

ABSTRACT
Arenavirus pathogens cause a wide spectrum of diseases in humans ranging from central nervous system disease to lethal hemorrhagic fevers with few treatment options. The reason why some arenaviruses can cause severe human diseases while others cannot is unknown. We find that the Z proteins of all known pathogenic arenaviruses, lymphocytic choriomeningitis virus (LCMV) and Lassa, Junin, Machupo, Sabia, Guanarito, Chapare, Dandenong, and Lujo viruses, can inhibit retinoic acid-inducible gene 1 (RIG-i) and Melanoma Differentiation-Associated protein 5 (MDA5), in sharp contrast to those of 14 other nonpathogenic arenaviruses. Inhibition of the RIG-i-like receptors (RLRs) by pathogenic Z proteins is mediated by the protein-protein interactions of Z and RLRs, which lead to the disruption of the interactions between RLRs and mitochondrial antiviral signaling (MAVS). The Z-RLR interactive interfaces are located within the N-terminal domain (NTD) of the Z protein and the N-terminal CARD domains of RLRs. Swapping of the LCMV Z NTD into the nonpathogenic Pichinde virus (PICV) genome does not affect virus growth in Vero cells but significantly inhibits the type I interferon (IFN) responses and increases viral replication in human primary macrophages. In summary, our results show for the first time an innate immune-system-suppressive mechanism shared by the diverse pathogenic arenaviruses and thus shed important light on the pathogenic mechanism of human arenavirus pathogens.
IMPORTANCE We show that all known human-pathogenic arenaviruses share an innate immune suppression mechanism that is based on viral Z protein-mediated RLR inhibition. Our report offers important insights into the potential mechanism of arenavirus pathogenesis, provides a convenient way to evaluate the pathogenic potential of known and/or emerging arenaviruses, and reveals a novel target for the development of broad-spectrum therapies to treat this group of diverse pathogens. More broadly, our report provides a better understanding of the mechanisms of viral immune suppression and host-pathogen interactions.
INTRODUCTION
Intracellular RNA viruses are recognized by a family of cytosolic RNA helicase proteins called retinoic acid-inducible gene 1 (RIG-i)-like receptors (RLRs) to activate the antiviral and inflammatory signals (1, 2). The RLR members include RIG-i, Melanoma Differentiation-Associated protein 5 (MDA5), and Laboratory of Genetics and Physiology 2 (LGP2) (3–5). RIG-i recognizes short double-stranded RNA (dsRNA) with 5′ triphosphate, while MDA5 recognizes long RNA duplexes (6). Upon ligand binding by the C-terminal domains (CTD) of RIG-i and MDA5, these proteins undergo conformational changes to activate the N-terminal CARD domains that mediate their interactions with the adaptor molecule mitochondrial antiviral signaling (MAVS)/IPS-1/virus-induced signaling adaptor (VISA)/Cardif to trigger the signaling cascades that consist of tumor necrosis factor (TNF) receptor-associated factors (TRAFs), TANK-binding kinase 1 (TBK1), and inhibitor-κB kinase ε (IKKε) to activate transcription factors NF-κB, interferon (IFN) regulatory factor 3 (IRF3), and IRF7, which induce the production of the type I IFNs and other cytokines (3). The RLR pathway is essential for host innate immunity to RNA viruses and is thus a major target of viral immune evasion mechanisms (4, 7). Influenza virus NS1 inhibits RIG-i activation by interacting with TRIM25 to prevent RIG-i ubiquitinylation (8). Paramyxovirus V protein binds and inhibits MDA5 (9). Ebola virus (EBOV) VP35 blocks RLR signaling through multiple mechanisms such as sequestering the RIG-i cofactor PKR activator (PACT), preventing the interactions of TBK1 and IKKε with IRFs, and inhibiting IRF7 activity (10–14). Arenaviral nucleoprotein (NP) strongly inhibits the production of type I IFNs through its DEDDH exoribonuclease (RNase) activity, possibly by degrading the immunostimulatory dsRNA substrates (15–20).
Arenaviruses are a diverse family of negative-strand enveloped RNA viruses with a bisegmented RNA genome, which encodes two proteins on each segment in an ambisense orientation—glycoprotein GPC and nucleoprotein NP on the S segment and L polymerase protein and the small matrix protein Z on the L segment (21). Arenaviruses can cause a wide spectrum of diseases in humans, with limited preventive or therapeutic options (22, 23). Lymphocytic choriomeningitis virus (LCMV) can cause neurologic diseases (24). Dandenong virus (DANV) was isolated from organ transplant patients who died of a febrile illness (25). Hemorrhagic fever (HF) arenaviruses, such as Lassa virus (LASV), Lujo virus (LUJV), Junin virus (JUNV), Machupo virus (MACV), Sabia virus (SABV), Guanarito virus (GTOV), and Chapare virus (CHPV), can cause multisystem organ failure and death. LASV causes endemic infection in many countries in West Africa, with an estimated 500,000 cases resulting in ∼5,000 deaths annually (26). Except for Candid#1, which is used as the JUNV vaccine in Argentina, no licensed vaccine for human usage is currently available. Therapeutic options are limited and depend mainly on supportive care. Ribavirin, a broad-spectrum antiviral compound, has shown some efficacy only when it has been administered at an early stage of viral infection when the symptoms are insidious (27). Transfusion of immune plasma has been used with some success to treat JUNV-caused Argentine HF (AHF) (28) but not Lassa hemorrhagic fever. It is unknown why other arenaviruses, e.g., Mobala virus (MOBV), Mopeia virus (MOPV), Ippy virus (IPPYV), Amapari virus (AMAV), and Pichinde virus (PICV), are not associated with human diseases even though they have been isolated from the same host species and belong to the same serogroups as the other arenaviral pathogens (23).
Arenavirus Z protein is a small 15-kDa protein with multiple functions (29), such as forming a matrix layer of virions (30, 31), mediating virus budding (32, 33), and regulating viral genome replication and transcription (34, 35). It has been reported that the Z protein of New World (NW) pathogenic arenaviruses, i.e., MACV, JUNV, SABV, and GTOV, but not of Old World (OW) pathogenic ones (LASV and LCMV) can bind RIG-i and inhibit IFN production (36). We report here a novel discovery: the Z proteins of all known human arenavirus pathogens, but not of nonpathogens, inhibit the RLRs by binding to RLRs and disrupting the RLR-MAVS interactions. The determinant for the RLR binding and inhibition has been mapped to the N-terminal domain (NTD) of pathogenic Z proteins. Swapping of a pathogenic Z NTD to the nonpathogenic Pichinde virus (PICV) genome does not affect viral growth in Vero cells but significantly inhibits the type I IFN responses and increases viral replication in human primary macrophages. Our report reveals a common innate immune-system-suppressive mechanism of all pathogenic arenaviruses that may provide important insights into arenavirus pathogenesis.
MATERIALS AND METHODS
Cell lines and viruses.
Human kidney epithelial cells 293T (Y. Liang laboratory) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 50 μg/ml penicillin-streptomycin. BHK21 baby hamster kidney cells (Y. Liang laboratory) and Vero African green monkey kidney cells (Y. Liang laboratory) were maintained in DMEM supplemented with 10% FBS and 50 μg/ml penicillin-streptomycin. BSRT7-5 cells, which are BHK21 cells stably expressing T7 RNA polymerase, were obtained from K. K. Conzelmann (Ludwig-Maximilians-Universität, Germany) and cultured in minimal essential medium (MEM) supplemented with 10% FBS, 1 μg/ml Geneticin, and 50 μg/ml penicillin-streptomycin. Recombinant PICVs were amplified in BHK21 cells, and the infectious virus titer was determined by plaque assay in Vero cells as described previously (37).
Isolation and culture of primary human MDMs (hMDMs).
Monocyte-derived macrophages (MDMs) were obtained and purified from human peripheral blood mononuclear cells (PBMCs), following the same protocol as that described by Pannetier et al. (38). Briefly, human PBMCs were freshly isolated from individual healthy-donor buffy coats (Memorial Blood Center, St. Paul, MN) after Ficoll-Hypaque gradient centrifugation. The CD14+ monocytes were isolated from the mononuclear fraction using a MACS CD14 isolation kit (Milteny Biotec) according to the manufacturer's instructions. MDMs were obtained by culturing monocytes in medium (RPMI medium supplemented with 100 U/ml l-glutamine, 100 μg/ml penicillin-streptomycin, and 1 mM sodium pyruvate) containing 10 U/ml recombinant human macrophage colony-stimulating factor (rhM-CSF) (PeproTech) for 7 days, during which medium with cytokines was replaced with fresh medium every 2 days (38). The purity of the MDM populations was determined to be >97% by flow cytometric analysis using fluorescein isothiocyanate (FITC)-conjugated anti-CD14 antibody (BD Biosciences).
Plasmids.
Expression plasmids for C-terminal hemagglutinin (HA)-tagged PICV and LASV Z proteins have been described previously (39). Expression plasmids for the other arenavirus Z proteins, with a C-terminal HA tag, were similarly constructed in the pCAGGS vector. The Z protein sequences of the following viruses (accession no.) were obtained from GenBank: DANV (ABY20731), LCMV Armstrong (AAX49343), LUJV (YP_002929492), CHPV (ABY87070), MACV (NP_899214.1), GTOV (NP_899220.1), JUNV (NP_899216.1), SABV (YP_089659.1), MOBV (YP_516228.1), MOPV (YP_170707.1), IPPY (YP_516232.1), AMAV (YP_001649217.1), Tacaribe virus (TCRV; NP_694847.1), Pirital virus (PIRV; YP_025092.1), Bear Canyon virus (BCNV; YP_001649224.1), Cupixi virus (CPXV; YP_001649219.1), Flexal virus (FLEV; (ACC94297.1), Latino virus (LATV; YP_001936025.1), Oliveros virus (OLVV; ABY59840.1), Tamiami virus (TAMV; YP_001911117.1), and White Water Arroyo virus (WWAV; ACD03599.1). Overlapping oligonucleotides spanning the ∼300-bp full-length Z open reading frame (ORF) with C-terminal HA sequences were chemically synthesized (Life Technologies), annealed, ligated, and inserted into the pCAGGS expression vector. The PICV-LASV and PICV-LCMV chimeric Z protein expression plasmids were constructed by similar methods using overlapping oligonucleotides. Each plasmid was confirmed by sequence analysis. Expression plasmids pEF-FLAG-RIG-i, pEF-FLAG-RIG-iN, pEF-FLAG-RIG-iC, pEF-FLAG-MDA-5, pEF-FLAG-MDA-5N, and pEF-FLAG-MDA-5C were obtained from Takashi Fujita (Kyoto University, Japan). Plasmids pMyc-MAVS, pFLAG-TBK1, pFLAG-IKKε, and pFLAG-IRF3/5D were obtained from Rongtuan Lin (McGill University, Canada). The pGST-RIG-iN plasmid was constructed by subcloning the amino acid (aa) 1 to 229 N-terminal region of RIG-i in frame with the glutathione S-transferase (GST) tag in the pGEX-4T-1 plasmid.
IFN-β-promoter-dependent LUC assay.
293T cells were transfected with either empty vector or an individual Z expression vector in different amounts (10 ng, 100 ng, and 1,000 ng), together with 100 ng of IFN-β–LUC vector, which expresses the firefly luciferase (Fluc) reporter gene from the IFN-β promoter, and 50 ng of a β-galactosidase (β-Gal)-expressing plasmid, for the purpose of transfection efficiency normalization. For RIG-i- or MDA5-mediated IFN induction, 100 ng of pEF-FLAG-RIG-iN (expressing the N-terminal domain of RIG-i) or pEF-FLAG-MDA-5N (expressing the N-terminal domain of MDA5) was included in the transfection. For Sendai virus (SeV)-induced IFN activation, the transfected 293T cells were infected with 100 hemagglutination units (HAU) ml−1 of SeV. Cell lysates were prepared at 24 h posttransfection or 16 h postinfection (hpi) for Fluc and β-Gal assays. Fluc activities were normalized to the β-Gal values as previously described (16). Each transfection was conducted in triplicate experiments and repeated in at least two independent experiments.
Coimmunoprecipitation (co-IP).
293T cells were transfected with 10 μg of HA-tagged Z expression plasmid and 10 μg of FLAG-tagged RIG-i, MDA5, or IKKε plasmid, with or without myc-tagged MAVS plasmid. Cell lysates were prepared at 48 h posttransfection in lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5% NP-40, 0.1% SDS, 10 μg/ml aprotinin, 20 μg/ml phosphoramidon, 1 μg/ml leupeptin, 50 μg/ml phenylmethylsulfonyl fluoride [PMSF], 0.2 mM sodium orthovanadate) and immunoprecipitated (IP) with either anti-FLAG or anti-HA antibody as previously described (40). After three washes, the precipitants were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. Standard Western blot analyses were conducted with anti-HA, anti-FLAG, and anti-myc antibodies. For endogenous coimmunoprecipitation (co-IP) assays, hMDMs were infected with the respective arenaviruses at a multiplicity of infection (MOI) of 5 for 24 h. The cell lysates were accordingly subjected to immunoprecipitation using rabbit anti-RIGi antibody (catalog no. 3743S; Cell Signaling, MA), rabbit anti-MDA5 antibody (catalog no. 5321S; Cell Signaling, MA), or normal rabbit IgG. After washing, the precipitants were separated by SDS-PAGE and analyzed by Western blotting with anti-RIGi, anti-MDA5, anti-FLAG, anti-HA, anti-Myc, and rabbit anti-Z antibody (kindly provided by N. Lopez, Centro de Virología Animal, Argentina).
GST pulldown.
GST and GST-RIG-iN proteins were purified from bacteria as previously described (41). For GST pulldown assays, the Z protein was generated by an in vitro translation reaction at 30°C for 90 min using a TNT quick-coupled transcription-translation rabbit reticulocyte system with Transcend biotinylated lysine-tRNA (Promega). The in vitro translation products were then incubated with purified GST or RIG-iN proteins bound to GST agarose beads at 4°C for 1 h followed by repeated washing with buffer (50 mM Tris [pH 8], 120 mM sodium chloride, 0.5% Triton X-100). The biotin-labeled Z proteins bound to the GST agarose beads were separated by protein electrophoresis on a 15% SDS-polyacrylamide gel and visualized by the use of streptavidin-horseradish peroxidase (HRP) followed by chemiluminescent detection (Promega).
Generation of recombinant PICV with NTD from LCMV Z protein and/or NP exoribonuclease mutation.
Recombinant PICVs were generated using a reverse genetics system that we previously developed (42). The LCMV Z NTD sequences and the NP RNase mutation (NP D380A) were introduced into the plasmids encoding the P18 L and S segments, respectively, by an overlapping-PCR method. To generate recombinant virus, the respective plasmids encoding P18 L and S segments of either the wild-type form or the mutant form were cotransfected into BSRT7-5 cells that stably express T7 RNA polymerase. Supernatants were collected at 48 h for plaque assaying on Vero cells. After plaque purification, viruses were amplified in BHK-21 cells. The sequences of the rescued mutant viruses were confirmed.
Growth curve analysis.
Cells were seeded in 6-well plates at 90% to 100% confluence and infected (in triplicate experiments) with viruses at MOI of 0.01 for 1 h at 37°C. After the cells were washed with phosphate-buffered saline (PBS), a fresh aliquot of cell-culture medium was added into the culture. At different time points postinfection, aliquots of the supernatant were harvested for plaque assaying on Vero cells.
Quantification of IFN-β production from arenavirus-infected hMDMs.
Human MDMs in triplicate experiments were treated with lipopolysaccharide (LPS; Sigma-Aldrich) (0.5 μg/ml) for 6 h or infected with the respective arenaviruses at MOI of 1 for 1 h. After PBS washing, a fresh aliquot of cell-culture medium was added to the culture. At 12, 24, and 48 hpi, supernatants were collected and quantified for IFN-β levels using a human IFN-β enzyme-linked immunosorbent assay (ELISA) kit (PBL Interferon Source, Piscataway, NJ), following the manufacturer's instructions.
Quantitative RT-PCR (qRT-PCR).
Total RNA was extracted from cells using TRIzol reagent (Life Technology, Carlsbad, CA) according to the manufacturer's instructions. Total RNA was cleared of possible plasmid DNA contamination by incubation for 30 min at 37°C with DNase I, which was inactivated by incubation at 85°C for 15 min. Reverse transcription (RT) was conducted using Superscript III reverse transcriptase (Life Technology, Carlsbad, CA) with oligo(dT) as the RT primer. Real-time quantitative PCR (qPCR), in duplicate experiments, was carried out in a 20-μl reaction mixture with gene-specific primers using PerfeCTa SYBR green DNA dye (Quanta Biosciences, Gaithersburg, MD). The qPCR primers were 5′-GAAGTGGACCTCTACGCTTTGG-3′ and 5′-TGATGCCATCCCGTAGGTCTGT-3′ for human PKR, 5′-GGCTGTTTACCAGACTCCGACA-3′ and 5′-CACAAAGCCTGGCAGCTCTCTA-3′ for human Mx1, 5′-AGGAAAGGTGCTTCCGAGGTAG-3′ and 5′-GGACTGAGGAAGACAACCAGGT-3′ for human OAS1, 5′-CTCTGAGCATCCTGGTGAGGAA-3′ and 5′-AAGGTCAGCCAGAACAGGTCGT-3′ for human interferon-stimulated gene 15 (ISG15), 5′-GCCTTGCTGAAGTGTGGAGGAA-3′ and 5′-ATCCAGGCGATAGGCAGAGATC-3′ for human IFIT1, 5′-GGAGCAGATTCTGAGGCTTTGC-3′ and 5′-GGATGAGGCTTCCAGACTCCAA-3′ for human IFIT2, 5′-GCCGCATTGACCATCTATGA-3′ and 5′-GCCAGGAGGTTCTCAACAATAG-3′ for human IFN-β1, and 5′-GAAGGTGAAGGTCGGAGTC-3′ and 5′-GAAGATGGTGATGGGATTTC-3′ for human glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The PCRs were incubated at 95°C for 3 min, followed by 40 cycles of amplification at 95°C for 10 s and 55°C for 30 s. The RNA levels of each gene from virus-infected cells were first normalized to that of GAPDH and then compared to mock-treated cell results and are shown as fold induction values (2−Δ[ΔC(t)]) in the graph. Data shown are the averages of the results of three independent experiments.
RESULTS
(i) The Z proteins of pathogenic arenaviruses but not nonpathogenic ones can block RLR-induced IFN-β activation.
In an attempt to identify viral antagonists of type I IFNs, we transfected 293T cells with an IFN-β promoter-directed luciferase plasmid (pIFNβ–LUC) and a plasmid expressing the RIG-i or MDA5 activation domain (RIG-iN or MDA5N) with or without a Z protein expression plasmid. We found that the Z protein of LASV, but not of nonpathogenic PICV, could strongly inhibit RIG-iN- and MDA5N-induced IFN-β activation in a dose-dependent manner (Fig. 1A). A previous report by Fan and colleagues shows that the Z proteins of NW arenaviruses (JUNV, MACV, GTOV, and SABV), but not of OW ones (LASV and LCMV), bind to RIG-i and inhibit type I IFN induction (36). Our data, however, show that LASV Z can also strongly inhibit RLR-induced IFN-β production. The discrepancies between the two studies may be due to the differences in the experimental systems, such as the methods used to induce and quantify the IFN-β induction, the Z expression levels, etc.
FIG 1.

The Z proteins of pathogenic arenaviruses but not nonpathogenic ones can block RLR-induced IFN-β activation. (A) LASV Z, but not PICV Z, inhibits RIG-iN- and MDA5N-induced IFN-β activation in a luciferase (LUC)-based promoter assay. 293T cells were transfected with two different concentrations (100 ng and 1,000 ng) of empty vector (V) or Z plasmids (LASV or PICV) together with IFN-β–LUC, β-Gal, RIG-iN, or MDA5N plasmids. neg, negative control (no RIG-iN or MDA5N transfection). (B) Expression of arenavirus Z proteins. Plasmids (0.1 or 1 μg) with HA-tagged Z protein from each of the arenaviruses shown on the figure were transfected into 293T cells in separate wells of 6-well plates. Cell lysates were separated in 12% SDS-PAGE and analyzed by Western blotting using anti-HA antibody. (C) The Z proteins from all pathogenic arenaviruses (LASV, DANV, LCMV, LUJV, CHPV, MACV, GTOV, JUNV, and SABV) but not nonpathogenic ones (MOBV, MOPV, IPPYV, AMAV, TCRV, PIRV, PICV, BCNV, CPXV, FLEV, LATV, OLVV, TAMV, and WWAV) inhibit the RIG-iN- and MDA5N-induced IFN-β activation. 293T cells were transfected with empty vector or 1,000 ng of Z plasmid for each arenavirus shown on the figure, IFN-β–LUC, β-Gal, RIG-iN, or MDA5N. Luciferase (LUC) activities were normalized to the β-Gal values as previously described (16). (D) The Z protein of LASV, LCMV, and MACV, but not of PICV, TCRV, or MOBV, can inhibit the Sendai virus-induced IFN-β1 expression. 293T cells were transfected with empty vector or 1,000 ng of Z plasmid for each arenavirus shown on the figure and infected with Sendai virus to induce the IFN expression. The levels of IFN-β1 mRNA were quantified by qRT-PCR and are shown as 2–Δ[ΔC(t)] values, which were obtained after normalization with GAPDH and in comparison to mock infection. (E) Dose-dependent IFN inhibition by LASV Z and NP in the LUC promoter assay. 293T cells were transfected with empty vector (V) or an expression plasmid of LASV NP or Z at the indicated amounts (10 ng, 100 ng, or 1,000 ng), together with IFN-β–LUC and β-Gal plasmids, and infected with Sendai virus. Mock infection was included as a negative control (neg). Statistical analysis was conducted using the Student t test. ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, no statistical significance.
Given the contrasting results seen with pathogenic LASV and nonpathogenic PICV Z proteins with respect to IFN inhibition, we decided to examine the Z proteins from an additional 21 arenaviruses with known sequences. We used overlapping oligonucleotides to generate entire Z open-reading frames (ORFs) and cloned each fused with a C-terminal HA tag into an expression vector. Each Z protein was readily expressed, as shown by Western blotting (Fig. 1B). Tested in the IFN-β-promoter assay, the Z proteins of all 9 causal agents of human diseases (LASV, DANV, LCMV, LUJV, CHPV, MACV, GTOV, JUNV, and SABV) strongly suppressed RLR-induced IFN-β activation (Fig. 1C). In sharp contrast, Z proteins of 14 other arenaviruses (Amapari virus [AMAV], Bear Canyon virus [BCNV], Cupixi virus [CPXV], Flexal virus [FLEX], Ippy virus [IPPYV], Latino virus [LATV], Mobala virus [MOBV], Mopeia virus [MOPV], Oliveros virus [OLVV], Pichinde virus [PICV], Pirital virus [PIRV], Tacaribe virus [TCRV], Tamiami virus [TAMV], and White Water Arroyo virus [WWAV]), which either are not known to be associated with human diseases or have unknown pathogenicity, completely lacked the ability to suppress RLR-induced IFN-β activation (Fig. 1C). These 23 Z proteins are from most if not all of the currently known arenaviruses that include all viral subtypes and clades, OW, NW clade A (NW-A), NW-B, NW-C, and NW-rec A/B (43). To complement the LUC reporter assay, we quantified the level of IFN-β1 mRNA by qRT-PCR. The Z protein of pathogenic arenaviruses (LASV, LCMV, and MACV), but not of nonpathogens (PICV, TCRV, and MOBV), could strongly inhibit Sendai virus-induced IFN-β1 expression in 293T cells (Fig. 1D). We also compared LASV Z to the known IFN antagonist NP with respect to suppression of Sendai virus-induced IFN production in the LUC reporter assay (Fig. 1E). The presence of either LASV Z or LASV NP led to significant inhibition of IFNs in plasmid transfections even at a low dose (10 ng) and to stronger inhibition at higher doses (Fig. 1E). Compared at the same plasmid dose, the presence of LASV NP led to a greater level of IFN inhibition than was seen with LASV Z, which may reflect their distinct inhibitory mechanisms, as NP mediates the inhibition via its RNase enzymatic activity whereas LASV Z does so through protein-protein interactions (see below). Taken together, our data for the first time reveal a unique feature shared by all confirmed pathogenic arenaviruses, i.e., Z-mediated inhibition of RLR-induced IFN-β activation, regardless of geographical distribution (OW or NW) or viral subgroup and independent of the disease symptoms (hemorrhagic fever or central nervous system disease) or severity.
(ii) LASV Z protein inhibits RLR signaling by interacting with the N-terminal CARD domains of RLRs.
To determine the molecular mechanism by which pathogenic arenavirus Z proteins inhibit the production pathway of type I IFNs, we examined the inhibitory activity of LASV Z in response to enforced expression of several known factors in the RLR pathway (44): RIG-iN, MDA5N, MAVS/IPS-1, TBK1, IKKε, and IRF3/5D (Fig. 2A). LASV Z could strongly inhibit the IFN-β induction by RIG-iN and MDA5N in a dose-dependent manner but not the IFN-β induction by any of downstream factors MAVS/IPS-1, TBK1, IKKε, and IRF3/5D, suggesting that LASV Z inhibits the RLR-dependent IFN pathway at a step(s) upstream of MAVS, most likely by targeting the RLRs themselves. We therefore determined whether LASV Z could interact with the RLRs by performing co-IP after expressing HA-tagged Z in 293T cells, with or without FLAG-tagged full-length RIG-i or MDA5. Indeed, LASV Z could be pulled down by anti-FLAG antibody when coexpressed with RIG-i or MDA5, demonstrating its specific association with RLRs (Fig. 2B). That LASV Z, but not PICV Z, interacts specifically with RIG-i and MDA5 was also confirmed by the inverse co-IP with an anti-HA antibody followed by Western blotting with an anti-FLAG antibody (Fig. 2C).
FIG 2.

LASV Z inhibits RLR signaling by interacting with RLRs at the N-terminal CARD domains. (A) LASV Z inhibits type I IFN activation induced by RIG-iN and MDA5N but not by MAVS, TBK1, IKKε, or IRF3/5D. 293T cells were transfected with empty vector (V) or increasing amounts of PICV or LASV Z plasmids (10, 100, and 1,000 ng), IFN-β–LUC, and plasmids expressing the respective IFN activators. neg, negative control (no RIG-iN or MDA5N transfection). Each sample was compared to the vector control by statistical analysis using the Student t test. *, P < 0.05; ***, P < 0.001; ns, no statistical significance. (B) LASV Z can interact with RIG-i and MDA5. HA-tagged ZLASV and FLAG-tagged full-length RIG-i or MDA5 were cotransfected into 293T cells. Cell lysates were immunoprecipitated with anti-FLAG antibody and detected by anti-HA or anti-FLAG. (C) LASV Z, but not PICV Z, can interact with RIG-i and MDA5 by inverse coimmunoprecipitation. 293T cells were cotransfected with HA-tagged LASV or PICV Z plasmid and FLAG-tagged full-length RIG-i or MDA5. The cell lysates, IgG, and anti-HA immunoprecipitates were detected by Western blotting with anti-FLAG and anti-HA antibodies. (D) LASV Z interacts with the N-terminal CARD domain, but not the C-terminal domain, of RIG-i. HA-tagged ZLASV was cotransfected with FLAG-tagged full-length RIG-i, RIG-iN, and RIG-iC, respectively. The Z proteins in the cell lysates, IgG, and anti-FLAG immunoprecipitates were detected by Western blotting with anti-HA and anti-FLAG antibodies. (E) LASV but not PICV Z directly binds to the N-terminal region of RIG-i. In vitro-translated and biotin-labeled LASV Z and PICV Z proteins were incubated with purified GST or GST-tagged RIG-iN domain (GST-RIG-iN) in vitro. After GST pulldown, the associated Z proteins were detected by Western blotting with streptavidin-HRP followed by chemiluminescent detection.
Both RIG-i and MDA5 harbor the N-terminal tandem CARD domains and a central ATPase-containing DExD/H box helicase domain (3). That LASV Z can strongly inhibit RIG-iN- and MDA5N-induced IFN production suggests that the Z-interactive region is located within the N-terminal CARD domains. Indeed, we demonstrated that LASV Z associated specifically with RIG-iN but not RIG-iC by co-IP (Fig. 2D). To determine whether LASV Z directly binds to RLR, we expressed and purified GST-tagged RIG-iN protein from bacteria. We then incubated the purified GST-RIG-iN or GST control protein with the biotinylated LASV or PICV Z protein that was generated by an in vitro transcription-translation system. After GST pulldown with glutathione-Sepharose 4B beads, the associated Z protein was detected by the use of streptavidin-horseradish peroxidase followed by chemiluminescent detection. As shown in Fig. 2E, LASV Z but not PICV Z specifically binds to GST-RIG-iN, demonstrating the direct protein-protein interaction between LASV Z and RIG-iN. Taking the results together, LASV Z protein inhibits RLR signaling by interacting directly with the RLR CARD domains.
(iii) Pathogenic but not nonpathogenic Z proteins can interact with RLRs and disrupt the RLR-MAVS binding.
We then examined whether the other pathogenic Z proteins could also interact with RLRs in a manner similar to that seen with LASV Z. Using the same co-IP experiment as described in Fig. 2B, we showed that the Z proteins of all other pathogenic arenaviruses (LUJV, MACV, LCMV, CHPV, DANV, GTOV, JUNV, and SABV), but not of nonpathogenic ones (PICV, TCRV, AMAV, IPPYV, MOBV, MOPV, and PIRV), could interact with RIG-i and MDA5 (Fig. 3A), correlating with their differential abilities to inhibit RLR-induced IFN-β production. We also showed that the interaction of pathogenic Z with RIG-i/MDA5 was not due to the potential aggregation-prone nature of the pathogenic Z protein, as LASV Z and LCMV Z did not associate with IKKε under the same co-IP assaying conditions (Fig. 3B).
FIG 3.

Pathogenic Z proteins interact with RLRs to disrupt the RLR-MAVS interaction. (A) Pathogenic but not nonpathogenic Z proteins can interact with RLRs. Each HA-tagged Z protein was transfected into 293T cells together with FLAG-tagged full-length RIG-i or MDA5. Cell lysates were immunoprecipitated with anti-FLAG antibody. Both cell lysates and immunoprecipitates were analyzed by Western blotting with anti-HA or anti-FLAG antibody (left panel). For GTOV, JUNV, SABV, and PICV Z proteins, cell lysates and immunoprecipitates with control IgG or anti-FLAG antibody were analyzed by Western blotting with anti-HA or anti-FLAG antibody (right panel). (B) LASV or LCMV Z does not interact with IKKε. 293T cells were cotransfected with HA-tagged LASV or LCMV Z together with FLAG-tagged IKKε plasmid. The cell lysates, IgG, and anti-FLAG immunoprecipitates were detected by Western blotting with anti-HA and anti-FLAG antibodies. (C) Interaction of pathogenic Z proteins with RLRs disrupts the RLR-MAVS association. 293T cells were transfected with individual Z plasmid, FLAG-tagged RIG-iN or MDA5N, and myc-tagged MAVS plasmids. Cell lysates were pulled down by anti-FLAG antibody and detected by anti-myc, anti-HA, and anti-FLAG antibodies.
RLRs are known to activate the downstream target MAVS/IPS-1 via CARD-CARD interactions (45). We hypothesized that pathogenic Z proteins bind to the RLR CARD to disrupt the RLR-MAVS association and thus block MAVS activation. To test this, we transfected cells with HA-tagged Z and FLAG-tagged RIG-iN or MDA5N, together with myc-tagged MAVS. We pulled down the RIG-i complex and the MDA5 complex using anti-FLAG antibody and detected the presence of Z and MAVS by Western blotting using anti-HA and anti-myc antibodies, respectively (Fig. 3C). As predicted, in cell lysates that contained the pathogenic Z proteins (LASV, LUJV, MACV, LCMV, CHPV, and DANV), the RIG-i or MDA5 complex included Z but not MAVS. In contrast, in cell lysates of viruses that contained nonpathogenic Z proteins (PICV, TCRV, AMAV, IPPYV, MOBV, MOPV, and PIRV), the RIG-i or MDA5 complex included MAVS but not Z (Fig. 3C). Taking the data together, we showed that pathogenic Z proteins interact specifically with RLRs to disrupt their association with MAVS.
(iv) Pathogenic Z NTD is a critical determinant for RLR binding and inhibition.
Arenavirus Z proteins share a Gly2 myristoylation site, the central RING domain, and the C-terminal late domain(s), but there are significant sequence variations, primarily at N and C termini (29). To define the sequence determinant of the pathogenic Z proteins for RLR inhibition, we generated PICV-LASV Z chimeric proteins, PICV-ZNLASV and PICV-ZCLASV, in which the N-terminal domain (NTD) and C-terminal domain (CTD) of PICV Z were replaced with the corresponding sequences from LASV Z (Fig. 4A). These Z protein constructs, all with HA tags, were readily expressed in the plasmid-transfected 293Tcells (Fig. 4A). PICV-ZNLASV, but not PICV-ZCLASV, could interact with RIG-i and MDA5 and disrupted the MAVS association (Fig. 4B), demonstrating that LASV Z NTD mediates the RLR interaction. Correspondingly, tested in the IFN-β-promoter assay, PICV-ZNLASV, but not PICV-ZCLASV, could inhibit the RIG-iN- and MDA5N-induced IFN-β activation (Fig. 4C), suggesting that LASV Z NTD is the determinant for the RLR inhibition. Similar results were obtained with the PICV-LCMV Z chimeric proteins. PICV-ZNLCMV, with the NTD derived from LCMV (Armstrong), but not PICV-ZCLCMV, could inhibit the RLR-dependent IFN-β activation (Fig. 4D). In addition, we compared the levels of Z-mediated RLR inhibition in 293T cells infected with Sendai virus (Fig. 4E) or transfected with purified virion RNAs (Fig. 4F), both known to induce IFN-β production via RLR. Pathogenic Z proteins (LASV and LCMV), as well as the chimeric PICV Z proteins with the NTD of LASV or LCMV (PICV-ZNLASV and PICV-ZNLCMV), significantly inhibited Sendai virus- and virion RNA-induced IFN-β activation, in contrast to PICV Z and the chimeric PICV Z with the CTD of LASV or LCMV (PICV-ZCLASV or PICV-ZCLCMV). Therefore, the ability to interact with and inhibit RLRs has been mapped to the pathogenic Z NTD. The discrepancy between our results and those reported in a previous study, which failed to detect Sendai virus-induced IFN-β activation by LCMV Z based on the results of chloramphenicol acetyltransferase (CAT) and green fluorescent protein (GFP) reporter assays (15), can be explained by the differences in the two experimental systems, such as different dosages of Sendai virus, different protein expression levels, and/or different cell lines or IFN-β promoter plasmids used in the experiments. Taken together, our results demonstrate that the NTD of the pathogenic Z proteins contains a critical determinant for binding and inhibiting RIG-i and MDA5.
FIG 4.

The N-terminal domain (NTD) of pathogenic Z is a critical determinant for RLR binding and inhibition. (A) Diagrams showing the NTD, central RING domain, and CTD of LASV Z, PICV Z, and the chimeric Z proteins. Expression of these Z proteins in the transfected 293T cells was detected by Western blotting using anti-HA. (B) PICV-ZNLASV but not PICV-ZCLASV can interact with RIG-i and MDA5 and disrupt RLR-MAVS association in a coimmunoprecipitation assay as described in the Fig. 3 legend. (C) PICV-ZNLASV but not PICV-ZCLASV can inhibit RIG-iN- and MDA5N-induced IFN-β activation in a LUC-based promoter assay. (D) PICV-ZNLCMV but not PICV-ZCLCMV can inhibit RIG-iN- and MDA5N-induced IFN-β activation in a LUC-based promoter assay. (E) The Z proteins with a pathogenic NTD (ZLASV, ZLCMV, and chimeric PICV-ZNLASV and PICV-ZNLCMV) can inhibit Sendai virus-induced IFN-β production. (F) The Z proteins with a pathogenic NTD (ZLASV, ZLCMV, and chimeric PICV-ZNLASV and PICV-ZNLCMV) can inhibit virion RNA-induced IFN-β production. Each sample was compared to the vector control by statistical analysis using the Student t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not statistically significant. (G) Sequence comparison of arenavirus Z NTDs. The available Z protein sequences were obtained from GenBank. Alignment of multiple sequences was conducted using ClustalW in MacVector 12 software. Names of pathogenic viruses are shown in bold.
(v) LCMV Z NTD leads to increased viral replication in human primary macrophages.
To demonstrate that the pathogenic Z NTD is indeed involved in the inhibition of RLR-dependent IFN signaling in the context of an authentic arenavirus infection, we introduced the 31-residue LCMV Z NTD into the PICV (P18 strain) genome by a reverse-genetics method (42). We first conducted co-IP experiments to confirm the endogenous Z-RLR interactions. Human monocyte-derived macrophages (hMDMs) (38) were infected with LCMV, rP18-ZNLCMV, rP18, or TCRV, at MOI of 5. The RIG-i and MDA5 immunoprecipitates contained the Z proteins from LCMV and rP18-ZNLCMV but not those from rP18 or TCRV (Fig. 5A), demonstrating that LCMV Z NTD allows the specific interaction with host RIG-i and MDA5. We then examined whether the LCMV Z NTD affects viral basic replication by conducting a growth curve analysis in the IFN-deficient Vero cells at MOI of 0.01. This rP18-ZNLCMV chimeric virus replicated with kinetics similar to those seen with the parental rP18 virus in Vero cells (Fig. 5B), demonstrating that NTD swapping does not affect the essential biological functions of the Z protein in the viral life cycle. We then compared levels of viral growth in hMDMs after infection at MOI of 1. LCMV and rP18-ZNLCMV grew to ∼105 PFU/ml at 24 hpi, a level roughly 1 log higher than that seen with TCRV or PICV (P2 and P18 strains) (Fig. 5C), suggesting that LCMV Z NTD confers a significant growth advantage for arenavirus infection in macrophages, the early target cells of arenavirus infection.
FIG 5.
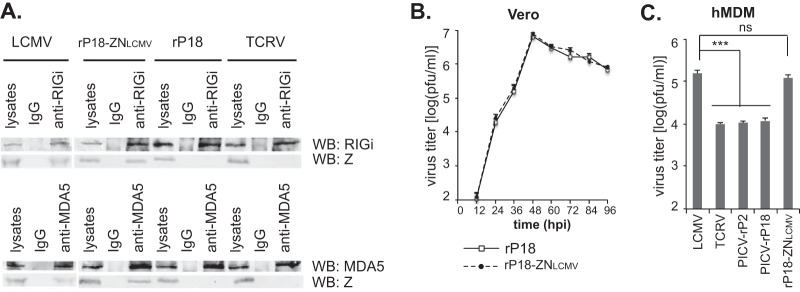
Introduction of LCMV Z NTD to the nonpathogenic PICV genome allows Z-RLR binding in the infected cells and increases viral replication in macrophages. (A) LCMV Z NTD mediates the interaction with endogenous RIG-i and MDA5 in virus-infected cells. The hMDMs were infected with the respective arenaviruses at MOI of 5. Cell lysates were immunoprecipitated with rabbit IgG, rabbit anti-RIGi, or rabbit anti-MDA5 antibodies and detected by anti-RIGi antibody, anti-MDA5 antibody, or anti-Z antibody, respectively. WB, Western blotting. (B) Similar viral growth kinetics of rP18-ZNLCMV and the parental rP18 in Vero cells infected at MOI of 0.01. (C) Viral titers at 24 hpi from hMDMs infected (MOI = 1) with LCMV, TCRV, PICV P2, PICV P18, or rP18-ZNLCMV. Results shown represent the averages of the results of three independent experiments, each performed using PBMCs from a different donor. Statistical analysis was conducted using the Student t test. **, P < 0.01; *** P < 0.001; ns, not statistically significant.
(vi) LCMV Z NTD leads to inhibition of IFN responses in human primary macrophages.
We then determined the role of LCMV Z NTD in the IFN inhibition upon viral infection of hMDMs. These hMDMs were treated with PBS (mock) or LPS or were infected (MOI = 1) with LCMV, TCRV, PICV (both strains P2 and P18), or rP18-ZNLCMV. At 12, 24, and 48 h postinfection (hpi), we quantified the IFN-β protein levels in the supernatants by ELISA (Fig. 6A). LPS treatment was included as a positive control and indeed strongly induced the production of IFN-β at 12 h. For nonpathogenic TCRV and PICV infections, IFN-β was detected at 12 hpi and reached high levels at 24 and 48 hpi. In contrast, LCMV and, remarkably, rP18-ZNLCMV, which contains only the 31-residue LCMV NTD in the backbone of PICV-P18 virus, induced very low levels of IFN-β at all time points tested, and the levels were significantly lower than those induced by TCRV and PICV at 24 and 48 hpi (Fig. 6A). We also compared the levels of IFN-β production from virally infected cells at different MOIs (MOI = 2 or 5) and observed similar results (Fig. 6B). In particular, significant differences were observed between cells infected with LCMV or rP18-ZNLCMV at a lower MOI (MOI = 2) and those infected with TCRV or PICV at a higher MOI (MOI = 5), suggesting that the nature of the Z NTD, rather than the level of viral proteins or RNAs, is the critical determinant for the differential levels of induction of IFNs. Corresponding to the IFN production, qRT-PCR analysis of some representative interferon-stimulated genes (ISGs) showed that PKR, Mx1, OAS1, ISG15, IFIT1, and IFIT2 were highly induced by TCRV and PICV infections but were barely induced by LCMV or rP18-ZNLCMV (Fig. 6C). Thus, LCMV Z NTD leads to effective inhibition of the RLR-dependent type I IFN responses in macrophages, which correlates with increased viral growth in these cells (Fig. 5C).
FIG 6.

Introduction of LCMV Z NTD into the nonpathogenic PICV genome reduces responses of type I IFNs in macrophages. (A) The levels of IFN-β released from human monocyte-derived macrophages (hMDMs) treated with LPS or PBS (mock) or infected with the respective viruses at MOI of 1 at the indicated times postinfection were measured by ELISA. (B) Comparison of the levels of IFN-β production from hMDMs infected with the respective arenaviruses at MOI of 2 or 5. The levels of IFN-β in the cell supernatants at 24 hpi were measured by ELISA. (C) The mRNA levels of ISGs (PKR, Mx1, OAS1, ISG15, IFIT1, and IFIT2) in virus-infected hMDMs at 24 hpi were quantified by qRT-PCR and are shown as 2–Δ[ΔC(t)] values, which were obtained after normalization with GAPDH and comparison to mock treatment. (D) Both pathogenic Z and NP RNase activities contribute to IFN suppression in arenavirus-infected human macrophages. The respective recombinant PICVs, with or without a functional NP RNase domain (NPm1 is an NP RNase mutant D380A) and with or without a pathogenic Z (ZNLCMV), were used to infect human macrophages at MOI of 1 for 24 h. The levels of IFN-β were quantified by ELISA. Results shown are the averages of the results of three independent experiments, each using PBMCs from a different donor. Statistical analysis was conducted using Student's t test. ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, not statistically significant.
As all arenaviruses encode a strong IFN antagonist NP, which inhibits the IFN induction through the conserved RNase domain (15–20), we wondered what the respective roles are of pathogenic Z and NP in the IFN suppression during an authentic arenavirus infection. We thus generated rPICVs encoding a pathogenic Z protein (ZNLCMV) in the backbone of an NP RNase catalytically inactive D380A mutant (NPm1). The respective rPICVs, with or without a pathogenic Z and with or without a functional NP RNase, were used to infect hMDMs at MOI of 1, and the IFN-β levels were quantified at 24 hpi by ELISA (Fig. 6D). The rPICV encoding both IFN antagonists (NP RNase and ZNLCMV), i.e., rP18-ZNLCMV, did not induce an appreciable level of IFNs compared to the mock infection control results, while the virus lacking both antagonists (rP18-NPm1) induced significant amount of IFNs, and the viruses lacking either inhibitory mechanism (rP18-ZNLCMV-NPm1 and rP18) produced intermediate levels of IFNs (Fig. 6D). Taken together, our data strongly suggest that pathogenic Z and NP RNases play distinct and physiologically relevant roles in IFN suppression during an authentic arenavirus infection.
DISCUSSION
In recent years, multiple novel arenaviruses have been identified from natural reservoirs (mainly rodents) and from human patients. Evaluation of their pathogenicity, however, is hampered by the lack of an animal model that can consistently show clinical signs after infection with arenavirus pathogens. For example, LCMV, as a low-risk pathogen, does not normally cause disease signs even in nonhuman primates except for a few selected strains (46). Some unique characteristics have been noted for arenavirus pathogens, such as the use of human TfR1 as an entry receptor by NW pathogenic arenaviruses (47) and the selective inhibition of macrophages by LASV but not nonpathogenic MOPV (38, 48). These findings are limited to a single pathogen or a specific subgroup of pathogens. Here we show for the first time a characteristic that is shared by all the confirmed pathogenic arenaviruses—the ability of the viral Z protein to inhibit RLR signaling. Our assessment of LCMV Z in RLR inhibition can predict its potential to cause disease in humans. On the other hand, the Z protein of WWAV, whose link with several fatalities in North America has not been confirmed, cannot inhibit RLR signaling, suggesting that WWAV may not be a human pathogen. This is consistent with a study by Reignier and colleagues in which analysis of the receptor usage by WWAV glycoprotein did not support the hypothesis of WWAV as a human pathogen (49). As this method of evaluation can be safely and quickly conducted with the knowledge of the Z protein sequence only and without the use of infectious virus, it allows early identification, precautions, and prevention with respect to emerging arenavirus pathogens and their infections.
We have shown that the differential RLR inhibition activities lie in the Z NTD, a region of 30 to 42 residues with highly variable primary sequences (Fig. 4G) and dynamic structures (50). The high degree of sequence variation in the Z NTD suggests that it is nonessential for basic viral replication, an interpretation which is supported by our results obtained using recombinant PICV containing LCMV Z NTD (Fig. 5B). Sequence comparison of Z NTDs did not reveal any obvious consensus residues required for disease pathogenicity (Fig. 4G). Future structural analysis of the pathogenic Z NTDs in complex with RLRs is required to understand the molecular basis of the inhibition.
Two viral IFN antagonists have been identified in arenaviruses so far—NP and pathogenic Z. Arenavirus NP can strongly inhibit the IFN production via an RNase domain that is conserved among all arenaviruses (15–20). The pathogenic Z protein binds and inhibits RLRs to suppress the IFN production (this study). Using recombinant PICVs (Fig. 6D), we have shown that both NP and pathogenic Z proteins contribute to effective IFN suppression in human macrophages during arenavirus infection. Taking the results at face value, the virus containing a pathogenic Z (rP18-ZNLCMV-NPm1) appeared to produce lower levels of IFNs than the one with a functional NP RNase (rP18), indicating that the pathogenic Z may be more efficient than the NP RNase in suppressing the IFN production from arenavirus-infected human macrophages. As macrophages are the early targets of arenavirus infection and represent the first line of host immunity, effective suppression of the type I IFN responses by pathogenic Z protein in macrophages is expected to have a major impact on viral pathogenicity. While both NP and Z can suppress IFN expression, the seemingly differential degrees of inhibition by NP and Z in plasmid-transfected 293T cells (Fig. 1E) and in virus-infected human macrophages (Fig. 6D) are noted. Further studies, therefore, are required to evaluate the relative contributions of pathogenic Z and NP to IFN suppression in different permissive cell types and at different stages of viral infection. It is also noteworthy that, although pathogenic arenaviruses encode both NP and Z proteins to inhibit production of IFNs, for some arenavirus-caused diseases, such as JUNV-caused Argentine HF (AHF), high levels of IFN-α have been detected in patient serum and correlate with disease severity (51). The sources of high levels of IFN-α have not been identified in vivo but are unlikely to be macrophages and monocytes (52). We speculate that arenavirus-induced IFN suppression may be regulated at multiple levels such as cell type, viral strain, and stage of infection and that type I IFNs have both protective and pathogenic roles in arenavirus pathogenesis (53–55). Thus, effective suppression of IFNs mediated by both NP and pathogenic Z in macrophages and monocytes early in the infection, when type I IFNs trigger the innate immunity to control virus infection, may be a critical determinant of viral pathogenicity, while a sustained elevation of levels of type I IFNs through viral and/or cell type-specific mechanisms later in infection in JUNV patients may lead to damaging effects.
The results of our study indicate that pathogenic Z NTD-mediated RLR inhibition may be a common pathogenic mechanism underlying the diverse arenavirus pathogens. While each arenavirus pathogen may have its unique pathogenesis leading to variable disease symptoms in humans, all of them encode a Z protein that can inhibit RLR signaling. We have shown that introduction of a pathogenic Z NTD into a nonpathogenic virus genome is sufficient to effectively inhibit the IFN responses and significantly increase viral replication in human primary macrophages (Fig. 5 and 6), the early target cells of arenavirus infection, which may partly explain arenavirus virulence and disease pathogenesis in vivo (56, 57). Nevertheless, arenavirus pathogens differ greatly in the degrees of disease severity and clinical manifestations (43). We propose that the pathogenic Z NTD is necessary for pathogenic arenaviruses to establish the potential to cause human diseases. Whether this potential leads to self-limiting infections or severe diseases with diverse clinical symptoms (neurological diseases or full-blown hemorrhagic fevers) depends on multiple other factors such as the virus strains, viral inoculum dose, infection route, host immune status, preexisting immunity, and genetic variations. Additional virulence and pathogenic mechanisms must contribute to the complex pathogenic processes of specific arenavirus pathogens (22, 43), such as the selective use of human TfR1 as an entry receptor by the NW pathogenic arenaviruses (47). Further studies are required to fully understand the relationships among these virulent factors and/or mechanisms in disease development by the diverse arenavirus pathogens.
In summary, we reveal for the first time that all the confirmed arenavirus pathogens, but not nonpathogens, share an immune-system-suppressive mechanism based on Z NTD-dependent RLR inhibition that blunts the IFN-mediated antiviral responses. Knowledge from these studies provides important insights into arenavirus pathogenesis as well as into the mechanisms of virus-mediated host immune suppression in general and may lead to the development of broad-spectrum therapies against known and emerging pathogenic arenaviruses by targeting the Z-RLR interaction.
ACKNOWLEDGMENTS
We thank T. Fujita (Kyoto University, Japan) and R. Lin (McGill University, Canada) for sharing plasmids, N. Lopez (Centro de Virología Animal, Argentina) for the rabbit anti-Z antibody, and the staff in A. Fernandez-Sesma's laboratory (Icahn School of Medicine at Mount Sinai Hospital, NY) for advice on isolating and culturing human primary macrophage.
This work was supported by NIH grants AI093680 to H.L. and AI083409 to Y.L.
REFERENCES
- 1.Takeuchi O, Akira S. 2009. Innate immunity to virus infection. Immunol Rev 227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stetson DB, Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Dixit E, Kagan JC. 2013. Intracellular pathogen detection by RIG-I-like receptors. Adv Immunol 117:99–125. doi: 10.1016/B978-0-12-410524-9.00004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerlier D, Lyles DS. 2011. Interplay between innate immunity and negative-strand RNA viruses: towards a rational model. Microbiol Mol Biol Rev 75:468–490, second page of table of contents. doi: 10.1128/MMBR.00007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loo YM, Gale M Jr. 2011. Immune signaling by RIG-I-like receptors. Immunity 34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fitzgerald ME, Rawling DC, Vela A, Pyle AM. 2014. An evolving arsenal: viral RNA detection by RIG-I-like receptors. Curr Opin Microbiol 20:76–81. doi: 10.1016/j.mib.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber M, Weber F. 20May2014, posting date RIG-I-like receptors and negative-strand RNA viruses: RLRly bird catches some worms. Cytokine Growth Factor Rev doi: 10.1016/j.cytogfr.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, Garcia-Sastre A. 2009. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. 2004. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A 101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luthra P, Ramanan P, Mire CE, Weisend C, Tsuda Y, Yen B, Liu G, Leung DW, Geisbert TW, Ebihara H, Amarasinghe GK, Basler CF. 2013. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 14:74–84. doi: 10.1016/j.chom.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cárdenas WB, Loo YM, Gale M Jr, Hartman AL, Kimberlin CR, Martinez-Sobrido L, Saphire EO, Basler CF. 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol 80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basler CF, Amarasinghe GK. 2009. Evasion of interferon responses by Ebola and Marburg viruses. J Interferon Cytokine Res 29:511–520. doi: 10.1089/jir.2009.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, Klenk HD, Palese P, Garcia-Sastre A. 2003. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol 77:7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prins KC, Cardenas WB, Basler CF. 2009. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J Virol 83:3069–3077. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martínez-Sobrido L, Zuniga EI, Rosario D, Garcia-Sastre A, de la Torre JC. 2006. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol 80:9192–9199. doi: 10.1128/JVI.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qi X, Lan S, Wang W, Schelde LM, Dong H, Wallat GD, Ly H, Liang Y, Dong C. 2010. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 468:779–783. doi: 10.1038/nature09605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hastie KM, Liu T, Li S, King LB, Ngo N, Zandonatti MA, Woods VL Jr, de la Torre JC, Saphire EO. 2011. Crystal structure of the Lassa virus nucleoprotein-RNA complex reveals a gating mechanism for RNA binding. Proc Natl Acad Sci U S A 108:19365–19370. doi: 10.1073/pnas.1108515108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang X, Huang Q, Wang W, Dong H, Ly H, Liang Y, Dong C. 2013. Structures of arenaviral nucleoproteins with triphosphate dsRNA reveal a unique mechanism of immune suppression. J Biol Chem 288:16949–16959. doi: 10.1074/jbc.M112.420521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hastie KM, King LB, Zandonatti MA, Saphire EO. 2012. Structural basis for the dsRNA specificity of the Lassa virus NP exonuclease. PLoS One 7:e44211. doi: 10.1371/journal.pone.0044211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hastie KM, Kimberlin CR, Zandonatti MA, Macrae IJ, Saphire EO. 2011. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc Natl Acad Sci U S A 108:2396–2401. doi: 10.1073/pnas.1016404108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buchmeier MJ, De La Torre JC, Peters CJ. 2007. Arenaviridae: the viruses and their replication, p 1791–1827 InKnipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 22.McLay L, Ansari A, Liang Y, Ly H. 2013. Targeting virulence mechanisms for the prevention and therapy of arenaviral hemorrhagic fever. Antiviral Res 97:81–92. doi: 10.1016/j.antiviral.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zapata JC, Salvato MS. 2013. Arenavirus variations due to host-specific adaptation. Viruses 5:241–278. doi: 10.3390/v5010241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonthius DJ. 2012. Lymphocytic choriomeningitis virus: an underrecognized cause of neurologic disease in the fetus, child, and adult. Semin Pediatr Neurol 19:89–95. doi: 10.1016/j.spen.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palacios G, Druce J, Du L, Tran T, Birch C, Briese T, Conlan S, Quan PL, Hui J, Marshall J, Simons JF, Egholm M, Paddock CD, Shieh WJ, Goldsmith CS, Zaki SR, Catton M, Lipkin WI. 2008. A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med 358:991–998. doi: 10.1056/NEJMoa073785. [DOI] [PubMed] [Google Scholar]
- 26.McCormick JB, Webb PA, Krebs JW, Johnson KM, Smith ES. 1987. A prospective study of the epidemiology and ecology of Lassa fever. J Infect Dis 155:437–444. doi: 10.1093/infdis/155.3.437. [DOI] [PubMed] [Google Scholar]
- 27.McCormick JB, King IJ, Webb PA, Scribner CL, Craven RB, Johnson KM, Elliott LH, Belmont-Williams R. 1986. Lassa fever. Effective therapy with ribavirin. N Engl J Med 314:20–26. [DOI] [PubMed] [Google Scholar]
- 28.Enria DA, Briggiler AM, Sanchez Z. 2008. Treatment of Argentine hemorrhagic fever. Antiviral Res 78:132–139. doi: 10.1016/j.antiviral.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fehling SK, Lennartz F, Strecker T. 2012. Multifunctional nature of the arenavirus RING finger protein Z. Viruses 4:2973–3011. doi: 10.3390/v4112973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neuman BW, Adair BD, Burns JW, Milligan RA, Buchmeier MJ, Yeager M. 2005. Complementarity in the supramolecular design of arenaviruses and retroviruses revealed by electron cryomicroscopy and image analysis. J Virol 79:3822–3830. doi: 10.1128/JVI.79.6.3822-3830.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salvato MS, Schweighofer KJ, Burns J, Shimomaye EM. 1992. Biochemical and immunological evidence that the 11 kDa zinc-binding protein of lymphocytic choriomeningitis virus is a structural component of the virus. Virus Res 22:185–198. doi: 10.1016/0168-1702(92)90050-J. [DOI] [PubMed] [Google Scholar]
- 32.Perez M, Craven RC, de la Torre JC. 2003. The small RING finger protein Z drives arenavirus budding: implications for antiviral strategies. Proc Natl Acad Sci U S A 100:12978–12983. doi: 10.1073/pnas.2133782100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strecker T, Eichler R, Meulen J, Weissenhorn W, Dieter Klenk H, Garten W, Lenz O. 2003. Lassa virus Z protein is a matrix protein and sufficient for the release of virus-like particles [corrected]. J Virol 77:10700–10705. doi: 10.1128/JVI.77.19.10700-10705.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cornu TI, de la Torre JC. 2002. Characterization of the arenavirus RING finger Z protein regions required for Z-mediated inhibition of viral RNA synthesis. J Virol 76:6678–6688. doi: 10.1128/JVI.76.13.6678-6688.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kranzusch PJ, Whelan SP. 2011. Arenavirus Z protein controls viral RNA synthesis by locking a polymerase-promoter complex. Proc Natl Acad Sci U S A 108:19743–19748. doi: 10.1073/pnas.1112742108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan L, Briese T, Lipkin WI. 2010. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J Virol 84:1785–1791. doi: 10.1128/JVI.01362-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lan S, McLay L, Aronson J, Ly H, Liang Y. 2008. Genome comparison of virulent and avirulent strains of the Pichinde arenavirus. Arch Virol 153:1241–1250. doi: 10.1007/s00705-008-0101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pannetier D, Faure C, Georges-Courbot MC, Deubel V, Baize S. 2004. Human macrophages, but not dendritic cells, are activated and produce alpha/beta interferons in response to Mopeia virus infection. J Virol 78:10516–10524. doi: 10.1128/JVI.78.19.10516-10524.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Danzy S, Kumar N, Ly H, Liang Y. 2012. Biological roles and functional mechanisms of arenavirus Z protein in viral replication. J Virol 86:9794–9801. doi: 10.1128/JVI.00385-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xing J, Wang S, Lin R, Mossman KL, Zheng C. 2012. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J Virol 86:3528–3540. doi: 10.1128/JVI.06713-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frangioni JV, Neel BG. 1993. Solubilization and purification of enzymatically active glutathione S-transferase (pGEX) fusion proteins. Anal Biochem 210:179–187. doi: 10.1006/abio.1993.1170. [DOI] [PubMed] [Google Scholar]
- 42.Lan S, McLay Schelde L, Wang J, Kumar N, Ly H, Liang Y. 2009. Development of infectious clones for virulent and avirulent pichinde viruses: a model virus to study arenavirus-induced hemorrhagic fevers. J Virol 83:6357–6362. doi: 10.1128/JVI.00019-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLay L, Liang Y, Ly H. 2014. Comparative analysis of disease pathogenesis and molecular mechanisms of New World and Old World arenavirus infections. J Gen Virol 95:1–15. doi: 10.1099/vir.0.057000-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takeuchi O, Akira S. 2008. MDA5/RIG-I and virus recognition. Curr Opin Immunol 20:17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 45.Johnson CL, Gale M Jr. 2006. CARD games between virus and host get a new player. Trends Immunol 27:1–4. doi: 10.1016/j.it.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 46.Zapata JC, Pauza CD, Djavani MM, Rodas JD, Moshkoff D, Bryant J, Ateh E, Garcia C, Lukashevich IS, Salvato MS. 2011. Lymphocytic choriomeningitis virus (LCMV) infection of macaques: a model for Lassa fever. Antiviral Res 92:125–138. doi: 10.1016/j.antiviral.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Radoshitzky SR, Kuhn JH, Spiropoulou CF, Albarino CG, Nguyen DP, Salazar-Bravo J, Dorfman T, Lee AS, Wang E, Ross SR, Choe H, Farzan M. 2008. Receptor determinants of zoonotic transmission of New World hemorrhagic fever arenaviruses. Proc Natl Acad Sci U S A 105:2664–2669. doi: 10.1073/pnas.0709254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lukashevich IS, Maryankova R, Vladyko AS, Nashkevich N, Koleda S, Djavani M, Horejsh D, Voitenok NN, Salvato MS. 1999. Lassa and Mopeia virus replication in human monocytes/macrophages and in endothelial cells: different effects on IL-8 and TNF-alpha gene expression. J Med Virol 59:552–560. [PMC free article] [PubMed] [Google Scholar]
- 49.Reignier T, Oldenburg J, Flanagan ML, Hamilton GA, Martin VK, Cannon PM. 2008. Receptor use by the Whitewater Arroyo virus glycoprotein. Virology 371:439–446. doi: 10.1016/j.virol.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Volpon L, Osborne MJ, Borden KL. 2008. NMR assignment of the arenaviral protein Z from Lassa fever virus. Biomol NMR Assign 2:81–84. doi: 10.1007/s12104-008-9090-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grant A, Seregin A, Huang C, Kolokoltsova O, Brasier A, Peters C, Paessler S. 2012. Junin virus pathogenesis and virus replication. Viruses 4:2317–2339. doi: 10.3390/v4102317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Groseth A, Hoenen T, Weber M, Wolff S, Herwig A, Kaufmann A, Becker S. 2011. Tacaribe virus but not Junin virus infection induces cytokine release from primary human monocytes and macrophages. PLoS Negl Trop Dis 5:e1137. doi: 10.1371/journal.pntd.0001137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trinchieri G. 2010. Type I interferon: friend or foe? J Exp Med 207:2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, Schreiber RD, de la Torre JC, Oldstone MB. 2013. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340:207–211. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG. 2013. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bergthaler A, Flatz L, Hegazy AN, Johnson S, Horvath E, Lohning M, Pinschewer DD. 2010. Viral replicative capacity is the primary determinant of lymphocytic choriomeningitis virus persistence and immunosuppression. Proc Natl Acad Sci U S A 107:21641–21646. doi: 10.1073/pnas.1011998107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McLay L, Lan S, Ansari A, Liang Y, Ly H. 2013. Identification of virulence determinants within the L genomic segment of the pichinde arenavirus. J Virol 87:6635–6643. doi: 10.1128/JVI.00044-13. [DOI] [PMC free article] [PubMed] [Google Scholar]