

Abstract
Ribosome biogenesis involves a large inventory of proteinaceous and RNA cofactors. More than 250 ribosome biogenesis factors (RBFs) have been described in yeast. These factors are involved in multiple aspects like rRNA processing, folding, and modification as well as in ribosomal protein (RP) assembly. Considering the importance of RBFs for particular developmental processes, we examined the complexity of RBF and RP (co-)orthologs by bioinformatic assignment in 14 different plant species and expression profiling in the model crop Solanum lycopersicum. Assigning (co-)orthologs to each RBF revealed that at least 25% of all predicted RBFs are encoded by more than one gene. At first we realized that the occurrence of multiple RBF co-orthologs is not globally correlated to the existence of multiple RP co-orthologs. The transcript abundance of genes coding for predicted RBFs and RPs in leaves and anthers of S. lycopersicum was determined by next generation sequencing (NGS). In combination with existing expression profiles, we can conclude that co-orthologs of RBFs by large account for a preferential function in different tissue or at distinct developmental stages. This notion is supported by the differential expression of selected RBFs during male gametophyte development. In addition, co-regulated clusters of RBF and RP coding genes have been observed. The relevance of these results is discussed.
Keywords: next generation sequencing, orthologous prediction, ribosome biogenesis, MACE, qRT-PCR, tomato
Introduction
Ribosome biogenesis is a complex biological process during which pre-ribosome particles are processed to mature functional ribosome subunits. The process is best described for Saccharomyces cerevisiae.1–3 Initially, the ribosomal RNA transcript (pre-rRNA) containing three of the four mature rRNAs is transcribed in the nucleolus priming the assembly of 90S pre-ribosomes. Subsequent cleavage of the 35S pre-rRNA leads to the separation of the small and the large ribosomal subunit maturation. This maturation of the ribosomal subunits occurs largely in the nucleus, and only the final rRNA processing steps take place in the cytoplasm.3,4 Here, both ribosomal subunits undergo further modifications and quality control events before final assembly to translational active ribosomes.5
The maturation of the ribosomal particles requires a large number of RNA and proteinaceous molecules. Small nucleolar RNAs (snoRNAs) base pairing with the pre-rRNA are required for ribosome biogenesis. Seventy-five snoRNAs have been identified in yeast as part of box C/D or box H/ACA snoRNPs,6 which catalyze the methylation and pseudouridylation of the rRNA, respectively.7 Besides snoRNAs, more than 250 proteins have been identified and assigned as ribosome biogenesis factors (RBFs) in yeast. These factors belong to protein families like RNA-helicases, GTPases, ATPases, RNA-binding proteins, endo- and exonucleases.2,3
However, information on plant ribosome biogenesis in general as well as on plant snoRNAs is rather sparse. More than 100 binding sites of snoRNAs on plant pre-rRNA have been predicted (http://bioinf.scri.ac.uk/cgi-bin/plant_snorna/home),8,9 but their functional relevance has not been experimentally approached yet. Similarly, not much is known about plant RBFs. Recently, two bioinformatic studies have provided the first insights into the putative inventory of plant RBFs, one focusing on the family of RNA helicases in Zea mays, Glycine max, Arabidopsis thaliana, and Oryza sativa10 and the other describing the evolutionary development of the RBF set known in yeast.11 The latter used 255 yeast RBFs to predict orthologs in plants including A. thaliana and Solanum lycopersicum.11 This study realized a significant, but not complete overlap of about 75% between the RBF inventory in yeast and plants. However, the work focused on the discovery of the RBF family,11 but did not evaluate the complexity caused by existence of co-orthologous genes in single species. The latter is important to explore whether plants have tissue- or developmental stage- specific RBFs or whether RBFs of a certain protein family might have taken on the function of factors not found in the plant genomes. For example, one of the two predicted A. thaliana orthologs of yeast nucleolin Nsr1 gene is abundantly expressed in all plant organs, while the second exhibits higher expression in flower buds.12,13 Similarly, the two A. thaliana orthologs of yeast Lsg1 show a differential functional activity.14
We analyzed the complexity of RBF families and ribosomal proteins (RPs) in 14 plants. We further examined the expression of the predicted RBFs and RPs in tomato, using available RNA-seq data in combination with quantitative next generation sequencing (NGS) by Massive Analysis of 3′-cDNA Ends (MACE)15 on leaves and anthers, considering the importance of RBFs for sexual reproduction.16–18 Additionally, the transcript profile of selected RBFs and RPs was investigated and compared by co-expression analysis. This allowed us to examine whether different (co-)orthologs exhibit a tissue- or developmental stage-specific expression, considering that several genetic studies have shown that mutations in specific genes involved in ribosome biogenesis are associated with developmental defects.19 We realized that most of the RBFs are expressed in different tissues, while for a subset of RBFs a tissue-specific expression can be concluded.
Materials and Methods
Ortholog search and clustering
The set of 255 yeast RBFs11 and of 129 ribosomal proteins of the large subunit (RPL) and 89 ribosomal proteins of the small subunit (RPS) sequences from A. thaliana (http://ribosome.med.miyazaki-u.ac.jp)20 were used for co-ortholog assignment followed by inspection of the domain architecture as described.21 For the ortholog search via OrthoMCL and InParanoid, we used 14 plant species including eudicots (A. thaliana, G. max, Lotus japonicus, Medicago truncatula, Populus trichocarpa, Solanum tuberosum, S. lycopersicum, Vitis vinifera), monocots (Brachypodium distachyon, O. sativa, Sorghum bicolor, Z. mays), the Bryopsida Physcomitrella patens, the Chlorophyceae Chlamydomonas reinhardtii, and the fungi S. cerevisiae as outgroup and bait. We searched for RBFs in the orthologous groups for all plants (Supplementary Table 1) and additional information restricted to tomato and A. thaliana is provided (Supplementary Tables 2 and 3). The number of RPL and RPS orthologs is deposited in Supplementary Table 4.
Plant material, RNA extraction, cDNA synthesis, and MACE
Total RNA was extracted from young leaves and anthers of eight-week-old tomato plants (cv Moneymaker) grown in a 16/8 hours day/night cycle (24–20 °C) in the greenhouse. Anthers were collected from flower buds of all developmental stages and pooled. Pollen cells from different developmental stages, namely tetrads, post-meiotic microspores, and mature pollen grains from open flowers, were isolated according to existing protocols.22 Total RNA was extracted using the NucleoSpin® RNA isolation kit (Macherey and Nagel) following manufacturer’s instructions. One microgram of RNA was reverse-transcribed using Revert AidTM RvTranscriptase (Fermentas) and cDNA synthesis was performed using Oligo dT12–18 primer. The MACE libraries were prepared and sequenced by GenXPro GmbH.15,23
NGS analysis pipeline
The analysis of the NGS data was performed with the flexible pipeline CRACPipe (Supplementary Method; Supplementary Figs. 1–3), which allows application to any species with available sequence information (genome or transcriptome) by screening NGS data with the BLAST-like Alignment Tool BLAT24 for adapter sequence removal, quality score determination by Fast Quality Control FastQC,25 and mapping with Sequence Search and Alignment by Hashing Algorithm 2 SSAHA2.26 The pipeline parses genomic data provided in GenBank format to internally construct an annotated genome divided in Watson and Crick strand27 containing information of the position, name and type of each feature. Supported feature types are mRNAs and non-protein coding RNAs, like tRNAs, rRNAs, snoRNAs, snRNAs, miRNAs and other ncRNAs.28 As output, mochi files are created for visualizing the results in the genome browser MochiView.29 The pipeline can be used via web page front end (https://www.uni-frankfurt.de/fb/fb15/institute/inst-3-mol-biowiss/AK-Schleiff/cracpipe/). For details see Supplementary Method.
NGS results
The MACE results are available at the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/; series: GSE56610). Twelve million reads (100 bp) with more than 14 nucleotides after linker removal have been obtained for both samples (Table 1). The cut-off resulted in the unique assignment of >97% of all reads. Assignment of reads to >22,000 genes and ∼30,000 regions (non-feature) without current annotation has been achieved.
Table 1.
Experimental overview for MACE analysis.
| LEAF | ANTHER | |
|---|---|---|
| Total read number | 11.762.204 | 12.687.296 |
| Total mapped reads | 11.534.512 | 12.411.606 |
| Percentage of unique mapped reads | 98% | 97% |
| Hits on watson strand | 6.235.294 | 6.386.089 |
| Hits on crick strand | 5.299.286 | 6.043.673 |
| Genesa | 22.894 | 24.539 |
| Non-feature | 29.666 | 30.540 |
Notes: Analysis of properties of MACE libraries (column 1) in leaf (column 2) or anther (column 3) sample.
In total, 29.345 genes have been assigned.
MACE is a digital gene expression method in which each read represents a cDNA molecule.15,30 By this, transcripts per million (TPM) is equivalent to reads per million, which is calculated by dividing the number of all reads mapped to an individual gene by all mapped reads normalized to a million reads. Furthermore, the cut-off for detection of a gene is the detection of a unique read for that gene.15,30 In turn, a gene is annotated as not expressed if no unique read was mapped. The latter was justified by the estimation of the coverage obtained (Supplementary Fig. 4).31,32 The analysis revealed that the call of “not expressed” has a reliability of 95% for the leaf dataset and of 99% for the dataset for anthers.
We used NoiSeq31 to simulate five replicates by a multinomial distribution and mapping probabilities were approximated from the available MACE library, where each feature had the chance to appear assigning a value bigger than 0. The five simulated replicates were used for differential expression analysis by the assumption that a high probability in NoiSeq correlates with a high probability that an observed difference in expression is significant.
Existing RNA-seq libraries containing expression values for the majority of tomato genes as well as Affymetrix microarray data were used to verify the quality of our MACE data. First, genes with low expression variance irrespective of developmental stages or experimental conditions were identified. We postulate that such genes are equally expressed under most experimental conditions and thus are best suited to judge the comparability of the two datasets generated. The variance of expression values for individual genes identified in Affymetrix studies of different tissues and developmental stages (91 samples; GEO, ArrayExpress) and after stress application (11 samples; GEO, TFGD; Supplementary Table 5) was determined. The 20 genes with the lowest variance were selected (Supplementary Table 6). The expression of these genes in our MACE datasets for leaves and anthers shows a linear relation of 0.97 (Supplementary Fig. 5). This confirms that the extracted TPM values for the two datasets are comparable. The R2 value for linear regression shown in Supplementary Figure 5 is 0.91.
Secondly, the RNA-seq data for leaves and flowers were directly compared to our MACE results. While we detected expression of a higher number of genes in leaves compared to the published NGS dataset,33 the comparison between our MACE data and NGS data (GEO: GSE33507) revealed a high correlation (rs = 0.74, P = 2 × 10−7) confirming the validity of the data used (Fig. 1).
Figure 1.
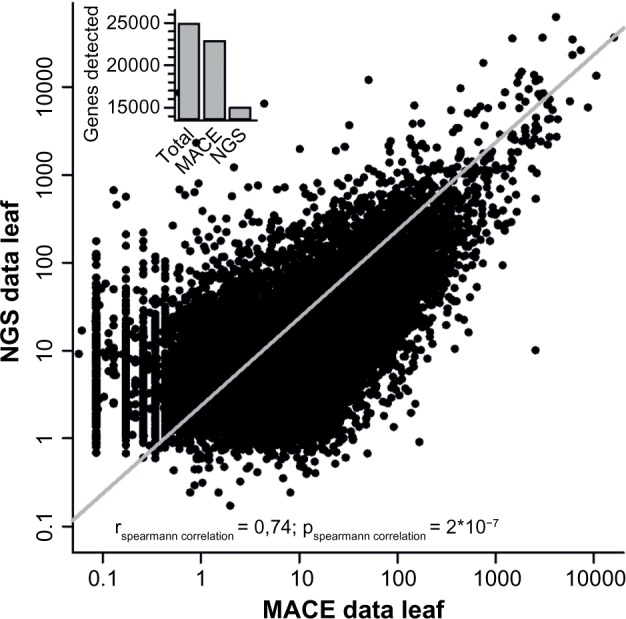
NGS and MACE comparison. Comparison of MACE and NGS data for leaf tissue. The line indicates the Spearman correlation for both leaf expression datasets using RNA-seq and MACE. The inset gives the number of genes assigned in the genome, detected by MACE and detected by NGS, and depicts the higher coverage achieved by MACE.
Cluster analysis of expression profiles of genes encoding RPs and RBFs
The expression values of the RPs and RBFs from NGS experiments of tomato plants cv. Moneymaker (Supplementary Tables 7 and 8)33 were used for cluster analysis. RPs and RBFs were independently clustered according to their expression profiles in different tomato tissues. For k-means clustering, Multi Experiment Viewer (MeV; http://www.tm4.org/mev.html)34 was used to determine six different clusters of each set of RPs and RBFs. The number of clusters required was determined as previously described (Supplementary Fig. 6).35 For k-means clustering, 1000 iterations have been performed and the Pearson correlation was used as distance.36 The accession numbers of the co-orthologs in the individual clusters are listed in Supplementary Table 9.
Quantitative real-time PCR
Gene expression of selected genes was determined using two biological replicas by real-time PCR on a Stratagene Mx3000P cycler (Agilent Technologies). The qPCR reaction (10 μL) consisted of gene-specific primers (Supplementary Table 10), PerfeCTa® SYBR® Green FastMix Low ROX™ (Quanta Biosciences), and the template. The cycling conditions were 95 °C/3 minutes followed by 95 °C/15 seconds, 60 °C/30 seconds, and 72 °C/30 seconds for 40 cycles. Primers were designed using PRIMER3 (www-genome.wi.mit.edu/cgi-bin/primer/primer3.cgi/). Data were analyzed using the 2−∆∆Ct method37 and presented as relative levels of gene expression, using ubiquitin (Solyc07g064130) and EF1α (Solyc06 g005060) genes as internal standards. Expression values are the mean of two biological replicates.
Results
The global assignment of co-orthologs to RBFs and RPs in plant genomes
We selected 14 plant genomes including the green algae C. reinhardtii, the moss P. patens, four monocots and eight dicots including the agriculturally relevant S. lycopersicum (tomato), and the model plant A. thaliana (Fig. 2A). Previously, orthologs to about 170 of 255 RBFs in yeast have been assigned in these 14 plant species and the highest number was found in the genomes of S. lycopersicum and A. thaliana with 173 and 174 genes, respectively (Supplementary Tables 1–3).11 Remarkably, only 116 RBFs have been found in all 14 plant genomes. The discovery rate of RBFs associated with different complexes is comparable. However, orthologs to RBFs of the exosome, the mitochondrial RNA processing (MRP) complex, and Trf4/Air2/Mtr4p polyadenylation (TRAMP) complex are under-represented in all plants compared to yeast (Fig. 2B). At least two co-orthologs are present in most of the species for approximately 20–40% of RBFs (Fig. 2C), while the same holds true for more than 50% of the RBFs in S. bicolor, G. max, and B. distachyon (Fig. 2C).
Figure 2.
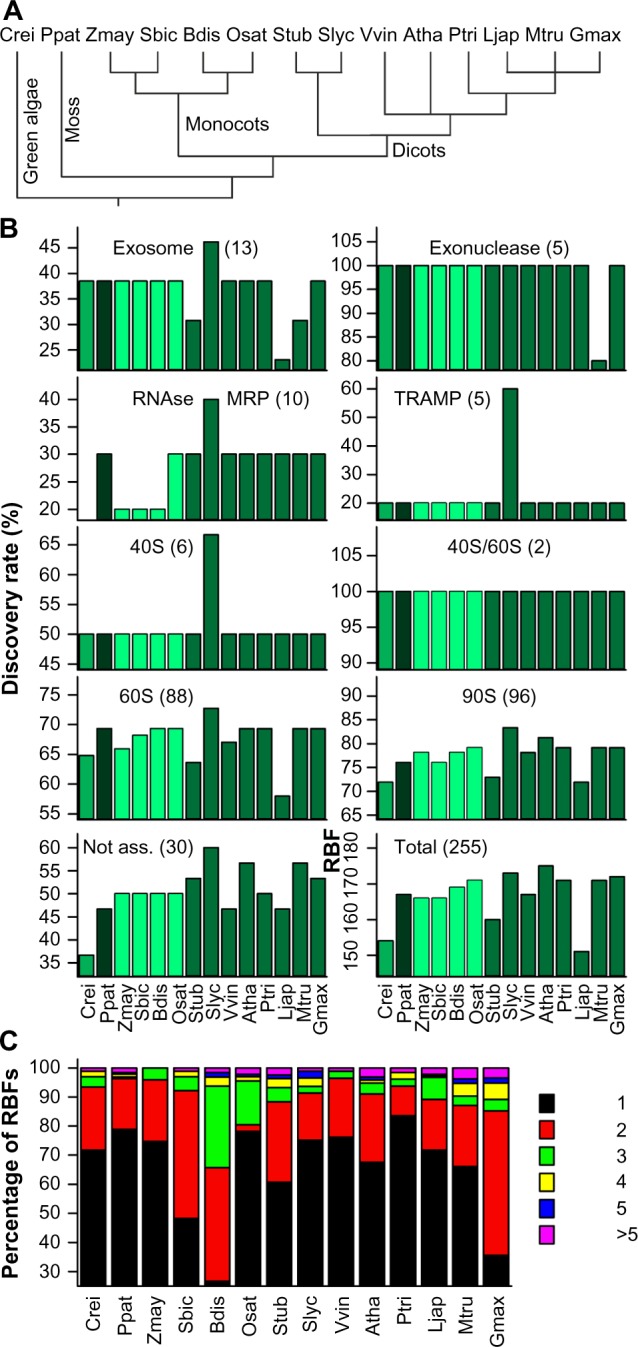
Prediction of RBF coding genes. (A) Phylogenetic relation of the 14 plant species used for the analysis. (B) Discovery rate of RBFs based on 255 yeast RBFs11 in the genomes of 14 plant species in percentage of the number found in yeast given for the different families. The different colors indicate the phylogenetic relation (green: green algae; black: moss; light green: monocots; dark green: eudicots). (C) The percentage of plant RBFs encoded by the indicated number of co-orthologs. Atha: A. thaliana; Bdis: B. distachyon; Crei: C. reinhardtii; Gmax: G. max; Ljap: L. japonicus; Mtru: M. truncatula; Osat: O. sativa; Ppat: P. patens; Ptri: P. trichocarpa; Sbic: S. bicolor; Slyc: S. lycopersicum; Stub: S. tuberosum; Vvin: V. vinifera; Zmay: Z. mays.
In parallel to the analysis of RBFs, we inspected the diversity of RP-coding genes based on the sequences assigned in A. thaliana.20 In most of the 14 plant species, the majority of the RPs are encoded by two or three co-orthologs (Fig. 3; Supplementary Table 4). Only in the algae C. reinhardtii they are encoded by one gene, while in P. patens, P. trichocarpa, Z. mays, and G. max, the majority is encoded by three or more genes. Comparing the number of co-orthologs found for RBFs (Fig. 2) and RPs (Fig. 3), we did not observe a correlation between the occurrence of co-orthologs of RBFs and RPs.
Figure 3.
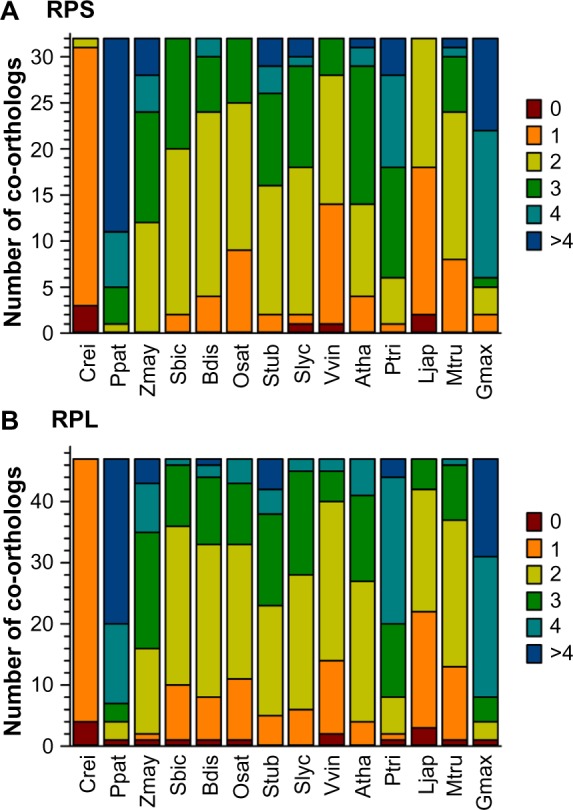
Prediction of RP coding genes. The distribution of the number of co-orthologs identified for RPSs (A) and RPLs (B) in the 14 analyzed plants as shown in Figure 1.
Sequence analysis of identified RBF co-orthologs in tomato
In S. lycopersicum and A. thaliana, more than one co-ortholog exists for about 24% and 27% of all RBFs, respectively. The total number of sequences assigned to the complexes is by large the same or even larger compared to yeast (Fig. 4A). In total, 249/260 sequences have been assigned as (co)-orthologs to RBFs in S. lycopersicum/A. thaliana, respectively. The extent of the occurrence of multiple orthologs varied among different complexes (Fig. 4B). With the exception of MRP and TRAMP in both species, and the exosome in S. lycopersicum, co-orthologs are found for RBFs of all complexes in all plants (Fig. 4B).
Figure 4.
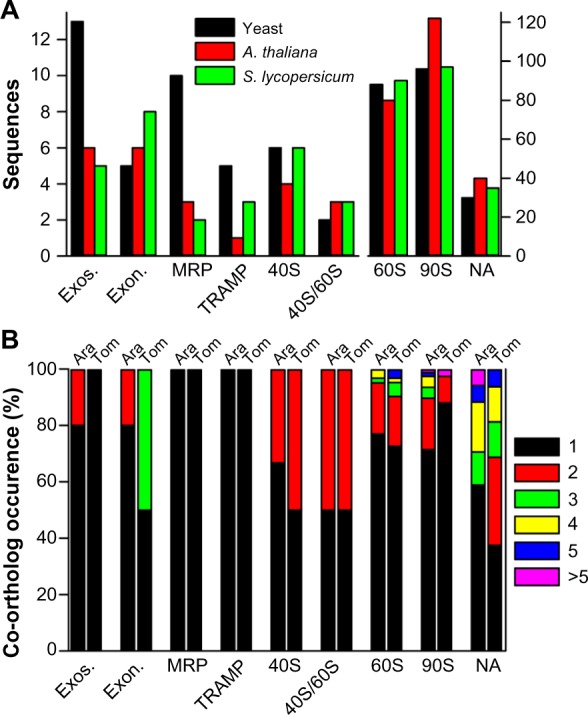
Prediction of co-orthologs to RBF coding genes. (A) Comparison of the total number of sequences in yeast (black bar), A. thaliana (red bar), and tomato (green bar). (B) The percentage of RBFs assigned to the specific complexes encoded by the number of co-orthologs shown on the right.
However, ortholog assignment is not necessarily proof for a function of the encoded proteins in the same process as the ancestral protein.38 To provide additional evidence for comparable functions between yeast RBFs and the proteins encoded by the S. lycopersicum genes, we categorized the predicted tomato RBF sequences based on (i) euKaryotic Orthologous Groups (KOG), (ii) their functional description, as well as (iii) Pfam domain presence and order (Supplementary Table 2). For six RBFs (DBP2, DRS2, FAP7, MDN1, YAR1, and YVH1), the co-orthologous sequences differ in at least one of the analyzed categories. Manual inspection of the corresponding alignments (Fig. 5; Supplementary Alignment S1 Supplementary Table 3) showed that one co-ortholog of DRS2 (Solyc01g011100) has a shorter N-terminal domain; one FAP7 co-ortholog (Solyc06g075310) lacks the C-terminal region; and the co-orthologs of MDN1 (Solyc04g072270) and YAR1 (Solyc04g008580) represent only a short fragment. The latter holds true for two of the co-orthologs assigned to YVH1 as well (Solyc12g021180, Solyc12g021190; Fig. 5). Solyc12g021180 encodes for a protein with a partial dual specificity phosphatase catalytic domain (DSPc) and a shorter C-terminal domain compared to the full-length protein, Solyc00g185750. However, Solyc12g021180 and Solyc12g021190 together match the unigene SGN-U584680 indicating that the two Solyc IDs correspond to a single gene (putative Solyc12g021185), which encodes for a protein with a full-length DSPc domain and C-terminus matching the protein model of Solyc00g185750 (Fig. 5). Collectively, this evaluation reveals that at least all sequences of DRS2 and FAP7 can be considered as co-orthologs most likely with similar function, while four sequences are probably non-functional RBFs. In addition, YVH1 might be represented by two co-orthologs considering Solyc12g021185 as functional gene. Summarizing, we assigned 245 putative functional RBF (co-)orthologs in the genome of S. lycopersicum to the 255 RBFs from yeast.
Figure 5.
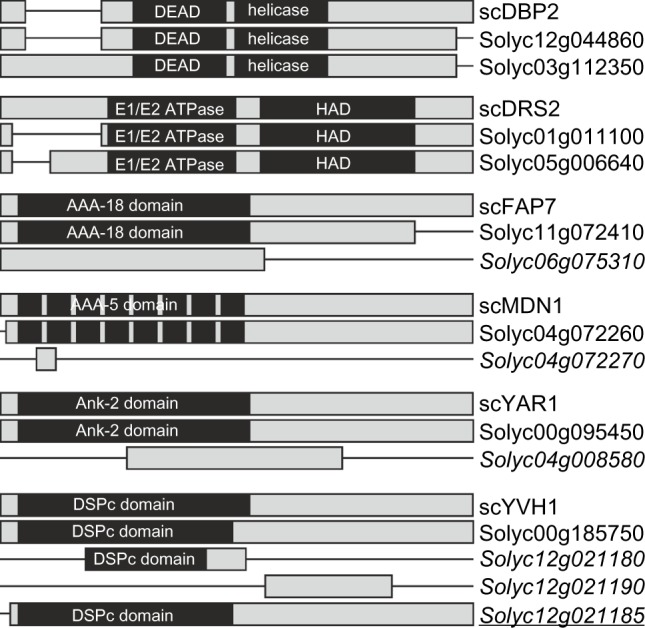
RBF co-orthologs with alterations in domain architecture. Alignments given in Supplemental Alignment 1 are shown as bar diagrams including the Pfam domains assigned. See Supplementary Table 3 for visualization of differences. Italics indicates the genes excluded from further analysis; italics and underlined indicates the gene found in unigene database, but not yet represented by a Solyc ID.
Expression of S. lycopersicum RBF and RP (co-) orthologs
The presence of multiple co-orthologs in plant genomes might indicate redundant or tissue- and/or developmental stage-specific functions for some co-orthologs. Thus, we examined the expression of all predicted RBFs and RPs in an existing expression dataset derived from RNA-seq analysis of S. lycopersicum cv. Moneymaker.33 From the 245 predicted RBF and 193 RP (co-)orthologs in tomato, 242 and 188 were expressed in at least one of the examined tissues, respectively (Supplementary Tables 7 and 8). However, transcripts for only 129 RBF and 166 RP (co-)orthologs were detected in open flowers without sepals (Fig. 6A). The low number of RBFs and RPs expressed in flowers containing reproductive tissues of Moneymaker33 was surprising, because rapidly developing tissues are expected to require massive ribosome biogenesis, particularly as mutants of RBFs have been found to be defective in pollen development.16,17,39 Thus, we performed NGS of RNA in leaves and anthers to re-evaluate the expression of RBFs in reproductive tissue. We focused on anthers, which are part of the male reproductive tissue and contain sporophytic tissues and gametophytic cells, including pollen corresponding to different developmental stages, ranging from meiotic to mature stage. In addition, we analyzed the transcript abundance in young expanding leaves for comparison.
Figure 6.

The overall expression profile of RBF and RP (co-)orthologs in tomato. (A) Tissues used for NGS RNA-seq analysis (left) and the number of RBFs for which expression is detected in a least one tissue (beside) or in the individual tissue (below). On the right, the tissues analyzed by MACE in this study are indicated. (B) Relation of the TPM expression value in leaves and anthers for all genes (black circles), for all RBF genes (red circles), and for all RP genes (yellow star). Gray line indicates identical expression in leaves and anthers, the long dashed gray line expression with two-fold change, and the short dashed gray line expression with four-fold change (45% genes with less than two-fold change; 64% with less than four-fold change of expression). Indicated are RBF genes not expressed (top), expressed only in leaves (left) or anther (right). Inset on the right shows the distribution of the expression difference between leaf and anther. The gray section indicates a pool of genes with significantly higher expression in anthers, and the red line shows the least square fit analysis to a Gaussian equation.
First, the NGS data for leaves and anther were evaluated (GEO: GSE56610). 21,509 genes have been identified by at least one read in both tissues. Here, we use the MACE approach, for which it was discussed that occurrence of one read is sufficient for the detection of a transcript (see Methods).15,30 Thus, in the two samples the expression of 25,924 of the 29,345 assigned genes was detected, which accounts for 88.3% of the total annotated genes. This is comparable to the detection of 83.6% of all annotated genes in a previous NGS study including different tissues and developmental stages of S. lycopersicum cv. Moneymaker.33 45% of all genes have similar transcript abundance (<2-fold change); while 36% show a >4-fold difference of expression in the two samples (Fig. 6B). 1,385 and 3,030 genes are expressed exclusively in leaves and anthers, respectively.
The genes with the highest expression in leaves (normalized to TPM) predominantly encode for proteins involved in photosynthesis (Table 2; Supplementary Table 11). Many of these genes show a high expression in anthers as well, which documents that anthers are a mixture of reproductive and sporophytic tissues. The genes with the highest expression in anthers are rather moderately expressed in leaves and encode for metabolic or regulatory proteins (Table 2; Supplementary Table 11). Inspecting the profile of differences in expression, one realizes that besides the 3,030 genes exclusively expressed in anthers, 1,455 genes have a 10-fold higher abundance in anthers than in leaves indicating strong preferential expression (Fig. 6B, inset; Supplementary Information 1).
Table 2.
Genes expressed highest in leaves and anthers.
| GENE | PROTEIN FUNCTION | RANK LEAVES | RANK ANTHER | |
|---|---|---|---|---|
| Highest TPM values in leaves | Solyc02g085950 | Ribulose bisphosphate carboxylase small chain 3B | 1 | 539 |
| Solyc07g066310 | psbR-homologue | 2 | 69 | |
| Solyc09g010800 | metallothionein LEMT1 | 3 | 9 | |
| Solyc07g047850 | chlorophyll a-b binding protein 4 | 4 | 33 | |
| Solyc10g006230 | chlorophyll a-b binding protein 7 | 5 | 116 | |
| Solyc03g034220 | Ribulose bisphosphate carboxylase small chain 2A | 6 | 21 | |
| Solyc07g064160 | chloroplastic thiamine thiazole synthase | 7 | 514 | |
| Solyc02g071030 | chlorophyll a-b binding protein 1B | 8 | 113 | |
| Solyc06g063370 | chlorophyll a-b binding protein 1A | 9 | 96 | |
| Solyc03g005760 | chlorophyll a-b binding protein 3C | 10 | 129 | |
| Highest TPM values in anther | Solyc01g009590 | carotenoid cleavage dioxygenase 1 | 1113 | 1 |
| Solyc01g086830 | seed storage/lipid transfer protein family protein | 1454 | 2 | |
| Solyc07g007250 | metallocarboxypeptidase inhibitor | 1511 | 3 | |
| Solyc09g008670 | threonine deaminase | 491 | 4 | |
| Solyc06g064480 | protein TAP1 | 2196 | 5 | |
| Solyc07g006380 | flower-specific gamma-purothionin protein | 2144 | 6 | |
| Solyc01g099540 | Anther specific NMT19-like | 4224 | 7 | |
| Solyc01g006400 | Hop-interacting cysteine-rich extensin-like protein-4 | 117 | 8 | |
| Solyc07g043420 | 2-oxoglutarate-dependent dioxygenase | 16 | 10 |
Notes: The TPM expression values are listed in Supplementary Table 4. The upper part contains the 10 most abundant genes in leaves and the lower part the 10 most abundant genes in anthers. The rank of the expressed genes is assigned concerning the descending TPM expression values obtained by MACE analysis.
We examined the expression of all genes coding for the RP (co-)orthologs and found that with the exception of three genes, all RPs are expressed in at least one of the two tissues (Fig. 6B; Supplementary Tables 7 and 8). RP coding genes are in general highly expressed in both tissues. RBF genes which showed tissue-specific expression had a low number of reads (median of 0.09TPM; Supplementary Table 7), indicating a low transcript abundance.
The three RBF coding genes for which no transcript was detected both in leaves and anthers are Solyc00g185750 coding for a co-ortholog of YVH1, Solyc00g095450 coding for YAR1, and Solyc05g018780 coding for RRB1 (Fig. 6B). Transcripts for these genes were not detected in the samples analyzed before by RNAseq33 or in a global analysis of the heat-induced transcriptome.36 This raises the question whether these genes are only induced under very specific stress conditions or are particularly expressed in a not yet analyzed tissue or cell type. Worth mentioning, the expressed co-orthologs of YAR1 code only for fragments of the protein (Fig. 5), and thus, expression of this RBF could not be demonstrated. In contrast, the transcripts of the newly assigned co-ortholog of YVH1 and the two additional co-orthologs of RRB1 are detected in anther and leaf (Supplementary Table 7). In summary, we detected transcripts for 239 (98%) assigned RBFs and for all RPs in anthers. The transcript abundance of most RP genes is slightly higher in leaves, while the transcript abundance of RBFs is globally comparable in both tissues (Fig. 6B).
The expression profile of predicted RPs and RBFs in tomato leaves and anthers
Nine RBF coding genes and 120 RP coding genes are either 2-fold higher or exclusively expressed in leaves. The RBF coding genes encode for factors possessing multiple co-orthologs with the exception of AIR2 (Table 3). Forty RBF genes and only one RP gene are more abundant (anther to leaf ratio >2) or exclusively expressed in anthers (Table 3). Nineteen genes represent the full set of co-orthologs coding for a certain RBF. Among the genes preferentially expressed in one of the tissues we found four factors represented by multiple co-orthologs, where one is highly expressed in leaves and another in anthers (NAP1, PTC3, TIF6, UBC9). Indeed, the NAP1 co-ortholog, which is highly expressed in anthers, was not detected in roots and leaves in a previous study (Supplementary Table 7).33 For the UBC9 co-ortholog, a high expression in roots, stem, and ripe fruits was reported, while the expression in leaves was found to be the lowest,33 which is consistent with our results (Supplementary Table 7).
Table 3.
RBF genes differentially expressed in leaves and anthers.
| GENE | CO | ACC. NUMBER | Log2 (TPMANTHER/TPMLEAF) | |
|---|---|---|---|---|
| Expressed higher in leaves | AIR2 | 1 | Solyc05g007090 | −1,93 |
| HRR25 | 11 | Solyc12g007280 | −Inf | |
| Solyc08g066870 | −2,57 | |||
| NAP1 | 5 | Solyc08g063000 | −1,34 | |
| NMD3 | 2 | Solyc11g011780 | −1,50 | |
| PTC3 | 3 | Solyc08g074230 | −Inf | |
| REI1 | 2 | Solyc08g006470 | −3,19 | |
| SMT3 | 4 | Solyc09g059970 | −1,84 | |
| TIF6 | 3 | Solyc12g010210 | −Inf | |
| UBC9 | 5 | Solyc02g093110 | −1,18 | |
| Expressed higher in anthers | ASC1 | 3 | Solyc12g040510 | 1,03 |
| Solyc06g069010 | 1,28 | |||
| Solyc03g119040 | 1,38 | |||
| BUD23 | 1 | Solyc06g072250 | 1,06 | |
| CBF5 | 1 | Solyc02g081810 | 1,80 | |
| CIA1 | 2 | Solyc09g009410 | 1,31 | |
| DIM1 | 1 | Solyc01g100430 | 1,32 | |
| EBP2 | 2 | Solyc01g006090 | 2,08 | |
| ERB1 | 1 | Solyc01g103740 | 1,43 | |
| ERP2 | 5 | Solyc12g015740 | 1,17 | |
| Solyc09g082770 | 1,26 | |||
| Solyc12g006090 | +Inf | |||
| FAL1 | 6 | Solyc07g040750 | 3,65 | |
| Solyc06g062800 | +Inf | |||
| FPR3 | 1 | Solyc01g091340 | 1,73 | |
| FPR4 | 1 | Solyc03g007170 | 1,59 | |
| GAR1 | 1 | Solyc10g078540 | 1,01 | |
| GRC3 | 1 | Solyc03g113610 | 1,76 | |
| JJJ1 | 2 | Solyc03g115140 | 1,13 | |
| Solyc03g115120 | 4,01 | |||
| KRI1 | 1 | Solyc07g064350 | 1,02 | |
| LSG1 | 1 | Solyc11g071910 | 5,91 | |
| NAP1 | 5 | Solyc06g062690 | 5,70 | |
| NOB1 | 1 | Solyc08g007990 | 1,18 | |
| NOP4 | 1 | Solyc01g109990 | 1,11 | |
| NOP58 | 2 | Solyc09g083080 | 1,12 | |
| POP1 | 1 | Solyc11g007800 | 1,14 | |
| PTC3 | 3 | Solyc12g042570 | 1,41 | |
| PWP2 | 1 | Solyc03g120080 | 3,22 | |
| RLI1 | 2 | Solyc08g075360 | +Inf | |
| RNA1 | 2 | Solyc01g079680 | 1,10 | |
| Solyc09g065190 | 1,25 | |||
| RRB1 | 4 | Solyc05g025630 | 1,82 | |
| RRP45 | 1 | Solyc05g047420 | 1,89 | |
| SNU13 | 2 | Solyc09g092080 | 1,03 | |
| TIF6 | 3 | Solyc12g015790 | 4,96 | |
| UBC9 | 5 | Solyc12g088680 | 1,82 | |
| UTP22 | 1 | Solyc02g085230 | 1,68 | |
| YTM1 | 2 | Solyc01g111030 | +Inf | |
| YVH1 | 3 | Solyc12g021180 | 1,48 |
To further support the observed differential expression of RBF coding genes based on MACE results, we randomly selected genes and analyzed their expression by qRT-PCR on mRNA isolated from anthers and leaves (Table 4). We included two factors represented by multiple co-orthologs into this analysis, namely RIL1 and ASC1. We obtained a qRT-PCR signal for each of the five genes coding for one of the two factors. This confirms the existence of a transcript for the multiple co-orthologs. For quantification, the expression values from qRT-PCR were normalized to either Solyc07g064130 coding for ubiquitin or Solyc06g005060 coding for EF1α. We observed by large the same ratios of expression levels in the two tissues as obtained by MACE analysis. Thus, we confirmed the leaf-specific expression of AIR2, which is only encoded by a single gene. Furthermore, we observed the anther-specific expression of one RIL1 coding gene. Remarkably, by qRT-PCR we also observed a higher expression for an additional gene coding for ASC1 in anthers (Table 4).
Table 4.
RBF expression comparison of MACE and qRT-PCR analysis in anther and leaf.
| NAME | GENE | qRT-PCR (NORMALIZED) | TPM-EXPRESSION VALUE | |||||
|---|---|---|---|---|---|---|---|---|
| NORM | LEAF | ANTHER | A/L | LEAF | ANTHER | A/L | ||
| RIL1 | Solyc08g075360.1 | EF1α | (6.4 ± 0.9)*10−3 | 0.15 ± 0.05 | 32 ± 7 | 0.0 | 0.44 | ∞ |
| Ubi | (3.06 ± 0.03)*10−4 | (1.2 ± 0.2)*10−2 | ||||||
| Solyc07g008340.2 | EF1α | (6.2 ± 0.9)*10−2 | (9 ± 2)*10−2 | 2 ± 1 | 47.9 | 69.0 | 1.44 | |
| Ubi | (2.8 ± 0.1)*10−3 | (8.2 ± 0.6)*10−3 | ||||||
| SSF1 | Solyc01g096800.2 | EF1α | 0.30 ± 0.04 | 0.19 ± 0.08 | 0.8 ± 0.1 | 13.2 | 14.9 | 1.13 |
| Ubi | (1.4 ± 0.4)*10−2 | (1.27 ± 0.01)*10−2 | ||||||
| LSG1 | Solyc11g071910.1 | EF1α | 0.2 ± 0.1 | 0.8 ± 0.2 | 8 ± 2 | 3.9 | 233.6 | 59.9 |
| Ubi | (1.3 ± 0.2)*10−2 | 0.14 ± 0.05 | ||||||
| PWP2 | Solyc03g120080.2 | EF1α | 0.3 ± 0.1 | 1.0 ± 0.2 | 10 ± 4 | 0.09 | 0.8 | 9.0 |
| Ubi | (1.1 ± 0.2)*10−2 | 0.19 ± 0.06 | ||||||
| ASC1 | Solyc12g040510.1 | EF1α | 0.3 ± 0.1 | 0.26 ± 0.06 | 0.9 ± 0.1 | 2.6 | 5.3 | 2.0 |
| Ubi | (2 ± 1)*10−2 | (2 ± 1)*10−2 | ||||||
| Solyc03g119040.2 | EF1α | 28.8 ± 0.9 | 19 ± 4 | 0.8 ± 0.1 | 73.6 | 192.2 | 2.6 | |
| Ubi | 1.02 ± 0.03 | 0.8 ± 0.1 | ||||||
| Solyc06g069010.2 | EF1α | (3.0 ± 0.6)*10−2 | 0.14 ± 0.02 | 4.5 ± 0.5 | 140.5 | 342.2 | 2.4 | |
| Ubi | (5 ± 2)*10−3 | (2.1 ± 0.9)*10−2 | ||||||
| RRP45 | Solyc05g047420.2 | EF1α | (4.5 ± 0.3)*10−2 | 0.17 ± 0.01 | 3.3 ± 0.7 | 0.13 | 0.48 | 3.7 |
| Ubi | (3.2 ± 0.2)*10−3 | (8.9 ± 0.1)*10−3 | ||||||
| KRE33 | Solyc04g051670.2 | EF1α | 0.84 ± 0.04 | 0.15 ± 0.03 | 0.4 ± 0.2 | 11.0 | 15.8 | 1.4 |
| Ubi | 0.05 ± 0.01 | 0.03 ± 0.01 | ||||||
| RRP5 | Solyc03g051900.2 | EF1α | 0.2 ± 0.1 | 0.15 ± 0.03 | 0.9 ± 0.3 | 2.7 | 4.2 | 1.6 |
| Ubi | 0.02 ± 0.01 | 0.03 ± 0.02 | ||||||
| NOP12 | Solyc08g076660.2 | EF1α | 0.8 ± 0.2 | 0.4 ± 0.2 | 0.7 ± 0.2 | 16.9 | 24.4 | 1.4 |
| Ubi | (2.9 ± 0.7)*10−2 | (2.3 ± 0.6)*10−2 | ||||||
| TSR1 | Solyc01g096760.2 | EF1α | 0.46 ± 0.06 | 0.45 ± 0.06 | 1.4 ± 0.5 | 14.1 | 23.4 | 1.7 |
| Ubi | 0.11 ± 0.03 | 0.2 ± 0.1 | ||||||
| UBA2 | Solyc01g109960.2 | EF1α | 0.2 ± 0.1 | 0.16 ± 0.01 | 1.2 ± 0.5 | 17.5 | 27.1 | 1.5 |
| Ubi | (1.1 ± 0.2)*10−2 | (2 ± 1)*10−2 | ||||||
| UTP20 | Solyc09g092240.2 | EF1α | 0.2 ± 0.1 | 0.2 ± 0.1 | 1.1 ± 0.3 | 5.8 | 7.8 | 1.3 |
| Ubi | (1.15 ± 0.06)*10−2 | (1.2 ± 0.1)*10−2 | ||||||
| AIR2 | Solyc05g007090.2 | EF1α | 0.28 ± 0.09 | 0.011 ± 0.009 | 0.05 ± 0.02 | 9.8 | 2.6 | 0.26 |
| Ubi | (1.37 ± 0.09)*10−2 | (8 ± 2)*10−4 | ||||||
| ENP1 | Solyc07g056550.2 | EF1α | 0.3 ± 0.1 | 0.3 ± 0.2 | 1.6 ± 0.7 | 7.1 | 7.0 | 1.0 |
| Ubi | (1.4 ± 0.7)*10−2 | (3 ± 1)*10−2 | ||||||
| DED1 | Solyc03g052980.2 | EF1α | 0.13 ± 0.02 | 0.10 ± 0.02 | 0.9 ± 0.2 | 76.8 | 66.8 | 0.9 |
| Ubi | (7 ± 3)*10−3 | (7 ± 2)*10−2 | ||||||
| REI1 | Solyc12g096450.1 | EF1α | 0.31 ± 0.01 | (1.2 ± 0.7)*10−2 | 0.08 ± 0.04 | 14.1 | 8.4 | 0.6 |
| Ubi | (3 ± 1)*10−2 | (4 ± 2)*10−3 | ||||||
We used the information on transcript abundance established previously33 to analyze the expression profiles of RBF and RP coding genes. The expression profiles of the genes encoding for RPs and RBFs have been assigned to six clusters (see Methods). While each cluster contains RPs or RBFs with distinct profiles, one cluster for each set (21 rp and 68 rbf) represents a collection of genes that did not match (extremely high error bars) the profiles of any of the other clusters (Fig. 7A; 21 RP genes and 68 RBF genes). Subsequently, the correlations between the profiles of RP and RBF genes have been determined excluding the above-mentioned cluster (Fig. 7B). We realized that two profiles are highly correlative (Fig. 7B, black), with a correlation value of 0.97. The genes of these two clusters show a moderate expression in roots and stem tissues; a lower expression in leaves, anthers, and flowers; and a higher but by large similar expression in all analyzed fruit samples. These genes encode for 15 RPLs, 12 RPSs, 8 90S RBFs, 7 60S RBFs, 1 40S RBF, 1 Exonuclease RBF, 1 Exosome RBF, 1 TRAMP complex RBF, and one not assigned RBF. This might point toward a specific functional relation between these RPs and RFBs, which is discussed below.
Figure 7.
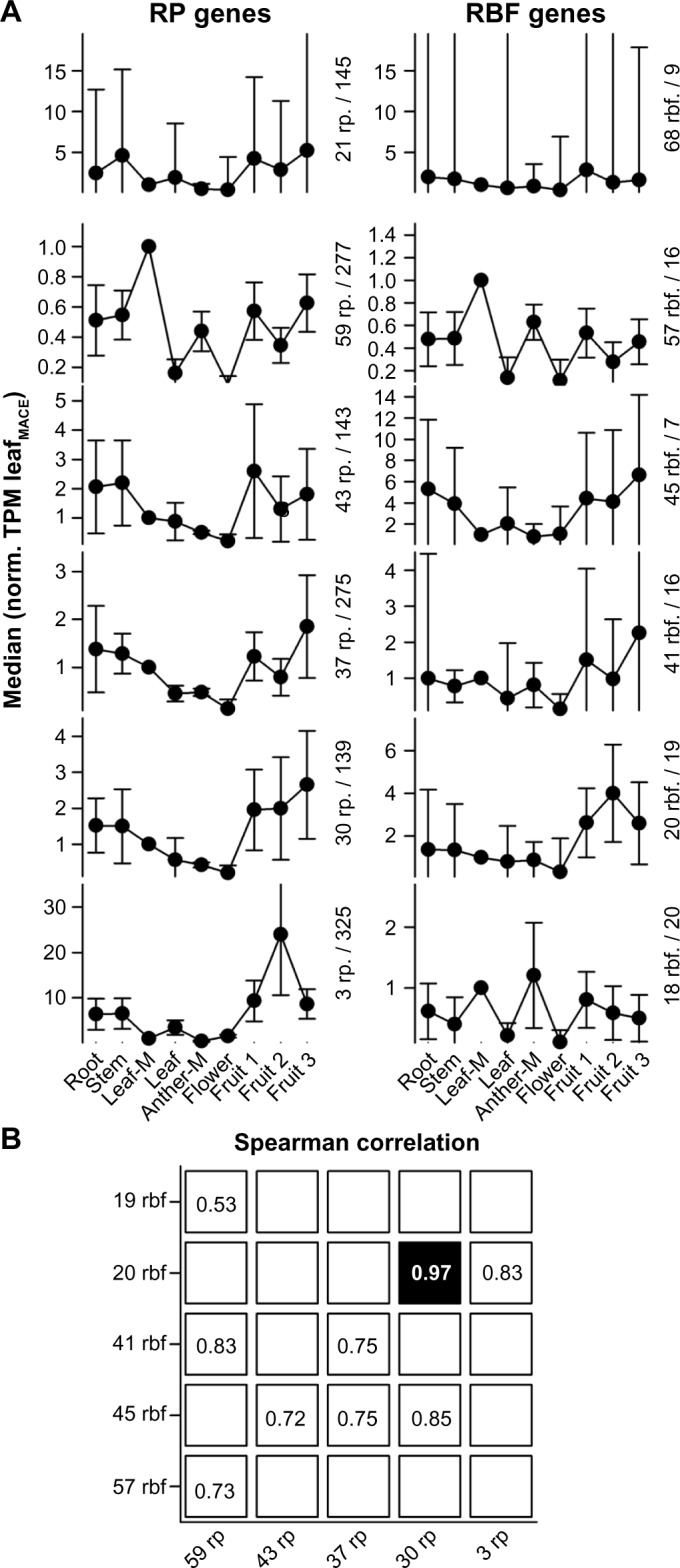
Clustering of RP and RBF genes based on their expression. (A) Mean of the expression values of clustered genes normalized to the TPM determined for MACE leaf sample. Clusters were generated by k-means analysis. Error bars indicate the standard deviation. The clusters are ordered according to the number of factors present (first value) and the median of the cluster for the leaves (second value) is indicated. The two clusters on the top represent the set of genes that could not be related to a specific profile. (B) The Spearman correlation between the cluster profiles of RPs and RBFs was calculated, and the highest value for each column and each row is shown. Only values with strong correlation (above 0.5) are highlighted.
Expression of predicted RBFs in pollen
The role of ribosomes in development in eukaryotes has been shown in genetic studies with mutations in RPs, ribosome assembly, and biogenesis factors.19 Furthermore, the importance of ribosome biogenesis in physiological gametophyte development has been documented.16,17,39 Our global analysis has focused on anthers of different developmental stages containing sporophytic tissues and gametophytic cells. Thus, the sample includes pollen ranging from meiotic to mature stage. The progression from proliferating microspores to terminally differentiated pollen grains is characterized by stage-specific gene transcriptional activation and repression as shown for A. thaliana pollen.40 Therefore, we examined the levels of the genes selected for qRT-PCR analysis in developing tomato pollen (Table 5). Indeed, we identified differential expression for 12 of the analyzed genes, which accounts for 64% of the studied genes.
Table 5.
Comparison of NGS and qRT-PCR analysis in pollen.
| NAME | GENE | qRT-PCR (NORMALIZED) | NORM | ||||
|---|---|---|---|---|---|---|---|
| NORM | TETRADS | POST-MITOTIC | MATURE | PM/T | M/T | ||
| RIL1 | Solyc08g075360.1 | EF1α | (6.3 ± 0.8)*10−2 | (8 ± 1)*10−2 | (1.2 ± 0.3)*10−2 | 1.26 ± 0.02 | 0.19 ± 0.01 |
| Ubi | 0.15 ± 0.05 | 0.19 ± 0.01 | (2.7 ± 0.2)*10−2 | ||||
| Solyc07g008340.2 | EF1α | (8.3 ± 0.4)*10−3 | (1.9 ± 0.3)*10−2 | (2.2 ± 0.2)*10−2 | 2.4 ± 0.3 | 2.2 ± 0.5 | |
| Ubi | (1.9 ± 0.3)*10−2 | (4 ± 1)*10−2 | (3.4 ± 0.6)*10−2 | ||||
| SSF1 | Solyc01g096800.2 | EF1α | (9 ± 1)*10−2 | 0.14 ± 0.05 | 0.16 ± 0.04 | 1.6 ± 0.3 | 1.7 ± 0.1 |
| Ubi | 0.16 ± 0.03 | 0.28 ± 0.09 | 0.27 ± 0.03 | ||||
| LSG1 | Solyc11g071910.1 | EF1α | 0.5 ± 0.2 | 0.4 ± 0.1 | 0.41 ± 0.06 | 0.8 ± 0.1 | 0.6 ± 0.1 |
| Ubi | 1.2 ± 0.4 | 0.9 ± 0.2 | 0.71 ± 0.03 | ||||
| PWP2 | Solyc03g120080.2 | EF1α | 0.9 ± 0.3 | 1.3 ± 0.6 | 0.7 ± 0.3 | 1.3 ± 0.1 | 0.7 ± 0.1 |
| Ubi | 3 ± 1 | 4.0 ± 1 | 2.2 ± 0.5 | ||||
| ASC1 | Solyc12g040510.1 | EF1α | 0.14 ± 0.03 | 0.57 ± 0.04 | 0.70 ± 0.01 | 0.33 ± 0.07 | 0.44 ± 0.05 |
| Ubi | 0.7 ± 0.2 | 0.2 ± 0.1 | 0.28 ± 0.05 | ||||
| Solyc03g119040.2 | EF1α | 2.08 ± 0.07 | 3.9 ± 0.3 | 1.57 ± 0.04 | 2.0 ± 0.2 | 0.7 ± 0.1 | |
| Ubi | 3.8 ± 0.5 | 8 ± 1 | 2.3 ± 0.2 | ||||
| Solyc06g069010.2 | EF1α | (2.8 ± 0.1)*10−2 | (7 ± 3)*10−2 | (6 ± 1)*10−2 | 2.8 ± 0.2 | 2.1 ± 0.1 | |
| Ubi | 0.117 ± 0.002 | 0.3 ± 0.1 | 0.24 ± 0.03 | ||||
| RRP45 | Solyc05g047420.2 | EF1α | (1.2 ± 0.4)*10−2 | (1.6 ± 0.5)*10−2 | (5.3 ± 0.7)*10−2 | 1.3 ± 0.2 | 4.1 ± 0.7 |
| Ubi | (5 ± 2)*10−2 | (6.2 ± 0.4)*10−2 | (19 ± 2)*10−2 | ||||
| KRE33 | Solyc04g051670.2 | EF1α | (2 ± 1)*10−2 | (1.7 ± 0.7)*10−2 | (1.8 ± 0.3)*10−2 | 0.7 ± 0.1 | 0.7 ± 0.1 |
| Ubi | 0.11 ± 0.05 | (9 ± 2)*10−2 | (7.5 ± 0.2)*10−2 | ||||
| RRP5 | Solyc03g051900.2 | EF1α | (3 ± 1)*10−2 | (6 ± 3)*10−2 | 0.17 ± 0.03 | 1.6 ± 0.5 | 5.4 ± 0.9 |
| Ubi | 0.13 ± 0.06 | 0.18 ± 0.07 | 0.64 ± 0.02 | ||||
| NOP12 | Solyc08g076660.2 | EF1α | (9 ± 2)*10−2 | (5 ± 0.2)*10−2 | (1.2 ± 0.6)*10−2 | 0.47 ± 0.08 | 0.15 ± 0.02 |
| Ubi | 0.4 ± 0.1 | 0.16 ± 0.08 | 0.06 ± 0.02 | ||||
| TSR1 | Solyc01g096760.2 | EF1α | (5.2 ± 0.7)*10−2 | 0.12 ± 0.04 | (6 ± 2)*10−2 | 2.3 ± 0.1 | 1.0 ± 0.2 |
| Ubi | 0.11 ± 0.03 | 0.27 ± 0.09 | 0.10 ± 0.03 | ||||
| UBA2 | Solyc01g109960.2 | EF1α | (5 ± 2)*10−2 | (2.2 ± 0.6)*10−2 | (2.7 ± 0.2)*10−2 | 0.39 ± 0.02 | 0.48 ± 0.01 |
| Ubi | 0.29 ± 0.09 | 0.109 ± 0.005 | 0.141 ± 0.005 | ||||
| UTP20 | Solyc09g092240.2 | EF1α | (2.7 ± 0.3)*10−2 | (1.8 ± 0.2)*10−2 | (2 ± 1)*10−3 | 0.6 ± 0.1 | 0.10 ± 0.03 |
| Ubi | 0.16 ± 0.01 | (8 ± 3)*10−2 | (2 ± 1)*10−2 | ||||
| AIR2 | Solyc05g007090.2 | EF1α | (1.37 ± 0.07)*10−2 | (4 ± 1)*10−3 | (1.1 ± 0.2)*10−2 | 0.37 ± 0.07 | 0.87 ± 0.09 |
| Ubi | (2.1 ± 0.2)*10−2 | (9 ± 4)*10−3 | (2.0 ± 0.4)*10−2 | ||||
| ENP1 | Solyc07g056550.2 | EF1α | (9 ± 3)*10−2 | 0.12 ± 0.05 | (6.8 ± 0.9)*10−2 | 1.3 ± 0.1 | 0.5 ± 0.3 |
| Ubi | 0.5 ± 0.2 | 0.6 ± 0.2 | 0.12 ± 0.02 | ||||
| DED1 | Solyc03g052980.2 | EF1α | (8.6 ± 0.1)*10−2 | (5 ± 2)*10−2 | (2.8 ± 0.1)*10−2 | 0.56 ± 0.02 | 0.52 ± 0.06 |
| Ubi | 0.17 ± 0.01 | 0.10 ± 0.04 | (8.4 ± 0.1)*10−2 | ||||
| REI1 | Solyc12g096450.1 | EF1α | (2.2 ± 0.2)*10−3 | (1.0 ± 0.3)*10−2 | (1.2 ± 0.5)*10−2 | 4.3 ± 0.5 | 4.9 ± 0.6 |
| Ubi | (1.3 ± 0.1)*10−2 | (5.2 ± 0.6)*10−2 | (6.0 ± 0.7)*10−2 | ||||
To evaluate the impact of expression in pollen on the transcript abundance observed for anthers, we calculated the ratio between the determined qRT-PCR values after normalization to the EF1 expression (Fig. 8). For two genes coding for ASC1 (Solyc12g040510) and RRP5 (Solyc03g051900), we observed a significantly higher transcript level in one of the pollen stages than in anthers. This suggests that the level observed in anthers represents the expression in pollen. Remarkably, both of the genes are not significantly higher expressed in anthers compared to the transcript level in leaves (Table 4). For six of the analyzed genes, we observed a comparable expression in pollen tissues and anthers. However, the gene that shows the highest expression in anthers compared to leaves does not show an enhanced expression in pollen compared to anthers (RIL1; Solyc08g075360; Fig. 8, Tables 4 and 5). This documents that the enhanced expression in sporophytic tissues and gametophytic cells compared to leaves cannot directly be related to expression in pollen.
Figure 8.
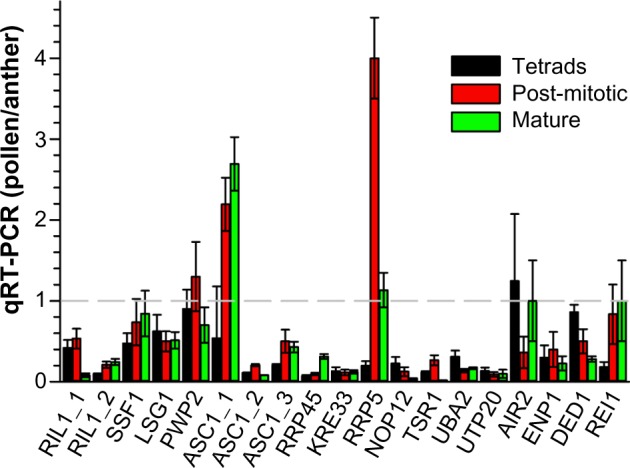
Comparison of expression in pollen and anthers. The ratio of the values listed in Tables 4 and 5 normalized to EF1α expression was calculated.
Discussion
RBF co-orthologs show a differential expression in tomato
We present the set of co-orthologs to RBFs and RPs in 14 plant genomes (Supplementary Tables 1–4). However, this analysis has two limits. On the one hand, additional information besides the assignment as co-ortholog is required to assign a gene to a specific functional pathway38; on the other hand, RBFs without orthologs in yeast cannot be detected by this approach. Nevertheless, the number of co-orthologs to yeast RBFs assigned in plant genomes demonstrates the conservation of the process and the corresponding proteins can be used as markers to investigate plant-specific pre-ribosomal complex compositions eg by proteomic studies.
With focus on tomato we predicted 245 co-orthologs to yeast RBFs. They represent orthologs to 173 of 255 RBFs described in yeast (Figs. 2, 4, and 5). With the exception of three genes (Solyc00g095450, Solyc00g185750, Solyc05g018780) all are found to be expressed in leaves and/or anthers (Fig. 6). Remarkably, based on existing results (GEO: GSE33507) we could not provide evidence for expression of YAR1, while the proposed YVH1 (Solyc12g021185) is expressed. Thus, based on ortholog search and expression analysis we found 243 co-orthologs in tomato to 172 RBFs in yeast.
Previously, several factors have been experimentally confirmed to participate in ribosome biogenesis in plants. Most of them belong to the above-described orthologs to yeast factors. In many cases, the nomenclature used matches the nomenclature proposed for yeast (Pwp2, Rrp5, Noc4, Enp1, and Nob117; Dim1A41; Rrp6L1 and Rrp6L242; Mtr443; Nuc112; eIf644; RID245; OsNug246; AtLsg114; Supplementary Table 12). In addition, AtFib1 and AtFib247 were assigned as co-orthologs of Nop1; AtSwa118 as ortholog of Utp15; AtRtl248 as ortholog of Rnt1; AtSwa239 as ortholog of Mak21; AtRH36/AtSwa316 as ortholog of Dbp8; AtXrn249 as one ortholog of Rat1/Xrn1; AtGigantus150 as ortholog of YNL035C; AtRH5751 as ortholog of Rok1; and AtNuc212 as co-ortholog of Nuc1 (Supplementary Table 12).
To the best of our knowledge, only two proteins that might be involved in ribosome biogenesis in plants have been described, for which no ortholog was detected in yeast, namely AtNufIP9 and Domino1.52 In tomato, these two factors are encoded by Solyc01g096920 and Solyc06g069770, respectively (Supplementary Table 12). Co-orthologs for Domino1 are present in all analyzed plant species. In contrast, the C-terminus of AtNufIP shares a high sequence homology with the C- terminal portion of Rsa1, which can be identified in proteins found in the genome of monocots and dicots,9 while orthologs to AtNufIP can only be identified in dicots most likely because of the very variable N-terminus of the protein (Supplementary Table 12). Combining this information with our ortholog search and expression analysis, the 245 tomato proteins can be assigned to ribosome biogenesis with high likelihood. These proteins represent 172 RBFs described in yeast.
In general, about 25% of all RBFs predicted in A. thaliana and tomato are encoded by at least two different genes, which applies for all complexes involved in ribosome biogenesis (Figs. 2–4). The genes coding for RBFs are more strongly expressed in anther compared to leaf (Fig. 6; Table 3), which is not surprising, considering that anther is a male reproductive organ undergoing rapid metabolic changes. However, we also observed genes that show a stronger expression in leaves. These are mostly RBFs represented by multiple co-orthologs (Table 3), which is a hint for a tissue-specific set of assembly factors for ribosome biogenesis. In-depth analysis of the expression pattern of tomato RBFs with multiple co-orthologs in different tissues revealed differential expression for all (Fig. 9). Thus, we can conclude that the co-orthologs to individual RBFs found in plants account for a preferential expression in distinct tissues or at different developmental stages. This is consistent with the observed variation of the number of RBFs to which co-orthologs could be assigned (Fig. 2B). We conclude that plant ribosome biogenesis requires the action of tissue- or developmental stage-specific factors, which is in agreement with reports showing tissue-specific developmental defects in some RBF mutants.16,17,19,39 On the other hand, considering the critical function of individual RBFs for a vital cellular process like ribosome biogenesis, we can assume that other proteins with similar functions might compensate for the factors not predicted in any plant, a hypothesis that needs to be experimentally challenged in the future. On the other hand, the absence of some factors in plants might be explained by differences in the processing pathway compared to yeast, since for example, not all cleavage sites on plant rRNA have been mapped so far.
Figure 9.
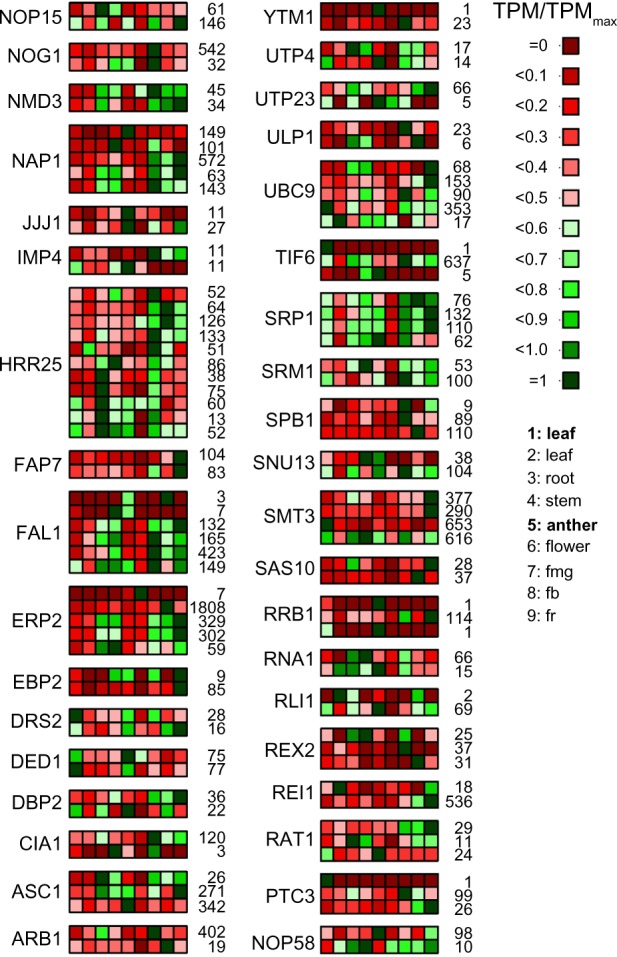
The expression profile of RBF encoded by multiple genes. The expression values (Supplementary Table 4) for genes coding for RBFs with multiple co-orthologs have been normalized to the individual maximal value given next to the panel and the expression profile is shown (scale on the right). The order of samples is indicated on the right, legend in bold indicates the MACE results; the rest of the samples are derived from the NGS data.33
Abbreviations: fmg: fruit mature green; fb: fruit breaker; fr: fruit ripe.
Correlation of RP and RBF gene expression
In yeast, it has been shown that many RPS but only a few RPL proteins are present in the 90S pre-ribosomal particle.53 In line, it is discussed that in eukaryotes the majority of the RPS of the body of the 40S subunit assemble co-transcriptionally.54 Interestingly, the cluster with 30 RPs represents co-orthologs of 12 RPS genes, 9 of which are classified as very early assembling (RPS3A, RPS4, RPS7, RPS8, RPS11, RPS13, RPS23, RPS24, RPS27).55 The other three RPS are considered as very late assembling (RPS17, RPS20, RPS26). The cluster with 20 RBFs contains genes encoding RBFs that are associated with the 90S particle (ENP2, FAL1, FYV7, HRR25, LCP5, NOC4, SAS10, TLR1/RRP36). Interestingly, this cluster of RBF genes has a similar expression profile to that of a cluster of 30 RPs (Fig. 7). Thus, these RBFs of the 90S particle and the RPSs might be functionally related. In line with such notion, yeast ENP2, FAL1, LCP5, and TLR1/RRP36 are required for processes leading to the 40S subunit.56–59
In turn, RLI1, present in the cluster with 20 RBFs, associates with 40S subunits and is involved in the ribosome quality control and recycling.60,61 This might link its function to the late assembly of RPS17, RPS20, and RPS26, representing the late assembling RPSs of the cluster with 30 RPs. Worth mentioning is that only one RIL co-ortholog shows the profile represented by the cluster with 20 RBFs (20 rbf).
Similarly, the RPLs of the cluster with 30 RPs suggest a regulation of the early processes of 60S maturation. In yeast, at least 10 of the 15 RPLs of this cluster have been identified in the pre-60S E1, the first pre-60S complex formed during 60S biogenesis (RPL5, RPL7A, RPL13A, RPL17, RPL18A, RPL26, RPL27, RPL30, RPL35, RPL36A).62 However, only for three of the six assigned 60S associated RBFs of the cluster with 20 RBFs (NIP7, NOP16, SPB1), a direct association with the early pre-60S E1 particle has been confirmed in yeast.63,64 In contrast, ARB1 was described as an export factor and thus is a late factor.65 Thus, the function of ARB1 might be related to the assembly of RPLs like RPL12, RPL23, and RPL37 present in the cluster with 30 RPs.
Summing up, it appears that co-orthologs of RBFs and RPs involved in early events of RP assembly are expressed in a similar manner. Furthermore, we observed that the highest populated clusters for each RP (Supplementary Table 13) and RBF (Supplementary Table 9) combine genes with maximum expression in young leaves (RP: 59 rp; RBF: 57 rbf). Interestingly, we also obtained one cluster of RBF factors, but not of RPs, which shows highest expression in anthers (19 rbf). This might suggest that a difference in tissue-specific ribosomes66,67 is not always dependent on the composition of RPs.
Supplementary Materials
Supplemental File 1. CRACPipe description.
Supplemental Table 1. RBF co-orthologs in 14 plant species.
Given is the complex the RBF associates to (column 1) the name of the factor in yeast (column 2) and the number of co-orthologs identified in 14 different plant species including Monocots, Eudicots, moss and green algae (columns 3–16): A. thaliana, S. lycopersicum, S. tuberosum, G. max, B. distachyon, L. japonicus, M. truncatula, O. sativa, P. patens, C. reinhardtii, P. trichocarpa, S. bicolor, V. vinifera and Z. mays.
Supplemental Table 2. RBF co-orthologs in tomato an A. thaliana.
Given is the complex the RBF associates to (column 1) the name of the factor in yeast (column 2), the number of co-orthologs identified in tomato (column 3) and A. thaliana (column 4), the accession numbers of the identified sequences in tomato (column 5) and A. thaliana (column 9), the KOG assignment (columns 6, 10), the description (columns 7, 11) and the Pfam domains and the order thereof (columns 8, 12).
Supplemental Table 3. RBF co-orthologs in tomato with difference in their annotations.
Shown are all RBFs for which co-orthologs have been identified in the tomato genome but contain a different functionalor domain assignnment. Given are the name of the factor, the accession number, the KOG entry, the description of the gene and the PFAM domain prediction. Yellow highlighted are the set of co-orthologues with at least one sequence which is not fully comparable to the others. (NF…no Pfam domain identified).
Supplemental Table 4. Ribosomal protein co-orthologs in 14 plant species.
Given is the complex the Ribosomal protein associates to (column 1) the name of the protein (column 2) and the number of co-orthologs identified in 14 different plant species including Monocots, Eudicots, moss and green algae (columns 3–16): A. thaliana, S. lycopersicum, S. tuberosum, G. max, B. distachyon, L. japonicus, M. truncatula, O. sativa, P. patens, C. reinhardtii, P. trichocarpa, S. bicolor, V. vinifera and Z. mays. The AGI (column 17) and Soly ID (column 18) of the co-orthologs is assigned to the ribosomal protein.
Supplemental Table 5. The Affymetrix chip datasets used for the meta analysis.
Given are the organism (column 1), if the dataset handles stress or tissues (column 2), the database (column 3), the series identifier (column 4), the number of samples (column 5) and a short description of the experiment (column 6).
Supplemental Table 6. The tomato genes with a variance in expression below 0.4.
Given is the gene ID (column 1), the gene description (column 2), the variance calculated for all tissue specific experiments (column 3), for all stress specific experiments (column 4), for all selected experiments (column 5). The columns 6 to 11 contain the information about maximum (column 6, 9), mean (column 7, 10) and minimum RMA values (column 8, 11).
Supplemental Table 7. Expression values for RBF coding genes.
Compared are the high throughput sequencing data of our MACE study (column 3–4; TPM) for vegetative (leaf; LeCo) and reproductive (anther; AnCo) tissue to the RNA-seq RPKM values of the study performed by the Tomato Genome Consortium (column 7–13) 20. the log2 fold changes and probability for differentially expressed genes comparing leaf and anther MACE libraries via NoiSeq18. In red are genes excluded by domain analysis.
Supplemental Table 8. Expression values for Ribosomal protein coding genes.
Compared are the high throughput sequencing data of our MACE study (column 3–4; TPM) for vegetative (leaf; LeCo) and reproductive (anther; AnCo) tissue to the RNA-seq RPKM values of the study performed by the Tomato Genome Consortium (column 7–13)20. In column 5 and 6 are the log2 fold changes and probability for differentially expressed genes comparing leaf and anther MACE libraries via NoiSeq18.
Supplemental Table 9. Clustering of ribosomal proteins concerning NGS expression data.
Clustered are the ribosomal proteins by k-means ending in 6 different clusters (columns 1–6). The ribosomal proteins were clustered concerning the different expression values of NGS data for specific tissues and developmental stages.
Supplemental Table 10. Primer for qRT-PCR of RBF co-orthologs in tomato.
Given is the Solyc ID for tomato (column 1) the name of the primer including the name of the factor in yeast (column 2 and the primer sequence (columns 3).
Supplemental Table 11. The TPM expression value of the 10 genes highest expressed in the according tissue.
Supplemental Table 12. Co-orthologs of RBFs experimentally approached in A. thaliana.
Supplemental Table 13. Clustering of ribosome biogenesis factors concerning NGS expression data.
Clustered are the ribosome biogenesis factor proteins by k-means ending in 6 different clusters (column 1–6). The ribosomal proteins were clustered concerning the different expression values of NGS data for specific tissues and developmental stages.
Supplemental Figure 1. Flowchart of CRACPipe.
Shown is a scheme of the pipeline workflow. The start (green) and endpoint (red) of the pipeline is shown as ellipses. First step is the splitting of the read preprocessing and genome annotation. The reads are preprocessed via BLAT and FastQC. Simultaneously, the annotated genome is prepared for mapping the reads onto the genome via SSAHA2. The mapped reads are weighted and statistical analyzed to create the output files. (Blue rectangle: script process; violet rectangle: external program; orange parallelogram: input/output; yellow rhomb: split process.
Supplemental Figure 2. Webinterface of CRACPipe.
Shown ist the web interface of CRACPipe. The logo of the program is shown on the top and the fields are ordered from top to bottom: E-mail address, input file, genome for mapping, kind of experiment (vivo, vitro, cultura, flag, RNA-seq, MACE), analysis method (Expressome, Genome), readtype (Solexa, Roche454, ABI SOLiD), reverse linker sequence, description. For submitting a job an e-mail address and a fastq file or zipped folder of fastq files containing short reads from NGS is required. A job can be given a short name (<=6 characters) and the type of experiment indicated (CRAC: in vivo, in vitro, in cultura; MACE; RNA-seq). For analyzing the NGS data the reference genome can either be submitted or selected from the available organisms (yeast, human, Arabidopsis, tomato, Anabaena).
Availability and requirements: Project name: CRACPipe; Project home page: https://www.uni-frankfurt.de/fb/fb15/english/institute/inst-3-mol-biowiss/AK-Schleiff/research/index.html; Operating system: Platform independent; Programming language: Python; Other requirements: MochiView v1.45 or higher; License: Academic Free (accessible after requesting username and password); Any restrictions to use by non-academics: No access allowed without negotiation with authors.
Supplemental Figure 3. Minimal read length determination.
(A) Shown is the percentage of all possible nucleotide patterns in the tomato genome (Y-axis) of a determined length (X-axis), when the threshold of minimal occurrences is variable (dark green…>0; light green…>1; green…>2; pale green…>3). (B) The MACE datasets for tomato are analyzed using different minimal pattern length parameter (X-axis) showing the percentage of unique mapped reads (blue) and all mapped reads (red).
Supplemental Figure 4. Saturation by sequencing depth.
Shown are the raw counts of MACE libraries from Anther (green) and Leaf (grey) correlated to the detected features (mRNAs) to quantify the sequencing depth via NoiSeq18. The saturation plot represents the number of newly detected features in the genome with more than a given number of counts. The mapped reads are used to approximate the needed number of reads for detecting new features and estimate the sequencing depth19. The detected features are shown as line plot and approximated from the real library size (black dot). The bars represent the new detections per million reads.
Supplemental Figure 5. The expression of the set of control genes.
Shown is the relation of the TPM expression value in leaves and anthers for the genes listed in Supplemental File S1, Table S6. The line represents the least square fit to the equation indicated.
Supplemental Figure 6. Determination of number of clusters for k-means clustering.
The line plot shows the correlation of number of clusters to within-cluster sum of distances for the set of RBFs and RPs. The k-means clustering was performed for the range of 1 to 120 clusters. Each clustering was performed 100 times to build the mean. The red dashed line represents the elbow of the curve and was used to define the number of clusters for the final clustering.
Acknowledgments
We would like to thank Oliver Mirus for constant help and support and Markus T. Bohnsack for support during pipeline development. We acknowledge funding by the Deutsche Forschungsgemeinschaft and by the EU/Marie Curie.
Glossary
Abbreviations
- csv
comma separated values
- gbk
GenBank
- gff
general feature format
- MRP
mitochondrial RNA processing
- NGS
next generation sequencing
- SAM
Sequence/Alignment Map
- TRAMP
Trf4/Air2/Mtr4p polyadenylation
- UTR
untranslated region
Footnotes
ACADEMIC EDITOR: J.T. Efird, Associate Editor
FUNDING: This is a manuscript of the SPOT-ITN consortium (www.spot-itn.eu). Funding was received from the SPOT-ITN Marie Curie EU project. Enrico Schleiff is also funded by grants from Deutsche Forschungsgemeinschaft (CRC-SFB902 B9 and the DFG Cluster of Excellence – Macromolecular Complexes in Action). The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE). Published by Libertas Academica. Learn more about this journal.
Author Contributions
ES conceptualized the project. ES, SF, SS and KDS headed the project. SS, JE and MK were involved in co-ortholog prediction, pipeline CRACPipe design, and NGS data analysis. SF and PP performed the plant experiments. SS, SF and ES performed data analysis and manuscript drafting. All authors approved the manuscript.
REFERENCES
- 1.Dez C, Tollervey D. Ribosome synthesis meets the cell cycle. Curr Opin Microbiol. 2004;7:631–7. doi: 10.1016/j.mib.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Henras AK, Soudet J, Gerus M, et al. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol Life Sci. 2008;65:2334–59. doi: 10.1007/s00018-008-8027-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woolford JL Jr Baserga SJ. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics. 2013;195:643–81. doi: 10.1534/genetics.113.153197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomson E, Tollervey D. The final step in 5.8S rRNA processing is cytoplasmic in Saccharomyces cerevisiae. Mol Cell Biol. 2010;30:976–84. doi: 10.1128/MCB.01359-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karbstein K. Quality control mechanisms during ribosome maturation. Trends Cell Biol. 2013;23:242–50. doi: 10.1016/j.tcb.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watkins NJ, Bohnsack MT. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA. 2012;3:397–414. doi: 10.1002/wrna.117. [DOI] [PubMed] [Google Scholar]
- 7.Bachellerie JP, Cavaille J, Huttenhofer A. The expanding snoRNA world. Biochimie. 2002;84:775–90. doi: 10.1016/s0300-9084(02)01402-5. [DOI] [PubMed] [Google Scholar]
- 8.Brown JW, Echeverria M, Qu LH, et al. Plant snoRNA database. Nucleic Acids Res. 2003;3:432–5. doi: 10.1093/nar/gkg009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodor J, Letelier I, Holuigue L, Echeverria M. Nucleolar RNPs: from genes to functional snoRNAs in plants. Biochem Soc Trans. 2010;38:672–6. doi: 10.1042/BST0380672. [DOI] [PubMed] [Google Scholar]
- 10.Xu R, Zhang S, Huang J, Zheng C. Genome-wide comparative in silico analysis of the RNA helicase gene family in Zea maysand Glycine max: a comparison with Arabidopsisand Oryza sativa. PLoS One. 2013;8:e78982. doi: 10.1371/journal.pone.0078982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ebersberger I, Simm S, Leisegang MS, et al. The evolution of the ribosome biogenesis pathway from a yeast perspective. Nucleic Acids Res. 2014;42:1509–23. doi: 10.1093/nar/gkt1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durut N, Abou-Ellail M, Pontvianne F, et al. A duplicated NUCLEOLIN gene with antagonistic activity is required for chromatin organization of silent 45S rDNA in Arabidopsis. Plant Cell. 2014;26:1330–44. doi: 10.1105/tpc.114.123893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pontvianne F, Matia I, Douet J, et al. Characterization of AtNUC-L1 reveals a central role of nucleolin in nucleolus organization and silencing of AtNUC-L2 gene in Arabidopsis. Mol Biol Cell. 2007;18:369–79. doi: 10.1091/mbc.E06-08-0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weis BL, Missbach S, Marzi J, Bohnsack MT, Schleiff E. The 60S associated ribosome biogenesis factor LSG1-2 is required for 40S maturation in Arabidopsis thaliana. Plant J. 2014;80(6):1043–56. doi: 10.1111/tpj.12703. [DOI] [PubMed] [Google Scholar]
- 15.Zawada AM, Rogacev KS, Müller S, et al. Massive analysis of cDNA Ends (MACE) and miRNA expression profiling identifies proatherogenic pathways in chronic kidney disease MACE and miRNA profiling in CKD. Epigenetics. 2013;9:161–72. doi: 10.4161/epi.26931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang CK, Huang LF, Huang JJ, Wu SJ, Yeh CH, Lu CA. A DEAD-box protein, AtRH36, is essential for female gametophyte development and is involved in rRNA biogenesis in Arabidopsis. Plant Cell Physiol. 2010;51:694–706. doi: 10.1093/pcp/pcq045. [DOI] [PubMed] [Google Scholar]
- 17.Missbach S, Weis BL, Martin R, Simm S, Bohnsack MT, Schleiff E. 40S ribosome biogenesis co-factors are essential for gametophyte and embryo development. PLoS One. 2013;8:e54084. doi: 10.1371/journal.pone.0054084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi DQ, Liu J, Xiang YH, Ye D, Sundaresan V, Yang WC. SLOW WALKER1, essential for gametogenesis in Arabidopsis, encodes a WD40 protein involved in 18S ribosomal RNA biogenesis. Plant Cell. 2005;17:2340–54. doi: 10.1105/tpc.105.033563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byrne ME. A role for the ribosome in development. Trends Plant Sci. 2009;14:512–9. doi: 10.1016/j.tplants.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 20.Nakao A, Yoshihama M, Kenmochi N. RPG: the ribosomal protein gene database. Nucleic Acids Res. 2004;32:D168–70. doi: 10.1093/nar/gkh004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paul P, Simm S, Blaumeiser A, et al. The protein translocation systems in plants – composition and variability on the example of Solanum lycopersicum. BMC Genomics. 2013;14:189. doi: 10.1186/1471-2164-14-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaturvedi P, Ischebeck T, Egelhofer V, Lichtscheidl I, Weckwerth W. Cell-specific analysis of the tomato pollen proteome from pollen mother cell to mature pollen provides evidence for developmental priming. J Proteome Res. 2013;12:4892–903. doi: 10.1021/pr400197p. [DOI] [PubMed] [Google Scholar]
- 23.Yakovlev IA, Lee Y, Rotter B, et al. Temperature-dependent differential transcriptomes during formation of an epigenetic memory in Norway spruce embryogenesis. Tree Genet Genomes. 2014;10:355–66. [Google Scholar]
- 24.Kent WJ. BLAT – the BLAST-like alignment tool. Genome Res. 2002;12:656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.James LR, Andrews S, Walker S, et al. High-throughput analysis of calcium signalling kinetics in astrocytes stimulated with different neurotransmitters. PLoS One. 2011;6:e26889. doi: 10.1371/journal.pone.0026889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ning Z, Cox AJ, Mullikin JC. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11:1725–9. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cartwright RA, Graur D. The multiple personalities of Watson and Crick strands. Biol Direct. 2011;6:7. doi: 10.1186/1745-6150-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eddy SR. Non-coding RNA genes and the modern RNA world. Nat Rev Genet. 2001;2:919–29. doi: 10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- 29.Homann OR, Johnson AD. MochiView: versatile software for genome browsing and DNA motif analysis. BMC Biol. 2010;8:49. doi: 10.1186/1741-7007-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asmann YW, Klee EW, Thompson EA, et al. 3′ tag digital gene expression profiling of human brain and universal reference RNA using Illumina Genome Analyzer. BMC Genomics. 2009;10:531. doi: 10.1186/1471-2164-10-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Consea A. Differential expression in RNA-seq: a matter of depth. Genome Res. 2011;21(12):2213–23. doi: 10.1101/gr.124321.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seesi SA, Tiagueu YT, Zelikovsky A, Măndoiu II. Bootstrap-based differential gene expression analysis for RNA-Seq data with and without replicates. BMC Genomics. 2014;15(suppl 8):S2. doi: 10.1186/1471-2164-15-S8-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomato Genome C. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485:635–41. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soukas A, Cohen P, Socci ND, Friedman JM. Leptin-specific patterns of gene expression in white adipose tissue. Genes Dev. 2000;14:963–80. [PMC free article] [PubMed] [Google Scholar]
- 35.Tibshirani R, Walther G, Hastie T. Estimating the number of clusters in a data set via the gap statistic. J R Stat Soc Series B Stat Methodol. 2001;63:411–23. [Google Scholar]
- 36.Fragkostefanakis S, Simm S, Paul P, Bublak D, Scharf KD, Schleiff E. Chaperone network composition in Solanum lycopersicumexplored by transcriptome profiling and microarray meta-analysis. Plant Cell Environ. 2014 doi: 10.1111/pce.12426. [DOI] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Koonin EV. Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet. 2005;39:309–38. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- 39.Li N, Yuan L, Liu N, et al. SLOW WALKER2, a NOC1/MAK21 homologue, is essential for coordinated cell cycle progression during female gametophyte development in Arabidopsis. Plant Physiol. 2009;151:1486–97. doi: 10.1104/pp.109.142414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Honys D, Twell D. Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol. 2004;5:R85. doi: 10.1186/gb-2004-5-11-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wieckowski Y, Schiefelbein J. Nuclear ribosome biogenesis mediated by the DIM1 A rRNA dimethylase is required for organized root growth and epidermal patterning in Arabidopsis. Plant Cell. 2012;24:2839–56. doi: 10.1105/tpc.112.101022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lange H, Holec S, Cognat V, et al. Degradation of a polyadenylated rRNA maturation by-product involves one of the three RRP6-like proteins in Arabidopsis thaliana. Mol Cell Biol. 2008;28:3038–44. doi: 10.1128/MCB.02064-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lange H, Sement FM, Gagliardi D. MTR4, a putative RNA helicase and exosome co-factor, is required for proper rRNA biogenesis and development in Arabidopsis thaliana. Plant J. 2011;68:51–63. doi: 10.1111/j.1365-313X.2011.04675.x. [DOI] [PubMed] [Google Scholar]
- 44.Kato Y, Konishi M, Shigyo M, Yoneyama T, Yanagisawa S. Characterization of plant eukaryotic translation initiation factor 6 (eIF6) genes: the essential role in embryogenesis and their differential expression in Arabidopsisand rice. Biochem Biophys Res Comm. 2010;397:673–8. doi: 10.1016/j.bbrc.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 45.Ohbayashi I, Konishi M, Ebine K, Sugiyama M. Genetic identification of Arabidopsis RID2 as an essential factor involved in pre-rRNA processing. Plant J. 2011;67:49–60. doi: 10.1111/j.1365-313X.2011.04574.x. [DOI] [PubMed] [Google Scholar]
- 46.Im CH, Hwang SM, Son YS, et al. Nuclear/nucleolar GTPase 2 proteins as a subfamily of YlqF/YawG GTPases function in pre-60S ribosomal subunit maturation of mono- and dicotyledonous plants. J Biol Chem. 2011;286:8620–32. doi: 10.1074/jbc.M110.200816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saez-Vasquez J, Caparros-Ruiz D, Barneche F, Echeverria M. Characterization of a crucifer plant pre-rRNA processing complex. Biochem Soc Trans. 2004;32:578–80. doi: 10.1042/BST0320578. [DOI] [PubMed] [Google Scholar]
- 48.Comella P, Pontvianne F, Lahmy S, et al. Characterization of a ribonuclease III-like protein required for cleavage of the pre-rRNA in the 3’ETS in Arabidopsis. Nucleic Acids Res. 2008;36:1163–75. doi: 10.1093/nar/gkm1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zakrzewska-Placzek M, Souret FF, Sobczyk GJ, Green PJ, Kufel J. Arabidopsis thalianaXRN2 is required for primary cleavage in the pre-ribosomal RNA. Nucleic Acids Res. 2010;38:4487–502. doi: 10.1093/nar/gkq172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gachomo EW, Jimenez-Lopez JC, Baptiste LJ, Kotchoni SO. GIGANTUS1 (GTS1), a member of Transducin/WD40 protein superfamily, controls seed germination, growth and biomass accumulation through ribosome-biogenesis protein interactions in Arabidopsis thaliana. BMC Plant Biol. 2014;14:37. doi: 10.1186/1471-2229-14-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsu YF, Chen YC, Hsiao YC, et al. AtRH57, a DEAD-box RNA helicase, is involved in feedback inhibition of glucose-mediated abscisic acid accumulation during seedling development and additively affects pre-ribosomal RNA processing with high glucose. Plant J. 2014;77:119–35. doi: 10.1111/tpj.12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lahmy S, Guilleminot J, Cheng CM, et al. DOMINO1, a member of a small plant-specific gene family, encodes a protein essential for nuclear and nucleolar functions. Plant J. 2004;39:809–20. doi: 10.1111/j.1365-313X.2004.02166.x. [DOI] [PubMed] [Google Scholar]
- 53.Grandi P, Rybin V, Bassler J, et al. 90S pre-ribosomes include the 35S pre-rRNA, the U3 snoRNP, and 40S subunit processing factors but predominantly lack 60S synthesis factors. Mol Cell. 2002;10:105–15. doi: 10.1016/s1097-2765(02)00579-8. [DOI] [PubMed] [Google Scholar]
- 54.Karbstein K. Inside the 40S ribosome assembly machinery. Curr Opin Chem Biol. 2011;15:657–63. doi: 10.1016/j.cbpa.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O’Donohue MF, Choesmel V, Faubladier M, Fichant G, Gleizes PE. Functional dichotomy of ribosomal proteins during the synthesis of mammalian 40S ribosomal subunits. J Cell Biol. 2010;190:853–66. doi: 10.1083/jcb.201005117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gerus M, Bonnart C, Caizergues-Ferrer M, Henry Y, Henras AK. Evolutionarily conserved function of RRP36 in early cleavages of the pre-rRNA and production of the 40S ribosomal subunit. Mol Cell Biol. 2010;30:1130–44. doi: 10.1128/MCB.00999-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kressler D, de la Cruz J, Rojo M, Linder P. Fal1p is an essential DEAD-box protein involved in 40S-ribosomal-subunit biogenesis in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:7283–94. doi: 10.1128/mcb.17.12.7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soltanieh S, Lapensee M, Dragon F. Nucleolar proteins Bfr2 and Enp2 interact with DEAD-box RNA helicase Dbp4 in two different complexes. Nucleic Acids Res. 2014;42:3194–206. doi: 10.1093/nar/gkt1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wiederkehr T, Pretot RF, Minvielle-Sebastia L. Synthetic lethal interactions with conditional poly(A) polymerase alleles identify LCP5, a gene involved in 18S rRNA maturation. RNA. 1998;4:1357–72. doi: 10.1017/s1355838298980955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van den Elzen AM, Schuller A, Green R, Seraphin B. Dom34-Hbs1 mediated dissociation of inactive 80S ribosomes promotes restart of translation after stress. EMBO J. 2014;33:265–76. doi: 10.1002/embj.201386123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yarunin A, Panse VG, Petfalski E, Dez C, Tollervey D, Hurt EC. Functional link between ribosome formation and biogenesis of iron-sulfur proteins. EMBO J. 2005;24:580–8. doi: 10.1038/sj.emboj.7600540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fatica A, Cronshaw AD, Dlakic M, Tollervey D. Ssf1p prevents premature processing of an early pre-60S ribosomal particle. Mol Cell. 2002;9:341–51. doi: 10.1016/s1097-2765(02)00458-6. [DOI] [PubMed] [Google Scholar]
- 63.Lebreton A, Rousselle JC, Lenormand P, et al. 60S ribosomal subunit assembly dynamics defined by semi-quantitative mass spectrometry of purified complexes. Nucleic Acids Res. 2008;36:4988–99. doi: 10.1093/nar/gkn469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Talkish J, Zhang J, Jakovljevic J, Horsey EW, Woolford JL., Jr Hierarchical recruitment into nascent ribosomes of assembly factors required for 27SB pre-rRNA processing in Saccharomyces cerevisiae. Nucleic Acids Res. 2012;40:8646–61. doi: 10.1093/nar/gks609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dong J, Lai R, Jennings JL, Link AJ, Hinnebusch AG. The novel ATP-binding cassette protein ARB1 is a shuttling factor that stimulates 40S and 60S ribosome biogenesis. Mol Cell Biol. 2005;25:9859–73. doi: 10.1128/MCB.25.22.9859-9873.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang J, Lan P, Gao H, Zheng L, Li W, Schmidt W. Expression changes of ribosomal proteins in phosphate- and iron-deficient Arabidopsis roots predict stress-specific alterations in ribosome composition. BMC Genomics. 2013;14:783. doi: 10.1186/1471-2164-14-783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xue S, Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat Rev Mol Cell Biol. 2012;13:355–69. doi: 10.1038/nrm3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental File 1. CRACPipe description.
Supplemental Table 1. RBF co-orthologs in 14 plant species.
Given is the complex the RBF associates to (column 1) the name of the factor in yeast (column 2) and the number of co-orthologs identified in 14 different plant species including Monocots, Eudicots, moss and green algae (columns 3–16): A. thaliana, S. lycopersicum, S. tuberosum, G. max, B. distachyon, L. japonicus, M. truncatula, O. sativa, P. patens, C. reinhardtii, P. trichocarpa, S. bicolor, V. vinifera and Z. mays.
Supplemental Table 2. RBF co-orthologs in tomato an A. thaliana.
Given is the complex the RBF associates to (column 1) the name of the factor in yeast (column 2), the number of co-orthologs identified in tomato (column 3) and A. thaliana (column 4), the accession numbers of the identified sequences in tomato (column 5) and A. thaliana (column 9), the KOG assignment (columns 6, 10), the description (columns 7, 11) and the Pfam domains and the order thereof (columns 8, 12).
Supplemental Table 3. RBF co-orthologs in tomato with difference in their annotations.
Shown are all RBFs for which co-orthologs have been identified in the tomato genome but contain a different functionalor domain assignnment. Given are the name of the factor, the accession number, the KOG entry, the description of the gene and the PFAM domain prediction. Yellow highlighted are the set of co-orthologues with at least one sequence which is not fully comparable to the others. (NF…no Pfam domain identified).
Supplemental Table 4. Ribosomal protein co-orthologs in 14 plant species.
Given is the complex the Ribosomal protein associates to (column 1) the name of the protein (column 2) and the number of co-orthologs identified in 14 different plant species including Monocots, Eudicots, moss and green algae (columns 3–16): A. thaliana, S. lycopersicum, S. tuberosum, G. max, B. distachyon, L. japonicus, M. truncatula, O. sativa, P. patens, C. reinhardtii, P. trichocarpa, S. bicolor, V. vinifera and Z. mays. The AGI (column 17) and Soly ID (column 18) of the co-orthologs is assigned to the ribosomal protein.
Supplemental Table 5. The Affymetrix chip datasets used for the meta analysis.
Given are the organism (column 1), if the dataset handles stress or tissues (column 2), the database (column 3), the series identifier (column 4), the number of samples (column 5) and a short description of the experiment (column 6).
Supplemental Table 6. The tomato genes with a variance in expression below 0.4.
Given is the gene ID (column 1), the gene description (column 2), the variance calculated for all tissue specific experiments (column 3), for all stress specific experiments (column 4), for all selected experiments (column 5). The columns 6 to 11 contain the information about maximum (column 6, 9), mean (column 7, 10) and minimum RMA values (column 8, 11).
Supplemental Table 7. Expression values for RBF coding genes.
Compared are the high throughput sequencing data of our MACE study (column 3–4; TPM) for vegetative (leaf; LeCo) and reproductive (anther; AnCo) tissue to the RNA-seq RPKM values of the study performed by the Tomato Genome Consortium (column 7–13) 20. the log2 fold changes and probability for differentially expressed genes comparing leaf and anther MACE libraries via NoiSeq18. In red are genes excluded by domain analysis.
Supplemental Table 8. Expression values for Ribosomal protein coding genes.
Compared are the high throughput sequencing data of our MACE study (column 3–4; TPM) for vegetative (leaf; LeCo) and reproductive (anther; AnCo) tissue to the RNA-seq RPKM values of the study performed by the Tomato Genome Consortium (column 7–13)20. In column 5 and 6 are the log2 fold changes and probability for differentially expressed genes comparing leaf and anther MACE libraries via NoiSeq18.
Supplemental Table 9. Clustering of ribosomal proteins concerning NGS expression data.
Clustered are the ribosomal proteins by k-means ending in 6 different clusters (columns 1–6). The ribosomal proteins were clustered concerning the different expression values of NGS data for specific tissues and developmental stages.
Supplemental Table 10. Primer for qRT-PCR of RBF co-orthologs in tomato.
Given is the Solyc ID for tomato (column 1) the name of the primer including the name of the factor in yeast (column 2 and the primer sequence (columns 3).
Supplemental Table 11. The TPM expression value of the 10 genes highest expressed in the according tissue.
Supplemental Table 12. Co-orthologs of RBFs experimentally approached in A. thaliana.
Supplemental Table 13. Clustering of ribosome biogenesis factors concerning NGS expression data.
Clustered are the ribosome biogenesis factor proteins by k-means ending in 6 different clusters (column 1–6). The ribosomal proteins were clustered concerning the different expression values of NGS data for specific tissues and developmental stages.
Supplemental Figure 1. Flowchart of CRACPipe.
Shown is a scheme of the pipeline workflow. The start (green) and endpoint (red) of the pipeline is shown as ellipses. First step is the splitting of the read preprocessing and genome annotation. The reads are preprocessed via BLAT and FastQC. Simultaneously, the annotated genome is prepared for mapping the reads onto the genome via SSAHA2. The mapped reads are weighted and statistical analyzed to create the output files. (Blue rectangle: script process; violet rectangle: external program; orange parallelogram: input/output; yellow rhomb: split process.
Supplemental Figure 2. Webinterface of CRACPipe.
Shown ist the web interface of CRACPipe. The logo of the program is shown on the top and the fields are ordered from top to bottom: E-mail address, input file, genome for mapping, kind of experiment (vivo, vitro, cultura, flag, RNA-seq, MACE), analysis method (Expressome, Genome), readtype (Solexa, Roche454, ABI SOLiD), reverse linker sequence, description. For submitting a job an e-mail address and a fastq file or zipped folder of fastq files containing short reads from NGS is required. A job can be given a short name (<=6 characters) and the type of experiment indicated (CRAC: in vivo, in vitro, in cultura; MACE; RNA-seq). For analyzing the NGS data the reference genome can either be submitted or selected from the available organisms (yeast, human, Arabidopsis, tomato, Anabaena).
Availability and requirements: Project name: CRACPipe; Project home page: https://www.uni-frankfurt.de/fb/fb15/english/institute/inst-3-mol-biowiss/AK-Schleiff/research/index.html; Operating system: Platform independent; Programming language: Python; Other requirements: MochiView v1.45 or higher; License: Academic Free (accessible after requesting username and password); Any restrictions to use by non-academics: No access allowed without negotiation with authors.
Supplemental Figure 3. Minimal read length determination.
(A) Shown is the percentage of all possible nucleotide patterns in the tomato genome (Y-axis) of a determined length (X-axis), when the threshold of minimal occurrences is variable (dark green…>0; light green…>1; green…>2; pale green…>3). (B) The MACE datasets for tomato are analyzed using different minimal pattern length parameter (X-axis) showing the percentage of unique mapped reads (blue) and all mapped reads (red).
Supplemental Figure 4. Saturation by sequencing depth.
Shown are the raw counts of MACE libraries from Anther (green) and Leaf (grey) correlated to the detected features (mRNAs) to quantify the sequencing depth via NoiSeq18. The saturation plot represents the number of newly detected features in the genome with more than a given number of counts. The mapped reads are used to approximate the needed number of reads for detecting new features and estimate the sequencing depth19. The detected features are shown as line plot and approximated from the real library size (black dot). The bars represent the new detections per million reads.
Supplemental Figure 5. The expression of the set of control genes.
Shown is the relation of the TPM expression value in leaves and anthers for the genes listed in Supplemental File S1, Table S6. The line represents the least square fit to the equation indicated.
Supplemental Figure 6. Determination of number of clusters for k-means clustering.
The line plot shows the correlation of number of clusters to within-cluster sum of distances for the set of RBFs and RPs. The k-means clustering was performed for the range of 1 to 120 clusters. Each clustering was performed 100 times to build the mean. The red dashed line represents the elbow of the curve and was used to define the number of clusters for the final clustering.