

Abstract
An efficient new molecular orbital (MO) basis algorithm is reported implementing the pair atomic resolution of the identity approximation (PARI) to evaluate the exact exchange contribution (K) to self-consistent field methods, such as hybrid and range-separated hybrid density functionals. The PARI approximation, in which atomic orbital (AO) basis function pairs are expanded using auxiliary basis functions centered only on their two respective atoms, was recently investigated by Merlot et al. [J. Comput. Chem.2013, 34, 1486]. Our algorithm is significantly faster than quartic scaling RI-K, with an asymptotic exchange speedup for hybrid functionals of (1 + X/N), where N and X are the AO and auxiliary basis dimensions. The asymptotic speedup is 2 + 2X/N for range separated hybrids such as CAM-B3LYP, ωB97X-D, and ωB97X-V which include short- and long-range exact exchange. The observed speedup for exchange in ωB97X-V for a C68 graphene fragment in the cc-pVTZ basis is 3.4 relative to RI-K. Like conventional RI-K, our method greatly outperforms conventional integral evaluation in large basis sets; a speedup of 19 is obtained in the cc-pVQZ basis on a C54 graphene fragment. Negligible loss of accuracy relative to exact integral evaluation is demonstrated on databases of bonded and nonbonded interactions. We also demonstrate both analytically and numerically that the PARI-K approximation is variationally stable.
1. Introduction
Modern Kohn–Sham density functional theory1,2 has become the most widely used electronic structure method. A crucial contribution to this success was Becke’s insight into the role of exact exchange in approximations to the adiabatic connection formula.3 This discovery led to the creation of the class of methods known as “hybrid” functionals that include some (usually empirically determined) weighted contribution from exact exchange,4−6 as exemplified by the widely used B3LYP functional. The use of hybrid functionals has yielded improved results for thermochemical properties,3 molecular geometries,7 and vibrational frequencies.8,9 More recently developed hybrid functionals such as M06-2X have yielded significant additional improvements in accuracy.10,11
The role of exact-exchange has since been further refined with the introduction of range-separated hybrid functionals,12,13 which hold particular promise for applications in the area of time-dependent density functional theory.14,15 Local functionals and standard hybrid functionals suffer from incorrect long-range behavior of charge transfer excited states due to self-interaction error of the transferred electron.16,17 Range-separated hybrid functionals partition the Coulomb operator into short and long-range components in order to smoothly increase the weight of exact exchange toward unity in the long-range limit, thus recovering the correct asymptotic behavior. The development of systematically optimized functionals such as the ωB97X-D functional18 led to significant improvements in accuracy over first generation range-separated hybrids. Further noteworthy improvements were recently reported in the ωB97X-V functional.19
Unfortunately, large basis sets are required to obtain the maximal accuracy which modern hybrid and range-separated hybrid functionals can provide. Many studies have demonstrated that a basis set of at least triple-ζ quality is required to obtain results reasonably close to the basis set limit with hybrid functionals.20−23 At least triple, and if possible quadruple-ζ basis sets are needed for accurate thermochemistry, while at least augmented triple-ζ basis sets are required for noncovalent interactions if no counterpoise corrections are performed.19 In this scenario, computation of exact-exchange typically consumes the vast majority of the computation time for a hybrid DFT calculation.
The K matrix required for the computation of exact exchange is computed via the expression:
| 1 |
The number of two-electron integrals appearing in (1) scales as the fourth power of molecular size. Schwarz-inequality-based integral screening algorithms can reduce this scaling to nearly O(M2) for sufficiently extended systems.24,25 In fact, linear scaling algorithms can be obtained by exploiting the density matrix sparsity that occurs in extended insulating systems, as is done in the SONX26 and LinK27,28 methods. However, such linear scaling behavior is in practice seldom obtained for small-gap systems and/or with larger basis sets which can lead to overlap ill-conditioning and resulting loss of density matrix sparsity.
Even with the employment of screening algorithms, the scaling of the evaluation of (1) remains fourth-order with respect to increasing the average number of basis functions per atom while molecule size is fixed. High-accuracy calculations in large basis sets are therefore most severely inhibited by the computational demands associated with exact exchange. With the introduction of still-more sophisticated treatments of electron correlation such as double-hybrid functionals,29−31 the need to compute efficiently with larger basis sets has only become more pressing.32
A variety of approaches have been used to improve the efficiency of constructing K in large basis sets, the most common of which is the application of the resolution of the identity (RI) approximation, also frequently referred to as the density fitting or DF approximation. In the RI approximation, products of orbital basis functions are further expanded in an auxiliary basis:
| 2 |
This reduces the dimensionality of the integral tensors that must be calculated from four to three. The use of the RI approximation to compute exact exchange was introduced by Früchtl,33 while the first direct implementation is due to Weigend.34 Unfortunately, without further approximation the RI-K algorithm scales fourth-order with molecular size, so performance deteriorates relative to conventional integral formation for extended systems. Hence, the application of RI-SCF (self-consistent field) has historically been limited to compact systems with large basis sets.33−35
It has been suggested that RI methods may be better described as “inner projection” methods, as in the absence of a complete auxiliary basis the RI approximation does not amount to an insertion of a resolution of the identity, but rather the insertion of an inner projection operator.36,37 The Cholesky decomposition (CD) of the two-electron integrals has been shown to be equivalent to the introduction of an inner projection operator onto the space spanned by the orthogonalized Cholesky vectors.38 In this sense, the Cholesky decomposition may be seen as generating an ideal auxiliary basis for the particular problem at hand.37 CD methods encompass many efficient algorithms. Since its introduction to the electronic structure community by Beebe and Linderberg,38 CD has been used to formulate a variety of efficient electronic structure algorithms, including the construction of the Fock matrix36,39 and the evaluation of the CCSD(T)40 and SOS-MP241 energies. CD methods are of great interest because they produce an auxiliary basis that does not depend on the model chemistry for which it was optimized, and thus can deliver consistent performance across a variety of theoretical methods.42 Additionally, CD methods allow for fine-grained control of the accuracy of the approximation simply by varying the decomposition threshold δ. This is in contrast to the RI method’s reliance on preoptimized auxiliary basis sets, which limits its accuracy to that attainable by the largest auxiliary basis set trained for the given theoretical method. However, as currently available RI auxiliary basis sets yield fitting errors in the tens of μHartree per atom for absolute energies,34,43−45 which is negligible compared to orbital basis set errors, the practical advantage provided by CD in terms of accuracy is often minimal.
CD methods have historically suffered from the inclusion of two-center functions in the resulting auxiliary basis, and therefore required the expensive evaluation of four-center integrals.42 The situation improves if the CD basis is restricted to contain only one-center auxiliary functions; examples of such methods include the one-center Cholesky decomposition42 (1C-CD) and the atomic Cholesky decomposition (aCD).46 The aCD and 1C-CD approaches have been used to obtain more compact sets of Cholesky vectors, with a size ≈5 times that of the orbital basis set found to yield acceptable accuracy.42,46 For comparison, the RI approximation typically requires an auxiliary basis set of 3–5 times the size of the orbital basis set to obtain accuracy in the μHartree range.34,43−45 Rigorous efficiency comparisons of RI and CD methods are scarce; in one such study, Weigend has shown that 1C-CD is outperformed significantly by RI-J.47 However, more recent work on so-called “method-specific” CDs, in which Hadamard products involving the two-electron integrals are decomposed instead of the two-electron integrals themselves, has been shown to require a drastically smaller auxiliary basis37 and has been found to be competitive with RI for the computation of exact exchange.47
The unfavorable scaling of RI-SCF can be mitigated through the use of several types of local approximations. The RI expansion coefficients are typically obtained in the Coulomb metric by minimizing the Coulomb repulsion of the density residual.48 The fitting coefficients thus obtained are highly nonlocal; the expansion of a product on a given center contains many numerically significant contributions from auxiliary functions on distant atomic centers. A root cause of this is local incompleteness of the auxiliary basis set, which leads to the inclusion of off-center auxiliary functions in order to obtain a better fit.46,49 The locality of the fit coefficients can be improved, while still retaining accuracy in the fit, by minimizing the residual repulsion based on an alternative local operator, a so-called “local metric”. Perhaps the simplest and most drastic local metric approach to density fitting is the overlap metric first introduced by Baerends et al.50 The overlap metric was subsequently employed by Vahtras et al. in SCF calculations and was found to give errors an order of magnitude larger than the Coulomb metric.51
In an attempt to combine the locality of the overlap metric with the accuracy of the Coulomb metric, Jung et al. introduced an attenuated Coulomb metric with rapid decay with interelectronic distance and similar RI errors to the full Coulomb metric.49 The attenuated Coulomb metric has subsequently been employed by Reine in conjunction with localized MOs to implement reduced-scaling RI-SCF.52 In an analogous development for CD methods, Aquilante et al. have shown that aCD methods yield a set of fit coefficients with spatial locality that increases as the decomposition threshold is decreased.46 This is a consequence of a more complete CD auxiliary basis reducing the need to include off-center auxiliary functions in order to represent on-center products.
An alternative ubiquitous approach to constructing K economically is a family of RI approximations employing local fitting domains. Instead of expanding each orbital basis function product using a fitting basis set spanning the entire molecule, a subset of the full fitting basis is chosen for each orbital product. There have been numerous applications of local fitting domains to RI-SCF.35,53−55 In the first development in this area, Polly et al. developed a linear scaling algorithm using local atomic fitting domains in conjunction with localized orbitals.53 Sodt and Head-Gordon have developed the ARI-K algorithm, wherein each pair of orbital basis functions is expanded using the auxiliary basis functions on the atoms in a certain radius around their parent atoms.35 Very recently, Mejía-Rodríguez et al. have developed the LDF-HF algorithm, which builds upon the work of Polly et al. with localization of the molecular orbitals at each SCF cycle in order to obtain very compact local fitting domains.56
Yet another unique approach to improving the locality and scaling of exchange matrix construction is the pseudospectral approach of Neese et al, in which one coordinate in the integrals from (1) is integrated by numerical quadrature.57 The locality of the grid points coupling to the AO basis is exploited to obtain a conditionally linear scaling algorithm. This approach has been shown to yield equal or superior speedups to RI-K with comparable accuracy.58 However, to our knowledge its performance has not been benchmarked relative to local density fitting methods.
The focus of this work is the construction of K using a drastically local form of the RI approximation, which we shall designate by the name used by Merlot et al. as the Pair Atomic Resolution of the Identity (PARI) approximation.54 In this approach, a given pair of orbital basis functions is expanded using only auxiliary basis functions on either of the parent atoms. This fitting approach was explored in early work by Baerends et al.59 and subsequently by several others.60−62 When used with Dunlap’s robust fit functional,63 the PARI approximation has been found to give surprisingly accurate results.54,55 However, it has been demonstrated that the use of the PARI approximation for the construction of J can lead to unphysical “attractive electron” states.54,55 Merlot et al. have explained this behavior on the basis of the loss of a positive semidefinite integral tensor, and have shown that this problem manifests for any local RI approximation employing the Dunlap functional. They attempted to correct this problem through local completion of the auxiliary basis using the Cholesky decomposition, but this approach resulted in the loss of all performance improvements derived from the PARI approximation.54 Hollman et al. have taken a different approach by computing subsets of the two-electron integrals analytically, and employing the PARI approximation for the remainder.55 We circumvent this variational stability issue entirely by using PARI-K alone, without PARI-J. We will analytically demonstrate that the use of PARI-K alone is variationally stable, and empirically demonstrate that PARI-K yields negligible errors compared to exact integral evaluation. The J matrix can in turn be constructed by one of several pre-existing efficient highly efficient algorithms.34,64
Merlot et al. have used the PARI approximation and the Dunlap functional63 to create a new algorithm, PARI-K, for the construction of K.54 These authors report an impressive speedups of between 5 and 7-fold for their PARI-K algorithm relative to the LinK algorithm,measured on systems of up to 42 atoms in a triple-ζ basis set. However, their algorithm involves contraction of three-center RI integrals over the full auxiliary basis with density-contracted fit coefficients:
| 3 |
The auxiliary index Q is coupled by sparsity to the AO-basis index μ as follows: for a given μ, all Q auxiliary basis functions on atoms with at least one basis function that has non-negligible overlap with μ is significant. The scaling of this term is thus asymptotically quadratic, just like direct evaluation of the two electron integrals, which is highly desirable. However, it suffers from the same unfavorable scaling of O(N4) with respect to basis set size for fixed molecular size, but with an improved prefactor based on the number of FLOPs required to sum over Q versus the FLOPs required to form a given two-electron integral directly. We shall demonstrate that dramatic improvements in performance can be obtained via an MO basis algorithm. In our algorithm, the two and three-center quantities evaluated separately in Merlot’s formulation are evaluated simultaneously in the MO basis. Unlike Merlot’s algorithm, the scaling of our algorithm remains fourth-order; however, we demonstrate by numerical experiments that our algorithm performs fewer operations even for very extended 1D systems. We obtain speedups of up to 19× over conventional K construction for systems of up to 3570 basis functions. We also demonstrate that auxiliary basis sets no larger than those used in conventional RI-SCF are required when the PARI-K approximation is used to obtain chemical accuracy for thermodynamic properties and intermolecular interactions, despite the drastically local nature of the approximation.
2. Theory
2.1. Variational Stability of RI-K and PARI-K
We shall abbreviate RI quantities using a tilde as follows:
| 4 |
Dunlap has shown that the following “robust” approximation of the two-electron integrals yields errors that are quadratic in the error resulting from the RI approximation on the individual products:63
| 5 |
Merlot et al. have shown that the use of this robust formulation in conjunction with local RI fitting domains and/or non-Coulomb fitting metrics results in a two-electron integral tensor that is not positive semidefinite.54 However, regardless of this complication, the following equality holds for the Dunlap formulation of any fitting method:63
| 6 |
The difference of the exact and fitted exchange energies may thus be written as
| 7 |
Transforming to the MO basis, we have
| 8 |
By an early result of Slater,65 each term in the above sum is strictly positive. The exchange energy thus can only increase by the application of robust fitting, and densities corresponding to negative eigenvalues of the two-electron integral matrix will be avoided by the SCF convergence algorithm. Local fit domains and non-Coulomb fitting metrics can therefore be applied to the computation of K without risk of variational instability.
We now prove a stronger result concerning the relationship between standard Coulomb-metric RI using a global fit domain and that of any local fit approximation. Denoting coefficients for the MO pair ij computed using global RI by CijG and those with local RI by Cij, we may express their difference by
| 9 |
We now consider the expansion of a single exchange integral using the Dunlap functional and local RI fit coefficients, using Iij to denote the three-center integral vector for this MO pair and V to denote the auxiliary basis metric:
The combination of the first two terms in the above expression yields the integral computed using global RI with the Coulomb metric. The next term vanishes by the inverse present in the Coulomb metric fit coefficients, and we thus obtain
| 12 |
The error term is non-negative, as V is positive semidefinite. We therefore see that the exchange energy computed with local RI methods is greater than or equal to both the exact and global Coulomb RI variants:
| 13 |
It is well-known that exchange integrals are bounded by their corresponding Coulomb integrals:66
| 14 |
This inequality holds when both integrals are expanded using a global auxiliary basis. Our treatment expands the Coulomb term in the global auxiliary basis, with the exchange term treated in the pair basis. Our above result proving that EKL ≥ EK therefore guarantees that the total electronic energy of the system cannot become negative.
2.2. Efficient MO Basis PARI-K Algorithm
Merlot et al. have applied Dunlap’s robust formulation in conjunction with the PARI approximation, in which orbital basis products between functions lying on the atoms A and B are expanded only using auxiliary basis functions on those two atoms:54
| 15 |
Using these two approximations, we shall now derive an efficient algorithm for the formation of the Hartree–Fock exchange matrix. We may write the expression for the exchange matrix in the following convenient form:
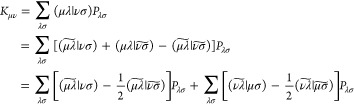 |
We then make the following definition:
| 16 |
to yield the resulting expression for the exchange matrix:
| 17 |
The problem of efficiently computing the exchange matrix now reduces to the efficient computation of L. We now introduce the RI approximation, presuming the use of the PARI fit coefficients in eq 15:
| 18 |
Grouping common terms yields
Reverting to the MO basis and rearranging yields an intelligently grouped expression:
| 19 |
Here the MO coefficient matrix is M. This expression is evaluated using the steps presented in Table 1.
Table 1. MO-Basis Algorithm for Exchange Matrix Formationa.
| step | operations | scaling | storage | |
|---|---|---|---|---|
| Before First SCF Iteration | ||||
| calculate (R|Q) | X2 | M2 | M2 | |
| [NB2]X̅ | M1 | M1 | ||
| For Each SCF Iteration | ||||
| loop over atomic batches of auxiliary functions Q | ||||
| o [NB2]X̅ | M2 | M1 | ||
| [NB2]X̅X | M2 | M1 | ||
| calculate integral batch (νσ|Q) | [NB2]X | M2 | M1 | |
| [NB2]X | M2 | M1 | ||
| o [NB2]X | M3 | M2 | ||
| oN [NBX] | M3 | M2 | ||
| N2 | M2 | M2 | ||
The second column gives the operation cost for each step in terms of [NB2] (number of significant orbital-basis function pairs, which is asymptotically linear in system size), X̅ (mean number of auxiliary basis functions per atom, independent of system size), X (number of auxiliary basis functions), o (number of occupied orbitals), and [NBX] (number of significant orbital-basis to aux-basis function pairs, which is also asymptotically linear).
Our algorithm is very similar to the LDF-HF algorithm of Mejía-Rodríguez et al.56 but without MO localization and with additional steps to account for the explicitly robust Dunlap formulation. Additionally, when all tensors cannot be held in memory, we batch over auxiliary functions rather than over molecular orbitals, allowing us to compute the three-center AO integrals only once per SCF cycle. For large systems, the step Lμν = ∑QiDiμQHi dominates the overall computation time. As we demonstrate for a series of linear alkanes, the onset of sparsity in the MO-contracted fit coefficients DiμQ is sufficiently slow even for linear systems that it is generally not economical to utilize sparsity in this step, and the algorithm is thus effectively fourth order scaling, but with a significantly smaller prefactor than RI-K. The two fourth-order steps in RI-K are given by
The formation of the B matrix is more expensive than its subsequent contraction by a factor of X/N. The fourth-order step in our algorithm has the same cost as the latter fourth order step of RI-K, and the asymptotic speedup for our algorithm relative to RI-K is thus 1 + X/N. It should be noted that our algorithm is capable of treating much larger systems on an economical time scale than the largest presented in this paper; for the purposes of this study, we were limited by the feasibility of timing the integral-driven code for comparison.
The Dunlap formulation converts first order error in the fitted products to second-order error in the approximate integral. We therefore may further economize our fit coefficients by using much looser screening criteria in their evaluation than in the overall calculation. Specifically, we may obtain the same effect as a given integral screening threshold by neglecting the fit coefficients of pairs whose integral estimates are less than the square root of said threshold. Thus, if the desired integral screening threshold for the calculation is 10–12, this level of accuracy can be preserved while setting the fit coefficients of products with integral estimates of approximately 10–6 uniformly to zero. Our results demonstrate that this approximation causes negligible loss of accuracy.
2.3. Additional Benefits for Range-Separated Hybrids
For range-separated functionals, both a short and long-range K build are required. For these functionals, we reuse the fit coefficients formed with the full Coulomb operator, and transfer the short and long-range operator dependence exclusively to the two and three-center integrals. Our expressions for each L matrix are therefore
It should be noted that a robust fit is still obtained due to the explicit use of Dunlap’s robust ansatz, and our results below demonstrate that the independent use of two sets of fit coefficients is unnecessary. To obtain similar accuracy with global RI-K using a single set of fit coefficients would require the explicit use of the Dunlap functional, at a significant performance penalty. We exploit this advantage to compress the evaluation of both exchange contributions into a single K build as follows:
As may be seen from the above equations, only steps which are nonrate determining are duplicated. In the limit of large systems, our algorithm will therefore outperform RI-K by an additional factor of 2 for range-separated hybrids, leading to a net speedup of 2(1 + X/N). We demonstrate that this limit is reached for relatively small systems.
3. Results
All calculations were performed with a development version of the Q-Chem program.67,68
3.1. Accuracy
Hollman and Merlot have evaluated the accuracy of the PARI approximation for HF and B3LYP—we now wish to assess its accuracy in the context of modern density functionals. The ωB97X-V functional was chosen due to its excellent and highly transferable performance for a variety of types of chemical interactions.19 Our calculations were performed in the aug-cc-pVTZ orbital basis set using the corresponding RI auxiliary basis for exchange.34 For an orbital basis set of this size, it is expected that the user will also wish to use an RI approach for the formation of the Coulomb matrix, and we therefore employ the RI-J algorithm in conjunction with PARI-K. In order to benchmark performance for thermochemical properties, a subset of the G3-05 test set69,70 was selected consisting of all compounds for which the auxiliary basis was supported. The errors for a variety of thermochemical properties are tabulated in Table 2. Larger than expected errors were found when employing the RI-J approximation for the calculation of electron affinities, so this approximation was not used for these table entries. An integral screening threshold of 10–12 was utilized, with the screening threshold for fit coefficients set to 10–6 as discussed above.
Table 2. Errors in Atomization Energies (AE), Ionization Potentials (IP), Electron Affinities (EA), and Proton Affinities (PA) Relative to No RI Approximation for a Subset of the G3-05 Test Seta.
| AE | IP | EA | PA | |
|---|---|---|---|---|
| mean absolute error | 0.03 | 0.0026 | 0.052 | 0.01 |
| mean signed error | –0.03 | 0.0004 | –0.052 | –0.009 |
| max error | 0.22 | 0.0108 | 0.17 | 0.09 |
All data is in kilocalories per mole.
In order to assess the errors introduced by the PARI-K approximation when applied to intermolecular interactions, we have also calculated counterpoise corrected binding energies for the S66 test set,71 which are given in Table 3.
Table 3. Errors in Counterpoise-Corrected Binding Energies for the S66 Set Relative to No RI Approximation, Decomposed by Interaction Typea.
| H | D | O | all | |
|---|---|---|---|---|
| mean absolute error | 0.01 | 0.02 | 0.01 | 0.01 |
| mean signed error | 0.01 | 0.02 | 0.01 | 0.01 |
| max error | 0.04 | 0.04 | 0.02 | 0.04 |
We use Řezàč’s original classification of H hydrogen-bonded, D dispersion, and O others.71 All data is in kilocalories per mole.
The results in Tables 2 and 3 demonstrate that PARI-K can be employed with the same size of auxiliary basis set used for standard RI-JK calculations with negligible loss of accuracy. It should be noted that the ωB97X-V functional scales the short-range exchange energy by a factor of 0.167 and the long-range by 1.0, thus reducing the error resulting from applying the PARI approximation relative to full exact exchange. Functionals with larger contributions from exact exchange will experience somewhat larger errors when used in conjunction with PARI-K.
3.2. Stability
All of our calculations were converged successfully to a threshold of 10–8. An initial guess for all systems was computed using the B97-D72 GGA functional in the same basis set. We were able to successfully converge all systems in our truncated G3 set, including the systems for which Merlot et al. and Hollman et al. observe convergence problems (hexafluorobenzene, chloro-pentafluorobenze, 3-butyn-2-one, and 2-butyn). As expected from our proof of variational stability, we do not observe the presence of any “attractive-electron” solutions, as these arise from the computation of J in the PARI approximation. The non positive-semidefinite integral tensor raises the possibility of the SCF encountering densities that lead to repulsive exchange interactions, but this is much less likely to pose a problem in practice as these densities will be actively avoided by the SCF optimizer. On the basis of our results, we assert that the PARI-K method may be reliably used with an appropriate guess.
3.3. Performance
We consider first the performance of our algorithm for a system to which it is “optimally suited”—a molecule of moderate spatial extent treated with a large basis set. The chosen example is a hydrogen-terminated three ring by six ring graphene lattice (shown in Figure 1) using the ωB97X-V functional with the cc-pVQZ basis set. Our method is compared against the following competing approaches implemented in Q-Chem: an integral-driven K build using the LinK algorithm27 (optimal for very large systems), the ARI-K algorithm35 (optimal for midsize systems in larger basis sets), and the RI-K algorithm34 (optimal for compact systems in very large basis sets). The results of this comparison are shown in Figure 2. Our method outperforms all examined alternatives, obtaining a 19 times speedup over the conventional integral-driven K build and a 2.7 times speedup over RI-K. The speedup relative to RI-K will increase with increasing system size; however, our RI-K code is at present unable to handle the memory requirements of jobs larger than those presented here. It should be noted that while the RI-K and ARI-K timings were produced by timing a single K build and scaling the result by 2, the integral-driven timings are for a single K build without scaling, as the short- and long-range K builds can be combined by transferring all of the operator dependence to the fundamental integrals prior to contraction and application of recurrence relations.
Figure 1.

Hydrogen-terminated 3 × 6 graphene lattice upon which QZ timings were performed.
Figure 2.
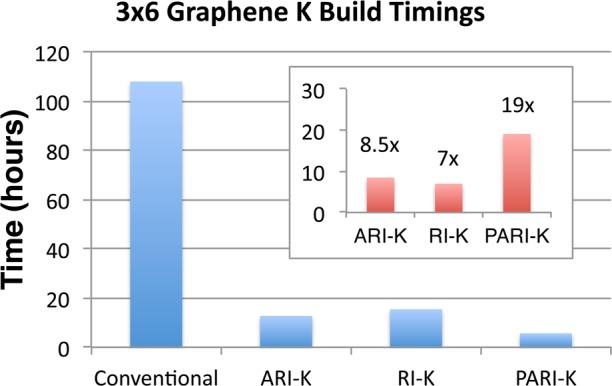
Wall time for the two K builds in the second SCF iteration for a hydrogen-terminated 3 × 6 graphene lattice with the range-separated ωB97X-V functional. (inset) Speedups relative to the integral-driven K build for the various RI methods. The first iteration is approximately 15% more expensive as of this writing due to initialization costs, but only for smaller systems. RI-K and ARI-K timings were calculated by timing one K build and scaling the result by 2, as the cost of the short and long-range K builds is essentially identical for these methods.
We now assess the performance of our algorithm as a function of system size in a smaller (TZ) basis set, where one would expect better performance from the lower-scaling integral-driven K build. We begin with acene-5 and extend the graphene sheet in two dimensions with additional chains of six rings along the y-axis. The results of this timing experiment are summarized in Figure 3. We are limited to a four by six aromatic lattice for RI-K due to the memory usage of our benchmark RI-K implementation. Our PARI-K implementation obtains a 6.4 times speedup relative to exact integrals for the largest lattice, a system with 2348 basis functions. This compares favorably with the speedups obtained by Merlot et al. for various smaller acenes. The superior efficiency of our MO algorithm thus appears to effectively compensate for its asymptotically higher scaling. Our algorithm outperforms RI-K by an increasing margin as the system size is increased, reaching a 3.4 times speedup for the largest lattice.
Figure 3.
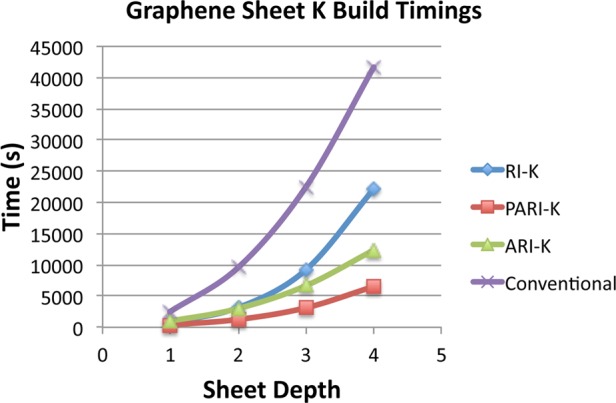
Wall time for the two K builds in the second SCF iteration for various N × 6 graphene lattices, performed in the cc-pVTZ basis. RI-K and ARI-K timings were calculated by timing one K build and scaling the result by 2, as the cost of the short and long-range K builds is essentially identical for these methods.
Our combined K build provides a dramatic advantage even for the smallest system in Figure 3: a single K build performed for the 1 × 6 graphene sheet requires 312 s, whereas the combined short and long-range K build takes only 371 s. It should be recalled that the performance of RI-K and ARI-K will be twice as good for functions that are not range-separated hybrids.
3.4. Sparsity and Performance Considerations for PARI Algorithms
We shall now demonstrate empirically that the loss of sparsity in the RI fit coefficients caused by contracting them with dense matrices leads to severe difficulties in practically attaining the low scaling that is expected for such local fit domains. We focus on the sparsity of the most important sparse tensor appearing in our algorithm, the MO-transformed RI fit coefficients DiμQ, as a function of system size. As can be seen from the sparsity of this tensor for a series of linear alkanes, shown in Figure 4, the onset of sparsity in Di is much slower than might be expected given the immense locality of the PARI fit coefficients. The tensor is approximately 83% sparse for the longest alkane chain. As a consequence of this slow onset, exploitation of sparsity does not become appreciably favorable even for very extended systems. This leads to the timings shown in Figure 5. Note that even as the sparsity of DiμQ increases, this gain for the sparse implementation is offset by increasing performance of the matrix-multiply routine called by the dense implementation as the matrix size grows. Thus, even for these ideal systems, the advantage gained by the formally lower-scaling algorithm which exploits sparsity is minimal. We acknowledge that a higher-performance sparse linear algebra implementation would yield a more favorable comparison. However, the tensor contraction expression in this step fundamentally takes the form of either a single matrix multiply in the dense case, or a series of what are essentially vector-matrix multiplies in the sparse case. The floating point performance difference is therefore bound to reflect the (usually large) difference in the performance of these two classes of operations for a given BLAS implementation.
Figure 4.
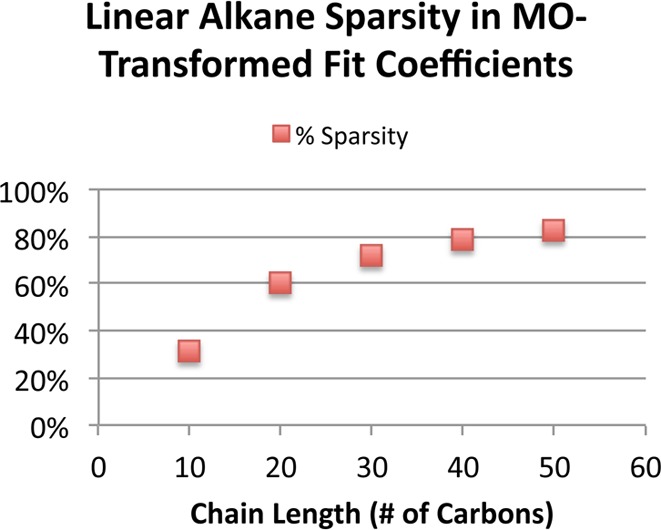
Sparsity in of DiμQ tensor for a series of linear alkanes using the cc-pVTZ basis set. Screening of the AO-basis RI fit coefficients was performed using a threshold of 10–6.
Figure 5.
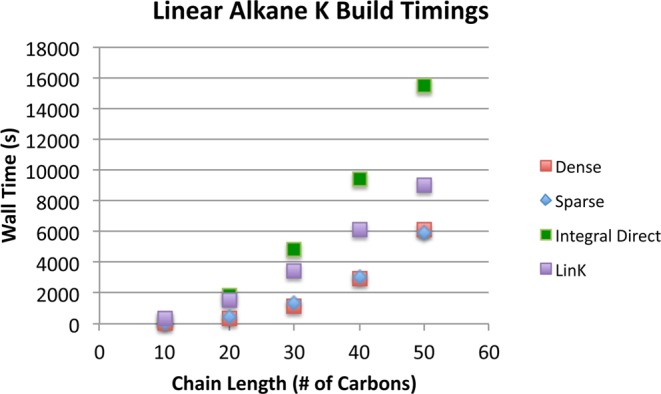
Wall time for a single second SCF iteration K build for several linear alkanes, performed in the cc-pVTZ basis. An integral threshold of 10–12 was used in conjunction with a fit coefficient threshold of 10–6.
It is important to recall that this unfortunate sparsity behavior is not a consequence of the MO-basis transformation per se; the density-matrix contracted fit coefficients appearing in the analogous step of Merlot’s algorithm will suffer the exact same slow onset of sparsity, assuming that density-matrix sparsity cannot be efficiently utilized. Rather, the lack of sparsity in the contracted fit coefficients may be viewed as a fundamental nonlocal behavior of the PARI approximation. Assuming a fully dense MO coefficient matrix (or density matrix for the analogous AO problem), an entry DiμQ will be nonzero if even a single orbital-basis AO function on the atom of Q has non-negligible overap with μ. This will hold for every single function Q on the given atom. The decay of Di is thus controlled exclusively by the most diffuse AO functions in the orbital basis and is slowed significantly by the fact that auxiliary functions are grouped in large atom blocks. Finally, we also note that our algorithm outperforms linear-scaling integral evaluation even for very long alkane chains, thus suggesting that widely held reservations about the use of RI-SCF methods due to their higher formal scaling may be somewhat misplaced.
Similar sparsity arguments are relevant to a discussion of the merits of an AO versus MO implementation. A reduction in the formal scaling of the algorithm can be obtained by utilizing the Gaussian product sparsity present in the AO basis representation of the relevant tensors, as is done in Merlot’s PARI-K algorithm. However, based on our data for linear alkanes, we believe that utilizing AO sparsity would actually increase the raw operation cost of this algorithm, even for very extended systems. We illustrate this point by considering the relative sizes of the MO-transformed three-center integrals and the original AO three-center integrals, accounting for sparsity. The ratio between these two quantities represents the ratio between the rate-determining step of our algorithm and an AO algorithm such as Merlot’s. This ratio reduces to a comparison the quantities oN and [NB2]. A plot of each of these quantities with respect to alkane length for the cc-pVTZ basis is shown in Figure 6. The permutational symmetry of the Gaussian product indices is accounted for in this figure. On the basis of the relative size of these quantities, we propose that the AO-basis algorithm, despite formally scaling lower than our method, will in fact require more operations even for very extended systems, in addition to suffering from a significant efficiency penalty due to the high performance of dense linear algebra operations. This serves as a reminder that the tremendous compactness of the occupied MO basis and the associated high efficiency of dense linear algebra libraries must be properly accounted for when assessing the efficacy of competing lower-scaling implementations. Many similar points about the relative merits of lower-scaling methods in contrast to lower-prefactor, higher-scaling competitors are stated eloquently in a review by Neese.73 On the basis of these concerns, it seems to us that the best path toward a lower-scaling PARI-K implementation is the use of localized occupied molecular orbitals.
Figure 6.
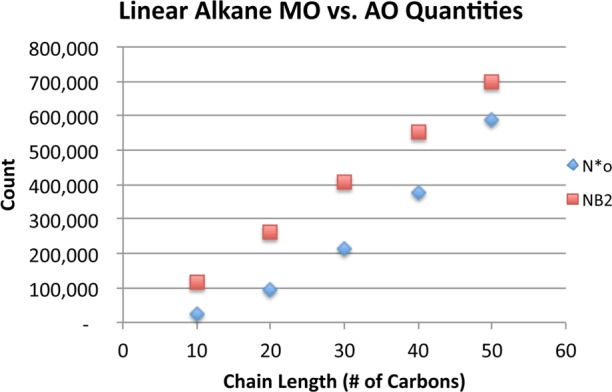
Comparison of varying dimensions of tensors in the rate-determining step for AO and MO algorithms. [NB2] was computed using a screening threshold of 10–12.
4. Conclusions
Our MO-based implementation of the PARI-K approximation has been shown to significantly accelerate large scale range-separated hybrid DFT calculations. Benchmark results for thermochemistry and intermolecular interactions indicate that impressive accuracy can be obtained with the modern ωB97X-V functional, while requiring an auxiliary basis set no larger than that used in conventional RI-HF. Our algorithm has been shown to out-perform commonly available alternatives for extended and compact systems in TZ and QZ basis sets.
Acknowledgments
Support for this work was provided through the Scientific Discovery through Advanced Computing (SciDAC) program funded by the U.S. Department of Energy, Office of Science, Advanced Scientific Computing Research, and Basic Energy Sciences. We also acknowledge partial support of this work from Q-Chem Inc through NIH SBIR grant no. GM096678. S.F.M. acknolwedges Patrick Merlot for helpful discussions and acknowledges Claire Manzer for support in all things.
The authors declare the following competing financial interest(s): M.H.G. and E.E. are part-owners of Q-Chem Inc.
Funding Statement
National Institutes of Health, United States
References
- Hohenberg P. Phys. Rev. 1964, 136, B864–B871. [Google Scholar]
- Kohn W.; Sham L. J. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar]
- Becke A. D. J. Chem. Phys. 1993, 98, 1372. [Google Scholar]
- Becke A. D. J. Chem. Phys. 1993, 98, 5648. [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. J. Chem. Phys. 1994, 98, 11623–11627. [Google Scholar]
- Becke A. D. J. Chem. Phys. 1997, 107, 8554. [Google Scholar]
- Bauschlicher C. W. Chem. Phys. Lett. 1995, 246, 40–44. [Google Scholar]
- Finley J.; Stephens P. THEOCHEM 1995, 357, 225–235. [Google Scholar]
- Wong M. W. Chem. Phys. Lett. 1996, 256, 391–399. [Google Scholar]
- Zhao Y.; Truhlar D. G. Theor. Chem. Acc. 2008, 120, 215. [Google Scholar]
- Peverati R.; Truhlar D. G. Philos. Trans. Royal Soc. A 2014, 372, 20120476. [DOI] [PubMed] [Google Scholar]
- Iikura H.; Tsuneda T.; Yanai T.; Hirao K. J. Chem. Phys. 2001, 115, 3540. [Google Scholar]
- Tawada Y.; Tsuneda T.; Yanagisawa S.; Yanai T.; Hirao K. J. Chem. Phys. 2004, 120, 8425–33. [DOI] [PubMed] [Google Scholar]
- Bauernschmitt R.; Ahlrichs R. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar]
- Hirata S.; Head-Gordon M. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar]
- Dreuw A.; Weisman J. L.; Head-Gordon M. J. Chem. Phys. 2003, 119, 2943. [Google Scholar]
- Dreuw A.; Head-Gordon M. J. Am. Chem. Soc. 2004, 126, 4007–16. [DOI] [PubMed] [Google Scholar]
- Chai J. D.; Head-Gordon M. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. Phys. Chem. Chem. Phys. 2014, 16, 9904–24. [DOI] [PubMed] [Google Scholar]
- Raymond K. S.; Wheeler R. A. J. Comput. Chem. 1999, 20, 207–216. [Google Scholar]
- Boese A. D.; Martin J. M. L.; Handy N. C. J. Chem. Phys. 2003, 119, 3005. [Google Scholar]
- Wang N. X.; Wilson A. K. J. Chem. Phys. 2004, 121, 7632–46. [DOI] [PubMed] [Google Scholar]
- Wang N. X.; Wilson A. K. Mol. Phys. 2005, 103, 345–358. [Google Scholar]
- Horn H.; Weiss H.; Haser M.; Ehrig M.; Ahlrichs R. J. Comput. Chem. 1991, 12, 1058–1064. [Google Scholar]
- Strout D. L.; Scuseria G. E. J. Chem. Phys. 1995, 102, 8448. [Google Scholar]
- Schwegler E.; Challacombe M. Theor. Chem. Acc. 2000, 104, 344–349. [Google Scholar]
- Ochsenfeld C.; White C. A.; Head-Gordon M. J. Chem. Phys. 1998, 109, 1663. [Google Scholar]
- Liang W. Z.; Shao Y. H.; Ochsenfeld C.; Bell A. T.; Head-Gordon M. Chem. Phys. Lett. 2002, 358, 43–50. [Google Scholar]
- Grimme S. J. Chem. Phys. 2006, 124, 034108. [DOI] [PubMed] [Google Scholar]
- Chai J.-D.; Head-Gordon M. J. Chem. Phys. 2009, 131, 174105. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Xu X.; Goddard W. A. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang I. Y.; Luo Y.; Xu X. J. Chem. Phys. 2010, 133, 104105. [DOI] [PubMed] [Google Scholar]
- Früchtl H. A.; Kendall R. A.; Harrison R. J.; Dyall K. G. Int. J. Quantum Chem. 1997, 64, 63–69. [Google Scholar]
- Weigend F. Phys. Chem. Chem. Phys. 2002, 4, 4285–4291. [Google Scholar]
- Sodt A.; Head-Gordon M. J. Chem. Phys. 2008, 128, 104106. [DOI] [PubMed] [Google Scholar]
- Koch H.; Sanchez de Meras A.; Pedersen T. B. J. Chem. Phys. 2003, 118, 9481. [Google Scholar]
- Boman L.; Koch H.; Sánchez de Merás A. J. Chem. Phys. 2008, 129, 134107. [DOI] [PubMed] [Google Scholar]
- Beebe N. H. F.; Linderberg J. Int. J. Quantum Chem. 1977, 12, 683–705. [Google Scholar]
- Aquilante F.; Pedersen T. B.; Lindh R. J. Chem. Phys. 2007, 126, 194106. [DOI] [PubMed] [Google Scholar]
- Koch H.; Sanchez de Meras A. J. Chem. Phys. 2000, 113, 508. [Google Scholar]
- Aquilante F.; Pedersen T. B. Chem. Phys. Lett. 2007, 449, 354–357. [Google Scholar]
- Aquilante F.; Lindh R.; Pedersen T. B. J. Chem. Phys. 2007, 127, 114107. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Haser M.; Patzelt H.; Ahlrichs R. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar]
- Weigend F.; Kohn A.; Hattig C. J. Chem. Phys. 2002, 116, 3175. [Google Scholar]
- Weigend F. J. Comput. Chem. 2008, 29, 167–75. [DOI] [PubMed] [Google Scholar]
- Aquilante F.; Gagliardi L.; Pedersen T. B.; Lindh R. J. Chem. Phys. 2009, 130, 154107. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Kattannek M.; Ahlrichs R. J. Chem. Phys. 2009, 130, 164106. [DOI] [PubMed] [Google Scholar]
- Whitten J. L. J. Chem. Phys. 1973, 58, 4496. [Google Scholar]
- Jung Y.; Sodt A.; Gill P. M. W.; Head-Gordon M. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 6692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerends E.; Ellis D.; Ros P. Chem. Phys. 1973, 2, 41–51. [Google Scholar]
- Vahtras O.; Almlöf J.; Feyereisen M. Chem. Phys. Lett. 1993, 213, 514–518. [Google Scholar]
- Reine S.; Tellgren E.; Krapp A.; Kjaergaard T.; Helgaker T.; Jansik B.; Host S.; Salek P. J. Chem. Phys. 2008, 129, 104101. [DOI] [PubMed] [Google Scholar]
- Polly R.; Werner H.-J.; Manby F. R.; Knowles P. J. Mol. Phys. 2004, 102, 2311–2321. [Google Scholar]
- Merlot P.; Kjæ rgaard T.; Helgaker T.; Lindh R.; Aquilante F.; Reine S.; Pedersen T. B. J. Comput. Chem. 2013, 34, 1486–96. [DOI] [PubMed] [Google Scholar]
- Hollman D. S.; Schaefer H. F.; Valeev E. F. J. Chem. Phys. 2014, 140, 064109. [DOI] [PubMed] [Google Scholar]
- Mejía-Rodríguez D.; Köster A. M. J. Chem. Phys. 2014, 141, 124114. [DOI] [PubMed] [Google Scholar]
- Neese F.; Wennmohs F.; Hansen A.; Becker U. Chem. Phys. 2009, 356, 98–109. [Google Scholar]
- Kossmann S.; Neese F. Chem. Phys. Lett. 2009, 481, 240–243. [Google Scholar]
- Baerends E.; Ellis D.; Ros P. Chem. Phys. 1973, 2, 41–51. [Google Scholar]
- Fonseca Guerra C.; Snijders J. G.; te Velde G.; Baerends E. J. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar]
- Watson M. A.; Handy N. C.; Cohen A. J. J. Chem. Phys. 2003, 119, 6475. [Google Scholar]
- Krykunov M.; Ziegler T.; van Lenthe E. Int. J. Quantum Chem. 2009, 109, 1676–1683. [Google Scholar]
- Dunlap B. THEOCHEM 2000, 529, 37–40. [Google Scholar]
- Sodt A.; Subotnik J. E.; Head-Gordon M. J. Chem. Phys. 2006, 125, 194109. [DOI] [PubMed] [Google Scholar]
- Slater J.Quantum Theory of Atomic Structure; McGraw-Hill: New York, 1960. [Google Scholar]
- Power J. D.; Pitzer R. M. Chem. Phys. Lett. 1974, 24, 478–483. [Google Scholar]
- Shao Y.; Molnar L. F.; Jung Y.; Kussmann J.; Ochsenfeld C.; Brown S. T.; Gilbert A. T. B.; Slipchenko L. V.; Levchenko S. V.; O’Neill D. P.; DiStasio R. A. Jr.; Lochan R. C.; Wang T.; Beran G. J. O.; Besley N. A.; Herbert J. M.; Lin C. Y.; Van Voorhis T.; Chien S. H.; Sodt A.; Steele R. P.; Rassolov V. A.; Maslen P. E.; Korambath P. P.; Adamson R. D.; Austin B.; Baker J.; Byrd E. F. C.; Dachsel H.; Doerksen R. J.; Dreuw A.; Dunietz B. D.; Dutoi A. D.; Furlani T. R.; Gwaltney S. R.; Heyden A.; Hirata S.; Hsu C.-P.; Kedziora G.; Khaliullin R. Z.; Klunzinger P.; Lee A. M.; Lee M. S.; Liang W.; Lotan I.; Nair N.; Peters B.; Proynov E. I.; Pieniazek P. A.; Rhee Y. M.; Ritchie J.; Rosta E.; Sherrill C. D.; Simmonett A. C.; Subotnik J. E.; Woodcock H. L. III; Zhang W.; Bell A. T.; Chakraborty A. K.; Chipman D. M.; Keil F. J.; Warshel A.; Hehre W. J.; Schaefer H. F. III; Kong J.; Krylov A. I.; Gill P. M. W.; Head-Gordon M. Phys. Chem. Chem. Phys. 2006, 8, 3172. [DOI] [PubMed] [Google Scholar]
- Shao Y.; Gan Z.; Epifanovsky E.; Gilbert A. T. B.; Wormit M.; Kussmann J.; Lange A. W.; Behn A.; Deng J.; Feng X.; Ghosh D.; Goldey M.; Horn P. R.; Jacobson L. D.; Kaliman I.; Khaliullin R. Z.; Kuś T.; Landau A.; Liu J.; Proynov E. I.; Rhee Y. M.; Richard R. M.; Rohrdanz M. A.; Steele R. P.; Sundstrom E. J.; Woodcock H. L. III; Zimmerman P. M.; Zuev D.; Albrecht B.; Alguire E.; Austin B.; Beran G. J. O.; Bernard Y. A.; Berquist E.; Brandhorst K.; Bravaya K. B.; Brown S. T.; Casanova D.; Chang C.-M.; Chen Y.; Chien S. H.; Closser K. D.; Crittenden D. L.; Diedenhofen M.; DiStasio R. A. Jr.; Do H.; Dutoi A. D.; Edgar R. G.; Fatehi S.; Fusti-Molnar L.; Ghysels A.; Golubeva-Zadorozhnaya A.; Gomes J.; Hanson-Heine M. W. D.; Harbach P. H. P.; Hauser A. W.; Hohenstein E. G.; Holden Z. C.; Jagau T.-C.; Ji H.; Kaduk B.; Khistyaev K.; Kim J.; Kim J.; King R. A.; Klunzinger P.; Kosenkov D.; Kowalczyk T.; Krauter C. M.; Lao K. U.; Laurent A.; Lawler K. V.; Levchenko S. V.; Lin C. Y.; Liu F.; Livshits E.; Lochan R. C.; Luenser A.; Manohar P.; Manzer S. F.; Mao S.-P.; Mardirossian N.; Marenich A. V.; Maurer S. A.; Mayhall N. J.; Neuscamman E.; Oana C. M.; Olivares-Amaya R.; O’Neill D. P.; Parkhill J. A.; Perrine T. M.; Peverati R.; Prociuk A.; Rehn D. R.; Rosta E.; Russ N. J.; Sharada S. M.; Sharma S.; Small D. W.; Sodt A.; Stein T.; Stück D.; Su Y.-C.; Thom A. J. W.; Tsuchimochi T.; Vanovschi V.; Vogt L.; Vydrov O.; Wang T.; Watson M. A.; Wenzel J.; White A.; Williams C. F.; Yang J.; Yeganeh S.; Yost S. R.; You Z.-Q.; Zhang I. Y.; Zhang X.; Zhao Y.; Brooks B. R.; Chan G. K. L.; Chipman D. M.; Cramer C. J.; Goddard W. A. III; Gordon M. S.; Hehre W. J.; Klamt A.; Schaefer H. F. III; Schmidt M. W.; Sherrill C. D.; Truhlar D. G.; Warshel A.; Xu X.; Aspuru-Guzik A.; Baer R.; Bell A. T.; Besley N. A.; Chai J.-D.; Dreuw A.; Dunietz B. D.; Furlani T. R.; Gwaltney S. R.; Hsu C.-P.; Jung Y.; Kong J.; Lambrecht D. S.; Liang W.-Z.; Ochsenfeld C.; Rassolov V. A.; Slipchenko L. V.; Subotnik J. E.; Van Voorhis T.; Herbert J. M.; Krylov A. I.; Gill P. M. W.; Head-Gordon M. Mol. Phys. 2014, 112, 1–32. [Google Scholar]
- Curtiss L. A.; Raghavachari K.; Redfern P. C.; Pople J. A. J. Chem. Phys. 2000, 112, 7374. [Google Scholar]
- Curtiss L. A.; Redfern P. C.; Raghavachari K. J. Chem. Phys. 2005, 123, 124107. [DOI] [PubMed] [Google Scholar]
- Řezáč J.; Riley K. E.; Hobza P. J. Chem. Theory Comput. 2011, 7, 2427–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimme S. J. Comput. Chem. 2006, 27, 1787–99. [DOI] [PubMed] [Google Scholar]
- Neese F. In Linear-Scaling Techniques in Computational Chemistry and Physics; Zalesny R., Papadopoulos M. G., Mezey P. G., Leszczynski J., Eds.; Springer: Netherlands, Dordrecht, 2011; pp 227–261. [Google Scholar]