

Abstract
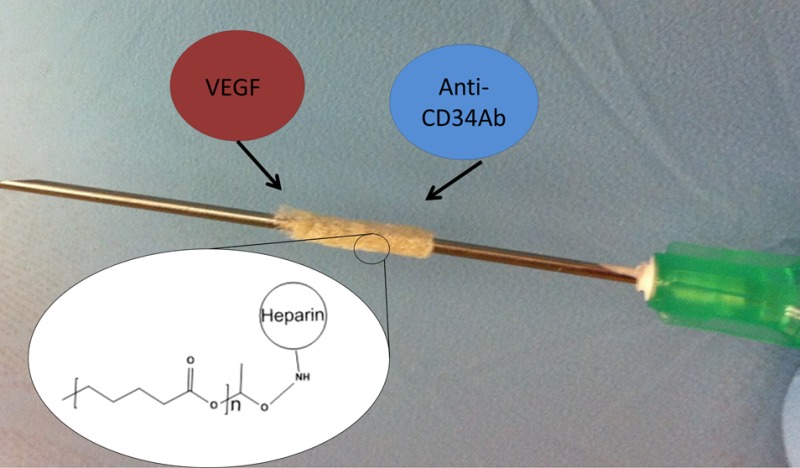
Surface modification of biodegradable vascular grafts is an important strategy to improve the in situ endothelialization of tissue engineered vascular grafts (TEVGs) and prevent major complications associated with current synthetic grafts. Important strategies for improving endothelialization include increasing endothelial cell mobilization and increased endothelial cell capture through biofunctionalization of TEVGs. The objective of this study was to assess two biofunctionalization strategies for improving endothelialization of biodegradable polyester vascular grafts. These techniques consisted of cross-linking heparin to graft surfaces to immobilize vascular endothelial growth factor (VEGF) or antibodies against CD34 (anti-CD34Ab). To this end, heparin, VEGF, and anti-CD34Ab attachment and quantification assays confirmed the efficacy of the modification strategy. Cell attachment and proliferation on these groups were compared to unmodified grafts in vitro and in vivo. To assess in vivo graft functionality, the grafts were implanted as inferior vena cava interpositional conduits in mice. Modified vascular grafts displayed increased endothelial cell attachment and activity in vivo, according to microscopy techniques, histological results, and eNOS expression. Inner lumen diameter of the modified grafts was also better maintained than controls. Overall, while both functionalized grafts outperformed the unmodified control, grafts modified with anti-CD34Ab appeared to yield the most improved results compared to VEGF-loaded grafts.
Introduction
Cardiovascular disease is the leading cause of mortality worldwide.1 To treat many of the conditions associated with cardiovascular disease, autologous vessels or synthetic grafts are often used. However, autologous vessels may be limited by existing conditions or previous surgeries.2,3 In synthetic grafts, complications include lack of growth potential, calcification from secondary graft failure, increased susceptibility to infection, and increased risk for thromboembolic events and stenosis.4,5 Tissue engineered vascular grafts (TEVGs) offer a potential strategy for overcoming these complications by providing a biodegradable scaffold for the autologous cells to attach, proliferate, and provide physiologic functionality. A scaffold that enables and encourages healthy vascular tissue growth while degrading over time would eliminate many of the complications associated with permanent, synthetic grafts. However, a primary mode of failure of small-diameter (<6 mm) TEVGs is graft stenosis due to neointimal hyperplasia and thrombosis.4,6−10 Thus, a successful TEVG must prevent thrombosis and intimal hyperplasia. Since the endothelial layer of blood vessels is crucial for maintaining vascular homeostasis, prevention of intimal hyperplasia, and thrombogenesis, the establishment of an endothelial cell (EC) monolayer that adequately covers the inner lumen of a TEVG is crucial to the graft’s long-term success.5,11,12 Rapid establishment of such a layer may alleviate the current challenges associated with biodegradable vascular grafts.
Establishing a monolayer of ECs on a TEVG can be accomplished via cell seeding and culturing before implantation. Grafts with a precultured endothelium before implantation perform well in vivo and demonstrate reduced complications traditionally associated with small-diameter vascular grafts.13−16 However, cell seeding of these grafts can be time-consuming, expensive, and clinically difficult.17
In an effort to expedite endothelialization and eliminate the challenges associated with cell seeding, researchers have investigated a variety of in situ endothelialization strategies.11,12 These strategies have largely focused on recruiting and promoting the attachment and proliferation of ECs and endothelial progenitor cells (EPCs) on the inner lumen of grafts after implantation. The exact role of EPCs in endothelialization is still under debate, but both early and late EPCs show positive effects on in vivo endothelialization of vascular prosthetics.18−20 Early EPCs may secrete angiogenic cytokines to support other EPCs and ECs, while late EPCs possess the potential for proliferation and EC colony formation.19,21 While identification methods for EPCs should still be standardized, a common marker of EPCs is CD34.
To take advantage of the in situ endothelialization potential of ECs and EPCs, we focus on two strategies of vascular graft modification: (1) antibody immobilization and (2) growth factor loading. Antibody immobilization strategies primarily function to improve cell attachment to graft surfaces, while vascular endothelial growth factor (VEGF) loading and subsequent elution may induce cell mobilization into the blood along with migration from neighboring tissues. A variety of specific and nonspecific molecules have been investigated to induce cell capture and attachment. One such biofunctional molecule, an antibody against CD34 (anti-CD34Ab), has been used to induce endothelialization of permanent vascular stents through the increased attachment of both ECs and EPCs.22−24 Including such an antibody may aid in the recruitment and attachment of neighboring ECs and EPCs. However, CD34+ vascular cells represent a small percentage of cells in circulation.19
To increase the available numbers of EPCs in circulation, it may be necessary to introduce a mobilizing factor. For example, VEGF may increase the fraction of EPCs in circulation.25 In addition, bound VEGF may influence EPC differentiation into mature EC-like phenotypes, while increasing the migration and proliferation of ECs.26−29 Besides its influence on EPCs, diffusion of VEGF may also induce the migration and proliferation of resident ECs from mature vessels across anastomotic sites.30,31 VEGF has been successfully delivered via scaffolds utilizing specific binding motifs present in heparin.32−34 Cross-linked heparin also protects the bioactivity of bound proteins, which may increase the efficacy of VEGF delivery.35 For example, heparin molecules cross-linked to a polycaprolactone scaffold mediated VEGF loading and diffusion to successfully promote increased angiogenesis over unmodified PCL scaffolds.33 In addition, heparin has antithrombotic properties conducive to minimizing thrombosis associated with the implantation of small-diameter vascular grafts, especially in localized dosages.36
We sought to expedite and improve the endothelialization of a biodegradable small-diameter vascular graft by assessing two coating strategies that utilized heparin-cross-linked surfaces to either load VEGF or immobilize anti-CD34Ab. By utilizing these biomolecules, we were particularly interested in studying which strategy was more conducive to the endothelialization of these biodegradable polymeric grafts. We examined whether an initial burst release of VEGF or surfaces modified with anti-CD34Ab would lead to more efficient and effective endothelialization of heparin-cross-linked vascular grafts. The effects of modified graft surfaces were characterized and tested by examining HUVEC and EPC attachment and proliferation in cell culture assays and an in vivo mouse model to assess endothelialization.
Materials and Methods
Graft Fabrication
The method of fabrication utilizes a solvent-casting technique described and characterized in previous studies.37,38 Briefly, 6.00 × 4.00 mm sections were cut from a 90:10 poly(glycolic-co-lactic acid) (PGLA) for in vitro assays and poly(glycolic acid) (PGA) polymer BIOFELT (Biomedical Structures, Warwick, RI) for in vivo tests. The PGLA sections were inserted into a polypropylene tube with an inner lumen diameter of 1.4 mm and a 21 g stainless steel needle was inserted into the opposite end of the tube to maintain the patency of the inner lumen of the graft. Then, a 40:60 copolymer poly(caprolactone-co-dl-lactic acid) (PCLLA) solution of 15% w/v in 1,4-dioxane was deposited into the polypropylene tubes to saturate the PGLA or PGA felt. Grafts were subsequently frozen at −20 °C for 30 min, followed by freeze-drying for 24 h to eliminate excess 1,4-dioxane solvent. After complete drying, grafts were stored at −20 °C until used.
Graft Modification Procedures
Modified grafts utilized heparin cross-linking to immobilize VEGF or anti-CD34Ab. We assessed the initial loading and retention of VEGF and anti-CD34Ab on heparin cross-linked surfaces.
Heparin Cross-Linking
Heparin cross-linking and quantification was adapted from a previously published method.33 Cross-linking chemistry is demonstrated in Figure 1. Before cross-linking, scaffolds were immersed in 0.05 M MES buffer (pH = 5.55) for 15 min. Scaffolds were subsequently submerged in a solution of 0.5 M ethyl-3-(3-(dimethylamino)propyl)-carbodiimide) (EDC), 0.5 M N-hydroxysuccinimide (NHS), and 1% w/v heparin in MES buffer. After incubation for 14 h, scaffolds were washed with distilled water to remove excess byproducts.
Figure 1.

Graft modification: EDC chemistry reaction for the cross-linking of heparin and subsequent loading and immobilization of VEGF and antibodies against CD34.
VEGF Loading
A sterile solution of VEGF was prepared in PBS at a concentration of 500 ng/mL, according to previously published methods.39 Scaffolds were incubated in the VEGF solution for 1 h, in sterile conditions, at room temperature. Following incubation, grafts underwent eight 5 min washes in sterile-filtered PBS solution to remove unbound VEGF.
Anti-CD34 Antibody Immobilization
For anti-CD34Ab coating, heparin cross-linked grafts were immersed in 10 μg/mL solutions of primary antibody against CD34 in PBS overnight at 4 °C in the dark. Grafts were then washed three times with PBS.
Surface Modification Characterization
Scanning Electron Microscopy
The topographies of the modified and unmodified grafts were visualized by a scanning electron microscope (SEM; Hitachi, Tokyo, Japan). Grafts (n = 5) were cut into sections 1 mm in length and fixed with 2% gluteraldehyde and underwent subsequent serial dehydration in ethanol. Samples were then allowed to dry and were subsequently mounted and sputter coated with carbon before SEM examination.
Toluidine Blue Staining Assay
Cross-linked heparin was confirmed via a toluidine blue stain assay. A 0.0005% (w/v) toluidine blue zinc chloride double salt solution was prepared in 0.001 N hydrochloric acid with 0.02% (w/v) sodium chloride. Heparin cross-linked and unmodified scaffolds were incubated in the toluidine solution overnight at room temperature. A deep purple hue on the surface of the scaffolds indicated the presence of heparin, while unmodified scaffolds remained white.
VEGF ELISA
To quantify VEGF attachment and release, a human VEGF ELISA kit (Sigma) was used according to manufacturer instructions. Briefly, standard VEGF curves were created according to manufacturer instructions and added to a 96-well plate coated with capturing antibodies (human VEGF-A). Samples for bound VEGF quantification were placed in the wells of a 96-well plate and served as the binding substrate for incubation with the 200 μL of biotinylated antihuman VEGF detection antibody (100 ng/mL). Next, 200 μL of streptavidin-horse radish-peroxidase solution was added to each well and the plates were incubated for 45 min at room temperature. Following this, 100 μL of tetramethylbenzidine (TMB) solution was added and plates were subsequently incubated for 30 min in the dark at room temperature. The reaction was stopped by adding 50 μL of 2 N H2SO4 “Stop” solution. The optical density (OD) of the resulting solutions was measured using a SpectraMax M5 plate reader at 450 nm with a reference wavelength of 650 nm. Values of VEGF immobilized on scaffolds were calculated from the standard curve. For VEGF release, scaffolds with bound VEGF were incubated in PBS at 37 °C with 65 rpm shaking. The PBS was collected at 1, 4, 24, and 40 h and replaced with fresh PBS. VEGF released into the solution was quantified using the previously described ELISA methods. In addition, nonspecific binding of VEGF was assessed by incubating VEGF with graft surfaces as described, except no cross-linking of heparin was performed.
Anti-CD34 Ab Fluorescence Assay and ELISA
To confirm antibody immobilization, antibody-modified and unmodified scaffolds were incubated at room temperature with 1% bovine serum albumin solution for 30 min at room temperature to prevent nonspecific binding. Scaffolds were then washed three times with PBS and a secondary antigoat IgG antibody conjugated with FITC was added at 10 μg/mL in PBS. Scaffolds were again washed three times with PBS. Successful antibody immobilization could be observed using fluorescent microscopy. To quantify antibody attachment, a Goat IgG ELISA kit (Alpha Diagnostic International, San Antonio, TX) was used. The procedure followed manufacturer instructions, substituting anti-CD34 antibodies instead of the IgG standards included with the kit. In addition, nonspecific binding of anti-CD34Ab was assessed by incubating anti-CD34Ab with graft surfaces as described, except no cross-linking of heparin was performed.
In Vitro Adhesion and Proliferation
In vitro cell culture assays were used to assess initial cell attachment and metabolic activity over time to assess differences between anti-CD34Ab- and VEGF-modified grafts compared to controls in 96-well tissue culture plates.
Human Umbilical Cord Vein Endothelial Cells (HUVEC)
HUVEC were obtained and cultured according to manufacturer’s instructions (Lonza, Basel, Switzerland). Grafts were cut to fit 96-well tissue culture plate and placed in the bottom of the wells. Culture plates with anti-CD34Ab-immobilized, heparinized control and unmodified control grafts were sterilized under ultraviolet (UV) irradiation for 1 h. Grafts intended for VEGF-modification were UV irradiated before loading with sterile solution VEGF. Cells were seeded in the wells at a density of 5 × 104 cells/well and incubated at 37 °C. To measure cell metabolic activity, an XTT assay was performed at 1.5 h, 1 day, 3 days, and 7 days after initial cell seeding. At each of these time points, cells also underwent Live/Dead staining and were counted via microscopy. Cell attachment numbers were defined by the total number of cells still adhered to graft surfaces after washing. Attachment percentage was calculated by comparison with the total cell numbers seeded on grafts, which was normalized to total cells attached to separate tissue culture polystyrene (TCPS) controls. Fold change in cell populations was calculated by dividing the final cell population count (day 7 time point) by the initial attachment number (1.5 h after seeding).
Endothelial Progenitor Cells (EPC)
Human EPCs were obtained and cultured according to the manufacturer’s instructions (CelProgen, San Pedro, CA). Methods for assessment were identical to HUVEC assays.
XTT Assay
XTT assays were performed according to the manufacturer protocols (Roche Diagnostics, Indianapolis, IN). In summary, each cell-containing well of the 96-well plates was washed with PBS. A total of 50 μL of XTT labeling mixture was added, along with 50 μL of culture medium. The plate was incubated at 37 °C for 4 h. Following incubation, the supernatant was transferred to a new plate. Absorbance of the supernatant was measured at 450 nm, with a 650 nm reference.
Real-Time Polymerase Chain Reaction
HUVECs and EPCs were cultured separately in six-well plates on grafts without modifications, with heparinization, with VEGF, or with anti-CD34Ab (n = 3). Cells were seeded at a density of 3 × 105 cells/well to ensure adequate RNA content for PCR analysis. RNA was extracted with an RNeasy kit (Qiagen, Dusseldorf, Germany) at 1, 3, and 7 days to be compared with initial RNA content isolated from cell samples immediately before seeding. Real-time PCR analysis was performed using a SYBR Green One-Step RT-PCR Kit (Qiagen). Reference numbers for primers are eNOS (NM_000603), VEGF (NM_001025366), and GAPDH (NM_001256799). The results were analyzed using the comparative threshold cycle method and normalized with GAPDH as an endogenous reference, and reported as relative values (ΔΔCT) to those of control.
In Vivo Implantation
All animal procedures were approved by the Nationwide Children’s Hospital Institutional Animal Care and Use Committee. An in vivo trial was performed in a manner adapted from a previous experiment we performed.37 Briefly, grafts (1 mm in diameter and 3 mm in length) were implanted in female mice 6–8 wks of age as inferior vena cava (IVC) interpositional grafts using microsurgical technique. Grafts with VEGF- (n = 10) or anti-CD34Ab-modified surfaces (n = 10), and unmodified surfaces (n = 10) were used. All grafts, after modification, were UV irradiated to sterilize them onsite before implantation. Mice were anesthetized and placed in the supine position, and an abdominal midline incision was made. The IVC was exposed, cross-clamped, and excised. Grafts were implanted using a 10–0 nylon suture for the proximal and distal anastomoses. Mice were recovered from surgery and maintained without antiplatelet or anticoagulation therapies.
Two weeks after the procedure, mice were anesthetized and sacrificed. After excision, grafts were fixed in 4% para-formaldehyde and embedded in paraffin for histology or embedded in optimal cutting temperature (OCT) compound (Tissue-Tek; Sakura Finetek, Torrance, CA, U.S.A.) for gene assay. Five micron thick sections were then stained with hematoxlyin and eosin (H&E) stain. Endothelial cells were identified with rabbit anti-CD31 (Abcam, MA, U.S.A.). Antibody binding was detected using biotinylated secondary antibodies, followed by binding of streptavidin-HRP. Color development was performed by a chromogenic reaction with 3,3-diaminobenzidine (Vector, CA, U.S.A.). Graft inner and outer diameters were measured using ImageJ software calculated from perimeter measurements. Nuclei were counterstained with hematoxylin. Explanted grafts frozen in OTC compound were sectioned into 20 sections, 30 μm each, using a Leica CM 1950 cryostat (Leica biosystems, Wetzlar, Germany). Excess OCT compound was removed by centrifugation in PBS. Total RNA was extracted and purified using the RNeasy mini kit (Qiagen) according to the manufacturer instructions. Reverse transcription was performed using High Capacity RNA-to-cDNA Kit (Applied Biosystems, CA, U.S.A.). All reagents and instrumentation for gene expression analysis were obtained from Applied Biosystems. Quantitative polymerase chain reaction (qPCR) was performed with a Step One Plus Real-Time PCR System using the TaqMan Universal PCR Master Mix Kit. Reference numbers for primers are eNOS (Mm00435217_ml), VEGF (Mm01281449_m1), and HPRT (HPRT; Mm00446968_m1). The results were analyzed using the comparative threshold cycle method and normalized with HPRT as an endogenous reference, and reported as relative values (ΔΔCT) to those of control. NIH guidelines for the care and use of laboratory animals (NIH publication #85–23 Rev. 1985) have been observed.
Statistical Analysis
Data were analyzed using analysis of variance single factor analysis with Student’s t-test or ANOVA assuming normal data distribution with a confidence of 95% (p < 0.05). Standard deviation error bars are reported on each figure along with relevant statistical relationships.
Results
Quantitative Assessment of Immobilized CD34 Antibodies and VEGF
The morphology of the grafts was analyzed via SEM images because biomaterial interactions can be influenced by nanometer-scale surface features.40,41 Figure 2 displays the acellular graft surfaces after the VEGF and anti-CD34Ab modifications. While the surface features of the experimental groups appear rougher than the control grafts, there is not a discernible visible difference between surface patterns on each of the modified surfaces. The process of heparinization of graft surfaces appears to introduce round grain formations onto the material. Heparin attachment was confirmed via toluidine blue assay. Anti-CD34Ab attachment was confirmed via secondary FITC-Ab attachment and ELISA. ELISA was also used to confirm successful VEGF attachment.
Figure 2.

Graft surfaces before and after modification: Control (A–C), anti-CD34Ab-modified (C–E), VEGF-modified (F–H), and heparin-only (I–K) are shown. Scale bars represent 100 μm (A, C, F, I), 40 μm (B, D, G, J), and 10 μm (C, E, H, K), respectively.
First analyzing loading efficiency of VEGF, ELISA results demonstrated that VEGF modifications produced 3.08 ± 0.33% VEGF loading efficiency. Anti-CD34Ab loading efficiency was 23.57 ± 0.62%. The elution rates of the VEGF from the heparin-cross-linked TEVG surfaces can be seen in Figure 3. In 24 h, 28.0 ± 2.9% of the VEGF remained on the VEGF-modified surfaces. Anti-CD34Ab retention after 24 h showed 99.3 ± 0.20% of the antibody remained on anti-CD34Ab-modified surfaces. Nonspecific adsorption of biofunctional molecules was also determined. After incubating biofunctional molecules without heparin/EDC cross-linking and subsequent thorough washing, only 1.56 ± 0.47% of the antibody was still adsorbed. In comparison, 34.08 ± 16.64% of total VEGF was found to be nonspecifically adsorbed.
Figure 3.

Persistence of biomolecules on graft surface: (A) VEGF and (B) anti-CD34Ab percent of initially loaded molecules remaining on TEVGs over various time points. Grafts were incubated in PBS at 37 °C, undergoing gentle shaking to investigate burst release of loaded biofunctional molecules within the first 48 h.
EC and EPC Response to Modified TEVG Surfaces
Total metabolic activity of HUVECs and EPCs results are shown in Figure 4. Anti-CD34Ab-modified grafts demonstrated a statistically significant increase of total HUVEC metabolic activity over control grafts on day 3, although there was no difference between anti-CD34Ab- and VEGF-modified grafts (p < 0.05). Otherwise, there was no discernible difference between total metabolic activity of HUVEC populations over the 7 days on the modified and unmodified grafts. For total EPC metabolic activity, both anti-CD34Ab-modified grafts demonstrated an increase over unmodified control and VEGF-modified TEVG surfaces at days 0 and 1 after initial cell seeding. At 7 days, only VEGF-modified grafts demonstrated a significant increase of total EPC metabolic activity compared to the control, although there was no difference between VEGF- and anti-CD34Ab-modified grafts. Total metabolic activity of attached cells experienced no differences between heparin-only and unmodified controls, except on day 3 when total HUVEC metabolic activity was decreased on heparin-only grafts compared to unmodified controls.
Figure 4.
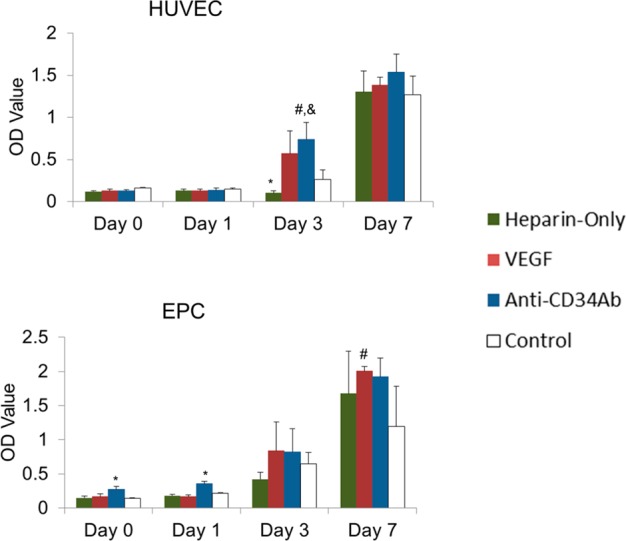
Total metabolic response to heparin-only, VEGF, and anti-CD34 modified grafts. (Top) HUVEC metabolic activity measured via relative absorbance of XTT media. (Bottom) EPC metabolic activity measured via relative absorbance of XTT media. Please note that n = 4; * represents statistical significance compared to all other groups within the time point, # represents statistical significance compared to the unmodified control of that time point, and & represents statistical significance compared to the heparin-only control of that time point (p < 0.05).
Anti-CD34Ab-coated grafts demonstrated higher initial HUVEC and EPC attachment than control and VEGF-modified grafts. Both VEGF- and anti-CD34Ab-modified grafts demonstrated a greater HUVEC population than the unmodified controls at day 1. On day 3, EPCs demonstrated greater cell numbers on anti-CD34Ab grafts than the control. Additionally, anti-CD34Ab-modified surfaces demonstrated a greater EPC population than both VEGF-modified and unmodified control grafts on day 7. Heparin-only controls demonstrated no differences compared to controls, other than a decrease in attached HUVEC populations on day 1. Figure 5 displays all Live/Dead counting results. Table 1 summarizes the initial attachment of cells to various graft surfaces, and Table 2 displays the proliferation of total cell populations attached to cell grafts after 7 days.
Figure 5.

Cell attachment and proliferation on heparin-only, VEGF, and anti-CD34 modified grafts. (Top) HUVEC population counted via Live/Dead. (Bottom) HUVEC population counted via Live/Dead. Please note that n = 4; * represents statistical significance compared to all other groups within the time point, % represents statistical significance compared to the heparin-only and unmodified control of that time point (p < 0.05).
Table 1. Initial Attachment Percentage of Cells on Graft Surfaces Normalized to Tissue Culture Polystyrenea.
| graft-type | HUVEC | EPC |
|---|---|---|
| control | 21.34 ± 10.24 | 28.06 ± 11.33 |
| heparin only | 22.24 ± 8.98 | 33.17 ± 6.50 |
| VEGF | 28.23 ± 7.32 | 41.07 ± 6.74 |
| anti-CD34Ab | 40.69 ± 10.69* | 53.51 ± 17.32* |
*Indicates statistical significance compared to control graft surfaces (p < 0.05).
Table 2. Fold Change of Cells over 7 Days.
| graft-type | HUVEC (fold change) | EPC (fold change) |
|---|---|---|
| control | 4.00 ± 1.93 | 5.22 ± 2.41 |
| heparin only | 4.13 ± 1.76 | 5.33 ± 1.11 |
| VEGF | 4.39 ± 0.50 | 5.23 ± 0.79 |
| anti-CD34Ab | 2.98 ± 0.61 | 7.02 ± 1.43 |
According to PCR results, as summarized in Figure 6, HUVECs attached to anti-CD34Ab grafts expressed an increased mRNA fold change in eNOS genes compared to other groups on day 1. On day 3, HUVECs on VEGF-modified grafts produced the most significantly increased fold change in VEGF gene expression compared to other grafts, while eNOS gene expression was significantly reduced. In addition, on day 3, both heparin-only and anti-CD34Ab grafts demonstrated higher fold change in VEGF expression of attached EPCs compared to VEGF-modified and unmodified grafts. EPCs attached to anti-CD34Ab grafts also demonstrated significantly higher expression of eNOS on day 3. There were no significant differences between graft surfaces on day 7.
Figure 6.

mRNA expression of cells on heparin-only, VEGF, and anti-CD34 modified grafts. (Top) HUVEC mRNA expression of VEGF (left) and eNOS (right). (Bottom) EPC mRNA expression of VEGF (left) and eNOS (right). Please note that n = 3; * represents statistical significance compared to all other groups within the time point, and represents statistical significance compared to the VEGF and unmodified control of that time point, % represents statistical significance compared to the heparin-only and unmodified control of that time point (p < 0.05).
In Vivo Assessment of Modified Grafts
All modified grafts demonstrated a larger inner lumen diameter compared to control grafts after two weeks of implantation. Luminal cross-sectioned examples of retrieved grafts can be seen in Figure 7. Overall, grafts modified with anti-CD34Ab resulted in a greater inner lumen diameter after two weeks of implantation compared to unmodified grafts and VEGF-modified grafts. Both VEGF- and anti-CD34Ab-modified grafts retained larger inner diameters compared to controls. Anti-CD34Ab-modified grafts also demonstrated greater outer diameter compared to unmodified control grafts. In addition, antibody-modified grafts maintained a smaller wall thickness compared to VEGF-modified and unmodified grafts. Wall thicknesses are compared in Table 3. Similarly, qPCR analysis of explanted anti-CD34Ab modified grafts demonstrated significantly higher gene expression of eNOS compared to control. VEGF grafts were not significantly different in terms of eNOS expression compared to explanted control graft samples. The three groups, when compared, did not demonstrate any significantly different levels of VEGF gene expression. Results are summarized and compared in Figure 8. CD31 staining (a marker for endothelial cells) demonstrated the formation of an endothelium in modified grafts, as shown in Figure 9. Endothelial formation was especially prominent in grafts modified with anti-CD34Ab.
Figure 7.
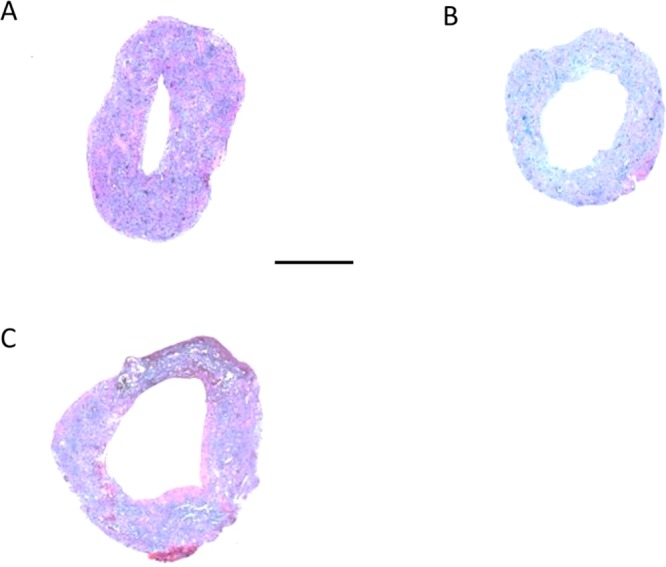
Cross sectional of TEVGs after implantation: (A) control, (B) VEGF, and (C) anti-CD34Ab. These representative cross sections demonstrate the visible difference in reduced diameter of the control (A) compared to the modified grafts (B, C) two weeks after implantation, as well as the tissue and extracellular matrix formation within the grafts. Scale bar represents 500 μm.
Table 3. Approximate Wall Thickness of Grafts Implanted within Mice after Two Weeksa.
| anti-CD34Ab | VEGF | control | |
|---|---|---|---|
| approximate wall thicknesses (mm) | 0.512 ± 0.182* | 0.574 ± 0.391 | 0.537 ± 0.232 |
*Indicates statistical significance compared to control graft surfaces (p < 0.05).
Figure 8.
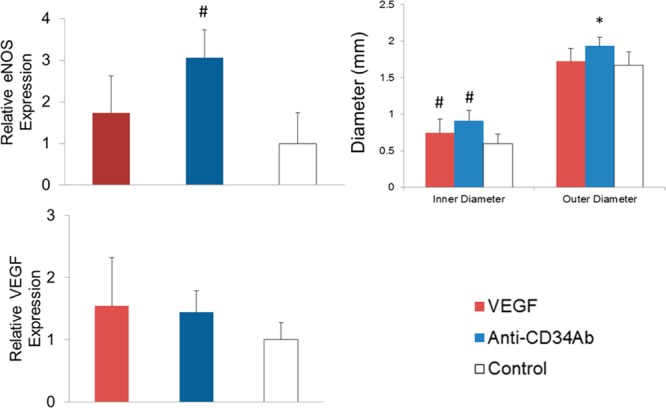
Biochemical and physical analysis of TEVGs. (Left) Relative eNOS expression of explanted samples. Anti-CD34Ab grafts resulted in increased eNOS expression in explanted tissues compared to explanted controls. (Right) Graft inner and outer lumen diameters after two weeks of implantation. VEGF and anti-CD34Ab grafts maintained in statistically significant greater inner diameter compared to unmodified controls. (Bottom) Comparing the four groups of modified and unmodified vascular grafts, there were no statistically significant differences in VEGF expression. Please note that n = 10; * represents statistical significance compared to all other groups, and # represents statistical significance compared to the control (p < 0.05).
Figure 9.

Endothelial cell staining of TEGVs. CD31 staining showing dark brown in images (indicated by arrows) of areas with endothelium formation and CD31 expression shown in (A) unmodified grafts and (B) VEGF, and (C) anti-CD34Ab modified grafts after two weeks of implantation in a mouse model. Anti-CD34Ab demonstrated increased more uniform CD31 staining. Scale bar represents 20 μm.
Discussion
The objective of this work was to contrast two strategies intended to improve the enhancement of vascular graft endothelialization. Specifically, we sought to determine if a burst release of VEGF from the graft surface or immobilized anti-CD34 antibodies would result in enhanced endothelialization.
Through the quantification of VEGF and anti-CD34 antibodies bound to the grafts, we were able to determine the loading efficiency and retention over time of these molecules on our biodegradable grafts. We found that VEGF experienced a burst release profile as expected from previous research.33 In addition, lower VEGF loading compared to anti-CD34Ab loading was expected based on these studies. It is possible that the highly specific heparin-binding domains of VEGF limits loading due to the specific orientation and presentation of binding sites presented by heparin.42 Because of these heparin-binding domains of VEGF, elution rates of VEGF from cross-linked heparin molecules in our study are similar to those that have been observed in other studies.33,48,49 In contrast to the VEGF elution, anti-CD34Ab concentrations did not significantly change over time, with minimal nonspecific adsorption and nearly all bound antibodies retained, indicating the antibodies are more permanently immobilized to the graft surface. This trend has been observed in similar research studies that hypothesize antibodies could be covalently linked due to aminolysis following EDC chemistry or may experience strong protein–protein interactions (such as van der Waals, hydrogen bonding, hydrophilic interactions, electrostatic interactions, etc.).43−47 Another cause of the immediate elution of VEGF is the larger percentage of nonspecifically bound VEGF compared to anti-CD34 antibodies (34.08 ± 16.64% vs 1.56 ± 0.47%).
Through our in vitro studies, we found that modifications of grafts produced a noticeable change in microscale graft topography. Such topographical roughness may influence cell attachment, as demonstrated in previous studies. However, to demonstrate whether or not heparinization alone (and the resulting addition of roughness) caused increased cell attachment compared to our unmodified control, we assessed cell populations attached to our grafts. Heparinized grafts, without the addition of VEGF or anti-CD34Ab, displayed no differences in cell attachment compared to unmodified, control grafts. Total metabolic activity of cells attached to such grafts were no different than control grafts either. In fact, after 1 day, total attached HUVECs numbered less than those on unmodified grafts, and after 3 days, total metabolic activity of HUVECs was decreased compared to controls. Conversely, HUVECs attached to such grafts expressed increased gene levels of eNOS and EPCs attached to these grafts expressed increased levels of VEGF on day 3, both compared to unmodified controls. Ultimately, the proliferation of cells on these grafts appeared no different than unmodified controls.
Our previous studies demonstrated that the small-diameter TEVG modified here possessed mechanical properties similar to native vessels and could be successfully implanted into a mouse model.37,38 Previous research has also indicated that two weeks is sufficient to predict vessel remodeling and demonstrate whether or not intimal hyperplasia will occur.50 Using a two week time point to determine acute endothelialization response, we modified our biodegradable polyester grafts and implanted them within mice. Though in vivo VEGF expression of tissues forming within the grafts was not statistically different between the groups, functionalized grafts demonstrated greater inner lumen diameter. Retention of inner lumen diameter at two weeks is a significant indication of lowered stenosis risks.51 Functionalized grafts also demonstrated endothelial cell activity through expression of CD31. CD31, an endothelial cell marker, was evident in both of the biofunctionalized graft groups, and staining for CD31 demonstrated good endothelial cell coverage of the inner lumen of the grafts. However, only anti-CD34Ab modified grafts demonstrated greater expression of eNOS compared to control grafts. Anti-CD34Ab grafts also demonstrated better retention of reduced wall thickness. These results may be related to the in vitro observation that anti-CD34Ab grafts demonstrated significantly higher HUVEC and EPC attachment. Higher initial cell attachment may result in earlier formation of a healthy endothelium which leads to less wall thickening and restenosis while maintaining inner lumen diameter within an in vivo environment.44,52,53 Interestingly, the effects of the modified grafts, at least in this experimental design, provided only transient or temporary advantages over control grafts in vitro. The most consistent results were in the initial attachment of cells to the antibody modified grafts. Combining this with the performance of anti-CD34Ab grafts in vivo potentially provides further support to the idea that expedited cell attachment may be one of the most important factors in improving in situ endothelialization
VEGF concentrations as little as 10 ng/mL can affect EC migration and proliferation.30 Given the small volume of media (200 μL) and the relatively large surface area (0.3165 cm2) of the wells of the 96-well plate, this threshold is easily attainable in vitro according to ELISA results. VEGF loading density onto biodegradable grafts was approximated to be 12.92 ± 2.42 ng/cm2 and, subsequently, ensured VEGF concentrations of greater than 10 ng/mL in vitro. Once implanted, the burst release of VEGF may have offered only acute benefits, leading to observably less endothelialization compared to anti-CD34Ab grafts. Still, tissue formed on VEGF-modified grafts did demonstrate increased CD31 expression when compared to the unmodified, control grafts. Such effects may be due to the recruitment and mobilization of ECs from neighboring tissues according to other studies.31,49 In fact, previous studies demonstrate that the endothelialization of unmodified implanted grafts is primarily due to migration of ECs over the anastomotic sites.18 Thus, VEGF may have acted locally to increase the mobilization of ECs from neighboring tissues to impart increased endothelialization over unmodified grafts rather than providing any systemic mobilization of EPCs.
Anti-CD34 antibodies have been shown to been a potent recruitment tool to increase both EC and EPC attachment, especially on permanent stents.22,44,47,54 While there are other CD34+ cells in whole blood circulation, previous research demonstrated that anti-CD34 antibodies effectively induced attachment of CD34+ EPCs at significantly higher rates than CD34+ hematopoietic stem cell populations, ostensibly due to higher antigen presentation.55,56 Our results supported the efficacy of anti-CD34 antibody recruitment in endothelial-like cell attachment to graft substrate and subsequent endothelial function. Such endothelial formation and function may have contributed to the thinner wall thickness of antibody-modified grafts, which may be indicative of reduced risk of restenosis. In conjunction with the results presented here, modification of biodegradable heparin-cross-linked vascular grafts with anti-CD34 antibodies, with or without other biofunctional molecules, may be a promising strategy for expediting and increasing graft surface endothelialization.
Overall, modified grafts demonstrated trends in great inner lumen diameter retention and eNOS expression, which is crucial for vascular homeostasis and can be used as an indicator for healthy endothelial function. Healthy endothelial formation was further confirmed through the staining of CD31 expression within the inner lumen of the explanted grafts, especially evident in those grafts modified with anti-CD34 antibodies.
Conclusions
The goal of this study was to determine if biofunctionalization of biodegradable vascular grafts could improve overall graft endothelialization and subsequently reduce stenosis after implantation. Biodegradable polyester vascular grafts were functionalized via a unique strategy of heparin-cross-linking to immobilize anti-CD34Ab or VEGF. Although in vitro data provided support only for transient increased endothelial activity or cell attachment, modified graft surfaces elicited better endothelial and endothelial-like cell attachment in vivo. It appears that heparin-cross-linked biodegradable polymer grafts modified with anti-CD34Ab modestly outperformed VEGF-modified grafts and significantly outperformed control grafts. Modified grafts promoted neotissue formation without major complications like thrombosis or stenosis. The performance of the modified, biodegradable vascular grafts appears to be a promising improvement to the in situ endothelialization of synthetic vascular grafts for tissue engineering.
Acknowledgments
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under the Award Number R01 AR061460 and through a seed grant from Children’s National Sheikh Zayed Institute for Pediatric Surgical Innovation and the A. James Clark School of Engineering at the University of Maryland. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Contributions
† These authors contributed equally to this paper (A.J.M. and N.H.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Lloyd-Jones D.; Adams R. J.; Brown T. M.; Carnethon M.; Dai S.; De Simone G.; Ferguson T. B.; Ford E.; Furie K.; Gillespie C.; Go A.; Greenlund K.; Haase N.; Hailpern S.; Ho P. M.; Howard V.; Kissela B.; Kittner S.; Lackland D.; Lisabeth L.; Marelli A.; McDermott M. M.; Meigs J.; Mozaffarian D.; Mussolino M.; Nichol G.; Roger V. L.; Rosamond W.; Sacco R.; Sorlie P.; Stafford R.; Thom T.; Wasserthiel-Smoller S.; Wong N. D.; Wylie-Rosett J. Circulation 2010, 121, e46–e215. [DOI] [PubMed] [Google Scholar]
- Veith F. J.; Moss C. M.; Sprayregen S.; Montefusco C. Surgery 1979, 85, 253–256. [PubMed] [Google Scholar]
- Darling R. C.; Linton R. F. Surgery 1972, 123, 472–479. [DOI] [PubMed] [Google Scholar]
- Wang X.; Lin P.; Yao Q.; Chen C. World J. Surg. 2007, 31, 682–689. [DOI] [PubMed] [Google Scholar]
- Chlupác J.; Filová E.; Bacáková L. Physiol. Res. 2009, 58, S119–39. [DOI] [PubMed] [Google Scholar]
- Haruguchi H.; Teraoka S. J. Artif. Organs 2003, 6, 227–235. [DOI] [PubMed] [Google Scholar]
- Zilla P.; Bezuidenhout D.; Human P. Biomaterials 2007, 28, 5009–5027. [DOI] [PubMed] [Google Scholar]
- Nieponice A.; Soletti L.; Guan J.; Hong Y.; Gharaibeh B.; Maul T. M.; Huard J.; Wagner W. R.; Vorp D. a. Tissue Eng., Part A 2010, 16, 1215–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino N.; McGillicuddy E.; Matsumura G.; Ichihara Y.; Naito Y.; Breuer C.; Shinoka T. J. Thorac. Cardiovasc. Surg. 2010, 139, 431–6436.e1–2.. [DOI] [PubMed] [Google Scholar]
- Kannan R. Y.; Salacinski H. J.; Butler P. E.; Hamilton G.; Seifalian A. M. J. Biomed. Mater. Res., Part B 2005, 74, 570–581. [DOI] [PubMed] [Google Scholar]
- De Mel A.; Jell G.; Stevens M. M.; Seifalian A. M. Biomacromolecules 2008, 9, 2969–2979. [DOI] [PubMed] [Google Scholar]
- Melchiorri A. J.; Hibino N.; Fisher J. P. Tissue Eng., Part B 2013, 19, 292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen N.; Lindblad B.; Ljungberg J.; Leide S.; Bergqvist D. Ann. Vasc. Surg. 1996, 10, 530–536. [DOI] [PubMed] [Google Scholar]
- Rupnick M. A.; Hubbard F. A.; Pratt K.; Jarrell B. E.; Williams S. K. J. Vasc. Surg. 1989, 9, 788–795. [DOI] [PubMed] [Google Scholar]
- Deutsch M.; Meinhart J.; Zilla P.; Howanietz N.; Gorlitzer M.; Froeschl A.; Stuempflen A.; Bezuidenhout D.; Grabenwoeger M. J. Vasc. Surg. 2009, 49, 352–362discussion 362. [DOI] [PubMed] [Google Scholar]
- Bordenave L.; Fernandez P.; Rémy-Zolghadri M.; Villars S.; Daculsi R.; Midy D. Clin. Hemorheol. Microcirc. 2005, 33, 227–234. [PubMed] [Google Scholar]
- Avci-Adali M.; Perle N.; Ziemer G.; Wendel H. P. Eur. Cell. Mater. 2011, 21, 157–176. [DOI] [PubMed] [Google Scholar]
- Hagensen M. K.; Vanhoutte P. M.; Bentzon J. F. Cardiovasc. Res. 2012, 95, 1–33. [DOI] [PubMed] [Google Scholar]
- Pearson J. D. J. Thromb. Haemost. 2009, 7, 255–262. [DOI] [PubMed] [Google Scholar]
- Yoder M. C. Cold Spring Harb. Perspect. Med. 2012, 2, a006692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschi K. K.; Ingram D. a; Yoder M. C. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1584–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa G.; Granada J. F.; Alviar C. L.; Tellez A.; Kaluza G. L.; Guilhermier M. Y.; Parker S.; Rowland S. M.; Kolodgie F. D.; Leon M. B.; Virmani R. JACC Cardiovasc. Interv. 2010, 3, 68–75. [DOI] [PubMed] [Google Scholar]
- Larsen K.; Cheng C.; Tempel D.; Parker S.; Yazdani S.; den Dekker W. K.; Houtgraaf J. H.; de Jong R.; Swager-ten Hoor S.; Ligtenberg E.; Hanson S. R.; Rowland S.; Kolodgie F.; Serruys P. W.; Virmani R.; Duckers H. J. Eur. Heart J. 2012, 33, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki J.; Serruys P. W.; van Beusekom H.; Ong A. T. L.; McFadden E. P.; Sianos G.; van der Giessen W. J.; Regar E.; de Feyter P. J.; Davis H. R.; Rowland S.; Kutryk M. J. B.. J. Am. Coll. Cardiol. 2005; Vol. 45, pp 1574–1579. [DOI] [PubMed] [Google Scholar]
- Chavakis E.; Dimmeler S. Antioxid. Redox Signaling 2011, 15, 967–980. [DOI] [PubMed] [Google Scholar]
- Wang Q. R.; Wang B. H.; Zhu W. B.; Huang Y. H.; Li Y.; Yan Q. Bone Marrow Res. 2011, 2011, 846096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y.; Yamamoto K.; Ando J.; Matsumoto K.; Matsuda T. Biochem. Biophys. Res. Commun. 2012, 423, 91–97. [DOI] [PubMed] [Google Scholar]
- Shen Y. H.; Shoichet M. S.; Radisic M. Acta Biomater. 2008, 4, 477–489. [DOI] [PubMed] [Google Scholar]
- Neufeld G.; Cohen T.; Gengrinovitch S.; Poltorak Z. FASEB J. 1999, 13, 9–22. [PubMed] [Google Scholar]
- Morales-Ruiz M.; Fulton D.; Sowa G.; Languino L. R.; Fujio Y.; Walsh K.; Sessa W. C. Circ. Res. 2000, 86, 892–896. [DOI] [PubMed] [Google Scholar]
- Shin Y. M.; Lee Y. Bin; Kim S. J.; Kang J. K.; Park J.-C.; Jang W.; Shin H. Biomacromolecules 2012, 13, 2020–2028. [DOI] [PubMed] [Google Scholar]
- Nillesen S. T. M.; Geutjes P. J.; Wismans R.; Schalkwijk J.; Daamen W. F.; Van Kuppevelt T. H. Biomaterials 2007, 28, 1123–1131. [DOI] [PubMed] [Google Scholar]
- Singh S.; Wu B. M.; Dunn J. C. Biomaterials 2011, 32, 2059–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C.; Markowicz M.; Pallua N.; Noah E. M.; Steffens G. Biomaterials 2008, 29, 66–74. [DOI] [PubMed] [Google Scholar]
- Brandner B.; Kurkela R.; Vihko P.; Kungl A. J. Biochem. Biophys. Res. Commun. 2006, 340, 836–839. [DOI] [PubMed] [Google Scholar]
- Andresen D. M.; Barker J. H.; Hjortdal V. E. Microsurgery 2002, 22, 265–272. [DOI] [PubMed] [Google Scholar]
- Melchiorri A. J.; Hibino N.; Brandes Z. R.; Jonas R. a; Fisher J. P. J. Biomed. Mater. Res., Part A 2013, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh J. D.; Nelson G. N.; Brennan M. P.; Mirensky T. L.; Yi T.; Hazlett T. F.; Tellides G.; Sinusas A. J.; Pober J. S.; Saltzman W. M.; Kyriakides T. R.; Breuer C. K. Biomaterials 2008, 29, 1454–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulf K.; Teske M.; Lo M.; Luderer F.; Schmitz K.-P.; Löbler M.; Sternberg K. J. Biomed. Mater. Res., Part B 2011, 98, 89–100. [DOI] [PubMed] [Google Scholar]
- Carpenter J.; Khang D.; Webster T. J. Nanotechnology 2008, 19, 505103. [DOI] [PubMed] [Google Scholar]
- Ranjan A.; Webster T. J. Nanotechnology 2009, 20, 305102. [DOI] [PubMed] [Google Scholar]
- Krilleke D.; Ng Y.-S. E.; Shima D. T. Biochem. Soc. Trans. 2009, 37, 1201–1206. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Wang Z.; Wang Z.; Feng S.; Xu H.; Zhao Q.; Wang S.; Fang J.; Qiao M.; Kong D. Colloids Surf., B 2011, 85, 32–39. [DOI] [PubMed] [Google Scholar]
- Chen J.; Cao J.; Wang J.; Maitz M. F.; Guo L.; Zhao Y.; Li Q.; Xiong K.; Huang N. J. Colloid Interface Sci. 2012, 368, 636–647. [DOI] [PubMed] [Google Scholar]
- Lin Q.; Ding X.; Qiu F.; Song X.; Fu G.; Ji J. Biomaterials 2010, 31, 4017–4025. [DOI] [PubMed] [Google Scholar]
- Liu S.; Liu T.; Chen J.; Maitz M.; Chen C.; Huang N. J. Biomed. Mater. Res., Part A 2013, 101, 1144–1157. [DOI] [PubMed] [Google Scholar]
- Yin M.; Yuan Y.; Liu C.; Wang J. J. Mater. Sci.: Mater. Med. 2009, 20, 1513–1523. [DOI] [PubMed] [Google Scholar]
- Liu S. L. S.; Chen J. C. J.; Chen C. C. C.; Huang N. H. N. IEEE Int. Nanoelectron. Conf., 3rd 2010, 70, 341–353. [Google Scholar]
- Zhou M.; Liu Z.; Wei Z.; Liu C.; Qiao T.; Ran F.; Bai Y.; Jiang X.; Ding Y. Artif. Organs 2009, 33, 230–239. [DOI] [PubMed] [Google Scholar]
- Hibino N.; Yi T.; Duncan D. R.; Rathore A.; Dean E.; Naito Y.; Dardik A.; Kyriakides T.; Madri J.; Pober J. S.; Shinoka T.; Breuer C. K. FASEB J. 2011, 25, 4253–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino N.; Yi T.; Duncan D. R.; Rathore A.; Dean E.; Naito Y.; Dardik A.; Kyriakides T.; Madri J.; Pober J. S.; Shinoka T.; Breuer C. K. FASEB J. 2011, 25, 4253–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avci-Adali M.; Stoll H.; Wilhelm N.; Perle N.; Schlensak C.; Wendel H. P. Pathobiology 2013, 80, 176–181. [DOI] [PubMed] [Google Scholar]
- Kong D.; Melo L. G.; Mangi A. A.; Zhang L.; Lopez-Ilasaca M.; Perrella M. A.; Liew C. C.; Pratt R. E.; Dzau V. J. Circulation 2004, 109, 1769–1775. [DOI] [PubMed] [Google Scholar]
- Lin G.; Finger E.; Gutierrez-Ramos J. C. Eur. J. Immunol. 1995, 25, 1508–1516. [DOI] [PubMed] [Google Scholar]
- Hatch A.; Hansmann G.; Murthy S. K. Langmuir 2011, 27, 4257–4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansmann G.; Plouffe B. D.; Hatch A.; von Gise A.; Sallmon H.; Zamanian R. T.; Murthy S. K. J. Mol. Med. (Berlin) 2011, 89, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]