

Abstract
5-Hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) form during active demethylation of 5-methylcytosine (5mC) and are implicated in epigenetic regulation of the genome. They are differentially processed by thymine DNA glycosylase (TDG), an enzyme involved in active demethylation of 5mC. Three modified Dickerson–Drew dodecamer (DDD) sequences, amenable to crystallographic and spectroscopic analyses and containing the 5′-CG-3′ sequence associated with genomic cytosine methylation, containing 5hmC, 5fC, or 5caC placed site-specifically into the 5′-T8X9G10-3′ sequence of the DDD, were compared. The presence of 5caC at the X9 base increased the stability of the DDD, whereas 5hmC or 5fC did not. Both 5hmC and 5fC increased imino proton exchange rates and calculated rate constants for base pair opening at the neighboring base pair A5:T8, whereas 5caC did not. At the oxidized base pair G4:X9, 5fC exhibited an increase in the imino proton exchange rate and the calculated kop. In all cases, minimal effects to imino proton exchange rates occurred at the neighboring base pair C3:G10. No evidence was observed for imino tautomerization, accompanied by wobble base pairing, for 5hmC, 5fC, or 5caC when positioned at base pair G4:X9; each favored Watson–Crick base pairing. However, both 5fC and 5caC exhibited intranucleobase hydrogen bonding between their formyl or carboxyl oxygens, respectively, and the adjacent cytosine N4 exocyclic amines. The lesion-specific differences observed in the DDD may be implicated in recognition of 5hmC, 5fC, or 5caC in DNA by TDG. However, they do not correlate with differential excision of 5hmC, 5fC, or 5caC by TDG, which may be mediated by differences in transition states of the enzyme-bound complexes.
Cytosine methylation by DNA methyltransferases1−4 to form 5-methylcytosine (5mC)5 is important in epigenetic regulation of the eukaryotic genome.6,7 The reconversion of 5mC to cytosine during active demethylation8−25 involves the stepwise oxidation of 5mC. Oxidation of 5mC to 5-hydroxymethylcytosine (5hmC)10,26,27 is accomplished by ten-eleven translocation (TET) dioxygenases15,28−30 and occurs in response to oxidative stress as a consequence of UV radiation.31 Further oxidation of 5hmC by TET dioxygenases forms 5-formylcytosine (5fC)27 and 5-carboxylcytosine (5caC).8,22,27−30,32 These have been detected in cellular DNA.32,33 Both 5fC and 5caC, but not 5hmC, are substrates for thymine DNA glycosylase (TDG),14 consistent with the greater abundance of 5hmC in mammalian tissues9 and implicating catalyzed base excision of oxidized 5mC derivatives in active demethylation.
The differential processing of 5fC and 5caC vs 5hmC by TDG could be mediated by their differential recognition in DNA. Recently, Raiber et al.555 reported that a DNA dodecamer containing three 5fC sites in an iterated CG repeat sequence exhibited 5fC-specific helical unwinding, due to specific changes in the geometry of the grooves and base pairs involving 5fC. DNA glycosylases may also exploit differential base pair opening rates as a basis for substrate recognition. For example, enhanced base pair opening rates at A:U base pairs facilitate the recognition of uracil by uracil DNA glycosylase (UDG).34 It has also been hypothesized that TDG recognizes wobble base pairing geometry at oxidized cytosines,17,19,23,35,36 as the imino tautomers of 5caC or 5fC may adopt wobble-like base pairs with the complementary G.20,36 However, calculations of the stabilities of the amino and imino tautomers of 5fC and 5caC at the nucleobase level have suggested that, when paired with G, both 5fC and 5caC, which are substrates for TDG,14 preferentially form Watson–Crick pairs.23 Alternatively, as proposed by Maiti et al.,14 the differential processing by TDG could be mediated by differences in the corresponding transition state catalytic complexes involving 5fC, 5caC, or 5hmC. Maiti et al.14 proposed that the preferential excision of 5fC and 5caC by TDG is facilitated by the presence of electron-withdrawing substituents at the C5 carbon for these two oxidized cytosines. This electron-withdrawing effect14,23 would be anticipated to stabilize developing negative charge in the transition state complex for base excision.
Here, we have incorporated 5hmC, 5fC, or 5caC into the 5′-T8X9G10-3′ sequence of the self-complementary Dickerson–Drew dodecamer (DDD),37,38 which contains the 5′-CG-3′ sequence associated with genomic cytosine methylation, forming DDDhm, DDDf, and DDDca duplexes (Chart 1), respectively. Importantly, the DDD is amenable to crystallographic37−42 and spectroscopic43−46 analyses. The characterization of the DDDhm, DDDf, and DDDca duplexes by thermal melting studies, measurements of base pair opening dynamics, crystallography, and NMR reveals lesion- and sequence-specific differences among 5hmC, 5fC, or 5caC in the 5′-T8X9G10-3′ sequence, which may be relevant to their recognition by TDG. Relative to 5hmC and 5fC, incorporation of 5caC increases the stability of the DDD. This is reflected in reduced base pair opening dynamics for DDDca, as compared to that for DDDhm and DDDf, at neighboring base pair A5:T8. Similar, but smaller, differences in base pair opening dynamics are observed at the oxidized base pair G4:X9, whereas minimal effects are observed at neighboring base pair C3:G10. No evidence for wobble base pairing interactions involving the oxidized cytosines is observed; each of these oxidized cytosines favors Watson–Crick base pairing. These sequence-specific differences in the DDD may be related to the recognition of these oxidized cytosines by TDG. However, they differ from the sequence-specific effects observed by Raiber et al.555 for an iterated CG repeat containing three 5fC sites. Moreover, they do not correlate with the differential ability of TDG to excise 5fC and 5caC vs 5hmC,14 which may be mediated by differences in the transition state complex for base excision.
Chart 1. (A) Structures of C, 5mC, 5hmC, 5fC, and 5caC and (B) Sequences and Numbering of the Nucleotides for DDD and Oxidized DDD Duplexesa.

a In solution, the two strands of the DDD exhibit pseudo-dyad symmetry. NMR resonances of symmetry-related nucleotides in the two strands are not individually observed. In the crystal, corresponding nucleotides from paired strands are not symmetry-related, and nucleotides are numbered individually. DDDm, DDDhm, DDDf, and DDDca refer to the DDD containing 5mC, 5hmC, 5fC, and 5caC, respectively.
Experimental Procedures
Oligodeoxynucleotide Synthesis
Oligodeoxynucleotides were synthesized by Midland Certified Reagent Co. (Midland, TX) and purified by anion-exchange HPLC. The DDDhm duplex was prepared with an Expedite 8909 DNA synthesizer (PerSeptive Biosystems) on a 1 μmol scale using ethylcyanide-protected 5-hydroxymethyl-dC, phenoxyacetyl-protected dA, 4-isopropyl-phenoxyacetyl-protected dG, acetyl-protected dC, and dT phosphoramidites and solid supports (Glen Research, Inc., Sterling, VA). The modified phosphoramidite was incorporated by removing the column from the synthesizer and sealing it with two syringes, one of which contained 250–300 μL of the manufacturer’s 1H-tetrazole activator solution (1.9–4.0% in CH3CN, v/v) and the other contained 250 μL of the modified phosphoramidite solution (15 mg in anhydrous CH3CN). The 1H-tetrazole and the phosphoramidite solutions were sequentially drawn through the column (1H-tetrazole first), and this procedure was repeated over 30 min. The column was washed with anhydrous CH3CN and returned to the synthesizer for capping, oxidation, and detritylation steps. The deprotection was accomplished with 30% NH4OH for 17 h at 75 °C.
Oligodeoxynucleotide Purification and Characterization
Oligodeoxynucleotides were purified by semipreparative HPLC at 260 nm (Atlantis, Waters Corporation, C18, 5 μm, 250 mm × 10.0 mm). The column was equilibrated either with 30 mM sodium phosphate (pH 7.0) (for DDDm, DDDhm, DDDca) or 0.1 M ammonium formate (pH 6.5) (for DDDf). The gradient was 1–15% CH3CN over 20 min, 15–80% CH3CN over 5 min, and 1% CH3CN over 5 min, at 4.5 mL/min. Oligodeoxynucleotides were desalted by passing over G-25 Sephadex (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Oligodeoxynucleotides were characterized by MALDI-TOF mass spectrometry (calcd for DDD [M – H]−m/z 3646.4, found m/z 3647.8; calcd for DDDm [M – H]−m/z 3660.5, found 3663.4; calcd for DDDhm [M – H]−m/z 3675.5, found 3679.7; calcd for DDDf [M – H]−m/z 3674.4, found 3673.2; calcd for DDDca [M – H]−m/z 3690.4, found 3693.1). Oligodeoxynucleotides were prepared in 100 mM NaCl, 50 μM Na2EDTA, in 10 mM sodium phosphate (pH 7.0), heated at 85 °C for 15 min, and annealed by cooling to room temperature. Duplex concentrations were determined by UV absorbance, using extinction coefficients calculated at 260 nm.47
Thermal Denaturation
The concentration of DNA was 1.2 μM. Measurements were conducted in 100 mM NaCl, 50 μM Na2EDTA, in 10 mM sodium phosphate (pH 7.0). The temperature was increased from 10 to 80 °C at 1 °C/min. Tm values were calculated from first-order derivatives of 260 nm absorbance vs temperature profiles.48
NMR
Spectra were obtained at 900 MHz using a 5 mm cryogenic probe (Bruker Biospin Inc., Billerica, MA). Oligodeoxynucleotides were prepared at a duplex concentration of 0.25 mM in 180 μL of 100 mM NaCl, 50 μM Na2EDTA, 11 mM NaN3, in 10 mM sodium phosphate (pH 7.0). The samples were exchanged with D2O and dissolved in 180 μL of 99.996% D2O to observe nonexchangeable protons. NOESY49 spectra were collected in 99.996% D2O to observe nonexchangeable protons. The temperature was 15 °C. TPPI quadrature detection was used, and data were collected at a mixing time of 250 ms. The relaxation delay was 2.0 s. Data were recorded with 2k real points in the t2 dimension and 1k real points in the t1 dimension. Spectra were zero-filled during processing to create a 2k × 2k matrix. Chemical shifts were referenced to the chemical shift of water at the corresponding temperature, with respect to 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS). To observe exchangeable protons, samples were prepared in 9:1 H2O/D2O. For observation of imino protons, spectra were recorded at 5, 15, 25, 35, 45, and 55 °C. NOESY spectra were collected at 5 °C with 70 or 250 ms mixing times and relaxation delay of 2.0 s. Water suppression was achieved by the Watergate pulse sequence.50 Data were processed with TOPSPIN (2.0.b.6, Bruker Biospin Inc., Billerica, MA).
Base Pair Opening
NMR data were collected at 500 MHz using a 5 mm cryogenic probe, at 15 °C. Samples were in 180 μL of 9:1 H2O/D2O containing 100 mM NaCl, 50 μM Na2EDTA, 11 mM NaN3, 1 mM triethanolamine, in 10 mM sodium phosphate (pH 8.9).51−55 The presence of triethanolamine enabled the pH of the sample to be monitored during the titration, in situ, by measuring the chemical shift difference between the two methylene groups.51 Magnetization transfer from water to the imino protons was followed by observation of the imino proton resonances after variable mixing times.56,666 Selective spin inversion of the water protons was achieved with a 2 ms 180° sinc pulse with 1000 points. To minimize effects of radiation damping during the mixing time, a 0.1 G cm–1 gradient was used. Water suppression was achieved by a binominal 1–1 echo sequence, jump and return,57 with flanking 1 ms smooth square shape gradients, 15 G cm–1. Sixteen values of the delay ranging form 1 ms to 15 s were used. Data were processed in TOPSPIN. Ammonia, pKa of 9.2 at 15 °C, was the proton acceptor.56 Data analyses were performed using PRISM (v. 6.0b, GraphPad Software, Inc., La Jolla, CA). Exchange rates were calculated using established methods.58,59 In order to determine rates of base pair opening, exchange rates were plotted against concentrations of the active form of the ammonia base catalyst. Equilibrium constants for base pair opening were calculated by fitting exchange rate data as a function of ammonia concentration.51
Crystallization and X-ray Diffraction
Crystals were grown at 18 °C over 8 to 16 days by hanging-drop vapor diffusion, using the nucleic acid mini-screen (Hampton Research, Aliso Viejo, CA). Droplets of 2 μL containing 1.2 mM duplex in precipitant solution were equilibrated against 0.75 μL of 35% MPD. The solution compositions are summarized in Table S1 in the Supporting Information. Single crystals were mounted in nylon loops and flash-frozen in liquid nitrogen.
For DDDhm, data were collected on the 19-ID beamline of the Structural Biology Center at the Advanced Photon Source (APS) of Argonne National Laboratory (ANL, Argonne, IL).60 The wavelength was 0.9794 Å. Initial indexing and scaling of diffraction images and further reflection merging was done using HKL3000.61 To ensure completeness of the data, two passes were collected. For DDDca, data were collected on the 24-IDC beamline of the Northeastern Collaborative Access Team (NE-CAT) at the APS (ANL). The wavelength was 0.97920 Å. Initial indexing and scaling of diffraction images, together with reflection merging, were done using XDS62,63 and SCALA64 in the CCP465 suite as part of the RAPD data collection strategy at NE-CAT. For DDDf, data were collected on the 21-IDD beamline of the Life-Sciences Collaborative Access Team (LS-CAT) at the APS (ANL). The wavelength was 1.000 Å. Initial indexing and scaling of diffraction images, together with reflection merging were done in HKL2000.66 Details are shown in Table 1.
Table 1. Crystal Data, Data Collection, and Refinement Statistics for the DDDhm, DDDf, and DDDca Duplexes.
| parameter | DDDhm | DDDf | DDDca |
|---|---|---|---|
| Crystal Data | |||
| space group | P212121 | P212121 | P212121 |
| Unit Cell | |||
| a (Å) | 25.61 | 25.09 | 24.25 |
| b (Å) | 41.34 | 41.47 | 41.34 |
| c (Å) | 64.32 | 65.69 | 66.41 |
| Data Collection | |||
| resolution range (Å) | 40–1.02 | 35–1.90 | 26–1.95 |
| no. of unique reflections | 37 637 | 5801 | 5113 |
| completeness (%) | 99.6 | 99.3 | 99.5 |
| in the outer shell (%) | 98.5 | 100 | 97.5 |
| Rmergea | 0.044 | 0.064 | 0.045 |
| in the outer shell | 0.979 | 0.738 | 0.619 |
| I/σ(I) | 52 | 60 | 16 |
| in the outer shell | 1.7 | 3.3 | 2.8 |
| Structure Refinement | |||
| resolution range (Å) | 40–1.02 | 35–1.90 | 26–1.95 |
| Rwork | 0.156 | 0.226 | 0.221 |
| Rfree | 0.178 | 0.245 | 0.267 |
| RMS Deviation | |||
| bond lengths (Å) | 0.014 | 0.011 | 0.009 |
| angle distances (deg) | 2.4 | 1.3 | 2.2 |
| no. of ions | 1 Mg2+ | ||
| no. of ligands | 3 | ||
| no. of water molecules | 178 | 17 | 13 |
Rmerge = ∑hkl ∑i|Ii – ⟨I⟩|/∑hkl ∑I|⟨I⟩|, where Ii is the intensity for the ith measurement of an equivalent reflection with indices h, k, and l.
Crystal Structure Determination and Refinement
Structures were determined by molecular replacement using the DDD as the search model (PDB ID code 436D).39 Molecular replacement searches were completed with MOLREP67,68 in the CCP4 suite.65 An initial model was checked and rebuilt in COOT.69 The model was rebuilt and further refined using REFMAC.70,71 Final models were refined against all reflections, except for 5% randomly selected reflections used for monitoring Rfree. The refinement statistics are presented in Table 1.
Data Deposition
Complete structure factors and data coordinates were deposited in the Protein Data Bank (http://pdb.org): PDB ID code 4I9V for DDDhm, 4QC7 for DDDf, and 4PWM for DDDca.
Results
Stabilities of the Duplexes Containing Oxidized Cytosines
The impact of placing 5hmC, 5fC, or 5caC site specifically into the 5′-CG-3′ sequence was investigated by incorporating each oxidized cytosine into the 5′-T8X9G10-3′ sequence of the DDD.37,38 The Tm values of the duplexes were obtained in 100 mM NaCl at pH 7. They were compared to both the unmodified DDD and also to the DDD containing 5mC in the 5′-T8X9G10-3′ sequence (DDDm). The Tm of the DDD duplex was 48 °C, the Tm of the DDDm duplex was 46 °C, the Tm of the DDDhm duplex was 48 °C, and the Tm of the DDDf duplex was 46 °C. These small differences in Tm suggested that the presence of 5mC or of the oxidized cytosines 5hmC or 5fC in the 5′-T8X9G10-3′ sequence did not greatly affect the Tm of the DDD. In contrast, the Tm of the DDDca duplex increased to 54 °C. NMR spectra of the exchangeable guanine N1H and thymine N3H imino protons were recorded from 5–55 °C (Figure 1). The resonances were assigned using standard methods.72 For the DDDca duplex, the G4 N1H proton remained sharp at 55 °C, consistent with the increased Tm value associated with the 5caC nucleobase in the 5′-T8X9G10-3′ sequence. At the neighbor A5:T8 base pair, the T8 N3H resonance remained detectable at 55 °C, although it exhibited broadening. At the neighbor C3:G10 base pair, the G3 N1H resonance remained detectable at 55 °C, also exhibiting broadening. The stabilizing effect extended two base pairs in each direction, also including the imino protons of base pairs G2:C11 and A6:T7. In contrast, for the DDDhm duplex at the oxidized G4:X9 base pair, the G4 N1H resonance was severely broadened at 55 °C. Likewise, the corresponding resonance in the DDDf duplex was severely broadened at 55 °C. At the neighboring base pair C3:G10, the G10 N1H resonance in the DDDf duplex broadened at 35 °C. The T8 N3H resonances broadened at 45 °C in the DDDf duplex and at 55 °C in the DDDhm duplex. The temperature dependence of line widths of the imino resonances is shown in Figure S1 of the Supporting Information.
Figure 1.

1H NMR of imino proton resonances as a function of temperature for (A) DDD, (B) DDDm, (C) DDDhm, (D) DDDf, and (E) DDDca. Data were collected at 900 MHz.
Base Pair Opening Dynamics
Magnetization transfer from water after variable times was followed by observation of the guanine N1H and thymine N3H resonances, at 15 °C. The imino proton exchange rates were measured in the absence and the presence of added ammonia base catalyst.44,51,56,666,59,73 The exchange with water follows a two-state model, where the base pair undergoes a conformational change from the closed to the open state, from which proton exchange occurs.56,73 The open base pair is exchange-competent because the proton is accessible to acceptors in solution. As described by Russu and co-workers,73−75 in the EX1 regime, the concentration of acceptors is sufficient for rapid exchange from the open state (kex,open ≫ kcl), so exchange occurs at each opening event and kex = kop. In the EX2 regime, where the concentration of base is low (kex,open ≪ kcl), the rate of exchange from the open state is proportional to the exchange rate and the concentration of the acceptor.73−75
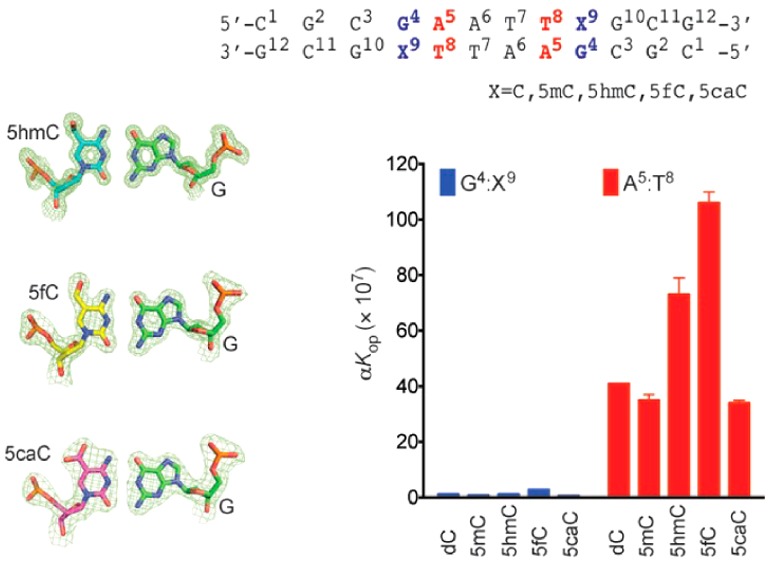
Figure 2 shows the results for the C3:G10, G4:X9, A5:T8, and A6:T7 base pairs of the DDDhm, DDDf, and DDDca duplexes. They were compared to both the unmodified DDD and to the DDDm duplexes. Plots of exchange rates as a function of ammonia concentration suggested that the EX1 regime73−75 was attained. Consistent with the results of Moe et al.,44 the rates of imino proton exchange were lower for G:C base pairs C3:G10 and G4:X9 and greater for A:T base pairs A5:T8 and A6:T7. At 15 °C, the oxidized cytosines differentially altered exchange rates of the imino protons of the C3:G10, G4:X9, A5:T8, and A6:T7 base pairs. The greatest effects were observed at the neighbor A5:T8 base pair. For the DDDhm and DDDf duplexes, the exchange rate of the A5:T8 base pair imino proton increased at all concentrations of ammonia (Figure 2). There was a 3-fold increased rate of base pair opening in the DDDhm duplex and a 5-fold increased rate of base pair opening in the DDDf duplex, with respect to the DDD duplex (Table 2). In contrast, for the DDDca duplex, the exchange rate of the A5:T8 imino proton was similar to those of the DDD and DDDm duplexes at all concentrations of ammonia. These differences were reflected in measurements of the respective equilibrium constants for base pair opening. For the DDDhm duplex, the equilibrium constant for base pair opening (αKop) at base pair A5:T8 was 7.3 × 108, and for the DDDf duplex, the equilibrium constant for base pair opening at A5:T8 was 1.1 × 109, differing from the DDD and DDDm duplexes (3.4 × 108 and 3.5 × 108, respectively). In contrast, for the DDDca duplex, the equilibrium constant for base pair opening of A5:T8 was 4.1 × 108, similar to that of the DDD and DDDm duplexes. The neighbor effect did not extend beyond the A5:T8 base pair. At base pair A6:T7, exchange rates as a function of ammonia concentration were comparable for all duplexes.
Figure 2.

Plots showing imino proton exchange rates obtained by monitoring magnetization from water as a function of ammonia base catalyst: (A) base pair C3:G10, (B) base pair G4:X9, (C) base pair A5:T8, and (D) base pair A6:T7 in the DDD (black), DDDm (green), DDDhm (blue), DDDf (red), and DDDca (pink) duplexes.
Table 2. Rate and Equilibrium Constants for DNA Base Pair Opening.
|
k0 (s–1)a |
|||||
|---|---|---|---|---|---|
| DDD | DDDm | DDDhm | DDDf | DDDca | |
| C3:G10 | 1.1 ± 0.06 | 1.0 ± 0.07 | 0.95 ± 0.05 | 0.71 ± 0.03 | 1.1 ± 0.06 |
| G4:C9 | 0.57 ± 0.03 | 0.56 ± 0.1 | 0.68 ± 0.05 | 0.39 ± 0.04 | 0.49 ± 0.04 |
| A5:T8 | 0.59 ± 0.03 | 0.73 ± 0.07 | 0.77 ± 0.03 | 0.79 ± 0.04 | 0.59 ± 0.03 |
| A6:T7 | 0.61 ± 0.03 | 0.43 ± 0.04 | 0.36 ± 0.01 | 0.27 ± 0.03 | 0.55 ± 0.03 |
|
kop (s–1) |
|||||
|---|---|---|---|---|---|
| DDD | DDDm | DDDhm | DDDf | DDDca | |
| C3:G10 | 45 ± 3 | 90 ± 12 | 42 ± 5 | 86 ± 18 | 64 ± 12 |
| G4:C9 | 8 ± 0.5 | 7 ± 0.6 | 16 ± 2 | 26 ± 2 | 4 ± 0.3 |
| A5:T8 | 40 ± 2 | 65 ± 2 | 110 ± 13 | 222 ± 53 | 58 ± 5 |
| A6:T7 | 36 ± 1 | 29 ± 1 | 45 ± 2 | 33 ± 3 | 44 ± 3 |
|
kcl (×10–7 s–1) |
|||||
|---|---|---|---|---|---|
| DDD | DDDm | DDDhm | DDDf | DDDca | |
| C3:G10 | 15 ± 1 | 25 ± 2 | 17 ± 1 | 21 ± 2 | 20 ± 1 |
| G4:C9 | 6.7 ± 0.2 | 9.4 ± 1 | 13 ± 0.8 | 9.3 ± 1 | 6.6 ± 0.1 |
| A5:T8 | 0.97 ± 0.04 | 1.9 ± 0.04 | 1.5 ± 0.05 | 2.1 ± 0.06 | 2.3 ± 0.05 |
| A6:T7 | 0.98 ± 0.09 | 1.1 ± 0.08 | 1.6 ± 0.08 | 1.1 ± 0.05 | 1.3 ± 0.01 |
|
Kop × 107 |
|||||
|---|---|---|---|---|---|
| DDD | DDDm | DDDhm | DDDf | DDDca | |
| C3:G10 | 2.9 ± 0.1 | 3.6 ± 0.1 | 2.5 ± 0.1 | 3.8 ± 0.1 | 3.2 ± 0.08 |
| G4:C9 | 1.2 ± 0.04 | 0.75 ± 0.04 | 1.2 ± 0.06 | 2.8 ± 0.1 | 0.6 ± 0.04 |
| A5:T8 | 41 ± 0.08 | 35 ± 2 | 73 ± 6 | 106 ± 4 | 34 ± 1 |
| A6:T7 | 37 ± 5 | 26 ± 3 | 29 ± 3 | 29 ± 2 | 34 ± 0.9 |
The observed exchange rate without an ammonia catalyst.
A smaller effect on base pair opening dynamics was observed at the G4:X9 base pair. For the DDDf duplex, the exchange rate of G4:X9 was greater at all concentrations of ammonia than that for the DDDhm, DDDca, DDDm, and DDD duplexes (kop = 26 s–1 in DDDf vs kop = 16, 4, 7, and 8 s–1 for DDDhm, DDDca, DDDm, and DDD, respectively). For the DDDf duplex, the equilibrium constant for base pair opening increased 3-fold, calculated as 2.8 × 107 vs 1.2 × 107, 7.5 × 106, 1.2 × 107, and 6.0 × 106, respectively, for the DDD, DDDm, DDDhm, and DDDca duplexes.
In contrast, base pair opening dynamics at the neighboring C3:G10 base pair were not affected by the presence of the oxidized cytosines in the DDDhm, DDDf, or DDDca duplex. Thus, the differences in base pair opening dynamics for the 5hmC, 5fC, and 5caC bases in the 5′-T8X9G10-3′ sequence of the DDD exhibit a pronounced sequence dependence, with the greatest effects being evident at the neighboring A5:T8 base pair. This is the base pair located in the 5′-direction with respect to the oxidized cytosine X9. The overall results are summarized in Table 2.
Structures of the DDDhm, DDDf, and DDDca Duplexes
The modified DDDhm, DDDf, and DDDca duplexes yielded diffraction-quality crystals. Crystals belonged to the orthorhombic P212121 space group. The crystal structures were determined using the unmodified DDD (PDB ID code 436D)39 as a search model for molecular replacement. Structures were refined using anisotropic B factors to a resolution of 1.02 Å for DDDhm and isotropic B factors to resolutions of 1.90 and 1.95 Å for DDDf and DDDca, respectively. Each of the structures was compared to that of the DDD.39 Overall, the structures were similar to the DDD,39 as indicated by comparative rmsd analyses, with rmsd values of 0.67, 0.46, and 0.49 Å for DDDhm, DDDf, and DDDca, respectively. Classical features of the DDD, including waters forming the minor groove spine of hydration,76 were conserved. The data and refinement statistics are provided in Table 1.
Figure 3 shows electron density and base pairing arrangements for the 5hmC:G, 5fC:G, and 5caC:G base pairs in the DDDhm, DDDf, and DDDca duplexes, respectively. Watson–Crick base pairing was evident, and the hydroxymethyl, formyl, or carboxyl moieties of the oxidized cytosines were oriented into the major groove. The formyl group of 5fC and the carboxyl group of 5caC were within hydrogen-bonding range of the N4 exocyclic amines of the oxidized cytosines. For the DDDhm duplex, electron density associated with the hydroxymethyl moiety of 5hmC suggested partial occupancy of two conformations. The major conformation refined with occupancy 0.8, and the minor conformation refined with occupancy 0.2. In the major conformation, the hydroxyl group hydrogen bonded with the terminal N1 ammonium moiety of a spermine and with G10O6 via an ordered water molecule (Figure S2 of the Supporting Information). In the minor conformation, the hydroxyl group was oriented toward the backbone phosphate and formed interactions with neighboring waters (Figure S3 of the Supporting Information). A hydrogen bond was also observed between the hydroxyl group at the modified cytosine X21 and an axially coordinated water (HOH 12) at a distance of 2.7 Å, with a further interaction to G22 N7 (2.8 Å). An additional hydrogen bond was observed between the X21 hydroxyl and G22O6 via water HOH 11 (3.0 Å distance from X21 to HOH 11 and 2.7 Å from HOH 11 to G22O6) (Figures S2 and S3 of the Supporting Information). Base stacking patterns for the DDDhm, DDDf, and DDDca duplexes were similar (Figure 4).
Figure 3.
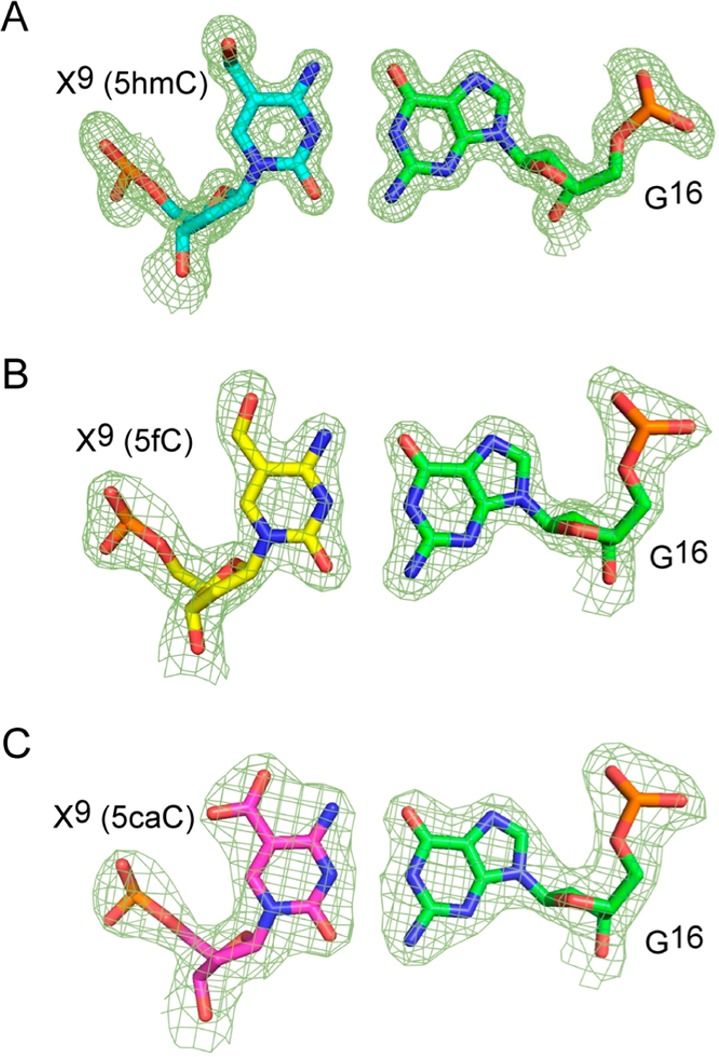
Fourier (2Fo – Fc) sum electron density contoured at the 1.0σ level (green meshwork) around the (A) 5hmC:G, (B) 5fC:G, and (C) 5caC:G base pairs showing Watson–Crick base pairing geometry.
Figure 4.
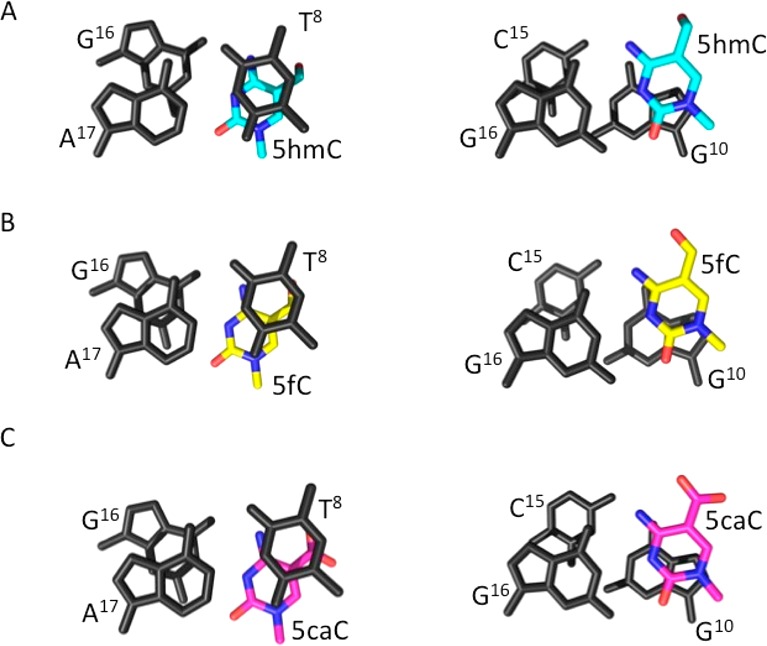
(A) DDDhm, (B) DDDf, and (C) DDDca structures, illustrating stacking interactions at oxidation sites.
The DDDhm, DDDf, and DDDca duplexes were also examined by NMR,43,45,46 using standard methods.77,78 The sequential base aromatic → deoxyribose anomeric NOEs were identified from C1 → G12 (Figure S4 of the Supporting Information). For the DDDhm, DDDf, and DDDca duplexes (and as well for the DDD and DDDm duplexes), the intensities of NOE cross-peaks between the purine H8 and pyrimidine H6 protons and the deoxyribose H1′ protons were of the same relative magnitudes as those between other bases in the sequence, indicating that the glycosyl bonds maintained the anti conformations. In all instances, the NOE connectivity of the purine N1H and pyrimidine N3H protons72 was obtained from G2:C11 → C3:G10 → G4:X9 → A5:T8 → A6:T7 (Figure 5). NOE cross-peaks from the oxidized base X9N4H1 and N4H2 protons to the complementary base G4 N1H proton were observed, as well as interactions to neighbor bases T8 N3H and G10 N1H, consistent with Watson–Crick geometry being favored, corroborating the crystallographic data (Figure 3). Significantly, evidence for intranucleotide hydrogen bonding involving the formyl group of 5fC or the carboxyl group of 5caC and the N4 exocyclic amine of 5fC or 5caC was evident in NMR spectra of the DDDf and DDDca duplexes, for which both of the X9N4 amino proton resonances shift downfield into the 7.8–8.8 ppm spectral range (Figure 5). The effect was most pronounced for the DDDca duplex. In contrast, for the DDD, DDDm, and DDDhm duplexes, one of the N4 amino protons shifts downfield, consistent with the maintenance of a Watson–Crick base pair, whereas the other remains in the 6.5–7.0 ppm spectral range, which is the anticipated result given that cytosine, 5mC, and 5hmC cannot form this hydrogen bond. Overall, the NMR data corroborated the crystallographic data, giving no indication of the presence of imino tautomers and suggesting that each of the oxidized cytosines participated in normal Watson–Crick base pairing when placed opposite guanine.
Figure 5.

NOESY spectra depicting resonances for the thymine and guanine imino protons and sequential NOE connectivity for the imino protons of the base pairs G2:C11 to A6:T7 for (A) DDD, (B) DDDm, (C) DDDhm, (D) DDDf, and (E) DDDca duplexes (lower panels). Expansion of the NOESY spectra for (A) DDD, (B) DDDm, (C) DDDhm, (D) DDDf, and (E) DDDca duplexes (upper panels), illustrating the conservation of Watson–Crick base pairing and base stacking at the modification sites: a, C9 or X9N4H1 → T8 N3H; b, C9 or X9N4H2 → T8 N3H; c, C9 or X9N4H1 → G10 N1H; d, C9 or X9N4H2 → G10 N1H; e, C9 or X9N4H1 → G4 N1H; f, A5 H2 → G4 N1H; and g, C9 or X9N4H2 → G4 N1H. (Indices m, hm, f, or ca refer to the base pairs in the modified duplexes, DDDm, DDDhm, DDDf, and DDDca, respectively.) Data were collected at 900 MHz.
Discussion
The 5hmC,10,26,27 5fC,27 and 5caC27,29,30 oxidation products of 5mC are intermediates in active demethylation8−25 and have potential roles in epigenetic regulation of cellular function.6,7 Their removal is orchestrated by glycosylase-mediated base excision repair; TDG is essential for active DNA demethylation.8 It has been reported that 5fC and 5caC, but not 5hmC, are substrates for thymine DNA glycosylase (TDG).14 Accordingly, it was of interest to determine whether these oxidized cytosines differentially alter duplex DNA and how such differences correlate with differences in excision of 5hmC, 5fC, and 5caC by TDG.14 The Dickerson–Drew dodecamer (DDD)37,38 provided a platform for conducting these studies. It contains the 5′-CG-3′ sequence associated with genomic cytosine methylation, and, most importantly, it is simultaneously amenable to crystallographic37−42 and spectroscopic43−46 analyses.
Stabilization of the DDD by 5caC
The presence of 5caC in the 5′-T8X9G10-3′ sequence stabilizes the DDD, as evidenced by the 6–8 °C increase in Tm for the DDDca as compared to the Tm values of the DDD and of the DDDm under the same conditions. NMR data for the base paired guanine N1H and thymine N3H imino protons (Figure 1) confirm this conclusion. For the DDDca duplex, at temperatures as high as 55 °C, the imino proton resonances of base pairs C3:G10, G4:X9, and A5:T8 remain detectable (Figure 1E). In contrast, for the DDDhm duplex and DDDf duplexes, the imino proton resonances of base pairs C3:G10, G4:X9, and A5:T8 broaden at temperatures above 35° (Figure 1C,D). While the inclusion of 5caC into the 5′-T8X9G10-3′ sequence in the DDD provides only a single data point for thermodynamic comparison, the observation that 5caC stabilizes the DDDca is consistent with calculations performed by Sumino et al.79 It also corroborates data obtained by the same group for the stabilities of 13-mers and 14-mers containing 5caC. This stabilization of the DDDca is not attributable to improved base stacking geometry of 5caC in DDDca because 5caC exhibits a base stacking geometry in the DDD that is similar to both 5hmC and 5fC (Figure 4). However, electronic dipole–dipole interactions associated with 5caC14,23 might enhance the thermodynamic stability of the DDDca duplex without disturbing the stacking geometry.
Sequence-Specific Base Pair Opening Dynamics of the Oxidized Duplexes
The imino proton exchange rates at base pairs C3:G10, G4:X9, and A5:T8 depend upon the identity of the cytosine oxidation product and exhibit sequence dependence. The greatest effects are observed for the neighbor base pair A5:T8, with a smaller effect at G4:X9 and minimal effect at the neighbor base pair C3:G10. Base pair A5:T8 is the 5′-neighbor with respect to the oxidized cytosine at position X9, whereas base pair C3:G10 is the 3′-neighbor with respect to the oxidized cytosine at position X9 (Chart 1). While the A5:T8 base pair of the DDD intrinsically exhibits enhanced exchange kinetics,44 the presence of either 5fC or 5hmC further enhances imino proton exchange rates at A5:T8, whereas the presence of 5caC does not (Figure 2), an observation that is consistent with the thermal stabilization of the duplex by 5caC as opposed to 5fC or 5hmC. Thus, for DDDf, base pair A5:T8 base pair opens with the frequency of kop = 222 s–1, five times faster than in the DDD. For the DDDhm duplex, base pair A5:T8 opens three times faster than in the DDD (kop = 110 s–1 vs kop = 40 s–1 in DDDhm and DDD, respectively).
Structures of Duplexes Containing 5hmC, 5fC, or 5caC
Evidence for wobble base pairing geometry at oxidized cytosines,17,19,23,35,36 arising from imino tautomers of 5caC or 5fC,20,36 is not observed. The results are consistent with calculations of the stabilities of the amino and imino tautomers of 5fC and 5caC at the nucleobase level, which have suggested that, when paired with G, both 5fC and 5caC preferentially form Watson–Crick pairs.23 Instead, each of the 5hmC, 5fC, and 5caC oxidation products favors Watson–Crick hydrogen-bonding interactions when located in the 5′-T8X9G10-3′ sequence (Figures 3–5).
A common structural feature of 5fC and 5caC in the DDD is formation of intranucleobase hydrogen bonds between the carbonyl oxygens of the formyl or carboxyl groups, respectively, and a cytosine N4H amino proton. This hydrogen bond had been observed between the exocyclic N4 amino group and the formyl oxygen at C5 of 5fC at the nucleoside level.27,80 The downfield shifts of both of the X9N4 amino proton resonances into the 7.8–8.8 ppm spectral range is evident in NMR spectra of the DDDf and DDDca duplexes (Figure 5) and is consistent with the formation of these hydrogen bonds. The NMR data corroborates the crystallographic structure data (Figure 3), which shows that the carbonyl oxygens of the formyl or carboxyl groups of 5fC or 5caC, respectively, and a cytosine N4H amino proton are within hydrogen-bonding distance. The effect in the NMR data is most pronounced for the DDDca duplex (Figure 5). In the crystallographic structure of the DDDca duplex (Figure 3), this hydrogen bond keeps the carboxyl group in plane with the oxidized cytosine. For the DDD, DDDm, and DDDhm duplexes, one of the N4 amino protons shifts downfield, consistent with the maintenance of a Watson–Crick base pair, whereas the other remains in the 6.5–7.0 ppm spectral range, which is the anticipated result given that cytosine, 5mC, and 5hmC cannot form this hydrogen bond.
Structure–Activity Relationships
DNA glycosylases35 typically employ an extrahelical base-flipping mechanism34,81−87 to position substrates for catalysis. Differences in the ability of TDG to excise 5fC, 5caC, or 5hmC from DNA14 could be mediated by differential recognition of these oxidized cytosine bases in DNA. Stivers et al.59,88−91 demonstrated that damage recognition by a different glycosylase, uracil DNA glycosylase (UDG), is facilitated by enhanced base pair opening rates for destabilized A:U base pairs.34 The present data reveal that site- and sequence-specific differences with regard to duplex stability and base pair opening dynamics are observed when the 5hmC, 5fC, and 5caC are placed into the DDDhm, DDDf, and DDDca dodecamers within the 5′-T8X9G10-3′ sequence. Neither the stabilization of the DDD by 5caC nor the differences in base pair opening dynamics correlate with differences in the excision of 5hmC, 5fC, and 5caC by TDG, as reported by Maiti et al.14 Both 5hmC and 5fC exhibit increased base pair opening rates at the neighboring A5:T8 base pair. However, only 5fC is excised by TDG. Moreover, 5caC, which also is excised by TDG, thermally stabilizes the DDDca and does not exhibit increased base pair opening kinetics at the 5′-neighbor A5:T8 base pair (Figure 2).
It has been proposed that the imino tautomers of 5caC or 5fC adopt wobble-like base pairing geometry with the complementary G, which might provide a basis for recognition by TDG.20,36 The present crystallographic and NMR data indicate that the 5hmC, 5fC, and 5caC bases each favor Watson–Crick base pairing in the DDD duplex. This argues against wobble base pairing involving imino tautomers of these oxidized cytosines as a primary mode of recognition by TDG. However, the presence of small amounts of the imino tautomers cannot be ruled out, nor can a shift from Watson–Crick base pairing to wobble pairing subsequent to enzyme binding. It has been proposed that the hydrogen bond between the exocyclic N4 amine and the formyl or carboxyl oxygen at C5 of the 5fC or the 5caC base might shift the equilibrium toward the imino tautomer92,93 and lead to protonation at N3 of the oxidized cytosine.36,94 Additionally, other factors such as electrostatic and steric contributions, which remain to be examined, might modulate the differential recognition of these oxidized cytosines by TDG.
Alternatively, differences in the ability of TDG to excise 5fC, 5caC, or 5hmC from DNA could be controlled by differences in the catalytic step of base excision, once the oxidized cytosine bases have been inserted into the active site of the glycosylase. Maiti et al.95 implied a role of the conserved Asn140 in the chemical step and of the conserved Arg275 in nucleotide flipping into the active site. In additional studies, Maiti et al.14 accounted for the differential excision ability of TDG with respect to 5hmC, 5fC, and 5caC by arguing that activity is greatest for oxidized cytosines possessing electron-withdrawing substituents at the C5 carbon, which stabilize developing negative charge in the transition state complex for base excision. Following their argument, 5fC is a good substrate and 5hmC is not.14 At neutral pH, 5caC exists as an anion with pKa values of 2.4 for the carboxyl and 4.3 for the N3 position,23 and catalysis is facilitated because the ionized carboxyl group lowers the pKa of cytosine and stabilization of the carboxyl by the exocyclic amine of cytosine creates an electron-withdrawing effect.14,23 Maiti et al.23 demonstrated that the excision ability of TDG with respect to 5fC is pH-independent but that the excision of 5caC is acid-catalyzed. Moreover, Zhang et al.25 found that TDG binds to 5caC with greater affinity than to 5fC, U, or T and proposed that residues Asn157, His151, and Tyr152 are involved in hydrogen bonds with the 5caC carboxyl group. Finally, the structure of TDG in complex with DNA containing a G:5hmU mismatch showed that TDG engages in hydrogen-bonding interactions with both 5hmU and 5caC.19
The present results are consistent with the proposal by Maiti et al.,14 in which the excision specificity of TDG for 5fC and 5caC vs 5hmC is dictated by differences in the enzyme–substrate complex transition state. Both 5fC and 5caC form hydrogen bonds between the carbonyl oxygens of their formyl or carboxyl groups, respectively, and a cytosine exocyclic N4H amino proton. The electron-withdrawing effect of the 5fC and 5caC substitutents14,23 should be enhanced by hydrogen bonding between the carbonyl oxygens of their formyl or carboxyl groups, respectively, and a cytosine exocyclic N4H amino proton. This would be anticipated to stabilize developing negative charge in the transition state complex for base excision.
Summary
The cytosine oxidation products 5hmC, 5fC, and 5caC exhibit differences in thermodynamics and base pair opening dynamics when placed into the 5′-T8X9G10-3′ sequence of the DDD, but these do not correlate with differences in the ability of TDG to excise these cytosine oxidation products.14 While TDG may exploit thermodynamic and base pair opening dynamics in the recognition of oxidized cytosines in DNA, differences in the transition state complexes for the base excision step may be rate-limiting with respect to the chemical step of base excision. Of course, the 5′-T8X9G10-3′ sequence is just one sequence, and it will be of interest to further examine the sequence dependence of these effects, particularly in light of the recent report from Raiber et al.555 showing that the presence of three 5fC sites in an iterated CG repeat sequence changes the geometry of the DNA grooves and base pairs containing the 5fC oxidation product. DNA glycosylases may exploit different mechanistic pathways toward base excision. The recognition of uracil by uracil DNA glycosylase (UDG) is reported to be facilitated by enhanced base pair opening rates at A:U base pairs.34 Interestingly, the 5mC DNA glycosylase DEMETER (DME) removes 5mC, 5hmC, and 5caC but has no activity for 5fC.96,97 Its inactivity toward 5fC also does not seem to be correlated with 5fC base pair opening rates, and it does not seem to correlate with the electron-withdrawing effect of the 5fC and 5caC substitutents.14,23
Acknowledgments
We thank Edward Hawkins (deceased), Dr. Plamen Christov, and Professor Carmelo J. Rizzo for help and guidance with synthesis of oligodeoxynucleotides.
Glossary
Abbreviations
- 5caC
5-carboxylcytosine
- 5fC
5-formylcytosine
- 5hmC
5-hydroxymethylcytosine
- 5mC
5-methylcytosine
- DDD
Dickerson–Drew dodecamer
- DME
DNA glycosylase DEMETER
- DSS
4,4-dimethyl-4-silapentane-1-sulfonic acid
- EDTA
ethylenediaminetetraacetic acid, sodium salt
- MPD
2-methyl-2,4-pentanediol
- NOE
nuclear Overhauser effect
- NOESY
two-dimensional nuclear Overhauser enhancement spectroscopy
- TDG
thymine DNA glycosylase
- TET
ten-eleven translocation dioxygenase
- UDG
uracil DNA glycosylase
Supporting Information Available
Tables S1: Crystallization conditions for the DDDhm, DDDf, and DDDca duplexes. Figure S1: Temperature dependence of line widths of the imino proton resonances of the DDD, DDDm, DDDhm, DDDf, and DDDca duplexes. Figure S2: Modification site of the DDDhm duplex displaying interactions between the modified 5hmC and 3′-flanking G22 through water molecules. Figure S3: Modification site of the DDDhm duplex displaying interactions between the modified 5hmC and 3′-flanking G22 through water molecules. Figure S4: Expanded plots from NOESY spectra, depicting sequential NOE connectivities of the DDD, DDDm, DDDhm, DDDf, and DDDca duplexes. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by NIH grants R01 CA-55678 (M.P.S.), R01 ES-019625 (B.F.E.), and P41 GM103403 (NE-CAT). Funding for NMR was supplied by NIH grants S10 RR-05805 and S10 RR-025677 and NSF grant DBI 0922862, the latter was funded by the American Recovery and Reinvestment Act of 2009 (Public Law 111-5). Vanderbilt University assisted with the purchase of NMR instrumentation. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-06CH11357.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bestor T. H. (1988) Cloning of a mammalian DNA methyltransferase. Gene 74, 9–12. [DOI] [PubMed] [Google Scholar]
- Bestor T.; Laudano A.; Mattaliano R.; Ingram V. (1988) Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol. 203, 971–983. [DOI] [PubMed] [Google Scholar]
- Pfeifer G. P.; Steigerwald S. D.; Grunwald S. (1989) The DNA methylation system in proliferating and differentiated cells. Cell Biophys. 15, 79–86. [DOI] [PubMed] [Google Scholar]
- Okano M.; Bell D. W.; Haber D. A.; Li E. (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. [DOI] [PubMed] [Google Scholar]
- Wyatt G. R. (1950) Occurrence of 5-methylcytosine in nucleic acids. Nature 166, 237–238. [DOI] [PubMed] [Google Scholar]
- Meissner A. (2010) Epigenetic modifications in pluripotent and differentiated cells. Nat. Biotechnol. 28, 1079–1088. [DOI] [PubMed] [Google Scholar]
- Feng S.; Jacobsen S. E.; Reik W. (2010) Epigenetic reprogramming in plant and animal development. Science 330, 622–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. C.; Zhang Y. (2010) Active DNA demethylation: many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 11, 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globisch D.; Munzel M.; Muller M.; Michalakis S.; Wagner M.; Koch S.; Bruckl T.; Biel M.; Carell T. (2010) Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One 5, e15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzel M.; Globisch D.; Carell T. (2011) 5-Hydroxymethylcytosine, the sixth base of the genome. Angew. Chem., Int. Ed. 50, 6460–6468. [DOI] [PubMed] [Google Scholar]
- Gu T. P.; Guo F.; Yang H.; Wu H. P.; Xu G. F.; Liu W.; Xie Z. G.; Shi L.; He X.; Jin S. G.; Iqbal K.; Shi Y. G.; Deng Z.; Szabo P. E.; Pfeifer G. P.; Li J.; Xu G. L. (2011) The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610. [DOI] [PubMed] [Google Scholar]
- Iqbal K.; Jin S. G.; Pfeifer G. P.; Szabo P. E. (2011) Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci. U.S.A. 108, 3642–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wossidlo M.; Nakamura T.; Lepikhov K.; Marques C. J.; Zakhartchenko V.; Boiani M.; Arand J.; Nakano T.; Reik W.; Walter J. (2011) 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun. 2, 241. [DOI] [PubMed] [Google Scholar]
- Maiti A.; Drohat A. C. (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem. 286, 35334–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y. F.; Li B. Z.; Li Z.; Liu P.; Wang Y.; Tang Q.; Ding J.; Jia Y.; Chen Z.; Li L.; Sun Y.; Li X.; Dai Q.; Song C. X.; Zhang K.; He C.; Xu G. L. (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti A.; Drohat A. C. (2011) Dependence of substrate binding and catalysis on pH, ionic strength, and temperature for thymine DNA glycosylase: insights into recognition and processing of G·T mispairs. DNA Repair 10, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortellino S.; Xu J.; Sannai M.; Moore R.; Caretti E.; Cigliano A.; Le Coz M.; Devarajan K.; Wessels A.; Soprano D.; Abramowitz L. K.; Bartolomei M. S.; Rambow F.; Bassi M. R.; Bruno T.; Fanciulli M.; Renner C.; Klein-Szanto A. J.; Matsumoto Y.; Kobi D.; Davidson I.; Alberti C.; Larue L.; Bellacosa A. (2011) Active DNA demethylation by thymine DNA glycosylase. Environ. Mol. Mutagen. 52, S14–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortellino S.; Xu J. F.; Sannai M.; Moore R.; Caretti E.; Cigliano A.; Le Coz M.; Devarajan K.; Wessels A.; Soprano D.; Abramowitz L. K.; Bartolomei M. S.; Rambow F.; Bassi M. R.; Bruno T.; Fanciulli M.; Renner C.; Klein-Szanto A. J.; Matsumoto Y.; Kobi D.; Davidson I.; Alberti C.; Larue L.; Bellacosa A. (2011) Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146, 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H.; Hong S.; Bhagwat A. S.; Zhang X.; Cheng X. (2012) Excision of 5-hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: its structural basis and implications for active DNA demethylation. Nucleic Acids Res. 40, 10203–10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H.; Zhang X.; Cheng X. (2012) Excision of thymine and 5-hydroxymethyluracil by the MBD4 DNA glycosylase domain: structural basis and implications for active DNA demethylation. Nucleic Acids Res. 40, 8276–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti A.; Noon M. S.; MacKerell A. D. Jr.; Pozharski E.; Drohat A. C. (2012) Lesion processing by a repair enzyme is severely curtailed by residues needed to prevent aberrant activity on undamaged DNA. Proc. Natl. Acad. Sci. U.S.A. 109, 8091–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J.; Wagner J. R. (2014) TET enzymatic oxidation of 5-methylcytosine, 5-hydroxymethylcytosine and 5-formylcytosine. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 764–765, 18–35. [DOI] [PubMed] [Google Scholar]
- Maiti A.; Michelson A. Z.; Armwood C. J.; Lee J. K.; Drohat A. C. (2013) Divergent mechanisms for enzymatic excision of 5-formylcytosine and 5-carboxylcytosine from DNA. J. Am. Chem. Soc. 135, 15813–15822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wossidlo M.; Arand J.; Sebastiano V.; Lepikhov K.; Boiani M.; Reinhardt R.; Scholer H.; Walter J. (2010) Dynamic link of DNA demethylation, DNA strand breaks and repair in mouse zygotes. EMBO J. 29, 1877–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Su L.; Wang Z.; Zhang S.; Guan J.; Chen Y.; Yin Y.; Gao F.; Tang B.; Li Z. (2012) The involvement of 5-hydroxymethylcytosine in active DNA demethylation in mice. Biol. Reprod. 86, 104. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S.; Heintz N. (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzel M.; Lischke U.; Stathis D.; Pfaffeneder T.; Gnerlich F. A.; Deiml C. A.; Koch S. C.; Karaghiosoff K.; Carell T. (2011) Improved synthesis and mutagenicity of oligonucleotides containing 5-hydroxymethylcytosine, 5-formylcytosine and 5-carboxylcytosine. Chemistry 17, 13782–13788. [DOI] [PubMed] [Google Scholar]
- Simmons J. M.; Muller T. A.; Hausinger R. P. (2008) Fe(II)/alpha-ketoglutarate hydroxylases involved in nucleobase, nucleoside, nucleotide, and chromatin metabolism. Dalton Trans. 5132–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M.; Koh K. P.; Shen Y.; Pastor W. A.; Bandukwala H.; Brudno Y.; Agarwal S.; Iyer L. M.; Liu D. R.; Aravind L.; Rao A. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S.; D’Alessio A. C.; Taranova O. V.; Hong K.; Sowers L. C.; Zhang Y. (2010) Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu C.; Wagner J. R.; Cadet J. (1996) Photosensitized oxidation of 5-methyl-2′-deoxycytidine by 2-methyl-1,4-naphthoquinone: characterization of 5-(hydroperoxymethyl)-2′-deoxycytidine and stable methyl group oxidation products. J. Am. Chem. Soc. 118, 11406–11411. [Google Scholar]
- Ito S.; Shen L.; Dai Q.; Wu S. C.; Collins L. B.; Swenberg J. A.; He C.; Zhang Y. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffeneder T.; Hackner B.; Truss M.; Munzel M.; Muller M.; Deiml C. A.; Hagemeier C.; Carell T. (2011) The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem., Int. Ed. 50, 7008–7012. [DOI] [PubMed] [Google Scholar]
- Raiber E.; Murat P.; Chirgadze D. Y.; Beraldi D.; Luisi B. F.; Balasubramanian S. (2015) 5-Formylcytosine alters the structure of the DNA double helix. Nat. Struct. Mol. Biol. 22, 44–49 10.1038/nsmb.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stivers J. T. (2004) Site-specific DNA damage recognition by enzyme-induced base flipping. Prog. Nucleic Acid Res. Mol. Biol. 77, 37–65. [DOI] [PubMed] [Google Scholar]
- Hardeland U.; Bentele M.; Lettieri T.; Steinacher R.; Jiricny J.; Schar P. (2001) Thymine DNA glycosylase. Prog. Nucleic Acid Res. Mol. Biol. 68, 235–253. [DOI] [PubMed] [Google Scholar]
- Hashimoto H.; Zhang X.; Cheng X. (2013) Selective excision of 5-carboxylcytosine by a thymine DNA glycosylase mutant. J. Mol. Biol. 425, 971–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing R.; Drew H.; Takano T.; Broka C.; Tanaka S.; Itakura K.; Dickerson R. E. (1980) Crystal structure analysis of a complete turn of B-DNA. Nature 287, 755–758. [DOI] [PubMed] [Google Scholar]
- Drew H. R.; Wing R. M.; Takano T.; Broka C.; Tanaka S.; Itakura K.; Dickerson R. E. (1981) Structure of a B-DNA dodecamer: conformation and dynamics. Proc. Natl. Acad. Sci. U.S.A. 78, 2179–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tereshko V.; Minasov G.; Egli M. (1999) The Dickerson–Drew B-DNA dodecamer revisited at atomic resolution. J. Am. Chem. Soc. 121, 470–471. [Google Scholar]
- Howerton S. B.; Sines C. C.; VanDerveer D.; Williams L. D. (2001) Locating monovalent cations in the grooves of B-DNA. Biochemistry 40, 10023–10031. [DOI] [PubMed] [Google Scholar]
- Kowal E. A.; Ganguly M.; Pallan P. S.; Marky L. A.; Gold B.; Egli M.; Stone M. P. (2011) Altering the electrostatic potential in the major groove: thermodynamic and structural characterization of 7-deaza-2′-deoxyadenosine:dT base pairing in DNA. J. Phys. Chem. B 115, 13925–13934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal E. A.; Lad R. R.; Pallan P. S.; Dhummakupt E.; Wawrzak Z.; Egli M.; Sturla S. J.; Stone M. P. (2013) Recognition of O6-benzyl-2′-deoxyguanosine by a perimidinone-derived synthetic nucleoside: a DNA interstrand stacking interaction. Nucleic Acids Res. 41, 7566–7576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare D. R.; Wemmer D. E.; Chou S. H.; Drobny G.; Reid B. R. (1983) Assignment of the non-exchangeble proton resonances of d(C-G-C-G-A-A-T-T-C-G-C-G) using two-dimensional nuclear magnetic resonance methods. J. Mol. Biol. 171, 319–336. [DOI] [PubMed] [Google Scholar]
- Moe J. G.; Russu I. M. (1990) Proton exchange and base-pair opening kinetics in 5′-d(CGCGAATTCGCG)-3′ and related dodecamers. Nucleic Acids Res. 18, 821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjandra N.; Tate S.-I.; Ono A.; Kainosho M.; Bax A. (2000) The NMR structure of a DNA dodecamer in an aqueous dilute liquid crystalline phase. J. Am. Chem. Soc. 122, 6190–6200. [Google Scholar]
- Singh S. K.; Szulik M. W.; Ganguly M.; Khutsishvili I.; Stone M. P.; Marky L. A.; Gold B. (2011) Characterization of DNA with an 8-oxoguanine modification. Nucleic Acids Res. 39, 6789–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaluzzi M. J.; Borer P. N. (2004) Revised UV extinction coefficients for nucleoside-5′-monophosphates and unpaired DNA and RNA. Nucleic Acids Res. 32, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marky L. A.; Breslauer K. J. (1987) Calculating thermodynamic data for transitions of any molecularity from equilibrium melting curves. Biopolymers 26, 1601–1620. [DOI] [PubMed] [Google Scholar]
- Bodenhausen G.; Wagner G.; Rance M.; Sorensen O. W.; Wuthrich K.; Ernst R. R. (1984) Longitudinal two-spin order in 2D exchange spectroscopy (NOESY). J. Magn. Reson. 59, 542–550. [Google Scholar]
- Piotto M.; Saudek V.; Sklenar V. (1992) Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 6, 661–665. [DOI] [PubMed] [Google Scholar]
- Chen C.; Russu I. M. (2004) Sequence-dependence of the energetics of opening of at basepairs in DNA. Biophys. J. 87, 2545–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Jiang L.; Michalczyk R.; Russu I. M. (2006) Structural energetics and base-pair opening dynamics in sarcin-ricin domain RNA. Biochemistry 45, 13606–13613. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Chen C.; Russu I. M. (2009) Dynamics and stability of individual base pairs in two homologous RNA–DNA hybrids. Biochemistry 48, 3988–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Weng X.; Russu I. M. (2010) Structural energetics of the adenine tract from an intrinsic transcription terminator. J. Mol. Biol. 397, 677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Weng X.; Russu I. M. (2011) Enhanced base-pair opening in the adenine tract of a RNA double helix. Biochemistry 50, 1857–1863. [DOI] [PubMed] [Google Scholar]
- Gueron M.; Leroy J. L. (1995) Studies of base pair kinetics by NMR measurement of proton exchange. Methods Enzymol. 261, 383–413. [DOI] [PubMed] [Google Scholar]
- Szulik M. W.; Voehler M.; Stone M. P. (2014) NMR analysis of base-pair opening kinetics in DNA. Curr. Prot. Nucl. Acid Chem. 59, 7.20.1–7.20.18 10.1002/0471142700.nc0720s59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plateau P.; Gueron M. (1982) Exchangeable proton NMR without base-line distortion, using new strong-pulse sequences. J. Am. Chem. Soc. 104, 7310–7311. [Google Scholar]
- Crenshaw C. M.; Wade J. E.; Arthanari H.; Frueh D.; Lane B. F.; Nunez M. E. (2011) Hidden in plain sight: subtle effects of the 8-oxoguanine lesion on the structure, dynamics, and thermodynamics of a 15-base pair oligodeoxynucleotide duplex. Biochemistry 50, 8463–8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J. B.; Stivers J. T. (2011) Dynamics of uracil and 5-fluorouracil in DNA. Biochemistry 50, 612–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum G.; Alkire R. W.; Evans G.; Rotella F. J.; Lazarski K.; Zhang R. G.; Ginell S. L.; Duke N.; Naday I.; Lazarz J.; Molitsky M. J.; Keefe L.; Gonczy J.; Rock L.; Sanishvili R.; Walsh M. A.; Westbrook E.; Joachimiak A. (2006) The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J. Synchrotron Radiat. 13, 30–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W.; Cymborowski M.; Otwinowski Z.; Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 62, 72–82. [DOI] [PubMed] [Google Scholar]
- (1994) The CCP4 suite: Programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Acta Crystallogr., Sect. A 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Vagin A. (1989) New translation and packing functions, in Newsletter on Protein Crystallography, Vol. 24, pp 117–121, Daresbury Laboratory. [Google Scholar]
- Vagin A.; Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025. [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Murshudov G. N.; Papiz M. Z. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321. [DOI] [PubMed] [Google Scholar]
- Vagin A. A.; Steiner R. A.; Lebedev A. A.; Potterton L.; McNicholas S.; Long F.; Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2184–2195. [DOI] [PubMed] [Google Scholar]
- Boelens R.; Scheek R. M.; Dijkstra K.; Kaptein R. (1985) Sequential assignment of imino- and amino-proton resonances in 1H NMR spectra of oligonucleotides by two-dimensional NMR spectroscopy. Application to a lac operator fragment. J. Magn. Reson. 62, 378–386. [Google Scholar]
- Russu I. M. (2004) Probing site-specific energetics in proteins and nucleic acids by hydrogen exchange and nuclear magnetic resonance spectroscopy. Methods Enzymol. 379, 152–175. [DOI] [PubMed] [Google Scholar]
- Every A. E.; Russu I. M. (2008) Influence of magnesium ions on spontaneous opening of DNA base pairs. J. Phys. Chem. B 112, 7689–7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. G.; Chen C. J.; Russu I. M. (2009) Structural energetics of a DNA–RNA hybrid containing a tract of dA–rU base pairs. J. Biomol. Struct. Dyn. 26, 900–900. [Google Scholar]
- Kopka M. L.; Fratini A. V.; Drew H. R.; Dickerson R. E. (1983) Ordered water structure around a B-DNA dodecamer. A quantitative study. J. Mol. Biol. 163, 129–146. [DOI] [PubMed] [Google Scholar]
- Patel D. J.; Shapiro L.; Hare D. (1987) DNA and RNA: NMR studies of conformations and dynamics in solution. Q. Rev. Biophys. 20, 35–112. [DOI] [PubMed] [Google Scholar]
- Reid B. R. (1987) Sequence-specific assignments and their use in NMR studies of DNA structure. Q. Rev. Biophys. 20, 2–28. [DOI] [PubMed] [Google Scholar]
- Sumino M.; Ohkubo A.; Taguchi H.; Seio K.; Sekine M. (2008) Synthesis and properties of oligodeoxynucleotides containing 5-carboxy-2′-deoxycytidines. Bioorg. Med. Chem. Lett. 18, 274–277. [DOI] [PubMed] [Google Scholar]
- Burdzy A.; Noyes K. T.; Valinluck V.; Sowers L. C. (2002) Synthesis of stable-isotope enriched 5-methylpyrimidines and their use as probes of base reactivity in DNA. Nucleic Acids Res. 30, 4068–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A. Y.; Scharer O. D.; Samson L.; Verdine G. L.; Ellenberger T. (1998) Crystal structure of a human alkylbase-DNA repair enzyme complexed to DNA: mechanisms for nucleotide flipping and base excision. Cell 95, 249–258. [DOI] [PubMed] [Google Scholar]
- Parikh S. S.; Mol C. D.; Hosfield D. J.; Tainer J. A. (1999) Envisioning the molecular choreography of DNA base excision repair. Curr. Opin. Struct. Biol. 9, 37–47. [DOI] [PubMed] [Google Scholar]
- Bruner S. D.; Norman D. P.; Verdine G. L. (2000) Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 403, 859–866. [DOI] [PubMed] [Google Scholar]
- Hollis T.; Ichikawa Y.; Ellenberger T. (2000) DNA bending and a flip-out mechanism for base excision by the helix-hairpin-helix DNA glycosylase, Escherichia coli AlkA. EMBO J. 19, 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tainer J. A. (2001) Structural implications of BER enzymes: dragons dancing—the structural biology of DNA base excision repair. Prog. Nucleic Acid Res. Mol. Biol. 68, 299–304. [DOI] [PubMed] [Google Scholar]
- Fromme J. C.; Banerjee A.; Verdine G. L. (2004) DNA glycosylase recognition and catalysis. Curr. Opin. Struct. Biol. 14, 43–49. [DOI] [PubMed] [Google Scholar]
- Fromme J. C.; Verdine G. L. (2004) Base excision repair. Adv. Protein Chem. 69, 1–41. [DOI] [PubMed] [Google Scholar]
- Cao C.; Jiang Y. L.; Stivers J. T.; Song F. (2004) Dynamic opening of DNA during the enzymatic search for a damaged base. Nat. Struct. Mol. Biol. 11, 1230–1236. [DOI] [PubMed] [Google Scholar]
- Krosky D. J.; Song F.; Stivers J. T. (2005) The origins of high-affinity enzyme binding to an extrahelical DNA base. Biochemistry 44, 5949–5959. [DOI] [PubMed] [Google Scholar]
- Parker J. B.; Bianchet M. A.; Krosky D. J.; Friedman J. I.; Amzel L. M.; Stivers J. T. (2007) Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature 449, 433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J. I.; Stivers J. T. (2010) Detection of damaged DNA bases by DNA glycosylase enzymes. Biochemistry 49, 4957–4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karino N.; Ueno Y.; Matsuda A. (2001) Synthesis and properties of oligonucleotides containing 5-formyl-2′-deoxycytidine: in vitro DNA polymerase reactions on DNA templates containing 5-formyl-2′-deoxycytidine. Nucleic Acids Res. 29, 2456–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H.; Tsuchiya H.; Karino N.; Ueno Y.; Matsuda A.; Harashima H. (2002) Mutagenicity of 5-formylcytosine, an oxidation product of 5-methylcytosine, in DNA in mammalian cells. J. Biochem. 132, 551–555. [DOI] [PubMed] [Google Scholar]
- Hashimoto H.; Liu Y.; Upadhyay A. K.; Chang Y.; Howerton S. B.; Vertino P. M.; Zhang X.; Cheng X. (2012) Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 40, 4841–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti A.; Morgan M. T.; Drohat A. C. (2009) Role of two strictly conserved residues in nucleotide flipping and N-glycosylic bond cleavage by human thymine DNA glycosylase. J. Biol. Chem. 284, 36680–36688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H.; Shin H.; Eichman B. F.; Huh J. H. (2014) Excision of 5-hydroxymethylcytosine by DEMETER family DNA glycosylases. Biochem. Biophys. Res. Commun. 446, 1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks S. C.; Fischer R. L.; Huh J. H.; Eichman B. F. (2014) 5-Methylcytosine recognition by Arabidopsis thaliana DNA glycosylases DEMETER and DML3. Biochemistry 53, 2525–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.