The Isobe–Nishikawa group has devoted their efforts to studying the asymmetric synthesis of tetrodotoxin and saxitoxin alkaloids since 1987.42,266-292 They first accomplished the synthesis of 5,11-dideoxytetrodotoxin in 1999.275,277,286 They then achieved the syntheses of 8,11-dideoxytetrodotoxin279,282 and 11-deoxytetrodotoxin (3a),278 in 2002. Following these successes, they completed the synthesis of tetrodotoxin (3) in 2003,281 reported an improved route in 2004,283,284 and finished the synthesis of chiriquitoxin (194) in 2014.287
Levoglucosenone (219) was used as the source of the chirality in all of these asymmetric syntheses (Fig. 40). The second-generation synthesis of 3 started with bromination of 219 followed by a regioselective Diels–Alder reaction with isoprene (220) to establish its central cyclohexane core.283 The resulting bromide 221 was then functionalized to provide 222, setting the stage for the introduction of the nitrogen-containing quaternary center by Overman rearrangement by thermolysis of the trichloroacetimidate of 222. Subsequent oxidation, ozonolysis of the olefin, and alkynylation of the resulting aldehyde converted 223 to 224. Oxidation of the acetylene group then yielded α-keto acid 225. Next, epoxide opening and protection of the hydroxyl groups gave 226. Finally, installation of the guanidine group via 227 provided tetrodotoxin (3) and 4,9-anhydrotetrodotoxin (228). Using a slightly revised route, the Isobe–Nishikawa group also successfully achieved the synthesis of chiriquitoxin (194).287 The key step was the aldol reaction of 229 and the camphor-derived iminolactone 230 to give 231. Subsequent deprotection and installation of the guanidine group yielded chiriquitoxin (194) and 4,9-anhydrochiriquitoxin.
Figure 40.

Isobe–Nishikawa's synthesis of tetrodotoxin (3) and chiriquitoxin (194).
The Nishikawa group also completed the synthesis of decarbamoyl α-saxitoxinol (237),293 a nontoxic natural analogue of saxitoxin, in 2011.290-292 Aziridine 232, prepared from (S)-Garner's aldehyde, was transformed into mesylate 233 (Fig. 41). Treating 233 with pyridinium tribromide initiated a bromocyclization reaction to give 234. The gem-dibromide group was then converted into enol acetate and reduced to yield alcohol 235. Installation of the second guanidine group provided 236, which was deprotected and cyclized to furnish 237.
Figure 41.
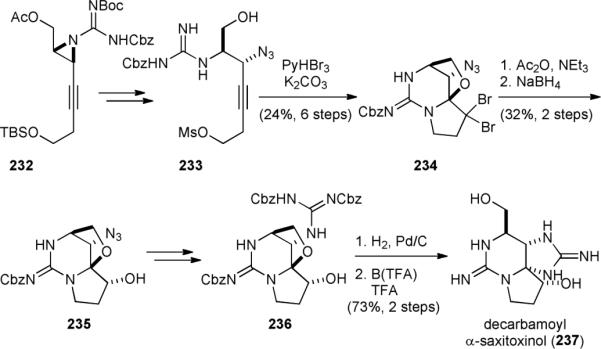
Nishikawa's synthesis decarbamoyl α-saxitoxinol (237).
4.4. Synthetic contributions from the Du Bois group
The Du Bois group has carried out a series of chemical and biological studies to provide better understanding of the reactivity and functions of the STXs/TTXs.39,294-303 They first developed a series of new rhodium-catalyzed nitrene C–H insertion reactions304,305 and used them to complete the synthesis of tetrodotoxin (3), saxitoxin (182a), and gonyautoxin 3 (182e) in 2003, 2006 and 2008, respectively.294-297 They then designed several saxitoxin-based probe reagents to study the functions of sodium ion channels.298-303
For the synthesis of 3, the DuBois group first used an intramolecular carbene C–H insertion reaction to construct the central cyclohexane ring and establish the C6 quaternary center.294 The rhodium-catalyzed C–H carbene insertion306,307 of 238 occurred exclusively at the congested tertiary ethereal position to give 239 after ketone reduction (Fig. 42). Subsequent enone reduction, diol protection, amidation, and C4a olefination yielded 240. Allylic oxidation of 240 occurred selectively at the C5 position to give an enone. Conjugate addition of vinyl cuprate followed by ketone reduction then afforded 241. Thermolysis of 241 induced the lactone formation and yielded 242. Next, installation of the carbamate group provided 243. The following olefin cleavage yielded 244, which underwent an intramolecular nitrene C–H amination304,305 at a highly hindered position to afford 245 selectively. Finally, installation of the guanidine group and deprotection completed the synthesis of 3.
Figure 42.

Du Bois’ synthesis of tetrodotoxin (3).
After accomplishing the asymmetric synthesis of tetrodotoxin (3), the Du Bois group turned their attention to the synthesis of saxitoxin (192a).295 They first prepared sulfamate 246 from (R)-glycerol acetonide (Fig. 43). An intramolecular C–H amination of 246 was then used to establish the aminodiol triad. Alkynylation of the resulting N,O-acetal 247 gave 248, which was transformed into guanidine 249. Cleavage of the oxathiazinane ring, reduction of the azido group, and formation of the guanidine group by a silver-promoted cyclization reaction yielded 250. Introduction of the carbamate group and ketohydroxylation308 of the olefin afforded 251. Interestingly, using osmium tetroxide as the catalyst for this (net) 4-electron oxidation reaction would result in a transannular cyclization through the alternative α-ketol tautomer to give a undesired bicyclic system. Finally, deprotection of 251 followed by alcohol oxidation furnished 192a.
Figure 43.
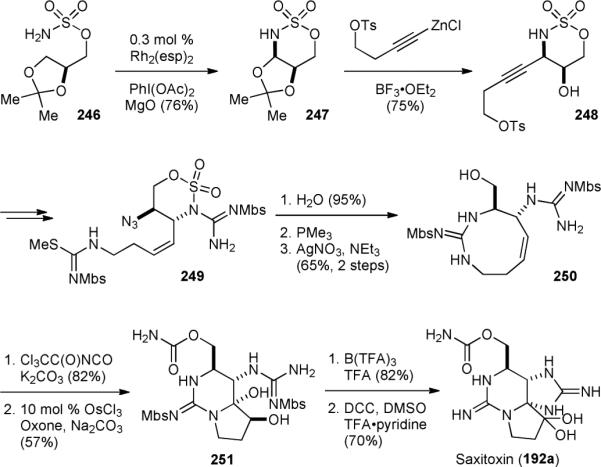
Du Bois’ first-generation synthesis of saxitoxin (192a).
The Du Bois group also reported a highly efficient second-generation synthesis of 192a in 2007.296 This new synthesis started with functionalization of N-Boc-l-serine methyl ester (252) to give nitrone 253 via protection, reduction, and hydroxylamine condensation (Fig. 44). Alkynylation of 253 with 254 gave 255, which was subjected to dimmide reduction and cleavage of the N–O linker to provide 256. The second guanidine group was then introduced to yield 257. Removal of the protecting groups and cyclization of the primary guanidine afforded 250, an advanced synthetic intermediate in their first-generation synthesis (Fig. 43). Impressively, this route allowed for the asymmetric synthesis of saxitoxin (192a) to be achieved in 14 steps and 6.5% overall yield from the commercially available serine derivative 242. Using this route, they have also prepared a series of STX derivatives for functional studies.298-303
Figure 44.
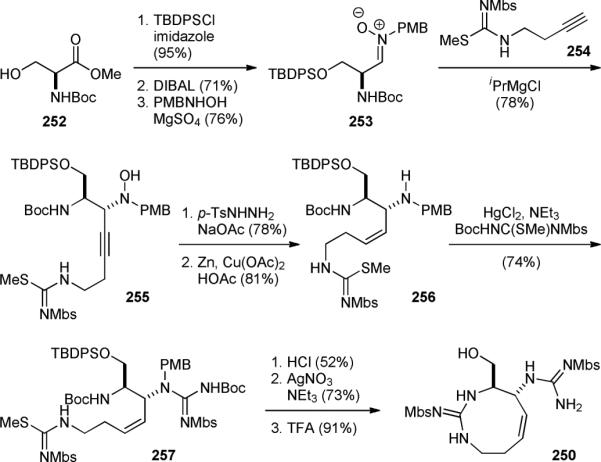
Du Bois’ second-generation synthesis of saxitoxin (192a).
The Du Bois group further reported a new approach for the synthesis of gonyautoxin III (192f), a sulfate-derivative of saxitoxin (192a), in 2008.297 Readily prepared from 252, aldehyde 258 was condensed with allylamine to promote a cyclization reaction to give 259 (Fig. 45). Subsequently, introduction of the guanidine group afforded 260 and set the stage for the key C–H amination reaction. Generation of the rhodium-nitrenoid of 260 led to aziridination of the pyrrole group and the formation of 261 after aziridine opening. Deacetoxylation and installation of the carbamate group then yield 262, which was dihydroxylated to afford 263. Selective protection of the less hindered hydroxyl group of 263 followed by alcohol oxidation gave 264. Finally, deprotection and sulfation furnished gonautoxin 3 (192f).
Figure 45.

Du Bois’ synthesis of gonyautoxin III (192f).
4.5. Synthetic contributions from the Sato group
The Sato group has also developed a unique chiron approach to synthesize tetrodotoxin (3).309-313 They used myo-inositol as the carbon source of the central cyclohexane ring to achieve the racemic synthesis in 2005,310 and D-glucose as the chiral building block to accomplish the asymmetric synthesis of 3 in 2008.311
In their asymmetric synthesis, glucose was first functionalized to give nitroalkene 265 (Fig. 46).311 Next, a conjugate addition of lithiated dithioacetal to 265 provided 266. Removal of the acetonide protecting group followed by treatment with a base promoted a Henry reaction affording the muco-inositol derivative 267. A Nef reaction was then used to convert the nitro group into a carbonyl group. The resulting ketone 268 was treated with lithiated methylene chloride to yield 269. Subsequent azidation proceeded through α-chloroepoxide 270 to provide aldehyde 271 with an inversion of the C8a stereochemistry. Subsequent Strecker reaction followed by nitrile reduction and alcohol protection afforded 272. The following Jones oxidation promoted the bridge formation and gave lactone 273. Installation of the guanidine group then provided 274. Finally, cyclization of the guanidine group afforded 4,9-anhydrotetrodotoxin (228), which slowly isomerized in acid to a 4:1 mixture of tetrodotoxin (3) and 4,9-anhydrotetrodotoxin (228).
Figure 46.

Sato's synthesis of tetrodotoxin (3).
4.6. Synthetic contributions from the Shinada–Ohfune group
The Shinada–Ohfune group reported an asymmetric synthesis of 5,6,11-trideoxytetrodotoxin (3i) in 2006.314-316 Their synthesis started with preparing 275 from (–)-quinic acid (Fig. 47). The phenylalanine group of 275 was then used to activate the adjacent ketone group and direct the cyanide addition to set the nitrogen-containing C8a quaternary center. Treatment of 275 with trimethylsilyl triflate promoted an intramolecular imine condensation reaction. Cyanide then attacked from the less hindered face of the bicyclic system to give 276 following oxidation of the resulting amine to imine. After hydrolytic removal of the amino acid auxiliary group, the C7 hydroxyl group was installed and the acetoxyl ester and cyano groups were reduced to provide 277. Subsequently, 277 was subjected to another Strecker reaction and the guanidine group was introduced to give 278. Oxidative cleavage of the olefin group occurred with concomitant hemiacetal formation to yield 279. Treatment of 279 with acid induced the guanidine cyclization and lactone formation to afford 3i as a 2:1 mixture of C4 diastereomers, together with 4,9-anhydro-5,6,11-trideoxytetrodotoxin (3m).
Figure 47.
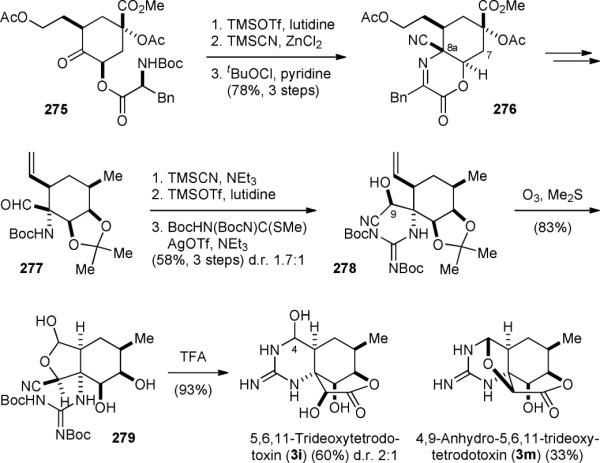
Shinada–Ohfune's synthesis of 5,6,11-trideoxytetrodotoxin (3i).
4.7. Synthetic contributions from the Nagasawa group
The Nagasawa group has also reported a series of synthetic and biochemical studies of saxitoxins (192).317-322 They used a 1,3-dipolar cycloaddition approach to complete the synthesis of decarbamoyloxysaxitoxin (192c), saxitoxin (192a) and gonyautoxin III (192f) in 2007, 2009 and 2010, respectively. The stereochemistry of the [3+2] cycloaddition reaction of nitroalkene 280 and nitrone 281 was controlled by the silyloxyl group of 281, which was derived from D-malic acid (Fig. 48). Epimerization of the C5 stereocenter and reduction of the nitro group gave 282. Protection of the hydroxylamine group, cleavage of the N–O bonds, and introduction of the guanidine group then provided pyrrolidine 283. Subsequently, cyclization of 283 and removal of the silyl protecting group produced cyclic guanidine 284. After sequential oxidation of 284 to give α-hydroxy ketone 285, the second guanidine group was installed to yield 286. The C11 hydroxyl group was next introduced by Rubbotom oxidation to afford 287. Finally, carbamate formation, deprotection, and sulfation furnished gonyautoxin III (192f).
Figure 48.

Nagasawa's synthesis of gonyautoxin III (192f).
4.8. Synthetic contributions from the Looper group
The Looper group have developed a series of new methods for the synthesis of aminioimidazole and guanidine.323-327 Using a novel metal-catalyzed sequential hydroamination approach,323,325 they further achieved theasymmetric synthesis of saxitoxin (192a) in 2011.328 The synthesis started with preparation of 288 following the method for the synthesis of 255 developed by Du Bois (Fig. 44).296 Subsequently, reduction of the hydroxylamine followed by installation of the carbamate group provided 289 without cyclic carbamate formation (Fig. 49). The two guanidine groups were then introduced to yield 290. Treating 290 with a catalytic amount of silver(I) acetate induced the first guanidine cyclization reaction. The 5-exo cyclization product 291 was subjected to a second guanidine cyclization reaction using silver(I) acetate and iodine to give 292. Reaction of 292 with silver(I) acetate and acetic acid promoted a carbonate cyclization to provide 293. This sequence of cyclization reactions could be carried out in one flask to deliver 293 directly from 290. Deprotection and mesylation of the hydroxyl group followed by cleavage of the cyclic carbamate group afforded 294 bearing the complete skeleton of saxitoxin. Finally, alcohol oxidation and deprotection gave 192a in only 14 steps from commercially available N-Boc-l-serine methyl ester (242).
Figure 49.

Looper's synthesis of saxitoxin (192a).
5. The crambescidin/batzelladine alkaloids
Ptilomycalin A (4a) is a structurally unique alkaloid discovered by Kusumi and Kashman from the Caribbean sponge Ptilocaulis spiculifer and the Red Sea sponge Hemimycale sp (Fig. 50).329-331 The molecular topology of 4a resembles a ship and its guanidine-containing pentacyclic core has been referred to as the “vessel” part and the spermidine-containing long aliphatic chain the “anchor” part.330 Kusumi and Kashman's NMR studies suggested that 4a can trap an anion between the “vessel” and “anchor” moieties and thus function as a phase-transfer molecule. Although ptilomycalin A (4a) contains one guanadium and two amino groups, 4a is a “strikingly non-polar substance”.330 The “vessel” part of 4a is structurally similar to the C2-symmetric chiral anion-receptor 295 developed by Lehn and De Mendoza.332 Murphy has carried out a series of crystallographic studies on 296 and 297, the meso-symmetric analogs of the “vessel” part of 4a, and found that 296 and 297 could effectively bind to a borate anion in different modes (Fig. 50).333 Taking inspirations from 4a, the Nagasawa group has developed a C2-symmetric chiral phase-transfer catalyst (298) for the asymmetric alkylation of the Schiff base of t-butyl glycinate and benzophenone.334
Figure 50.
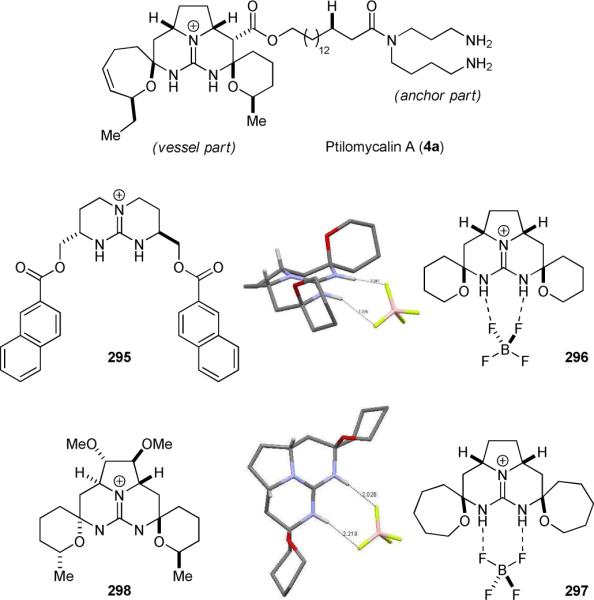
The structure of ptilomycalin A (4a) and its anion-recognition analogs.
The tricyclic part of the core of 4a is structurally similar to ptilocaulin (299) (Fig. 51).335,336 However, in contrast to 299, 4a has an endocyclic instead of an exocyclic guanidine group. The synthesis of 299 has been achieved by the Snider,337-340 Roush,341,342 Uyehara,343,344 Hassner,345,346 Asaoka,347 Cossy,348 Schmalz,349 and Livinghouse groups.350 A series of ptilomycalin congeners have also been discovered and named as crambescidins (4, 4b–g, and 4m,n),21,351-356 crambidine (300),21 celeromycalin (4h),357 fromiamycalin (4i),357 and neofolitispates (4j–l)358 (Fig. 51). Crambescidins are named based on their molecular weights. For example, crambescidin 816 (4) has a molecular weight of 816 daltons. The batzelladines (301) and dehydrobatzelladine (302) contain the same central guanadium tricyclic core as 4 whereas a different “anchor” group.359-365 The crambescins (303) have an even simpler monocyclic or bicyclic core.356,366-369
Figure 51.

Structures of the crambescidin/batzelladine family of alkaloids.
The crambescidin/batzelladine family of alkaloids has shown various useful biological properties. Interestingly, crambescidin 816 (4) binds to the voltage-gated calcium ion channel selectively while crambescins (303) binds to the potassium ion channels preferentially.370 The crambescidins (4's) and batzelladines (301) also show anticancer,371,372 antiviral,363,373,374 antifungal,375 and antimalarial376 activities. Based on the observation that the “vessel” part of 4 exists in a dynamic equilibrium between the spiroaminal and the imine forms under high pH, Overman proposed that 4 binds to their protein targets covalently through its imine form.377
5.1. Synthetic contributions from the Overman group
The Overman group has developed a tethered Biginelli reaction as a central strategy for the synthesis of the crambescidin/batzelladine alkaloids.377-390 They first achieved the asymmetric synthesis of ptilomycalin A (4a) in 1995.379 To construct the pentacyclic core of 4a, amino alcohol 304 was reacted with potassium isocyanate to form a urea (Fig. 52). Its olefin group was then cleaved to promote a cyclization to give ureido aldehyde 305. Biginelli condensation of 305 with β-ketoester 306 provided 307 with good diastereoselectivity (d.r. 7.5:1). After removal of the silyl protecting group, treatment of the resulting secondary alcohol with a catalytic amount of acid induced a spirocyclization to yield 308. The primary hydroxyl group was then oxidized and the urea group was methylated to afford 309. Reaction of aldehyde 309 with Grignard reagent 310 followed by alcohol oxidation gave ketone 311. Desilylation followed by treatment with ammonia then established the central guanidinium functionality. The spiroaminal formation also occurred under the reaction conditions to provide 312, the “vessel part” of ptilomycalin A (4a). Finally, introduction of the spermidine chain (the “anchor part”) and epimerization of the C14 stereocenter concluded the synthesis of 4a.
Figure 52.

Overman's synthesis of ptilomycalin A (4a).
The Overman group later used a more streamlined route to synthesize a series of crambescidin alkaloids. They first accomplished the synthesis of 13,14,15-isocrambescidin 800 (4n) in 1999.380 They then achieved the synthesis of crambescidin 657 (4d), neofolitispate 2 (4k), and carmbescidin 800 (4e) in 2000.381 They subsequently completed the synthesis of crambescidin 359 (4b) and crambescidin 431 (4c) in 2005.377 For the synthesis of 4n, the carbon framework of the 7-membered cyclic spiroaminal and the guanidine group were introduced at an early stage. They first constructed cyclic guanidine 313 using the same method as in their ptilomycalin A (4a) synthesis. Dihydroxylation of 313 followed by cleavage of the resulting diol gave pyrrolidine 314 (Fig. 53). Subsequent Biginelli condensation of 314 with 306 under Knoevenagel conditions provided exocyclic guanidine 315. A good level of diastereoselectivity (7:1) was obtained when 2,2,2-trifluoroethanol was used as the solvent. The C13 stereochemistry of 315 is consistent with 4n and opposite to that of 307. Deprotection of 315 followed by an acid-promoted spirocyclization completed the skeleton of 4n. Subsequent epimerization of the C14 stereocenter of 316 and introduction of the amide side-chain furnished 4n.
Figure 53.
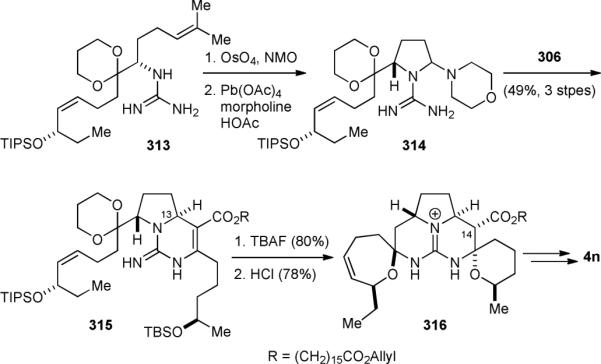
Overman's synthesis of 13,14,15-isocrambescidin 800 (4n).
The Overman group also completed the synthesis of crambidine (300) in 2005.382 The synthesis of 300 started with desilylation of 315, a synthetic intermediate of 4n. Desilylation of 315 followed by treatment with a weak acid induced the spirocyclization of the seven-membered spiroaminal without the formation of the six-membered spiroaminal (Fig. 54). The resulting 317 was then oxidized to give a chromatographically separable mixture of 318 and 319. Whereas the pentacyclic core 318 is the major isomer in deuterated chloroform, the tetracyclic core 319 exists predominantly in deuterated methanol, acetonitrile, and tetrahydrofuran. Consistently, acetylation of the primary alcohol of 319 is significantly slower in deuterated chloroform than in acetonitrile or pyridine. This mixture of 318 and 319 could also be obtained by oxidation of 315 followed by sequential desilylation. Subsequent installation of the hydroxyspermidine group furnished 300.
Figure 54.
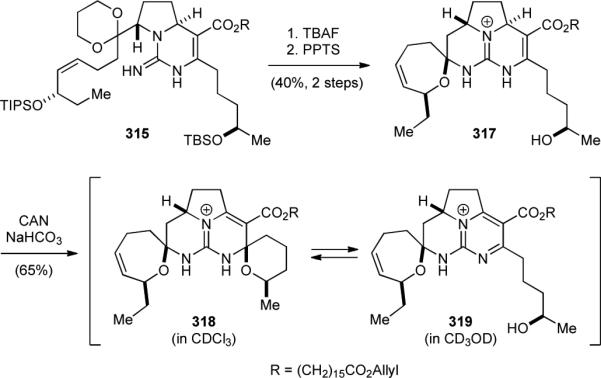
Overman's synthesis of crambidine (300).
Additionally, the Overman group completed the asymmetric synthesis of batzelladine D (301d) in 1999.384 They also revised the structure of batzelladine F (301f) and completed its asymmetric synthesis in 2001.385,388,389 They further reported the synthesis of dehydrobatzelladine C (302) in 2004.387 They found through synthetic studies that the originally proposed length of the alkyl chains and the relative stereochemistry of 301f were both incorrect. The tethered Biginelli reaction was used again to construct both tricyclic cores of 301f. Specifically, Biginelli condensation of guanidine 320 with 321 provided the tricyclic core 322 (Fig. 55). Decarboxylation of 322 followed by hydrogenation, and introduction of an acetoacetate group gave 323. Biginelli condensation of the highly functionalized 323 with guanidine 324 yielded 325. Finally, cyclization and hydrogenation of 325 completed the synthesis of 301f.
Figure 55.
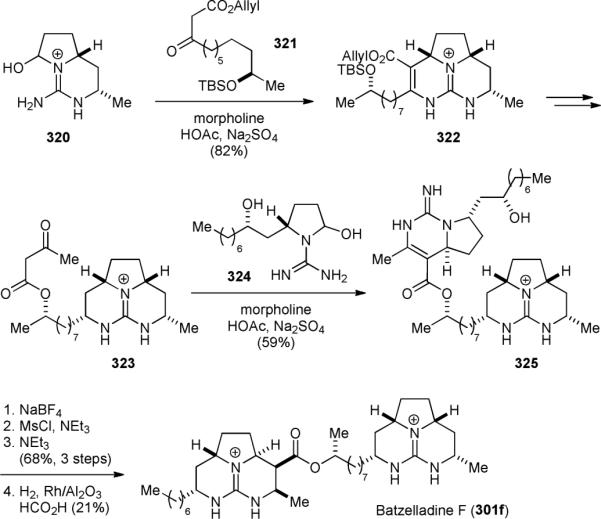
Overman's synthesis of batzelladine F (301f).
5.2. Synthetic contributions from the Snider group
The first biomimetic synthesis of the ptilomycalin A (4a) core was reported by the Snider group in 1994.391,392 They assembled the polyketide framework of 4a by a Knoevenagel condensation of 326 and 327 (Fig. 56). Reaction of the resulting double Michael acceptor 328 with O-methylisourea gave 329 as a 4:1 mixture of C10/C13 diastereomers. Subsequent treatment with ammonia provided the central tricyclic guanidinium core 330 as a 1:1 mixture of C10/C13 diastereomers. Removal of the silyl protecting groups under acidic conditions induced the formation of the seven-membered spiroaminal to yield 331. The formation of the six-membered spiroaminal was achieved under basic conditions to give the ptilomycalin core skeleton 332, which could be hydrolyzed back to 331.
Figure 56.
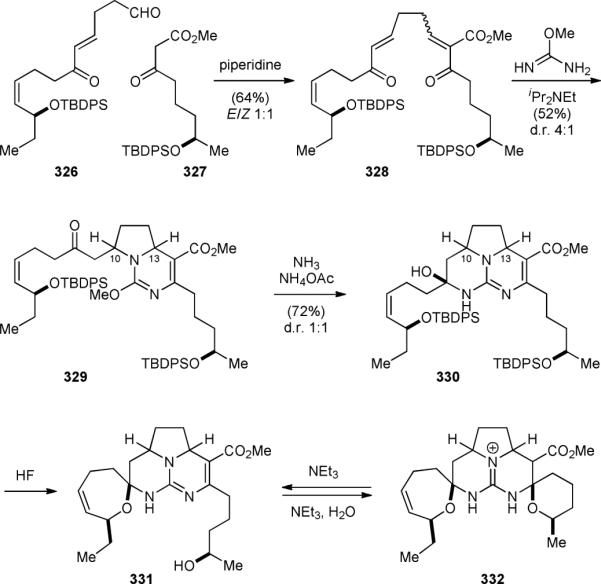
Snider's biomimetic synthesis of ptilomycalin A (4a) core.
The Snider group later used this flexible approach to achieve the first synthesis of a batzelladine alkaloid in 1998.393-395 Impressively, batzelladine E (301e) was obtained in only nine steps. Their synthetic studies also indicated that the relative stereochemistries of the tricyclic core of batzelladines A, D and F (301a,d,f) were originally misassigned.393,395
5.3. Synthetic contributions from the Murphy group
The Murphy group also used a biomimetic double Michael addition/bis-spirocyclization approach to synthesize the pentacyclic core skeleton of ptilomycalin A (4a).333,396-399 Unlike Snider who used O-methylisourea as the guanidine surrogate, Murphy used guanidine directly to construct the central core skeleton of 4a by a double Michael addition reaction. The Murphy group also independently found that the originally proposed relative stereochemistry of the tricyclic core of batzelladine F (301f) was incorrect.400-403 They completed the biomimetic synthesis of crambescidin 359 (4b) in 2003.404,405 The synthesis of 4b started with the Wittig reaction of 326 and 333 (Fig. 57). Condensation of the resulting bis-enone 334 with guanidine provided the tricyclic core of 4b. Subsequently, one of the silyl protecting groups was selectively removed upon treatment with acid. This operation also induced the formation of the six-membered spiroaminal. Finally, desilylation of 335 followed by another spiro-cyclization under acidic conditions provided 4b.
Figure 57.
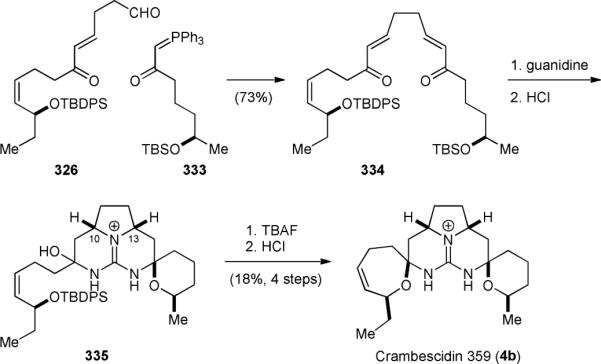
Murphy's biomimetic synthesis of crambescidin 359 (4b).
5.4. Synthetic contributions from the Rao group
Rao and co-workers reported a chiral synthon approach for the synthesis of the batzelladine core skeleton in 1995.406 The commercially available azetidinone derivative 336 was used as the source of chirality (Fig. 58). Coupling of 336 with the 2-(1,3-dioxolan-2-yl)ethyl Grignard reagent followed by hydrolytic cleavage of the β-lactam ring gave 337. The removal of the acetal protecting group and the oxidation of the resulting aldehyde were accomplished in one step by treating 337 with the Jones reagent. Subsequent Lawesson thionation gave 338. The remaining polyketide framework was then introduced by Eschenmoser sulfide contraction to provide 339. Next, reduction of the enone group of 339, introduction of the C30 nitrogen atom by a Mitsunobu reaction, and manipulations of the protecting groups afforded 340. The C32 nitrogen atom was introduced by another Mitsunobu reaction. Treatment of 341 with acid removed the amino protecting groups. The cyclic urea was then introduced to yield 342. Finally, cyclic guanidine formation via the O-methylurea of 342 provided 343, the originally proposed core structure of batzelladine A (301a).
Figure 58.
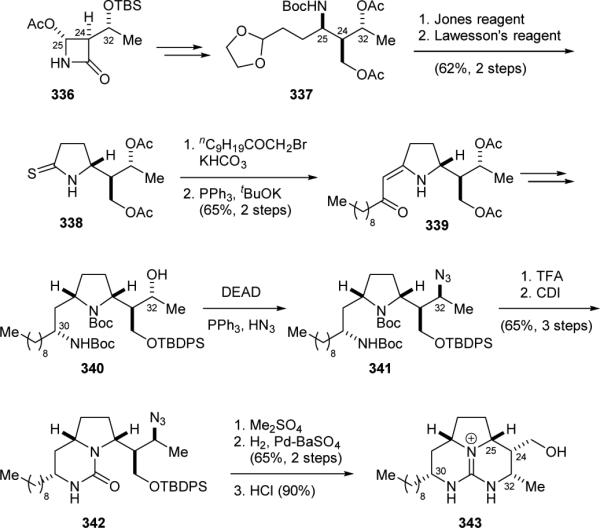
Rao's synthesis of the originally proposed tricyclic core of batzelladine A (301a).
5.5. Synthetic contributions from the Hiemstra group
The Hiemstra group reported a racemic approach for the synthesis of the tricyclic core skeleton of ptilomycalin A (4a) in 1996.407-409 The overall strategy is similar to that developed by the Rao group.406 Coupling of 344 and 345 gave 346 via an N-acyliminium ion (Fig. 59). The Eschenmoser condensation of the thiolactam of 346 and bromoacetophenone gave 347. Subsequent sulfide contraction provided 348. After adjustment of the oxidation state and protection of the amino group, the resulting 349 was converted to a mixture of mono- and bis-ketals. Finally, guanidination and bis-cyclization yielded 350.
Figure 59.
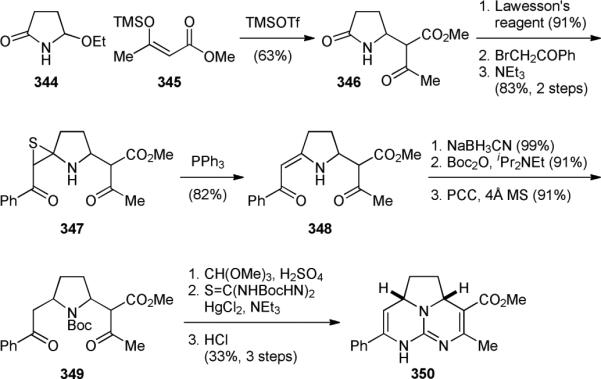
Hiemstra's synthesis of the tricyclic core of ptilomycalin A (4a).
5.6. Synthetic contributions from the Nagasawa group
The Nagasawa group reported a successive nitrone-olefin [3+2] cycloaddition approach for the synthesis of the core skeleton of ptilomycalin A in 2000.410,411 They then applied the [3+2] cycloaddition approach to the syntheses of crambescidin 359 (4b)412 and batzelladines A, D, and K (301a,d,k)413-419 during 2002–2010. The anion-binding ability of this cage-like pentacyclic scaffold has also inspired them to design a C2-symmetric phase-transfer catalyst for the asymmetric alkylation of glycinate Schiff bases.334
The synthesis of the bicyclic core of batzelladine A (301a) started with reacting nitrone 351 with olefin 352 to give 353 (Fig. 60).416 The bulky silyloxy group of 351 was used to control the stereochemistry of the nitrone–olefin [3+2] cycloaddition reaction. The ester group of 353 was then reduced and the silyl protecting group removed to yield 354. Selective protection of the primary alcohol followed by deoxygenation of the secondary alcohol and cleavage of the N–O linkage gave 355. Subsequently, installation of the guanidine group yielded 356, which was cyclized to generate the bicyclic core 357. The rhuthenium-catalyzed oxidation of 357 proceeded through an iminium intermediate to provide enal 358. Lindgren oxidation of 358 gave the corresponding carboxylic acid, which was converted into a methyl ester to facilitate the purification process. After thiolate-mediated ester hydrolysis and amide coupling with 359, the bicyclic guanidinium core 360 was obtained upon removal of the hydroxyl protecting group.
Figure 60.
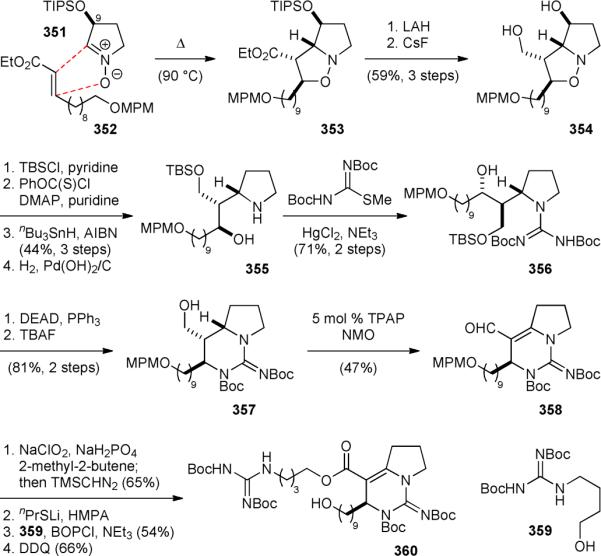
Nagasawa's synthesis of the bicyclic core of batzelladine A (301a).
The synthesis of the tricyclic guanidinium core of 301a began with the nitrone–olefin [3+2] cycloaddition reaction of 351 and 1-undecene (Scheme 61).416 Deoxygenation of the resulting 361 afforded 362, which was oxidized to provide nitrone 363 with good regioselectivity. The [3+2] cycloaddition reaction of 363 and methyl crotonate yielded 364 selectively. After N–O cleavage, the guanidine group was introduced to afford 365. Coupling of the two fragments 360 and 365 gave 366. Deprotection and cyclic guanidine formation via a Mitsunobu reaction completed the synthesis of 301a.
5.7. Synthetic contributions from the Gin group
The Gin group has made significant contributions to the field of the synthesis of crambescidin/batzelladine alkaloids. They first used the Eschenmoser sulfide contraction approach to synthesize the bicyclic core skeleton of batzelladine A (301a) and established its absolute stereochemistry in 2001.420 They then developed an elegant diastereoselective [4+2] annulation strategy and completed the syntheses of batzelladine D (301d),421 batzelladine A (301a),422 and crambidine (300)423 in 2005, 2006, and 2010, respectively.
The synthesis of crambidine (300) features a [4+2] annulation of a thioimidate with a vinyl carbodiimide, and a hydroamination of an alkyne with a 2-aminopyrimidine nucleophile.423 To prepare the carbodiimide fragment, alkyne 367 was acylated and subjected to an azide conjugate addition to give β-azidoacrylate 368 as a mixture of E/Z-isomers (Fig. 62). After the Staudinger reduction and in situ condensation of the resulting enamine with siloxymethyl isocyanate, the two isomers converged to yield (E)-carbodiimide 369. The preparation of the imine fragment started with coupling 370 and 371 to provide pyrrolidinone 372. Lawesson thionation followed by S-methylation gave thioimidate 373. Reaction of carbodiimide 369 and thioimidate 373 provided bicyclic pyrimidine 374. The siloxymethyl protecting group was then removed and the resulting 2-aminopyrimidine was subjected to a gold(III)-catalyzed intramolecular alkyne hydroamination reaction to afford tricyclic pyrimidium core (Z)-375 with good stereoselectivity. Treating 375 with acid induced the spiroaminal formation to yield 376. Finally, deprotection of 376 and coupling with 377 completed the synthesis of 300.
Figure 62.

Gin's synthesis of crambidine (300).
5.8. Synthetic contributions from the P.A. Evans group
The Evans group developed a unique radical cyclization strategy for the synthesis of the core skeleton of batzelladines in 2001,424 and subsequently completed the synthesis of batzelladine D (301d) in 2007.425 This convergent synthesis started with the treatment of 378 with octanyl cuprate followed by lithium trimethylsulfoxonium ylide to give anti-diol 379 (Fig. 63). Carbonate formation then yielded 380, which was subjected to a rhodium-catalyzed allylic amination reaction to give 381 with excellent regio- and stereoselectivities. The dihydropyrimidinone nucleophile was prepared by a Bignelli condensation. Hydrosilylation of 381 followed by transesterification, and conversion of the hydroxyl group to an azido group using the Mitsunobu method provided 382. Whereas the platinum-catalyzed hydrosilylation of 381 could also deliver the desire product smoothly, poor regioselectivity or reactivity was observed with hydroboration and other hydrometallation reactions of 381. Tamao–Fleming oxidation of 382 followed by conversion of the resulting alcohol into an iodide afforded 383, setting the stage for the key radical cyclization reaction. Treatment of 383 with tributyltin hydride, triethylborane, and oxygen initiated a radical cyclization reaction to provide the bicyclic core 384. In contrast, attempts to initiate the radical cyclization with AIBN led to competitive azide reduction. After removing the chiral sulfonyl group of 384, the cyclic urea was methylated to afford 385. Reduction of bis-azide 385 gave a diamine that cyclized under the reaction conditions to give the tricyclic core of batzelladines. Finally, installation of the acyclic guanidinium furnished 301d. Remarkably, starting from the chiral bis-epoxide 378, 301d could be obtained in 14 steps and 10% overall yield.
Figure 63.

Evan's synthesis of batzelladine D (301d).
5.9. Synthetic contributions from the Elliott group
The Elliott group reported an asymmetric synthesis of the methyl ester of batzelladine C (301c) in 2009.426-429 Similar to the Rao, Hiemstra, and Gin groups, they also used the Eschenmoser sulfide contraction reaction to derivatize the γ-lactam ring in preparing alkylidenepyrrolidine 387 from 386 (Fig. 64). They then constructed the bicyclic thiourea 388 by condensing 387 with hexanal and trimethylsilyl isothiocyante, a reaction previously developed by Kishi for the synthesis of saxitoxin (192a).259 Subsequently, conversion of the thiourea group into a guanidine group provided 389, which was transformed into 390 using the iodocyclization method developed by Gin for the synthesis of batzelladine D (301d).421 Finally, dehalogenation of 390 afforded 391, the methyl ester of 301c.
Figure 64.
Elliott's synthesis of “batzelladine C methyl ester” (391).
5.10. Synthetic contributions from the Wolfe group
The Wolfe group has developed a series of palladium-catalyzed olefin carboamination reactions430 and demonstrated their synthetic utility by applying the iterative carboamination reactions to the synthesis of merobatzelladine B (398) in 2012.431 Briefly, reaction of 392 with bromovinyltrimethylsilane in the presence of a catalytic amount of palladium gave pyrrolidine 393 with complete stereochemical control (Fig. 65). Subsequent protodesilylation, deprotection of the pyrrolidine, and urea formation yielded 394. The second carboamination reaction also proceeded well and the coupling of 394 with (Z)-1-bromobutene gave 395 with excellent stereoselectivity. Conversion of cyclic urea 395 into cyclic guanidine 396 followed by hydrogenation, Mitsunobu cyclization, and deprotection completed the synthesis of merobatzelladine B (397).
Figure 65.
Wolfe's synthesis of merobatzelladine B (397).
6. Outlook
The biological properties associated with the guanidine-containing natural products have made them valuable tools for studying protein functions. In addition, their complex chemical structures have inspired the development of many synthetic methods. While the special chemical reactivity of many cyclic guanidine-containing alkaloids remains poorly understood, significant progress has been made over the past decades. We anticipate that the utilities of this unique class of small molecules will continue to be unveiled through the chemical biological studies enabled by the newly developed chemical tools.
Figure 61.
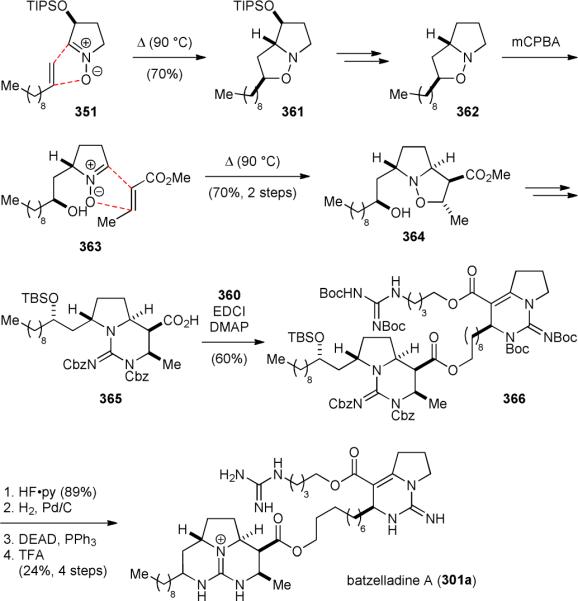
Nagasawa's synthesis of batzelladine A (301a).
Acknowledgments
Financial Support was provided by NIH (NIGMS R01-GM079554), the Welch Foundation (I-1596), and UT Southwestern. C.C. is a Southwestern Medical Foundation Scholar in Biomedical Research.
Biography
 Yuyong Ma was born in Xinyang, Henan, China in 1986. He received his B.S. degrees from Huazhong University of Science and Technology in 2008 working in the laboratory of Professor Jin Nie. Upon completion of his doctoral studies at Shanghai Institute of Organic Chemistry under the direction of Professor Biao Yu, he joined the laboratory of Professor Chuo Chen to study natural product synthesis.
Yuyong Ma was born in Xinyang, Henan, China in 1986. He received his B.S. degrees from Huazhong University of Science and Technology in 2008 working in the laboratory of Professor Jin Nie. Upon completion of his doctoral studies at Shanghai Institute of Organic Chemistry under the direction of Professor Biao Yu, he joined the laboratory of Professor Chuo Chen to study natural product synthesis.
 Saptarshi De was born in Kolkata, India in 1985. After receiving his M.S. degree from Indian Institute of Technology (IIT) Kanpur, India in 2008, he moved to USA to pursue doctoral studies at Wayne State University under the guidance of Professor James H. Rigby. He is currently studying natural product synthesis as a postdoctoral research fellow at UT Southwestern Medical Center in Professor Chuo Chen's laboratory.
Saptarshi De was born in Kolkata, India in 1985. After receiving his M.S. degree from Indian Institute of Technology (IIT) Kanpur, India in 2008, he moved to USA to pursue doctoral studies at Wayne State University under the guidance of Professor James H. Rigby. He is currently studying natural product synthesis as a postdoctoral research fellow at UT Southwestern Medical Center in Professor Chuo Chen's laboratory.
 Chuo Chen was born in Taipei, Taiwan, ROC in 1973. He worked in the laboratory of Professor Tien-Yau Luh as an undergraduate student at National Taiwan University and obtained his B.S. degree in 1995. He then studied natural product synthesis under the direction of Professor Matthew D. Shair at Harvard University, where he received his Ph.D. degree in 2001. He then joined Professor Stuart L. Schreiber's laboratory at Harvard University as a Howard Hughes Medical Institute (HHMI) postdoctoral fellow. He is presently an associate professor and a Southwestern Medical Foundation Scholar in Biomedical Research at UT Southwestern Medical Center. His current research interest includes natural product synthesis, synthetic methodology, and chemical biology.
Chuo Chen was born in Taipei, Taiwan, ROC in 1973. He worked in the laboratory of Professor Tien-Yau Luh as an undergraduate student at National Taiwan University and obtained his B.S. degree in 1995. He then studied natural product synthesis under the direction of Professor Matthew D. Shair at Harvard University, where he received his Ph.D. degree in 2001. He then joined Professor Stuart L. Schreiber's laboratory at Harvard University as a Howard Hughes Medical Institute (HHMI) postdoctoral fellow. He is presently an associate professor and a Southwestern Medical Foundation Scholar in Biomedical Research at UT Southwestern Medical Center. His current research interest includes natural product synthesis, synthetic methodology, and chemical biology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Coles MP. Chem. Commun. 2009:3659. doi: 10.1039/b901940e. [DOI] [PubMed] [Google Scholar]
- 2.Greenhill JV, Lue P. Prog. Med. Chem. 1993;30:203. doi: 10.1016/s0079-6468(08)70378-3. [DOI] [PubMed] [Google Scholar]
- 3.Leow D, Tan C-H. Synlett. 2010:1589. [Google Scholar]
- 4.Selig P. Synthesis. 2013;45:703. [Google Scholar]
- 5.Rozas I, Alkorta I, Elguero J. Struct. Chem. 2008;19:923. [Google Scholar]
- 6.Gallivan JP, Dougherty DA. Proc. Natl. Acad. Sci., U.S.A. 1999;96:9459. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blanco F, Kelly B, Alkorta I, Rozas I, Elguero J. Chem. Phys. Lett. 2011;511:129. [Google Scholar]
- 8.Berlinck RG. S. Nat. Prod. Rep. 1999;16:339. [Google Scholar]
- 9.Berlinck RG. S. Nat. Prod. Rep. 2002;19:617. doi: 10.1039/a901981b. [DOI] [PubMed] [Google Scholar]
- 10.Berlinck RGS, Kossuga MH. Nat. Prod. Rep. 2005;22:516. doi: 10.1039/b209227c. [DOI] [PubMed] [Google Scholar]
- 11.Berlinck RGS, Burtoloso ACB, Kossuga MH. Nat. Prod. Rep. 2008;25:919. doi: 10.1039/b507874c. [DOI] [PubMed] [Google Scholar]
- 12.Berlinck RGS, Burtoloso ACB, Trindade-Silva AE, Romminger S, Morais RP, Bandeira K, Mizuno CM. Nat. Prod. Rep. 2010;27:1871. doi: 10.1039/c0np00016g. [DOI] [PubMed] [Google Scholar]
- 13.Berlinck RGS, Trindade-Silva AE, Santos MF. C. Nat. Prod. Rep. 2012;29:1382. doi: 10.1039/c2np20071f. [DOI] [PubMed] [Google Scholar]
- 14.Katritzky AR, Rogovoy BV. Arkivoc. 2005;(4):49. [Google Scholar]
- 15.Suhs T, Konig B. Mini-Rev. Org. Chem. 2006;3:315. [Google Scholar]
- 16.Ishikawa T. Chem. Pharm. Bull. 2010;58:1555. doi: 10.1248/cpb.58.1555. [DOI] [PubMed] [Google Scholar]
- 17.Rauws TRM, Maes BU. W. Chem. Soc. Rev. 2012;41:2463. doi: 10.1039/c1cs15236j. [DOI] [PubMed] [Google Scholar]
- 18.Kinnel RB, Gehrken H-P, Scheuer PJ. J. Am. Chem. Soc. 1993;115:3376. [Google Scholar]
- 19.Cutignano A, Bifulco G, Bruno I, Casapullo A, Gomez-Paloma L, Riccio R. Tetrahedron. 2000;56:3743. [Google Scholar]
- 20.Woodward RB. Pure Appl. Chem. 1964;9:49. [Google Scholar]
- 21.Berlinck RGS, Braekman JC, Daloze D, Bruno I, Riccio R, Ferri S, Spampinato S, Speroni E. J. Nat. Prod. 1993;56:1007. doi: 10.1021/np50097a004. [DOI] [PubMed] [Google Scholar]
- 22.Hoffmann H, Lindel T. Synthesis. 2003;35:1753. [Google Scholar]
- 23.Jacquot DEN, Lindel T. Curr. Org. Chem. 2005;9:1551. [Google Scholar]
- 24.Du H, He Y, Sivappa R, Lovely CJ. Synlett. 2006:965. [Google Scholar]
- 25.Köck M, Grube A, Seiple IB, Baran PS. Angew. Chem. Int. Ed. 2007;46:6586. doi: 10.1002/anie.200701798. [DOI] [PubMed] [Google Scholar]
- 26.Weinreb SM. Nat. Prod. Rep. 2007;24:931. doi: 10.1039/b700206h. [DOI] [PubMed] [Google Scholar]
- 27.Arndt H-D, Riedrich M. Angew. Chem. Int. Ed. 2008;47:4785. doi: 10.1002/anie.200801793. [DOI] [PubMed] [Google Scholar]
- 28.Heasley B. Eur. J. Org. Chem. 2009:1477. [Google Scholar]
- 29.Forte B, Malgesini B, Piutti C, Quartieri F, Scolaro A, Papeo G. Mar. Drugs. 2009;7:705. doi: 10.3390/md7040705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shenvi RA, O'Malley DP, Baran PS. Acc. Chem. Res. 2009;42:530. doi: 10.1021/ar800182r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaich T, Baran PS. J. Org. Chem. 2010;75:4657. doi: 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]
- 32.Al-Mourabit A, Zancanella MA, Tilvi S, Romo D. Nat. Prod. Rep. 2011;28:1229. doi: 10.1039/c0np00013b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Ma Z, Wang X, De S, Ma Y, Chen C. Chem. Commun. 2014;50:8628. doi: 10.1039/c4cc02290d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garg NK, Stoltz BM. Chem. Commun. 2006:3769. doi: 10.1039/b605929e. [DOI] [PubMed] [Google Scholar]
- 35.Mollica A, Locatelli M, Stefanucci A, Pinnen F. Molecules. 2012;17:6083. doi: 10.3390/molecules17056083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Llewellyn LE. Nat. Prod. Rep. 2006;23:200. doi: 10.1039/b501296c. [DOI] [PubMed] [Google Scholar]
- 37.Wiese M, D'Agostino PM, Mihali TK, Moffitt MC, Neilan BA. Mar. Drugs. 2010;8:2185. doi: 10.3390/md8072185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cusick KD, Sayler GS. Mar. Drugs. 2013;11:991. doi: 10.3390/md11040991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thottumkara AP, Parsons WH, Du Bois J. Angew. Chem. Int. Ed. 2014;53:5760. doi: 10.1002/anie.201308235. [DOI] [PubMed] [Google Scholar]
- 40.Koert U. Angew. Chem. Int. Ed. 2004;43:5572. doi: 10.1002/anie.200461097. [DOI] [PubMed] [Google Scholar]
- 41.Chau J, Ciufolini MA. Mar. Drugs. 2011;9:2046. doi: 10.3390/md9102046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishikawa T, Isobe M. Chem. Rec. 2013;13:286. doi: 10.1002/tcr.201200025. [DOI] [PubMed] [Google Scholar]
- 43.Moczydlowski EG. Toxicon. 2013;63:165. doi: 10.1016/j.toxicon.2012.11.026. [DOI] [PubMed] [Google Scholar]
- 44.Heys L, Moore CG, Murphy PJ. Chem. Soc. Rev. 2000;29:57. [Google Scholar]
- 45.Aron ZD, Overman LE. Chem. Commun. 2003:253. doi: 10.1039/b309910e. [DOI] [PubMed] [Google Scholar]
- 46.Nagasawa K, Hashimoto Y. Chem. Rec. 2003;3:201. doi: 10.1002/tcr.10064. [DOI] [PubMed] [Google Scholar]
- 47.Al Mourabit A, Potier P. Eur. J. Org. Chem. 2001:237. [Google Scholar]
- 48.Foley LH, Büchi G. J. Am. Chem. Soc. 1982;104:1776. [Google Scholar]
- 49.Stout EP, Wang Y-G, Romo D, Molinski TF. Angew. Chem. Int. Ed. 2012;51:4877. doi: 10.1002/anie.201108119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stout EP, Morinaka BI, Wang Y-G, Romo D, Molinski TF. J. Nat. Prod. 2012;75:527. doi: 10.1021/np300051k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muñoz J, Moriou C, Gallard J-F, Marie PD, Al-Mourabit A. Tetrahedron Lett. 2012;53:5828. [Google Scholar]
- 52.Pöverlein C, Breckle G, Lindel T. Org. Lett. 2006;8:819. doi: 10.1021/ol0526219. [DOI] [PubMed] [Google Scholar]
- 53.Reyes JCP, Romo D. Angew. Chem. Int. Ed. 2012;51:6870. doi: 10.1002/anie.201200959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nguyen TNT, Tepe JJ. Curr. Med. Chem. 2009;16:3122. doi: 10.2174/092986709788803015. [DOI] [PubMed] [Google Scholar]
- 55.Annoura H, Tatsuoka T. Tetrahedron Lett. 1995;36:413. [Google Scholar]
- 56.Xu Y.-z., Yakushijin K, Horne DA. J. Org. Chem. 1997;62:456. doi: 10.1021/jo9619746. [DOI] [PubMed] [Google Scholar]
- 57.Sosa ACB, Yakushijin K, Horne DA. J. Org. Chem. 2000;65:610. doi: 10.1021/jo991277o. [DOI] [PubMed] [Google Scholar]
- 58.Portevin B, Golsteyn RM, Pierré A, De Nanteuil G. Tetrahedron Lett. 2003;44:9263. [Google Scholar]
- 59.Wan Y, Hur W, Cho CY, Liu Y, Adrian FJ, Lozach O, Bach S, Mayer T, Fabbro D, Meijer L, Gray NS. Chem. Biol. 2004;11:247. doi: 10.1016/j.chembiol.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 60.Papeo G, Posteri H, Borghi D, Varasi M. Org. Lett. 2005;7:5641. doi: 10.1021/ol052266m. [DOI] [PubMed] [Google Scholar]
- 61.He Q, Chen W, Qin Y. Tetrahedron Lett. 2007;48:1899. [Google Scholar]
- 62.Papeo G, Frau MAG-Z, Borghi D, Varasi M. Tetrahedron Lett. 2005;46:8635. [Google Scholar]
- 63.Patel J, Pelloux-Léon N, Minassian F, Vallée Y. Tetrahedron Lett. 2006;47:5561. [Google Scholar]
- 64.Mukherjee S, Sivappa R, Yousufuddin M, Lovely CJ. Org. Lett. 2010;12:4940. doi: 10.1021/ol1020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stien D, Anderson GT, Chase CE, Koh Y.-h., Weinreb SM. J. Am. Chem. Soc. 1999;121:9574. [Google Scholar]
- 66.Feldman KS, Saunders JC. J. Am. Chem. Soc. 2002;124:9060. doi: 10.1021/ja027121e. [DOI] [PubMed] [Google Scholar]
- 67.Hale KJ, Domostoj MM, Tocher DA, Irving E, Scheinmann F. Org. Lett. 2003;5:2927. doi: 10.1021/ol035036l. [DOI] [PubMed] [Google Scholar]
- 68.Domostoj MM, Irving E, Scheinmann F, Hale KJ. Org. Lett. 2004;6:2615. doi: 10.1021/ol0490476. [DOI] [PubMed] [Google Scholar]
- 69.Davis FA, Deng J. Org. Lett. 2005;7:621. doi: 10.1021/ol047634l. [DOI] [PubMed] [Google Scholar]
- 70.Trost BM, Dong G. J. Am. Chem. Soc. 2006;128:6054. doi: 10.1021/ja061105q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Endeshaw MM, Bayer A, Hansen LK, Gautun OR. Eur. J. Org. Chem. 2006:5249. [Google Scholar]
- 72.Ichikawa Y, Yamaoka T, Nakano K, Kotsuki H. Org. Lett. 2007;9:2989. doi: 10.1021/ol0709735. [DOI] [PubMed] [Google Scholar]
- 73.Yoshimitsu T, Ino T, Tanaka T. Org. Lett. 2008;10:5457. doi: 10.1021/ol802225g. [DOI] [PubMed] [Google Scholar]
- 74.Yoshimitsu T, Ino T, Futamura N, Kamon T, Tanaka T. Org. Lett. 2009;11:3402. doi: 10.1021/ol9012684. [DOI] [PubMed] [Google Scholar]
- 75.Wehn PM, Du Bois J. Angew. Chem. Int. Ed. 2009;48:3802. doi: 10.1002/anie.200806292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dickson DP, Wardrop DJ. Org. Lett. 2009;11:1341. doi: 10.1021/ol900133v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hama N, Matsuda T, Sato T, Chida N. Org. Lett. 2009;11:2687. doi: 10.1021/ol900799e. [DOI] [PubMed] [Google Scholar]
- 78.Movassaghi M, Siegel DS, Han S. Chem. Sci. 2010;1:561. doi: 10.1039/c0sc00351d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Han S, Siegel DS, Morrison KC, Hergenrother PJ, Movassaghi M. J. Org. Chem. 2013;78:11970. doi: 10.1021/jo4020112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Menjo Y, Hamajima A, Sasaki N, Hamada Y. Org. Lett. 2011;13:5744. doi: 10.1021/ol2023054. [DOI] [PubMed] [Google Scholar]
- 81.Kano T, Sakamoto R, Akakura M, Maruoka K. J. Am. Chem. Soc. 2012;134:7516. doi: 10.1021/ja301120z. [DOI] [PubMed] [Google Scholar]
- 82.Duspara PA, Batey RA. Angew. Chem. Int. Ed. 2013;52:10862. doi: 10.1002/anie.201304759. [DOI] [PubMed] [Google Scholar]
- 83.Wiese KJ, Yakushijin K, Horne DA. Tetrahedron Lett. 2002;43:5135. [Google Scholar]
- 84.Poullennec KG, Romo D. J. Am. Chem. Soc. 2003;125:6344. doi: 10.1021/ja034575i. [DOI] [PubMed] [Google Scholar]
- 85.Wang S, Romo D. Angew. Chem. Int. Ed. 2008;47:1284. doi: 10.1002/anie.200703998. [DOI] [PubMed] [Google Scholar]
- 86.Chung R, Yu E, Incarvito CD, Austin DJ. Org. Lett. 2004;6:3881. doi: 10.1021/ol0490532. [DOI] [PubMed] [Google Scholar]
- 87.Feldman KS, Skoumbourdis AP. Org. Lett. 2005;7:929. doi: 10.1021/ol0500113. [DOI] [PubMed] [Google Scholar]
- 88.Zöllinger M, Mayer P, Lindel T. Synlett. 2007:2756. [Google Scholar]
- 89.Imaoka T, Iwamoto O, Noguchi K.-i., Nagasawa K. Angew. Chem. Int. Ed. 2009;48:3799. doi: 10.1002/anie.200806233. [DOI] [PubMed] [Google Scholar]
- 90.Hewlett NM, Tepe JJ. Org. Lett. 2011;13:4550. doi: 10.1021/ol201741r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Walker RP, Faulkner DJ, Engen DV, Clardy J. J. Am. Chem. Soc. 1981;103:6772. [Google Scholar]
- 92.Keifer PA, Schwartz RE, Koker MES, Hughes RG, Jr., Rittschof D, Rinehart KL. J. Org. Chem. 1991;56:2965. [Google Scholar]
- 93.Kobayashi J, Tsuda M, Ohizumi Y. Experientia. 1991;47:301. doi: 10.1007/BF01958167. [DOI] [PubMed] [Google Scholar]
- 94.Eder C, Proksch P, Wray V, van Soest RWM, Ferdinandus E, Pattisina LA. Sudarsono J. Nat. Prod. 1999;62:1295. doi: 10.1021/np990071f. [DOI] [PubMed] [Google Scholar]
- 95.Appenzeller J, Tilvi S, Martin M-T, Gallard J-F, El-bitar H, Dau ETH, Debitus C, Laurent D, Moriou C, Al-Mourabit A. Org. Lett. 2009;11:4874. doi: 10.1021/ol901946h. [DOI] [PubMed] [Google Scholar]
- 96.Kubota T, Araki A, Yasuda T, Tsuda M, Fromont J, Aoyama K, Mikami Y, Wälchli MR, Kobayashi J. Tetrahedron Lett. 2009;50:7268. [Google Scholar]
- 97.Tilvi S, Moriou C, Martin M-T, Gallard J-F, Sorres J, Patel K, Petek S, Debitus C, Ermolenko L, Al-Mourabit A. J. Nat. Prod. 2010;73:720. doi: 10.1021/np900539j. [DOI] [PubMed] [Google Scholar]
- 98.Kobayashi J, Tsuda M, Murayama T, Nakamura H, Ohizumi Y, Ishibashi M, Iwamura M, Ohta T, Nozoe S. Tetrahedron. 1990;46:5579. [Google Scholar]
- 99.Williams DH, Faulkner DJ. Tetrahedron. 1996;52:5381. [Google Scholar]
- 100.Endo T, Tsuda M, Okada T, Mitsuhashi S, Shima H, Kikuchi K, Mikami Y, Fromont J, Kobayashi J. J. Nat. Prod. 2004;67:1262. doi: 10.1021/np034077n. [DOI] [PubMed] [Google Scholar]
- 101.Kinnel RB, Gehrken H-P, Swali R, Skoropowski G, Scheuer PJ. J. Org. Chem. 1998;63:3281. [Google Scholar]
- 102.Kobayashi J, Suzuki M, Tsuda M. Tetrahedron. 1997;53:15681. [Google Scholar]
- 103.Buchanan MS, Carroll AR, Addepalli R, Avery VM, Hooper JNA, Quinn RJ. J. Org. Chem. 2007;72:2309. doi: 10.1021/jo062007q. [DOI] [PubMed] [Google Scholar]
- 104.Kato T, Shizuri Y, Izumida H, Yokoyama A, Endo M. Tetrahedron Lett. 1995;36:2133. [Google Scholar]
- 105.Grube A, Köck M. Angew. Chem. Int. Ed. 2007;46:2320. doi: 10.1002/anie.200604076. [DOI] [PubMed] [Google Scholar]
- 106.Kobayashi H, Kitamura K, Nagai K, Nakao Y, Fusetani N, van Soest RWM, Matsunaga S. Tetrahedron Lett. 2007;48:2127. [Google Scholar]
- 107.Haber M, Carbone M, Ilan M, Gavagnin M. Arkivoc. 2010;(2):233. [Google Scholar]
- 108.Urban S, Leone P. d. A., Carroll AR, Fechner GA, Smith J, Hooper JNA, Quinn RJ. J. Org. Chem. 1999;64:731. doi: 10.1021/jo981034g. [DOI] [PubMed] [Google Scholar]
- 109.Yasuda T, Araki A, Kubota T, Ito J, Mikami Y, Fromont J, Kobayashi J. J. Nat. Prod. 2009;72:488. doi: 10.1021/np800645q. [DOI] [PubMed] [Google Scholar]
- 110.Nishimura S, Matsunaga S, Shibazaki M, Suzuki K, Furihata K, van Soest RWM, Fusetani N. Org. Lett. 2003;5:2255. doi: 10.1021/ol034564u. [DOI] [PubMed] [Google Scholar]
- 111.Grube A, Immel S, Baran PS, Köck M. Angew. Chem. Int. Ed. 2007;46:6721. doi: 10.1002/anie.200701935. [DOI] [PubMed] [Google Scholar]
- 112.Zhang H, Khalil Z, Conte MM, Plisson F, Capon RJ. Tetrahedron Lett. 2012;53:3784. [Google Scholar]
- 113.Grube A, Köck M. Org. Lett. 2006;8:4675. doi: 10.1021/ol061317s. [DOI] [PubMed] [Google Scholar]
- 114.Buchanan MS, Carroll AR, Quinn RJ. Tetrahedron Lett. 2007;48:4573. [Google Scholar]
- 115.Chang S.-j., McNally D, Shary-Tehrany S, Hickey SMJ, Boyd RH. J. Am. Chem. Soc. 1970;92:3109. [Google Scholar]
- 116.Verevkin SP, Emel'yanenko VN, Pimerzin AA, Vishnevskaya EE. J. Phys. Chem. A. 2011;115:12271. doi: 10.1021/jp203650d. [DOI] [PubMed] [Google Scholar]
- 117.Enoki N, Furusaki A, Suehiro K, Ishida R, Matsumoto T. Tetreahedron Lett. 1983;24:4341. [Google Scholar]
- 118.Jamison TF, Shambayati S, Crowe WE, Schreiber SL. J. Am. Chem. Soc. 1994;116:5505. [Google Scholar]
- 119.Jamison TF, Shambayati S, Crowe WE, Schreiber SL. J. Am. Chem. Soc. 1997;119:4353. [Google Scholar]
- 120.Paquette LA, Sun L-Q, Friedrich D, Savage PB. J. Am. Chem. Soc. 1997;119:8438. [Google Scholar]
- 121.Overman LE, Rogers BN, Tellew JE, Trenkle WC. J. Am. Chem. Soc. 1997;119:7159. [Google Scholar]
- 122.Bélanger G, Hong F-T, Overman LE, Rogers BN, Tellew JE, Trenkle WC. J. Org. Chem. 2002;67:7880. doi: 10.1021/jo026282y. [DOI] [PubMed] [Google Scholar]
- 123.Katz JD, Overman LE. Tetrahedron. 2004;60:9559. [Google Scholar]
- 124.Lanman BA, Overman LE, Paulini R, White NS. J. Am. Chem. Soc. 2007;129:12896. doi: 10.1021/ja074939x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Starr JT, Koch G, Carreira EM. J. Am. Chem. Soc. 2000;122:8793. [Google Scholar]
- 126.Breder A, Chinigo GM, Waltman AW, Carreira EM. Angew. Chem. Int. Ed. 2008;47:8514. doi: 10.1002/anie.200803284. [DOI] [PubMed] [Google Scholar]
- 127.Chinigo GM, Breder A, Carreira EM. Org. Lett. 2011;13:78. doi: 10.1021/ol102577q. [DOI] [PubMed] [Google Scholar]
- 128.Breder A, Chinigo GM, Waltman AW, Carreira EM. Chem. Eur. J. 2011;17:12405. doi: 10.1002/chem.201101862. [DOI] [PubMed] [Google Scholar]
- 129.Claus RE, Schreiber SL. Org. Synth. 1986;64:150. [Google Scholar]
- 130.Koenig SG, Leonard KA, Löwe RS, Austin DJ. Tetrahedron Lett. 2000;41:9393. [Google Scholar]
- 131.Koenig SG, Miller SM, Leonard KA, Löwe RS, Chen BC, Austin DJ. Org. Lett. 2003;5:2203. doi: 10.1021/ol0344063. [DOI] [PubMed] [Google Scholar]
- 132.Dilley AS, Romo D. Org. Lett. 2001;3:1535. doi: 10.1021/ol015864j. [DOI] [PubMed] [Google Scholar]
- 133.Dransfield PJ, Wang S, Dilley A, Romo D. Org. Lett. 2005;7:1679. doi: 10.1021/ol0473602. [DOI] [PubMed] [Google Scholar]
- 134.Dransfield PJ, Dilley AS, Wang S, Romo D. Tetrahedron. 2006;62:5223. [Google Scholar]
- 135.Wang S, Dilley AS, Poullennec KG, Romo D. Tetrahedron. 2006;62:7155. [Google Scholar]
- 136.Zancanella MA, Romo D. Org. Lett. 2008;10:3685. doi: 10.1021/ol801289b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang Y-G, Morinaka BI, Reyes JCP, Wolff JJ, Romo D, Molinski TF. J. Nat. Prod. 2010;73:428. doi: 10.1021/np900638e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lovely CJ, Du H, Dias HV. R. Org. Lett. 2001;3:1319. doi: 10.1021/ol015687m. [DOI] [PubMed] [Google Scholar]
- 139.Lovely CJ, Du H, He Y, Dias HV. R. Org. Lett. 2004;6:735. doi: 10.1021/ol036403w. [DOI] [PubMed] [Google Scholar]
- 140.Sivappa R, Hernandez NM, He Y, Lovely CJ. Org. Lett. 2007;9:3861. doi: 10.1021/ol0711568. [DOI] [PubMed] [Google Scholar]
- 141.Kawasaki I, Sakaguchi N, Fukushima N, Fujioka N, Nikaido F, Yamashita M, Ohta S. Tetrahedron Lett. 2002;43:4377. [Google Scholar]
- 142.Kawasaki I, Sakaguchi N, Khadeer A, Yamashita M, Ohta S. Tetrahedron. 2006;62:10182. [Google Scholar]
- 143.Baran PS, Zografos AL, O'Malley DP. J. Am. Chem. Soc. 2004;126:3726. doi: 10.1021/ja049648s. [DOI] [PubMed] [Google Scholar]
- 144.Baran PS, O'Malley DP, Zografos AL. Angew. Chem. Int. Ed. 2004;43:2674. doi: 10.1002/anie.200453937. [DOI] [PubMed] [Google Scholar]
- 145.Baran PS, Li K, O'Malley DP, Mitsos C. Angew. Chem. Int. Ed. 2006;45:249. doi: 10.1002/anie.200503374. [DOI] [PubMed] [Google Scholar]
- 146.Northrop BH, O'Malley DP, Zografos AL, Baran PS, Houk KN. Angew. Chem. Int. Ed. 2006;45:4126. doi: 10.1002/anie.200600514. [DOI] [PubMed] [Google Scholar]
- 147.O'Malley DP, Li K, Maue M, Zografos AL, Baran PS. J. Am. Chem. Soc. 2007;129:4762. doi: 10.1021/ja069035a. [DOI] [PubMed] [Google Scholar]
- 148.Yamaguchi J, Seiple IB, Young IS, O'Malley DP, Maue M, Baran PS. Angew. Chem. Int. Ed. 2008;47:3578. doi: 10.1002/anie.200705913. [DOI] [PubMed] [Google Scholar]
- 149.O'Malley DP, Yamaguchi J, Young IS, Seiple IB, Baran PS. Angew. Chem. Int. Ed. 2008;47:3581. doi: 10.1002/anie.200801138. [DOI] [PubMed] [Google Scholar]
- 150.Su S, Seiple IB, Young IS, Baran PS. J. Am. Chem. Soc. 2008;130:16490. doi: 10.1021/ja8074852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Seiple IB, Su S, Young IS, Lewis CA, Yamaguchi J, Baran PS. Angew. Chem. Int. Ed. 2010;49:1095. doi: 10.1002/anie.200907112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Seiple IB, Su S, Young IS, Nakamura A, Yamaguchi J, Jørgensen L, Rodriguez RA, O'Malley DP, Gaich T, Köck M, Baran PS. J. Am. Chem. Soc. 2011;133:14710. doi: 10.1021/ja2047232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Su S, Rodriguez RA, Baran PS. J. Am. Chem. Soc. 2011;133:13922. doi: 10.1021/ja206191g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.D'Auria M, Racioppi R. J. Photochem. Photobiol., A. 1998;112:145. [Google Scholar]
- 155.Reynolds DW, Harirchian B, Chiou H-S, Marsh BK, Bauld NL. J. Phys. Org. Chem. 1989;2:57. [Google Scholar]
- 156.Leber PA, Baldwin JE. Acc. Chem. Res. 2002;35:279. doi: 10.1021/ar010100p. [DOI] [PubMed] [Google Scholar]
- 157.Kishi YJ. Nat. Prod. 1979;42:549. [Google Scholar]
- 158.Nakatsubo F, Cocuzza AJ, Keeley DE, Kishi Y. J. Am. Chem. Soc. 1977;99:4835. doi: 10.1021/ja00456a056. [DOI] [PubMed] [Google Scholar]
- 159.Nakatsubo F, Fukuyama T, Cocuzza AJ, Kishi Y. J. Am. Chem. Soc. 1977;99:8115. doi: 10.1021/ja00456a056. [DOI] [PubMed] [Google Scholar]
- 160.Fukuyama T, Nakatsubo F, Cocuzza AJ, Kishi Y. Tetreahedron Lett. 1977;18:4295. [Google Scholar]
- 161.Birman VB, Jiang X-T. Org. Lett. 2004;6:2369. doi: 10.1021/ol049283g. [DOI] [PubMed] [Google Scholar]
- 162.Garrido-Hernandez H, Nakadai M, Vimolratana M, Li Q, Doundoulakis T, Harran PG. Angew. Chem. Int. Ed. 2005;44:765. doi: 10.1002/anie.200462069. [DOI] [PubMed] [Google Scholar]
- 163.Ding H, Roberts AG, Harran PG. Angew. Chem. Int. Ed. 2012;51:4340. doi: 10.1002/anie.201200205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ding H, Roberts AG, Harran PG. Chem. Sci. 2012;4:303. doi: 10.1039/C2SC21651E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Myers AG, Yang BH, David KJ. Tetrahedron Lett. 1996;37:3623. [Google Scholar]
- 166.Cernak TA, Gleason JL. J. Org. Chem. 2008;73:102. doi: 10.1021/jo701866g. [DOI] [PubMed] [Google Scholar]
- 167.Hudon J, Cernak TA, Ashenhurst JA, Gleason JL. Angew. Chem. Int. Ed. 2008;47:8885. doi: 10.1002/anie.200803344. [DOI] [PubMed] [Google Scholar]
- 168.Bultman MS, Ma J, Gin DY. Angew. Chem. Int. Ed. 2008;47:6821. doi: 10.1002/anie.200801969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Namba K, Kaihara Y, Yamamoto H, Imagawa H, Tanino K, Williams RM, Nishizawa M. Chem. Eur. J. 2009;15:6560. doi: 10.1002/chem.200900622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fukahori Y, Takayama Y, Imaoka T, Iwamoto O, Nagasawa K. Chem. Asian J. 2012;8:244. doi: 10.1002/asia.201200820. [DOI] [PubMed] [Google Scholar]
- 171.Namba K, Inai M, Sundermeier U, Greshock TJ, Williams RM. Tetrahedron Lett. 2010;51:6557. doi: 10.1016/j.tetlet.2010.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Feldman KS, Nuriye AY. Org. Lett. 2010;12:4532. doi: 10.1021/ol1018322. [DOI] [PubMed] [Google Scholar]
- 173.Feldman KS, Nuriye AY, Li J. J. Org. Chem. 2011;76:5042. doi: 10.1021/jo200740b. [DOI] [PubMed] [Google Scholar]
- 174.Tan X, Chen C. Angew. Chem. Int. Ed. 2006;45:4345. doi: 10.1002/anie.200601208. [DOI] [PubMed] [Google Scholar]
- 175.Lu J, Tan X, Chen C. J. Am. Chem. Soc. 2007;129:7768. doi: 10.1021/ja072844p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ma Z, Lu J, Wang X, Chen C. Chem. Commun. 2011;47:427. doi: 10.1039/c0cc02214d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wang X, Ma Z, Lu J, Tan X, Chen C. J. Am. Chem. Soc. 2011;133:15350. doi: 10.1021/ja207386q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wang X, Wang X-L, Tan X, Lu J, Cormier KW, Ma Z, Chen C. J. Am. Chem. Soc. 2012;134:18834. doi: 10.1021/ja309172t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ma Z, Wang X, Wang X, Rodriguez RA, Moore CE, Gao S, Tan X, Ma Y, Rheingold AL, Baran PS, Chen C. Science. 2014;346:219. doi: 10.1126/science.1255677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wang X, Chen C. Tetrahedron. 2014 doi: 10.1016/j.tet.2014.10.027. DOI: 10.1016/j.tet.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bellville DJ, Wirth DD, Bauld NL. J. Am. Chem. Soc. 1981;103:718. [Google Scholar]
- 182.Bauld NL, Pabon R. J. Am. Chem. Soc. 1983;105:633. [Google Scholar]
- 183.Reynolds DW, Bauld NL. Tetrahedron. 1986;42:6189. [Google Scholar]
- 184.Bauld NL. Tetrahedron. 1989;45:5307. [Google Scholar]
- 185.Yueh W, Bauld NL. J. Am. Chem. Soc. 1995;117:5671. [Google Scholar]
- 186.Julliard M, Chanon M. Chem. Rev. 1983;83:425. [Google Scholar]
- 187.Zeitler K. Angew. Chem. Int. Ed. 2009;48:9785. doi: 10.1002/anie.200904056. [DOI] [PubMed] [Google Scholar]
- 188.Yoon TP, Ischay MA, Du J. Nat. Chem. 2010;2:527. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]
- 189.Ischay MA, Yoon TP. Eur. J. Org. Chem. 2012:3359. [Google Scholar]
- 190.Yoon TP. ACS Catal. 2013;3:895. doi: 10.1021/cs400088e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Teplý F. Collect. Czech. Chem. Commun. 2011;76:859. [Google Scholar]
- 192.Narayanam JMR, Stephenson CR. J. Chem. Soc. Rev. 2011;40:102. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]
- 193.Tucker JW, Stephenson CR. J. J. Org. Chem. 2012;77:1617. doi: 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]
- 194.Xuan J, Xiao W-J. Angew. Chem. Int. Ed. 2012;51:6828. doi: 10.1002/anie.201200223. [DOI] [PubMed] [Google Scholar]
- 195.Prier CK, Rankic DA, MacMillan DWC. Chem. Rev. 2013;113:5322. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ischay MA, Anzovino ME, Du J, Yoon TP. J. Am. Chem. Soc. 2008;130:12886. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]
- 197.Du J, Yoon TP. J. Am. Chem. Soc. 2009;131:14604. doi: 10.1021/ja903732v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ischay MA, Lu Z, Yoon TP. J. Am. Chem. Soc. 2010;132:8572. doi: 10.1021/ja103934y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ischay MA, Ament MS, Yoon TP. Chem. Sci. 2012;3:2807. doi: 10.1039/C2SC20658G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Du J, Skubi KL, Schultz DM, Yoon TP. Science. 2014;344:392. doi: 10.1126/science.1251511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hurtley AE, Cismesia MA, Ischay MA, Yoon TP. Tetrahedron. 2011;67:4442. doi: 10.1016/j.tet.2011.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lin S, Ischay MA, Fry CG, Yoon TP. J. Am. Chem. Soc. 2011;133:19350. doi: 10.1021/ja2093579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lu Z, Yoon TP. Angew. Chem. Int. Ed. 2012;51:10329. doi: 10.1002/anie.201204835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lu Z, Shen M, Yoon TP. J. Am. Chem. Soc. 2011;133:1162. doi: 10.1021/ja107849y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Du J, Espelt LR, Guzei IA, Yoon TP. Chem. Sci. 2011;2:2115. doi: 10.1039/C1SC00357G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Parrish JD, Ischay MA, Lu Z, Guo S, Peters NR, Yoon TP. Org. Lett. 2012;14:1640. doi: 10.1021/ol300428q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Finefield JM, Sherman DH, Kreitman M, Williams RM. Angew. Chem. Int. Ed. 2012;51:4802. doi: 10.1002/anie.201107204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kohmoto S, Kashman Y, McConnell OJ, Rinehart KL, Jr., Wright A, Koehn F. J. Org. Chem. 1988;53:3116. [Google Scholar]
- 209.Morris SA, Andersen RJ. Tetrahedron. 1990;46:715. [Google Scholar]
- 210.Fahy E, Potts BCM, Faulkner DJ, Smith K. J. Nat. Prod. 1991;54:564. [Google Scholar]
- 211.Wright AE, Pomponi SA, Cross SS, McCarthy P. J. Org. Chem. 1992;57:4772. [Google Scholar]
- 212.Capon RJ, Rooney F, Murray LM, Collins E, Sim ATR, Rostas JAP, Butler MS, Carroll AR. J. Nat. Prod. 1998;61:660. doi: 10.1021/np970483t. [DOI] [PubMed] [Google Scholar]
- 213.Garg NK, Sarpong R, Stoltz BM. J. Am. Chem. Soc. 2002;124:13179. doi: 10.1021/ja027822b. [DOI] [PubMed] [Google Scholar]
- 214.Garg NK, Caspi DD, Stoltz BM. J. Am. Chem. Soc. 2004;126:9552. doi: 10.1021/ja046695b. [DOI] [PubMed] [Google Scholar]
- 215.Caspi DD, Garg NK, Stoltz BM. Org. Lett. 2005;7:2513. doi: 10.1021/ol050952f. [DOI] [PubMed] [Google Scholar]
- 216.Garg NK, Caspi DD, Stoltz BM. Synlett. 2006:3081. [Google Scholar]
- 217.Garg NK, Caspi DD, Stoltz BM. J. Am. Chem. Soc. 2005;127:5970. doi: 10.1021/ja050586v. [DOI] [PubMed] [Google Scholar]
- 218.Mandal D, Yamaguchi AD, Yamaguchi J, Itami K. J. Am. Chem. Soc. 2011;133:19660. doi: 10.1021/ja209945x. [DOI] [PubMed] [Google Scholar]
- 219.Feldman KS, Ngernmeesri P. Org. Lett. 2005;7:5449. doi: 10.1021/ol0522081. [DOI] [PubMed] [Google Scholar]
- 220.Feldman KS, Ngernmeesri P. Org. Lett. 2010;12:4502. doi: 10.1021/ol1018008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Feldman KS, Ngernmeesri P. Org. Lett. 2011;13:5704. doi: 10.1021/ol202535f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Feldman KS, Ngernmeesri P. Synlett. 2012;23:1882. doi: 10.1055/s-0031-1290692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ikoma M, Oikawa M, Sasaki M. Tetrahedron Lett. 2008;49:7197. [Google Scholar]
- 224.Oikawa M, Ikoma M, Sasaki M. Eur. J. Org. Chem. 2011:4654. [Google Scholar]
- 225.Yang C-G, Wang J, Jiang B. Tetrahedron Lett. 2002;43:1063. [Google Scholar]
- 226.Yang C-G, Liu G, Jiang B. J. Org. Chem. 2002;67:9392. doi: 10.1021/jo026450m. [DOI] [PubMed] [Google Scholar]
- 227.Huntley RJ, Funk RL. Org. Lett. 2006;8:4775. doi: 10.1021/ol0617547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wang B, Qin H, Zhang F, Jia Y. Tetrahedron Lett. 2014;55:1561. [Google Scholar]
- 229.Schantz EJ, Mold JD, Stanger DW, Shavel J, Riel FJ, Bowden JP, Lynch JM, Wyler RS, Riegel B, Sommer H. J. Am. Chem. Soc. 1957;79:5230. [Google Scholar]
- 230.Schantz EJ, Ghazarossian VE, Schnoes HK, Strong FM, Springer JP, Pezzanite JO, Clardy J. J. Am. Chem. Soc. 1975;97:1238. doi: 10.1021/ja00838a045. [DOI] [PubMed] [Google Scholar]
- 231.Bordner J, Thiessen WE, Bates HA, Rapoport H. J. Am. Chem. Soc. 1975;97:6008. doi: 10.1021/ja00854a009. [DOI] [PubMed] [Google Scholar]
- 232.Shimizu Y, Alam M, Oshima Y, Fallon WE. Biochem. Biophys. Res. Commun. 1975;66:731. doi: 10.1016/0006-291x(75)90571-9. [DOI] [PubMed] [Google Scholar]
- 233.Oshima Y, Fallon WE, Shimizu Y, Noguchi T, Hashimoto Y. Bull. Jpn. Soc. Sci. Fish. (Nippon Suisan Gakkaishi) 1976;42:851. [Google Scholar]
- 234.Shimizu Y, Buckley LJ, Alam M, Oshima Y, Fallon WE, Kasai H, Miura I, Gullo VP, Nakanishi K. J. Am. Chem. Soc. 1976;98:5414. doi: 10.1021/ja00433a072. [DOI] [PubMed] [Google Scholar]
- 235.Oshima Y, Buckley LJ, Alam M, Shimizu Y. Comp. Biochem. Physiol. C. 1977;57:31. [Google Scholar]
- 236.Hsu CP, Marchand A, Shimizu Y, Sims GG. J. Fish. Res. Board Can. 1979;36:32. [Google Scholar]
- 237.Fuhrman FA, Fuhrman GJ, Mosher HS. Science. 1969;165:1376. doi: 10.1126/science.165.3900.1376. [DOI] [PubMed] [Google Scholar]
- 238.Brown GB, Kim YH, Küntzel H, Mosher HS, Fuhrman GJ, Fuhrman FA. Toxicon. 1977;15:115. doi: 10.1016/0041-0101(77)90030-7. [DOI] [PubMed] [Google Scholar]
- 239.Yotsu-Yamashita M, Kim YH, Dudley SC, Jr., Choudhary G, Pfahnl A, Oshima Y, Daly JW. Proc. Natl. Acad. Sci., U.S.A. 2004;101:4346. doi: 10.1073/pnas.0400368101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Yokoo A. J. Chem. Soc. Jpn. (Nippon Kagaku Zasshi) 1950;71:590. [Google Scholar]
- 241.Tsuda K, Kawamura M. J. Pharm. Soc. Jpn. (Yakugaku Zasshi) 1952;72:771. [Google Scholar]
- 242.Goto T, Kishi Y, Takahashi S, Hirata T. Tetrahedron Lett. 1963;4:2105. [Google Scholar]
- 243.Tsuda K, Ikuma S, Kawamura M, Tachikawa R, Sakai K, Tamura C, Amakasu O. Chem. Pharm. Bull. 1964;12:1357. doi: 10.1248/cpb.12.634. [DOI] [PubMed] [Google Scholar]
- 244.Yasumoto T, Yotsu M, Murata M, Naoki H. J. Am. Chem. Soc. 1988;110:2344. [Google Scholar]
- 245.Yotsu-Yamashita M, Schimmele B, Yasumoto T. Biosci. Biotechnol. Biochem. 1999;63:961. doi: 10.1271/bbb.63.961. [DOI] [PubMed] [Google Scholar]
- 246.Yotsu-Yamashita M, Goto A, Nakagawa T. Chem. Res. Toxicol. 2005;18:865. doi: 10.1021/tx050015g. [DOI] [PubMed] [Google Scholar]
- 247.Kudo Y, Yasumoto T, Konoki K, Cho Y, Yotsu-Yamashita M. Mar. Drugs. 2012;10:655–667. doi: 10.3390/md10030655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kudo Y, Finn J, Fukushima K, Sakugawa S, Cho Y, Konoki K, Yotsu-Yamashita M. J. Nat. Prod. 2014;77:1000. doi: 10.1021/np401097n. [DOI] [PubMed] [Google Scholar]
- 249.Kim YH, Brown GB, Mosher HS. Science. 1975;189:151. doi: 10.1126/science.1138374. [DOI] [PubMed] [Google Scholar]
- 250.Yotsu M, Yasumoto T, Kim YH, Naoki H, Kao CY. Tetrahedron Lett. 1990;31:3187. [Google Scholar]
- 251.Sato S, Kodama M, Ogata T, Saitanu K, Furuya M, Hirayama K, Kakinuma K. Toxicon. 1997;35:137. doi: 10.1016/s0041-0101(96)00003-7. [DOI] [PubMed] [Google Scholar]
- 252.Sheumack DD, Howden ME, Spence I, Quinn RJ. Science. 1978;199:188. doi: 10.1126/science.619451. [DOI] [PubMed] [Google Scholar]
- 253.Ritson-Williams R, Yotsu-Yamashita M, Paul VJ. Proc. Natl. Acad. Sci., U.S.A. 2006;103:3176. doi: 10.1073/pnas.0506093103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kishi Y, Aratani M, Fukuyama T, Nakatsubo F, Goto T, Inoue S, Tanino H, Sugiura S, Kakoi H. J. Am. Chem. Soc. 1972;94:9217. doi: 10.1021/ja00781a038. [DOI] [PubMed] [Google Scholar]
- 255.Kishi Y, Fukuyama T, Aratani M, Nakatsubo F, Goto T, Inoue S, Tanino H, Sugiura S, Kakoi H. J. Am. Chem. Soc. 1972;94:9219. doi: 10.1021/ja00781a039. [DOI] [PubMed] [Google Scholar]
- 256.Kishi Y, Nakatsubo F, Aratani M, Goto T, Inoue S, Kakoi H, Sugiura S. Tetrahedron Lett. 1970;11:5127. doi: 10.1016/s0040-4039(00)96956-9. [DOI] [PubMed] [Google Scholar]
- 257.Kishi Y, Nakatsubo F, Aratani M, Goto T, Inoue S, Kakoi H. Tetrahedron Lett. 1970;11:5129. doi: 10.1016/s0040-4039(00)96957-0. [DOI] [PubMed] [Google Scholar]
- 258.Taguchi H, Yazawa H, Arnett JF, Kishi Y. Tetrahedron Lett. 1977;18:627. [Google Scholar]
- 259.Tanino H, Nakata T, Kaneko T, Kishi Y. J. Am. Chem. Soc. 1977;99:2818. doi: 10.1021/ja00450a079. [DOI] [PubMed] [Google Scholar]
- 260.Kishi Y. Heterocycles. 1980;14:1477. [Google Scholar]
- 261.Hannick SM, Kishi Y. J. Org. Chem. 1983;48:3833. [Google Scholar]
- 262.Hong CY, Kishi Y. J. Am. Chem. Soc. 1992;114:7001. [Google Scholar]
- 263.Jacobi PA, Brownstein A, Martinelli M, Grozinger K. J. Am. Chem. Soc. 1981;103:239. [Google Scholar]
- 264.Jacobi PA, Martinelli MJ, Polanc S. J. Am. Chem. Soc. 1984;106:5594. [Google Scholar]
- 265.Martinelli MJ, Brownstein AD, Jacobi PA, Polanc S. Croat. Chem. Acta. 1986;59:267. [Google Scholar]
- 266.Isobe M, Nishikawa T, Pikul S, Goto T. Tetrahedron Lett. 1987;28:6485. [Google Scholar]
- 267.Isobe M, Nishikawa T, Fukami N, Goto T. Pure Appl. Chem. 1987;59:399. [Google Scholar]
- 268.Isobe M, Fukuda Y, Nishikawa T, Chabert P, Kawai T, Goto T. Tetrahedron Lett. 1990;31:3327. [Google Scholar]
- 269.Yamamoto N, Isobe M. Tetrahedron. 1993;49:6581. [Google Scholar]
- 270.Yamamoto N, Isobe M. Chem. Lett. 1994;23:2299. [Google Scholar]
- 271.Yamamoto N, Nishikawa T, Isobe M. Synlett. 1995:505. [Google Scholar]
- 272.Bamba M, Nishikawa T, Isobe M. Tetrahedron Lett. 1996;37:8199. [Google Scholar]
- 273.Bamba M, Nishikawa T, Isobe M. Synlett. 1998:371. [Google Scholar]
- 274.Nishikawa T, Ohyabu N, Yamamoto N, Isobe M. Tetrahedron. 1999;55:4325. [Google Scholar]
- 275.Nishikawa T, Asai M, Ohyabu N, Yamamoto N, Isobe M. Angew. Chem. Int. Ed. 1999;38:3081. doi: 10.1002/(sici)1521-3773(19991018)38:20<3081::aid-anie3081>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 276.Nishikawa T, Asai M, Ohyabu N, Yamamoto N, Fukuda Y, Isobe M. Tetrahedron. 2001;57:3875. [Google Scholar]
- 277.Asai M, Nishikawa T, Ohyabu N, Yamamoto N, Isobe M. Tetrahedron. 2001;57:4543. [Google Scholar]
- 278.Nishikawa T, Asai M, Isobe M. J. Am. Chem. Soc. 2002;124:7847. doi: 10.1021/ja0265153. [DOI] [PubMed] [Google Scholar]
- 279.Nishikawa T, Urabe D, Yoshida K, Iwabuchi T, Asai M, Isobe M. Org. Lett. 2002;4:2679. doi: 10.1021/ol026177a. [DOI] [PubMed] [Google Scholar]
- 280.Nishikawa T, Urabe D, Yoshida K, Iwabuchi T, Asai M, Isobe M. Pure Appl. Chem. 2003;75:251. [Google Scholar]
- 281.Ohyabu N, Nishikawa T, Isobe M. J. Am. Chem. Soc. 2003;125:8798. doi: 10.1021/ja0342998. [DOI] [PubMed] [Google Scholar]
- 282.Nishikawa T, Urabe D, Yoshida K, Iwabuchi T, Asai M, Isobe M. Chem. Eur. J. 2004;10:452. doi: 10.1002/chem.200305111. [DOI] [PubMed] [Google Scholar]
- 283.Nishikawa T, Urabe D, Isobe M. Angew. Chem. Int. Ed. 2004;43:4782. doi: 10.1002/anie.200460293. [DOI] [PubMed] [Google Scholar]
- 284.Urabe D, Nishikawa T, Isobe M. Chem. Asian J. 2006;1:125. doi: 10.1002/asia.200600038. [DOI] [PubMed] [Google Scholar]
- 285.Satake Y, Nishikawa T, Hiramatsu T, Araki H, Isobe M. Synthesis. 2010:1992. [Google Scholar]
- 286.Adachi M, Imazu T, Isobe M, Nishikawa T. J. Org. Chem. 2013;78:1699. doi: 10.1021/jo302773f. [DOI] [PubMed] [Google Scholar]
- 287.Adachi M, Imazu T, Sakakibara R, Satake Y, Isobe M, Nishikawa T. Chem. Eur. J. 2014;20:1247. doi: 10.1002/chem.201304110. [DOI] [PubMed] [Google Scholar]
- 288.Satake Y, Adachi M, Tokoro S, Yotsu-Yamashita M, Isobe M, Nishikawa T. Chem. Asian J. 2014;9:1922. doi: 10.1002/asia.201402202. [DOI] [PubMed] [Google Scholar]
- 289.Nakazaki A, Ishikawa Y, Sawayama Y, Yotsu-Yamashita M, Nishikawa T. Org. Biomol. Chem. 2014;12:53. doi: 10.1039/c3ob42017e. [DOI] [PubMed] [Google Scholar]
- 290.Sawayama Y, Nishikawa T. Angew. Chem. Int. Ed. 2011;50:7176. doi: 10.1002/anie.201102494. [DOI] [PubMed] [Google Scholar]
- 291.Sawayama Y, Nishikawa T. Synlett. 2011:651. [Google Scholar]
- 292.Sawayama Y, Nishikawa T. J. Synth. Org. Chem. Jpn. (Yuki Gosei Kagaku Kyokaishi) 2012;70:1178. [Google Scholar]
- 293.Onodera H, Satake M, Oshima Y, Yasumoto T, Carmichael WW. Nat. Toxins. 1997;5:146. doi: 10.1002/1522-7189(1997)5:4<146::AID-NT4>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 294.Hinman A, Du Bois J. J. Am. Chem. Soc. 2003;125:11510. doi: 10.1021/ja0368305. [DOI] [PubMed] [Google Scholar]
- 295.Fleming JJ, Du Bois J. J. Am. Chem. Soc. 2006;128:3926. doi: 10.1021/ja0608545. [DOI] [PubMed] [Google Scholar]
- 296.Fleming JJ, McReynolds MD, Du Bois J. J. Am. Chem. Soc. 2007;129:9964. doi: 10.1021/ja071501o. [DOI] [PubMed] [Google Scholar]
- 297.Mulcahy JV, Du Bois J. J. Am. Chem. Soc. 2008;130:12630. doi: 10.1021/ja805651g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Andresen BM, Du Bois J. J. Am. Chem. Soc. 2009;131:12524. doi: 10.1021/ja904179f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Ondrus AE, Lee H.-l. D., Iwanaga S, Parsons WH, Andresen BM, Moerner WE, Du Bois J. Chem. Biol. 2012;19:902. doi: 10.1016/j.chembiol.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Walker JR, Novick PA, Parsons WH, McGregor M, Zablocki J, Pande VS, Du Bois J. Proc. Natl. Acad. Sci., U.S.A. 2012;109:18102. doi: 10.1073/pnas.1206952109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Parsons WH, Du Bois J. J. Am. Chem. Soc. 2013;135:10582. doi: 10.1021/ja4019644. [DOI] [PubMed] [Google Scholar]
- 302.Hoehne A, Behera D, Parsons WH, James ML, Shen B, Borgohain P, Bodapati D, Prabhakar A, Gambhir SS, Yeomans DC, Biswal S, Chin FT, Du Bois J. J. Am. Chem. Soc. 2013;135:18012. doi: 10.1021/ja408300e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Eberlin LS, Mulcahy JV, Tzabazis A, Zhang J, Liu H, Logan MM, Roberts HJ, Lee GK, Yeomans DC, Du Bois J, Zare RN. J. Am. Chem. Soc. 2014;136:6401. doi: 10.1021/ja501635u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Zalatan DN, Du Bois J. Top. Curr. Chem. 2010;292:347. doi: 10.1007/128_2009_19. [DOI] [PubMed] [Google Scholar]
- 305.Roizen JL, Harvey ME, Du Bois J. Acc. Chem. Res. 2012;45:911. doi: 10.1021/ar200318q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem. Rev. 2010;110:704. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]
- 307.Davies HML, Morton D. Chem. Soc. Rev. 2011;40:1857. doi: 10.1039/c0cs00217h. [DOI] [PubMed] [Google Scholar]
- 308.Murahashi S-I, Naota T, Hanaoka H. Chem. Lett. 1993;22:1767. [Google Scholar]
- 309.Sato K.-i., Kajihara Y, Nakamura Y, Yoshimura J. Chem. Lett. 1991;20:1559. [Google Scholar]
- 310.Sato K.-i., Akai S, Sugita N, Ohsawa T, Kogure T, Shoji H, Yoshimura J. J. Org. Chem. 2005;70:7496. doi: 10.1021/jo050342t. [DOI] [PubMed] [Google Scholar]
- 311.Sato K.-i., Akai S, Shoji H, Sugita N, Yoshida S, Nagai Y, Suzuki K, Nakamura Y, Kajihara Y, Funabashi M, Yoshimura J. J. Org. Chem. 2008;73:1234. doi: 10.1021/jo701655v. [DOI] [PubMed] [Google Scholar]
- 312.Akai S, Seki H, Sugita N, Kogure T, Nishizawa N, Suzuki K, Nakamura Y, Kajihara Y, Yoshimura J, Sato K.-i. Bull. Chem. Soc. Jpn. 2010;83:279. [Google Scholar]
- 313.Akimoto T, Masuda A, Yotsu-Yamashita M, Hirokawa T, Nagasawa K. Org. Biomol. Chem. 2013;11:6642. doi: 10.1039/c3ob41398e. [DOI] [PubMed] [Google Scholar]
- 314.Ohtani Y, Shinada T, Ohfune Y. Synlett. 2003:619. [Google Scholar]
- 315.Umezawa T, Hayashi T, Sakai H, Teramoto H, Yoshikawa T, Izumida M, Tamatani Y, Hirose T, Ohfune Y, Shinada T. Org. Lett. 2006;8:4971. doi: 10.1021/ol062098d. [DOI] [PubMed] [Google Scholar]
- 316.Umezawa T, Shinada T, Ohfune Y. Chem. Lett. 2010;39:1281. [Google Scholar]
- 317.Iwamoto O, Koshino H, Hashizume D, Nagasawa K. Angew. Chem. Int. Ed. 2007;46:8625. doi: 10.1002/anie.200703326. [DOI] [PubMed] [Google Scholar]
- 318.Iwamoto O, Shinohara R, Nagasawa K. Chem. Asian J. 2009;4:277. doi: 10.1002/asia.200800382. [DOI] [PubMed] [Google Scholar]
- 319.Iwamoto O, Nagasawa K. Org. Lett. 2010;12:2150. doi: 10.1021/ol1006696. [DOI] [PubMed] [Google Scholar]
- 320.Shinohara R, Akimoto T, Iwamoto O, Hirokawa T, Yotsu-Yamashita M, Yamaoka K, Nagasawa K. Chem. Eur. J. 2011;17:12144. doi: 10.1002/chem.201101058. [DOI] [PubMed] [Google Scholar]
- 321.Iwamoto O, Akimoto T, Nagasawa K. Pure Appl. Chem. 2012;84:1445. [Google Scholar]
- 322.Tsuchiya S, Cho Y, Konoki K, Nagasawa K, Oshima Y, Yotsu-Yamashita M. Org. Biomol. Chem. 2014;12:3016. doi: 10.1039/c4ob00071d. [DOI] [PubMed] [Google Scholar]
- 323.Giles RL, Sullivan JD, Steiner AM, Looper RE. Angew. Chem. Int. Ed. 2009;48:3116. doi: 10.1002/anie.200900160. [DOI] [PubMed] [Google Scholar]
- 324.Giles RL, Nkansah RA, Looper RE. J. Org. Chem. 2010;75:261. doi: 10.1021/jo902326d. [DOI] [PubMed] [Google Scholar]
- 325.Gainer MJ, Bennett NR, Takahashi Y, Looper RE. Angew. Chem. Int. Ed. 2011;50:684. doi: 10.1002/anie.201006087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Looper RE, Haussener TJ, Mack JB. C. J. Org. Chem. 2011;76:6967. doi: 10.1021/jo201264j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Yang M, Odelberg SJ, Tong Z, Li DY, Looper RE. Tetrahedron. 2013;69:5744. doi: 10.1016/j.tet.2013.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Bhonde VR, Looper RE. J. Am. Chem. Soc. 2011;133:20172. doi: 10.1021/ja2098063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Kashman Y, Hirsh S, McConnell OJ, Ohtani I, Kusumi T, Kakisawa H. J. Am. Chem. Soc. 1989;111:8925. [Google Scholar]
- 330.Ohtani I, Kusumi T, Kakisawa H, Kashman Y, Hirsh S. J. Am. Chem. Soc. 1992;114:8472. [Google Scholar]
- 331.Bensemhoun J, Bombarda I, Aknin M, Vacelet J, Gaydou EM. J. Nat. Prod. 2007;70:2033. doi: 10.1021/np070340z. [DOI] [PubMed] [Google Scholar]
- 332.Echavarren A, Galan A, Lehn JM, Mendoza JD. J. Am. Chem. Soc. 1989;111:4994. [Google Scholar]
- 333.Murphy PJ, Williams HL, Hibbs DE, Hursthouse MB, Abdul Malik KM. Chem. Commun. 1996:445. [Google Scholar]
- 334.Kita T, Georgieva A, Hashimoto Y, Nakata T, Nagasawa K. Angew. Chem. Int. Ed. 2002;41:2832. doi: 10.1002/1521-3773(20020802)41:15<2832::AID-ANIE2832>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 335.Harbour GC, Tymiak AA, Rinehart KL, Jr., Shaw PD, Hughes R, Jr., Mizsak SA, Coats JH, Zurenko GE, Li LH, Kuentzel SL. J. Am. Chem. Soc. 1981;103:5604. [Google Scholar]
- 336.Tavares R, Daloze D, Braekman JC, Hajdu E, Soest RW. M. V. J. Nat. Prod. 1995;58:1139. [Google Scholar]
- 337.Snider BB, Faith WC. Tetrahedron Lett. 1983;24:861. [Google Scholar]
- 338.Snider BB, Faith WC. J. Am. Chem. Soc. 1984;106:1443. [Google Scholar]
- 339.Ruben RL, Snider BB, Hobbs FW, Jr., Confalone PN, Dusak BA. Invest. New Drugs. 1989;7:147. doi: 10.1007/BF00170851. [DOI] [PubMed] [Google Scholar]
- 340.Yu M, Pochapsky SS, Snider BB. J. Org. Chem. 2008;73:9065. doi: 10.1021/jo801956w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Roush WR, Walts AE. J. Am. Chem. Soc. 1984;106:721. [Google Scholar]
- 342.Walts AE, Roush WR. Tetrahedron. 1985;41:3463. [Google Scholar]
- 343.Uyehara T, Furuta T, Kabasawa Y, Yamada J-I, Kato T. J. Chem. Soc. Chem. Commun. 1986:539. [Google Scholar]
- 344.Uyehara T, Furuta T, Kabawawa Y, Yamada J, Kato T, Yamamoto Y. J. Org. Chem. 1988;53:3669. [Google Scholar]
- 345.Hassner A, Murthy KS. K. Tetrahedron Lett. 1986;27:1407. [Google Scholar]
- 346.Murthy KSK, Hassner A. Isr. J. Chem. 1991;31:239. [Google Scholar]
- 347.Asaoka M, Sakurai M, Takei H. Tetrahedron Lett. 1990;31:4759. [Google Scholar]
- 348.Cossy J, BouzBouz S. Tetrahedron Lett. 1996;37:5091. [Google Scholar]
- 349.Schellhaas K, Schmalz H-G, Bats JW. Chem. Eur. J. 1998;4:57. [Google Scholar]
- 350.Shen K, Livinghouse T. Synlett. 2010:247. [Google Scholar]
- 351.Jares-Erijman EA, Sakai R, Rinehart KL. J. Org. Chem. 1991;56:5712. [Google Scholar]
- 352.Jares-Erijman EA, Ingrum AL, Carney JR, Rinehart KL, Sakai R. J. Org. Chem. 1993;58:4805. [Google Scholar]
- 353.Tavares R, Daloze D, Braekman JC, Hajdu E, Muricy G, Soest RW. M. V. Biochem. Syst. Ecol. 1994;22:645. [Google Scholar]
- 354.Braekman JC, Daloze D, Tavares R, Hajdu E, Soest RW. M. V. J. Nat. Prod. 2000;63:193. doi: 10.1021/np990403g. [DOI] [PubMed] [Google Scholar]
- 355.Chang LC, Whittaker NF, Bewley CA. J. Nat. Prod. 2003;66:1490. doi: 10.1021/np030256t. [DOI] [PubMed] [Google Scholar]
- 356.Bondu S, Genta-Jouve G, Leirόs M, Vale C, Guigonis J-M, Botana LM, Thomas OP. RSC Adv. 2012;2:2828. [Google Scholar]
- 357.Palagiano E, De Marino S, Minale L, Riccio R, Zollo F, Iorizzi M, Carré JB, Debitus C, Lucarain L, Provost J. Tetrahedron. 1995;51:3675. [Google Scholar]
- 358.Venkateswarlu Y, Reddy MVR, Ramesh P, Venkateswara Rao J. Indian J. Chem., Sect. B. 1999;38:254. [Google Scholar]
- 359.Patil AD, Kumar NV, Kokke WC, Bean MF, Freyer AJ, Brosse CD, Mai S, Truneh A, Carte B. J. Org. Chem. 1995;60:1182. [Google Scholar]
- 360.Patil AD, Freyer AJ, Taylor PB, Carté B, Zuber G, Johnson RK, Faulkner DJ. J. Org. Chem. 1997;62:1814. [Google Scholar]
- 361.Patil AD, Freyer AJ, Offen P, Bean MF, Johnson RK. J. Nat. Prod. 1997;60:704. doi: 10.1021/np960652u. [DOI] [PubMed] [Google Scholar]
- 362.Gallimore WA, Kelly M, Scheuer PJ. J. Nat. Prod. 2005;68:1420. doi: 10.1021/np050149u. [DOI] [PubMed] [Google Scholar]
- 363.Hua H-M, Peng J, Dunbar DC, Schinazi RF, Andrews A. G. d. C., Cuevas C, Garcia-Fernandez LF, Kelly M, Hamann MT. Tetrahedron. 2007;63:11179. [Google Scholar]
- 364.Laville R, Thomas OP, Berrué F, Marquez D, Vacelet J, Amade PJ. Nat. Prod. 2009;72:1589. doi: 10.1021/np900244g. [DOI] [PubMed] [Google Scholar]
- 365.Takishima S, Ishiyama A, Iwatsuki M, Otoguro K, Yamada H, Õmura S, Kobayashi H, van Soest RWM, Matsunaga S. Org. Lett. 2009;11:2655. doi: 10.1021/ol9006794. [DOI] [PubMed] [Google Scholar]
- 366.Berlinck RGS, Braekman JC, Daloze D, Hallenga K, Ottinger R, Bruno I, Riccio R. Tetrahedron Lett. 1990;31:6531. [Google Scholar]
- 367.Jares-Erijman EA, Ingrum AA, Sun F, Rinehart KL. J. Nat. Prod. 1993;56:2186. doi: 10.1021/np50102a025. [DOI] [PubMed] [Google Scholar]
- 368.Snider BB, Shi Z. J. Org. Chem. 1992;57:2526. [Google Scholar]
- 369.Snider BB, Shi Z. J. Org. Chem. 1993;58:3828. [Google Scholar]
- 370.Martín V, Vale C, Bondu S, Thomas OP, Vieytes MR, Botana LM. Chem. Res. Toxicol. 2013;26:169. doi: 10.1021/tx3004483. [DOI] [PubMed] [Google Scholar]
- 371.Aron ZD, Pietraszkiewicz H, Overman LE, Valeriote F, Cuevas C. Bioorg. Med. Chem. Lett. 2004;14:3445. doi: 10.1016/j.bmcl.2004.04.071. [DOI] [PubMed] [Google Scholar]
- 372.Rubiolo JA, López-Alonso H, Roel M, Vieytes MR, Thomas O, Ternon E, Vega FV, Botana LM. Br. J. Pharmacol. 2014;171:1655. doi: 10.1111/bph.12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Bewley CA, Ray S, Cohen F, Collins SK, Overman LE. J. Nat. Prod. 2004;67:1319. doi: 10.1021/np049958o. [DOI] [PubMed] [Google Scholar]
- 374.Olszewski A, Sato K, Aron ZD, Cohen F, Harris A, McDougall BR, Robinson WE, Jr., Overman LE, Weiss GA. Proc. Natl. Acad. Sci., U.S.A. 2004;101:14079. doi: 10.1073/pnas.0406040101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Rubiolo JA, Ternon E, López-Alonso H, Thomas OP, Vega FV, Vieytes MR, Botana LM. Mar. Drugs. 2013;11:4419. doi: 10.3390/md11114419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Lazaro JEH, Nitcheu J, Mahmoudi N, Ibana JA, Mangalindan GC, Black GP, Howard-Jones AG, Moore CG, Thomas DA, Mazier D, Ireland CM, Concepcion GP, Murphy PJ, Diquet B. J. Antibiot. 2006;59:583. doi: 10.1038/ja.2006.78. [DOI] [PubMed] [Google Scholar]
- 377.Aron ZD, Overman LE. J. Am. Chem. Soc. 2005;127:3380. doi: 10.1021/ja042875+. [DOI] [PubMed] [Google Scholar]
- 378.Overman LE, Rabinowitz MH. J. Org. Chem. 1993;58:3235. [Google Scholar]
- 379.Overman LE, Rabinowitz MH, Renhowe PA. J. Am. Chem. Soc. 1995;117:2657. [Google Scholar]
- 380.Coffey DS, McDonald AI, Overman LE, Stappenbeck F. J. Am. Chem. Soc. 1999;121:6944. [Google Scholar]
- 381.Coffey DS, McDonald AI, Overman LE, Rabinowitz MH, Renhowe PA. J. Am. Chem. Soc. 2000;122:4893. [Google Scholar]
- 382.Overman LE, Rhee YH. J. Am. Chem. Soc. 2005;127:15652. doi: 10.1021/ja055464h. [DOI] [PubMed] [Google Scholar]
- 383.Franklin AS, Ly SK, Mackin GH, Overman LE, Shaka AJ. J. Org. Chem. 1999;64:1512. doi: 10.1021/jo981971o. [DOI] [PubMed] [Google Scholar]
- 384.Cohen F, Overman LE, Sakata SK. L. Org. Lett. 1999;1:2169. doi: 10.1021/ol991269u. [DOI] [PubMed] [Google Scholar]
- 385.Cohen F, Overman LE. J. Am. Chem. Soc. 2001;123:10782. doi: 10.1021/ja017067m. [DOI] [PubMed] [Google Scholar]
- 386.Cohen F, Collins SK, Overman LE. Org. Lett. 2003;5:4485. doi: 10.1021/ol0357999. [DOI] [PubMed] [Google Scholar]
- 387.Collins SK, McDonald AI, Overman LE, Rhee YH. Org. Lett. 2004;6:1253. doi: 10.1021/ol0498141. [DOI] [PubMed] [Google Scholar]
- 388.Cohen F, Overman LE. J. Am. Chem. Soc. 2006;128:2594. doi: 10.1021/ja0574320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Cohen F, Overman LE. J. Am. Chem. Soc. 2006;128:2604. doi: 10.1021/ja057433s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Nilsson BL, Overman LE. J. Org. Chem. 2006;71:7706. doi: 10.1021/jo061199m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Snider BB, Shi Z. Tetrahedron Lett. 1993;34:2099. [Google Scholar]
- 392.Snider BB, Shi Z. J. Am. Chem. Soc. 1994;116:549. [Google Scholar]
- 393.Snider BB, Chen J, Patil AD, Freyer AJ. Tetrahedron Lett. 1996;37:6977. [Google Scholar]
- 394.Snider BB, Chen J. Tetrahedron Lett. 1998;39:5697. [Google Scholar]
- 395.Snider BB, Busuyek MV. J. Nat. Prod. 1999;62:1707. doi: 10.1021/np990312j. [DOI] [PubMed] [Google Scholar]
- 396.Murphy PJ, Williams HL, Hursthouse MB, Abdul Malik KM. J. Chem. Soc., Chem. Commun. 1994:119. [Google Scholar]
- 397.Murphy PJ, Williams HL. J. Chem. Soc., Chem. Commun. 1994:819. [Google Scholar]
- 398.Murphy PJ, Lloyd WH, Hibbs DE, Hursthouse MB, Abdul Malik KM. Tetrahedron. 1996;52:8315. [Google Scholar]
- 399.Black GP, Coles SJ, Hizi A, Howard-Jones AG, Hursthouse MB, McGown AT, Loya S, Moore CG, Murphy PJ, Smith NK, Walshe ND. A. Tetrahedron Lett. 2001;42:3377. [Google Scholar]
- 400.Black GP, Murphy PJ, Thornhill AJ, Walshe NDA, Zanetti C. Tetrahedron. 1999;55:6547. [Google Scholar]
- 401.Black GP, Murphy PJ, Walshe NDA, Hibbs DE, Hursthouse MB, Abdul Malik KM. Tetrahedron Lett. 1996;37:6943. [Google Scholar]
- 402.Black GP, Murphy PJ, Walshe ND. A. Tetrahedron. 1998;54:9481. [Google Scholar]
- 403.Bennett EL, Black GP, Browne P, Hizi A, Jaffar M, Leyland JP, Martin C, Oz-Gleenberg I, Murphy PJ, Roberts TD, Thornhill AJ, Vale SA. Tetrahedron. 2013;69:3061. [Google Scholar]
- 404.Moore CG, Murphy PJ, Williams HL, McGown AT, Smith NK. Tetrahedron Lett. 2003;44:251. [Google Scholar]
- 405.Moore CG, Murphy PJ, Williams HL, McGown AT, Smith NK. Tetrahedron. 2007;63:11771. [Google Scholar]
- 406.Rao AVR, Gurjar MK, Vasudevan J. J. Chem. Soc. Chem. Commun. 1995:1369. [Google Scholar]
- 407.Louwrier S, Ostendorf M, Tuynman A, Hiemstra H. Tetrahedron Lett. 1996;37:905. [Google Scholar]
- 408.Louwrier S, Ostendorf M, Boom A, Hiemstra H, Speckamp WN. Tetrahedron. 1996;52:2603. [Google Scholar]
- 409.Louwrier S, Tuynman A, Hiemstra H. Tetrahedron. 1996;52:2629. [Google Scholar]
- 410.Nagasawa K, Georgieva A, Nakata T. Tetrahedron. 2000;56:187. [Google Scholar]
- 411.Georgieva A, Hirai M, Hashimoto Y, Nakata T, Ohizumi Y, Nagasawa K. Synthesis. 2003:1427. [Google Scholar]
- 412.Nagasawa K, Georgieva A, Koshino H, Nakata T, Kita T, Hashimoto Y. Org. Lett. 2002;4:177. doi: 10.1021/ol0168263. [DOI] [PubMed] [Google Scholar]
- 413.Nagasawa K, Koshino H, Nakata T. Tetrahedron Lett. 2001;42:4155. [Google Scholar]
- 414.Nagasawa K, Ishiwata T, Hashimoto Y, Nakata T. Tetrahedron Lett. 2002;43:6383. [Google Scholar]
- 415.Ishiwata T, Hino T, Koshino H, Hashimoto Y, Nakata T, Nagasawa K. Org. Lett. 2002;4:2921. doi: 10.1021/ol026303a. [DOI] [PubMed] [Google Scholar]
- 416.Shimokawa J, Shirai K, Tanatani A, Hashimoto Y, Nagasawa K. Angew. Chem. Int. Ed. 2004;43:1559. doi: 10.1002/anie.200353200. [DOI] [PubMed] [Google Scholar]
- 417.Shimokawa J, Ishiwata T, Shirai K, Koshino H, Tanatani A, Nakata T, Hashimoto Y, Nagasawa K. Chem. Eur. J. 2005;11:6878. doi: 10.1002/chem.200500852. [DOI] [PubMed] [Google Scholar]
- 418.Shimokawa J, Iijima Y, Hashimoto Y, Chiba H, Tanaka H, Nagasawa K. Heterocycles. 2007;72:145. [Google Scholar]
- 419.Sekine M, Iijima Y, Iwamoto O, Nagasawa K. Heterocycles. 2010;80:395. [Google Scholar]
- 420.Durón SG, Gin DY. Org. Lett. 2001;3:1551. doi: 10.1021/ol015848m. [DOI] [PubMed] [Google Scholar]
- 421.Arnold MA, Durón SG, Gin DY. J. Am. Chem. Soc. 2005;127:6924. doi: 10.1021/ja0519029. [DOI] [PubMed] [Google Scholar]
- 422.Arnold MA, Day KA, Durón SG, Gin DY. J. Am. Chem. Soc. 2006;128:13255. doi: 10.1021/ja063860+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Perl NR, Ide ND, Prajapati S, Perfect HH, Durón SG, Gin DY. J. Am. Chem. Soc. 2010;132:1802. doi: 10.1021/ja910831k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Evans PA, Manangan T. Tetrahedron Lett. 2001;42:6637. [Google Scholar]
- 425.Evans PA, Qin J, Robinson JE, Bazin B. Angew. Chem. Int. Ed. 2007;46:7417. doi: 10.1002/anie.200700840. [DOI] [PubMed] [Google Scholar]
- 426.Elliott MC, Long MS. Tetrahedron Lett. 2002;43:9191. [Google Scholar]
- 427.Elliott MC, Long MS. Org. Biomol. Chem. 2004;2:2003. doi: 10.1039/b404679j. [DOI] [PubMed] [Google Scholar]
- 428.Davies CD, Elliott MC, Hill-Cousins J, Khan M.-u. A., Maqbool T, Wood JL. Synlett. 2008:2028. [Google Scholar]
- 429.Butters M, Davies CD, Elliott MC, Hill-Cousins J, Kariuki BM, Ooi L.-l., Wood JL, Wordingham SV. Org. Biomol. Chem. 2009;7:5001. doi: 10.1039/b914744f. [DOI] [PubMed] [Google Scholar]
- 430.Schultz DM, Wolfe JP. Synthesis. 2012;44:351. doi: 10.1055/s-0031-1289668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Babij NR, Wolfe JP. Angew. Chem. Int. Ed. 2012;51:4128. doi: 10.1002/anie.201201001. [DOI] [PMC free article] [PubMed] [Google Scholar]