

ABSTRACT
Stress granules (SGs) are cytoplasmic storage sites containing translationally silenced mRNPs that can be released to resume translation after stress subsides. We previously showed that poliovirus 3C proteinase cleaves the SG-nucleating protein G3BP1, blocking the ability of cells to form SGs late in infection. Many other viruses also target G3BP1 and inhibit SG formation, but the reasons why these functions evolved are unclear. Previously, we also showed a link between G3BP1-induced SGs and protein kinase R (PKR)-mediated translational control, but the mechanism of PKR interplay with SG and the antiviral consequences are unknown. Here, we show that G3BP1 exhibits antiviral activity against several enteroviruses, whereas truncated G3BP1 that cannot form SGs does not. G3BP1-induced SGs are linked to activation of innate immune transcriptional responses through NF-κB and JNK. The G3BP1-induced SGs also recruit PKR and other antiviral proteins. We show that the PXXP domain within G3BP1 is essential for the recruitment of PKR to SGs, for eIF2α phosphorylation driven by PKR, and for nucleating SGs of normal composition. We also show that deletion of the PXXP domain in G3BP1 compromises its antiviral activity. These findings tie PKR activation to its recruitment to SGs by G3BP1 and indicate that G3BP1 promotes innate immune responses at both the transcriptional and translational levels and integrates cellular stress responses and innate immunity.
IMPORTANCE Stress granules appear during virus infection, and their importance is not well understood. Previously, it was assumed that they were nonfunctional artifacts associated with cellular stress. PKR is a well-known antiviral protein; however, its regulation in cells is not well understood. Our work links cellular stress granules with activation of PKR and other innate immune pathways through the activity of G3BP1, a critical stress granule component. The ability of stress granules and G3BP1 to activate PKR and other innate immune transcriptional responses indicates that G3BP1 is an antiviral protein. This work helps to refine a longstanding paradigm indicating stress granules are inert structures and explains why G3BP1 is subverted by many viruses to promote a productive infection.
INTRODUCTION
Stress granules (SGs) are macromolecular triage centers for mRNAs that contain translationally silenced messenger RNPs (mRNPs). Stress granules contain many translation initiation factors, 40S ribosomal subunits, mRNAs, and RNA-binding proteins, which form in response to cellular stresses that inhibit protein synthesis (1, 2). Stress responses are induced during virus infection and are countered by viruses to maximize replication efficiency. Indeed, several examples of viral SG disruption and viral subversion of SG proteins have been described (3–6), indicating stress granules may play antiviral roles against these viruses. Ras-GTPase-activating protein (SH3 domain) binding protein 1 (G3BP1) is a stress granule-resident protein that nucleates stress granule assembly and is also inactivated or coopted by many viruses to promote productive infection (2, 7–10). We previously described a mechanism of SG disassembly during poliovirus (PV) infection wherein PV 3C protease cleaves G3BP1, separating the protein interaction domains from the RNA interaction domains (10).
Viruses may block the formation of SGs or disassemble them for several specific reasons. First, SG disassembly and release of sequestered translation components may allow protein synthesis to resume so the viral genome can be translated. Second, key SG proteins, including G3BP1, may be coopted by viruses for alternate uses supporting replication or virion assembly (5, 6, 9, 11). Finally, SGs have been recently implicated as platforms to amplify the innate immune response. For example, influenza A virus (IAV)-induced SGs colocalize with several innate immune proteins, including RIG-I, MDA5, and protein kinase R (PKR) (12). Furthermore, SG assembly is inhibited by the IAV protein NS1, which also prevents innate immune signaling and the induction of beta interferon (IFN-β). Encephalomyocarditis virus (EMCV) viral proteinase 3C cleaves G3BP1 in HeLa cells and prevents SG assembly, similar to poliovirus, and concurrently suppresses IFN-β induction (7). However, no study has delineated whether SGs and innate immunity occur in parallel or are interdependent.
On the other hand, recruitment of innate immune proteins to SGs does not invariably activate the innate immune response. MDA5, an RNA-sensing component of the innate immune response, localizes to SGs, but that localization is not required for mediating induction of IFN-β during infection with mengovirus, which is a strain of EMCV (13). These disparate results indicate the importance of studying individual SG-resident proteins and characterizing unique innate immune responses to each virus. This approach may clarify general versus virus-specific mechanisms mediated by SGs and/or G3BP1 within the innate immune response.
G3BP1 is critically important for SG assembly. Depletion of G3BP1 inhibits SG formation in response to several stressors (10, 12, 14), and overexpression of G3BP1 nucleates SG formation independently of additional stressors (2, 14, 15). We previously reported that large G3BP1-induced SGs, but not small granules, are capable of triggering eIF2α phosphorylation through PKR, resulting in translational repression (15). Together with data indicating G3BP1 is targeted by many viruses, these properties suggested that G3BP1 is an antiviral protein important in innate immunity. This work shows that G3BP1 is an antiviral protein that activates the transcriptional arm of the innate immune response through NF-κB and JNK transcription and release of certain cytokines. G3BP1 is also involved in recruitment and activation of PKR at SGs, indicating that G3BP1 regulates protein synthesis through PKR as part of a broad innate immune response. Strikingly, close proximity of G3BP1 and PKR within cells coincides with PKR activation, and the G3BP1 PXXP domain is required for PKR activation and the antiviral activity of G3BP1. These data indicate G3BP1 mediates cross talk between stress responses and innate immunity and is an antiviral protein itself.
MATERIALS AND METHODS
Plasmids and cloning.
All the expression constructs containing G3BP1 were derived from human G3BP1. G3BP1 deletions were generated using domain boundaries defined previously (16) and were made as both C-terminally green fluorescent protein (GFP)-λN- and N-terminally T7-tagged constructs (15). C-terminally G3BP1–GFP-λN-tagged constructs are referred to as GFP-tagged below. Each deletion maintained the first 6 amino acids of wild-type (wt) G3BP1 to ensure similar protein stabilities of deletion mutants (17). Deletion mutants were produced with the following primers containing 5′-phosphorylated ends using promenade PCR: ΔNTF2 rev, 5′-AGG CTT CTC CAT CAC CAT-3′; ΔNTF2 fw, 5′-GGG TTT GTC ACT GAG CC-3′; ΔAcidic rev, 5′-ACC AAA GAC CTC ATC TTG G-3′; ΔAcidic fw, 5′-ACT GCC CCT GAG GAT G-3′; ΔPXXP rev, 5′-TTC TTC TAA TAC TGG CTC AGG-3′; ΔPXXP fw, 5′-CAA CTC TTC ATT GGC AAC C-3′. N-terminally GFP-tagged G3BP1 was previously described (16). G3BP1Q326E was also previously described (10).
Wild-type and K64E and K296W mutant expression constructs for human PKR (hPKR) were subcloned from the previously described pcDNA6 (18) into pcDNA-cCherry to produce C-terminally mCherry-tagged PKR constructs (hPKR-mCherry). Other mutations within the two RNA-binding domains of PKR (PKR 4X contains K60A/64E and K150A/154E) were produced in accordance with previous studies demonstrating that these mutations disrupt RNA binding (19). The mutations were generated using site-directed mutagenesis with standard procedures. The luciferase transcriptional reporter plasmids were previously described (20, 21) or commercially available (SABiosciences). The NF-κB–GFP reporter is commercially available (SABiosciences).
Virus.
Transfected HeLa cells were infected with poliovirus type 1 (strain Mahoney) at a multiplicity of infection (MOI) of 10. Transfected mouse embryonic fibroblasts (MEFs) in 96-well plates were infected in duplicate with either enterovirus 70 (EV70), coxsackievirus B3 (CVB3) or CVB5, or vesicular stomatitis virus (VSV) strain Indiana at an MOI of 0.1 or 10, as indicated. The virus was allowed to absorb for approximately 2 h in 2% fetal bovine serum (FBS)-Dulbecco's modified Eagle's medium (DMEM) prior to washing the wells and adding fresh medium. At 72 h postinfection (p.i.), the supernatants were removed and 50% tissue culture infectious dose (TCID50) assays were performed to quantify the virus titers. TCID50 assays were performed on each sample in 96-well plates in duplicate on permissive cells appropriate for each virus (Vero for EV70 and VSV and HEK293 for CVB3 and CVB5). Each assay was repeated at least three times. For CVB5 infections, cells were infected at an MOI of 0.1 essentially as described above. At the indicated time points, cells and supernatant were collected and analyzed for IFN-β mRNA, and TCID50 assays were performed. CVB3-Discosoma sp. red fluorescent protein (dsRed) was kindly provided by Lindsay Whitton (Scripps Research Institute, La Jolla, CA) (22). Experiments with CVB3-dsRed were performed in HeLa cells using an MOI of 5. The cells were infected as described above for 7.5 h before immunofluorescence microscopy.
Cell culture and transfections.
Cells were cultured under standard conditions of 10% FBS in DMEM. G3BP1 knockout (KO) MEFs were a kind gift from Sophy Martin and Jamal Tazi (CNRS, France) (23). U2OS cells stably transfected with peGFP-C1-G3BP1 were produced as previously described and kindly provided by Nancy Kedersha (Brigham and Women's Hospital, Boston, MA) (24). For analysis of G3BP1-induced stress granules, G3BP1 expression constructs were transfected using Fugene HD under conditions optimized for SG induction, as previously described (Promega) (15). Briefly, cells were transfected in 2% FBS-DMEM overnight and harvested the following day for analysis. For transfection of PKR constructs, cells were harvested within 20 h posttransfection to avoid toxic effects of PKR overexpression. All mouse embryonic fibroblast and poly(I·C) transfections of HeLa and U2OS cells were performed with the Neon electroporation device (Life Technologies) in accordance with the manufacturer's instructions. Cells were typically transfected in the evening, cultured in antibiotic-free medium overnight, and harvested for analysis the following day. This procedure typically yielded greater than 70% transfection efficiency.
Stress conditions.
Arsenite was used at 500 μM for 30 min at 37°C. Poly(I·C) (125 ng) was transfected into 0.5 × 105 HeLa or U2OS cells with the Neon device according to the manufacturer's instructions, and cells were plated on glass coverslips. The cells were harvested 6 h after transfection for immunofluorescence (IF) analysis, which coincided with SG assembly and preceded induction of apoptosis.
Immunofluorescence microscopy.
Both epifluorescence and deconvolution microscopy (IF) were performed in the Integrated Microscopy Core at Baylor College of Medicine. Epifluorescence microscopy was performed using a Nikon TE2000 microscope, and deconvolution microscopy experiments were performed using an Applied Precision DeltaVision image restoration microscope with conservative deconvolution algorithms. Microscopy was performed essentially as described previously (15). For experiments in which imaging z-stacks and high resolution were not required (i.e., proximity ligation assays [PLA]), epifluorescence microscopy was used, while deconvolution was used to image all other experiments. The antibodies used for microscopy experiments were as follows: goat anti-T7 antibody, 1:500 (Bethyl); anti-OAS2, 1:200 (sc271117; Santa Cruz); anti-RNase L, 1:200 (sc22870; Santa Cruz); rabbit anti-PKR, 1:200 (3949; ProScience); mouse anti-GFP (Santa Cruz); rabbit anti-eIF4G (25); rabbit anti- poliovirus 3C (a kind gift of Bruce Korant); and rabbit anti-eIF3a, 1:200 (Cell Signaling). Primary antibodies were bound overnight at 4°C. All secondary antibodies (Molecular Probes) were used at 1:1,000 for 30 min at 25°C. All images were processed using Adobe Photoshop CS4.
Proximity ligation assays.
PLA (Sigma) were performed according to the manufacturer's instructions. The PLA signal was detected with far-red fluorophores. The assay depends on recognizing epitopes of interest with two antibodies that are amplified by rolling-circle amplification when the epitopes lie within a restricted 35- to 40-nm distance, thereby generating small spots distinguishable in epifluorescence microscopy. The antibodies used for the analysis were mouse anti-GFP, 1:5,000 (Santa Cruz); rabbit anti-PKR, 1:2,000 (PSc); mouse anti-G3BP1, 1:2,000 (BD Biosciences); and goat anti-Tia1 (Santa Cruz). Antibody binding was performed at room temperature for 2 to 3 h, followed by extensive washing before detection.
Quantification.
The PLA was quantified from at least 20 cells in three fields. The PLA signal was counted manually. For quantification of eIF2α phosphorylation, microscope parameters were calibrated relative to arsenite-treated control cells containing SGs. At least 20 cells from 10 fields or more were imaged. More cells were counted where smaller changes were observed, depending on the magnitude of the effect. The intensity of eIF2α phosphorylation was calibrated to arsenite-stressed cells and scored as present or absent. Cells containing SGs or PXXP granules were scored as large SGs using a threshold of 300-nm diameter (with a 300- to 500-nm diameter range for large granules). SG sizes were determined using Image J software. G3BP1 was imaged in cells containing GFP, and the SG status was recorded so that GFP-positive cells could be binned according to the SG status. For experiments with CVB3-dsRed, at least 50 cells in three fields were counted and scored for red and green signals.
Pearson's correlation coefficients were calculated using Image J2 on areas of interest. At least 30 stress granules in 2 fields were quantified from deconvoluted images for each measurement. The results were similar regardless of whether individual SGs or entire cells were used for analysis.
Quantitative-PCR (qPCR) analysis.
Cells from the CVB5 experiment were infected and harvested as described above. mRNA was isolated using the RNeasy RNA isolation kit (Qiagen), followed by cDNA synthesis with a Vilo cDNA synthesis kit (Life Technologies). IFN-β and β-actin mRNA levels were quantified with Sybr green master mix using a ViiA7 system (Applied Biosystems). The primer sequences are as follows: β-actin fw, 5′-AAG GCC AAC CGT GAA AAG AT-3′; β-actin rev, 5′-GTG GTA CGA CCA GAG GCA TAC-3′; IFN-β fw, 5′-ATG ACC AAC AAG TGT CTC CTC C-3′; IFN-β rev, 5′-GTC CAT GGA AAG AGC TGT AGT G-3′.
Immunoblotting.
Proteins were separated on 10% SDS-PAGE and transferred to nitrocellulose membranes in accordance with standard procedures. The membranes were blocked with 5% bovine serum albumin (BSA) in TBST (20 mM Tris-HCl, pH 7.5, 500 mM NaCl, 0.1% Tween 20), and the primary antibody (rabbit anti-G3BP1 at 1:4,000) was incubated for 3 h at room temperature (10). Secondary horseradish peroxidase (HRP)-conjugated antibody (Bio-Rad donkey anti-rabbit, 1:5,000) was incubated for 1 h at room temperature in TBST prior to detection with enhanced-chemiluminescence (ECL) reagents (Thermo).
Transcription assays.
HeLa cells were grown in 24-well plates and cotransfected as described above with luciferase reporter constructs and G3BP1 expression constructs. The following morning, cells were harvested in passive lysis buffer and analyzed with luciferase assay substrate (Promega), in accordance with standard procedures. The NF-κB transcription assay used a luciferase reporter with a region of the Ig(κ) promoter encompassing the NF-κB regulatory region ((21); the AP1 reporters were previously described (20). The ISRE and IRF3 reporters were obtained from SABiosciences.
Cytokine array.
G3BP1 KO MEFs were transfected as described above, and the supernatant was collected approximately 18 h after transfection. The supernatant was applied to a cytokine array measuring the levels of 62 cytokines (Raybiotech, Inc.). The cytokine array uses HRP as a readout, so multiple exposures and internal controls were used to compile the results presented here.
RESULTS
G3BP1 and G3BP1-induced SGs restrict enterovirus infection.
We have previously demonstrated that G3BP1 is a substrate for poliovirus 3C proteinase, and cleavage of G3BP1 at the glutamine-glycine bond (amino acids 326 to 327) (Fig. 1A) results in disassembly of SGs in HeLa cells (10, 26). Transient expression of the Q326E cleavage-resistant mutant of G3BP1 (G3BP1Q326E) in HeLa cells also reduced PV replicative output in the cell population 7-fold (10) and correlated with SG maintenance. We also previously demonstrated that assembly of large G3BP1-induced SGs led to translation inhibition through PKR-mediated eIF2α phosphorylation (15), which we anticipated would inhibit virus replication. To directly examine the role of SGs in inhibiting PV replication, we followed the production of viral protein 3C protease in individual infected cells containing SG using immunofluorescence microscopy. Cells expressing G3BP1Q326E for 18 to 24 h exhibit two phenotypes, with approximately half forming SGs and half without SGs, despite significant G3BP1 expression levels. Infected cells with large G3BP1-induced SGs were devoid of detectable 3C proteinase (3Cpro) expression (Fig. 1B). Further, cells expressing G3BP1 that did not form SGs displayed delayed kinetics of development of cytopathic effects (Fig. 1B, arrows), whereas expression of GFP alone had no effect on virus replication, as indicated by high expression levels of viral 3C protease, cell rounding, and cytopathic effects. These results indicate that G3BP1-induced SGs in cells strongly repress enterovirus replication, but they also suggest that G3BP1 can inhibit viral replication independently of SG assembly.
FIG 1.

G3BP1 promotes antiviral activity. (A) Domain map of G3BP1 indicating the borders of each domain used throughout this study and the 3Cpro cleavage site. RGG represents the RGG domain containing repeats of the arginine-glycine-glycine peptide motif, while RRM represents the RNA recognition motif within G3BP1. (B) HeLa cells were transfected with either GFP or the Q326E mutant of G3BP1, which is not cleaved by poliovirus 3C protease, and cells were infected with poliovirus at an MOI of 10 (10). At 6 h p.i., the cells were fixed and stained with antibodies directed against G3BP1 or poliovirus 3C protease. Standard epifluorescence microscopy was used for analysis. Arrows mark cells that show both G3BP1 and 3Cpro expression, but do not contain granules. (C) G3BP1 KO MEFs were transfected with the indicated proteins; GFP-G3BP1 and G3BP1-GFP refer to N-terminally and C-terminally tagged proteins, respectively. G3BP1ΔNTF2-GFP expression (G3BP1ΔNTF2) does not form SGs. The cells were then infected with the indicated viruses at an MOI of either 10 or 0.1. Supernatants were collected at 24 or 48 h, respectively, and virus titers were determined using TCID50 assays. The error bars indicate standard deviations.
These experiments utilized G3BP1 overexpression in HeLa cells, which contain endogenous G3BP1 and have impaired innate immunity due to expression of human papillomavirus (HPV) E6 and E7. HPV E6 and E7 proteins inhibit NF-κB activity in cervical epithelial cells and downregulate expression of the interferon-responsive genes (27, 28). Furthermore, HPV-16 E6 binds and inactivates IRF3, a major mediator of the interferon response; inhibits Toll-like receptor 9 activity (29); and interferes with PKR activation (30). Therefore, we sought to investigate whether antiviral properties of G3BP1 could be measured in other cells with more normal innate immune capacity.
To address this question, we employed G3BP1 KO MEFs (31) transiently transfected with either GFP alone, G3BP1 fused in two orientations relative to GFP (G3BP1-GFP and GFP-G3BP1), or a truncated G3BP1 lacking the N-terminal NTF2-like domain (G3BP1ΔNTF2) (Fig. 1A). Since poliovirus does not infect mouse cells due to lack of the CD155 virus receptor, G3BP1 KO MEFs expressing these transgenes were infected with three other human enteroviruses related to poliovirus—EV70, CVB3, and CVB5—as well as the unrelated negative-strand RNA virus VSV. Progeny virus in supernatants from these infections was quantified using TCID assays. Infections were performed as both single-cycle (MOI = 10) and multiple-cycle (MOI = 0.1) infections.
Rescue of G3BP1 expression (in either orientation with respect to GFP) in knockout MEFs induced SGs in only 20 to 30% of cells, which is roughly half the rate at which SGs are produced by expression in HeLa cells (reference 15 and data not shown). The truncated form of G3BP1 lacking the NTF2-like N-terminal domain does not induce SGs, as previously observed (14, 16). In both single-cycle and multiple-cycle infections, G3BP1 expression caused a potent antiviral effect ranging between 1- and 3-log-unit reduction of virus output compared to the GFP control in all three enteroviruses tested (Fig. 1C). The expression levels of transgene proteins in these experiments were similar (data not shown). G3BP1 inhibited enterovirus replication by 1 to 2 log units. The magnitude of the inhibitory effect differed between the two orientations of GFP-tagged G3BP1 constructs, with G3BP1-GFP producing a more potent antiviral effect (Fig. 1C). Multiple-cycle infections scored a stronger antiviral activity of G3BP1, indicating a cumulative antiviral effect (Fig. 1C). Further, truncated G3BP1ΔNTF2, which cannot induce SGs, failed to restrict virus replication. In contrast, VSV replication was only slightly affected by G3BP1 transgene expression, indicating a specificity of the G3BP1-mediated antiviral effect for plus-strand enteroviruses.
G3BP1 promotes activation of the innate immune transcriptional responses through NF-κB and JNK.
To investigate the breadth of innate immune activation by G3BP1 and stress granules, we considered whether innate immune and stress-responsive transcriptional programs are activated in concert with SG assembly. We employed a candidate approach to investigate whether G3BP1 could activate expression from several gene promoters associated with innate immune and stress responses. We tested NF-κB, AP1, Stat1, and IRF3 luciferase reporters for activation following induction of stress granules by G3BP1 expression. Interestingly, we found that both NF-κB and JNK were activated by G3BP1-GFP expression, while the Stat1 reporter was slightly activated and the IRF3 reporter was not activated (Fig. 2A). However, GFP alone was not capable of initiating any of these transcriptional pathways. The NF-κB transcriptional reporter was tested with increasing levels of G3BP1-GFP DNA transfection into cells to test for dose responsiveness to G3BP1 expression. Indeed NF-κB luciferase reporter transcription increased with G3BP1 and correlated with increasing percentages of cells bearing SGs (Fig. 2B). Together, these results indicate that a specific transcriptional response results from G3BP1 expression and implicate G3BP1 in regulation of NF-κB- and AP1-responsive target genes.
FIG 2.

NF-κB and JNK transcription are activated by G3BP1-GFP expression. (A) Luciferase assays were conducted in HeLa cells transfected with either GFP, wild-type G3BP1-GFP, or G3BP1ΔNTF2-GFP (G3BP1ΔNTF2) with reporter constructs driven by the indicated transcriptional response promoters. Fluc, firefly luciferase; RLuc, Renilla luciferase; RLU, relative light units. (B) The NF-κB-luciferase reporter was transfected with increasing concentrations of either G3BP1-GFP or GFP control expression plasmids, as indicated, followed by luciferase assays. (C) Dot blot analysis of 62 cytokine proteins was conducted on conditioned medium from G3BP1 KO MEFs expressing either GFP, G3BP1-GFP, GFP-G3BP1, or G3BP1ΔNTF2-GFP (G3BP1ΔNTF2), as indicated in Fig. 1. A summary of the cytokines elevated during G3BP1 expression is shown. Arb., arbitrary. (D) Cytokines elevated when either G3BP1 or the G3BP1ΔNTF2-GFP (G3BP1ΔNTF2) mutant was expressed. The error bars indicate standard deviations.
To determine whether cytokine expression positively correlates with G3BP1 expression in G3BP1 KO MEFs and whether it could be a result of enhanced innate immune transcription, we performed dot blot analysis for a panel of 62 cytokines from the supernatant of cells expressing G3BP1. Most cytokines, including IFN-γ, did not change significantly, but several cytokines, including interleukin 17 (IL-17), MCP-5, MIP-3a, MIP-3b, and SCF, were elevated with G3BP1 expression but not with GFP or the G3BP1ΔNTF2 mutant (Fig. 2C). These results suggest that full-length G3BP1, either within SGs or through an SG-independent mechanism, can augment the expression of some cytokines during infection. Our dot blot data also indicated that a subset of cytokines, including IL-13, IGFBP-6, leptin R, ICFBP-5, and CD30, are upregulated during expression of either full-length G3BP1 or the G3BP1ΔNTF2 mutant, but not with GFP alone (Fig. 2D). These results further indicate that G3BP1 can activate responses through SG-independent mechanisms.
Other innate immune proteins are recruited to G3BP1-induced SGs.
Previous studies indicated that other innate immune proteins, including OAS and RNase L, colocalize with influenza A virus-induced stress granules, suggesting that virus-induced SGs may regulate interferon-induced activities at multiple levels (12). To test if antiviral effects promoted by G3BP1 may include these pathways, we used an indirect immunofluorescence assay (IFA) to investigate whether OAS2 and RNase L concentrate in G3BP1-induced SGs similarly to virus-induced SGs. Indeed, when G3BP1-induced SGs formed in HeLa, G3BP1 KO, and U2OS cells, both OAS2 and RNase L also colocalized with the granules. However, the degree of colocalization with SGs was cell type dependent, and it appeared stronger in HeLa cells than in U2OS cells and MEFs, particularly with OAS2 (Fig. 3). Not all constituents of stress granules induced by oxidative stress, heat shock, or poliovirus infection are shared (32), suggesting that the degree of recruitment of OAS2 and RNase L to SGs may correlate with their activity. These results indicate that several innate immune proteins can be recruited to some extent to SGs, raising the possibility that activation of multiple innate immune pathways could be amplified with SG formation.
FIG 3.
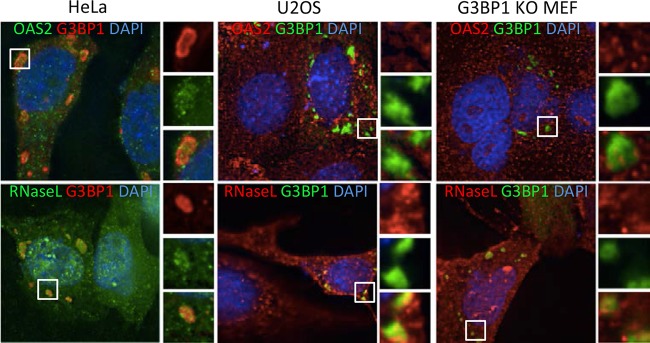
Innate immune proteins localize to G3BP1-induced SGs in a cell line-dependent manner. HeLa and U2OS cells and G3BP1 KO MEFs were transfected with plasmids expressing G3BP1, and the cells were fixed and stained with antibodies against either OAS2 or RNase L. In HeLa cells, G3BP1 (red) was expressed from pcDNA 3.1 HisC-G3BP1 and detected with anti-T7 antibodies. In KO MEFs and U2OS cells, G3BP1-GFP was expressed (green). DAPI (4′,6-diamidino-2-phenylindole) is shown in blue. The insets represent the boxed areas.
PKR partially localizes to G3BP1-induced SGs.
A significant fraction of transfected cells in our system contain G3BP1-induced stress granules, and we previously showed that assembly of large G3BP1-induced SGs triggers PKR activation. Therefore, we considered whether G3BP1-induced SGs could recruit PKR, similar to findings reported for virus-induced SGs (12, 33). When G3BP1 was expressed in HeLa cells, G3BP1 KO MEFs, and U2OS cells and stained for PKR, we found that PKR strongly colocalizes with G3BP1-induced stress granules in all cell types (Fig. 4A). Since PKR activation has been associated with increased innate immune- and stress-responsive transcriptional programs (18, 34) that are elevated with G3BP1 expression (Fig. 2), we focused further on PKR recruitment to SGs.
FIG 4.
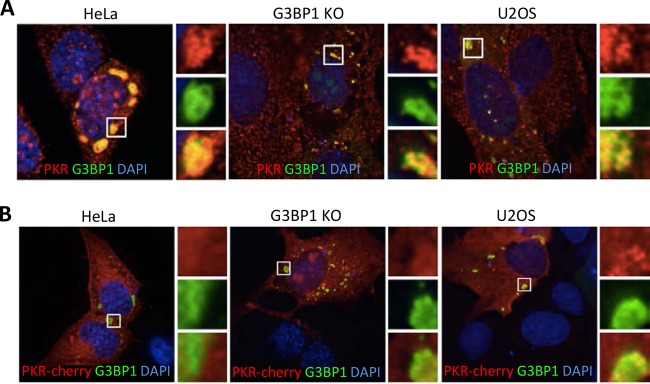
PKR colocalizes with G3BP1-induced stress granules. (A) HeLa cells, G3BP1 KO MEFs, and U2OS cells were transfected with G3BP1-GFP (green) constructs and stained for PKR (red). DAPI-stained nuclei are visible in blue. (B) PKR-mCherry (red) was expressed in the indicated cells, together with G3BP1-GFP (green). The images were captured using deconvolution microscopy. The insets represent the boxed areas.
Since antibody staining of PKR indicated that it is concentrated in G3BP1-induced stress granules, we considered whether mCherry-tagged PKR would also strongly colocalize with G3BP1-induced stress granules. Indeed, tagged PKR also colocalized with G3BP1-induced stress granules; however, its recruitment appeared much less complete, as only a portion of mCherry-PKR entered SGs (Fig. 4B). This suggested that recruitment of tagged PKR differed from that of endogenous PKR or that antibody-based IFA reported localization of only a subset of endogenous PKR. These data also imply that PKR could be recruited to G3BP1-induced SGs and activated at the stress granule, thus mediating the antiviral effects we observed for G3BP1 (Fig. 1 and 2).
The differences between endogenous PKR staining and use of mCherry-tagged PKR prompted us to investigate localization of PKR to G3BP1-induced SGs using other commercial PKR antibodies with different, defined epitopes. All three antibodies we used (labeled SC, PSc, and BD) reported ample PKR signal in immunofluorescence microscopy but yielded different localization results. The PKR antibody recognizing an N-terminal epitope within its first RNA-binding domain (PSc) displayed strong colocalization of PKR with G3BP1-induced stress granules (Fig. 5A). Another antibody recognizing an epitope in the extreme C terminus of PKR (Prosci3947) also strongly colocalized with G3BP1-induced SGs (data not shown). Conversely, an antibody specific for the second RNA-binding domain of PKR (BD) reported weak colocalization with SGs, and antibody specific for the kinase domain (SC) reported little or no colocalization of PKR with SGs in both HeLa cells and G3BP1 KO MEFs (Fig. 5A). The specificity of the antibodies was verified by small interfering RNA (siRNA) knockdown of PKR, which strongly reduced the IFA signal of BD and PSc in HeLa cells (Fig. 5B) and U2OS cells (data not shown), and loss of the PKR band in Western blot analysis (Fig. 5C). Together, these results suggest that a pool of PKR can colocalize with G3BP1-induced granules, but determining the extent of endogenous colocalization is dependent on variations in epitope availability. The central and kinase regions of PKR may be concealed in SGs, whereas the N terminus is available for antibody binding.
FIG 5.

PKR antibodies variably report PKR in G3BP1-induced SGs. (A) G3BP1 was expressed from either the pcDNA 3.1 HisC-G3BP1 (SC) construct or pG3BP1-GFP (PSc and BD), and PKR was stained with three different antibodies (SC, PSc, and BD, as indicated) in either HeLa or G3BP1 KO cells as represented in the indicated colors. The antibody epitopes on PKR are as follows: BD, second RNA-binding domain; SC, kinase domain; PSc, N-terminal RNA-binding domain. The BD antibody does not recognize mouse PKR protein. The insets represent the boxed areas. (B) Antibody specificities for the PSc and BD antibodies were demonstrated in IFA after control or PKR-specific siRNAs were used to deplete PKR signal using IF in HeLa cells. (C) Western blot analysis was performed on HeLa cells treated with either control siRNA or PKR-specific siRNA.
G3BP1 is in close proximity to PKR complexes during stress and PKR activation.
Since PKR becomes activated after G3BP1 expression and mediates translational repression (15), we considered whether we could probe potential G3BP1 and PKR interactions in situ by employing PLA (35, 36). The procedure is used to score where transient protein interactions take place in cells. It has several advantages over immunoprecipitation techniques used to assess protein-protein interactions. First, PLA depends on two interacting proteins residing within 40 nm of each other. Second, due to the distance-dependent ligation of DNA to induce rolling-circle amplification of fluorescently labeled DNA, the assay has a very large linear range, and protein complexes are easily quantified (35, 36). Finally, PLA can detect changes in protein interactions or proximity in their cellular context and can capture labile and transient interactions. Employing this assay, we were able to detect PLA foci indicating very close proximity and/or interacting complexes between G3BP1 and PKR, which occurred under unstressed conditions with endogenous expression levels. The PLA foci increased under arsenite stress where SGs were present in HeLa cells (Fig. 6A). Similar results were found in U2OS cells stably expressing low levels of GFP-G3BP1 (Fig. 6B). Consistent with the results in HeLa cells, PLA signal increased during arsenite stress in U2OS cells and correlated with formation of G3BP1-containing stress granules. One possibility is that PKR could be recruited to and activated within stress granules, generating a signal that is then transduced to the translation apparatus via eIF2α phosphorylation. To investigate this, we activated endogenous PKR using poly(I·C) transfection in both HeLa and U2OS cells and then performed PLA to detect changes in G3BP1-PKR proximity foci. Poly(I·C) caused a 3- to 4-fold increase in G3BP1 and PKR PLA foci in both HeLa cells and U2OS cells expressing G3BP1 (Fig. 6C and D). This is consistent with mobilization or concentration of G3BP1 and PKR in SGs, since SGs are induced by poly(I·C) treatment (Fig. 6D). Together, these results suggest that G3BP1 and PKR are present in shared or overlapping subsets of protein complexes and that they may regulate each other's activities.
FIG 6.

PKR-G3BP1 proximity PLA foci increase with stress and PKR activation. Proximity ligation assays were used to assess close colocalization and/or interaction of G3BP1 and PKR in cells. (A and B) HeLa cells (A) or U2OS cells stably expressing GFP-G3BP1 (green) (B) were untreated or stressed with 500 μM arsenite (Ars) for 30 min before processing for PLA. (C and D) Untransfected HeLa cells and U2OS cells stably expressing GFP-G3BP1 (green) (D) were transfected with poly(I·C) to activate PKR, and PLA was performed. (E) PLA was performed on HeLa cells expressing G3BP1-GFP using antibodies against G3BP1 and GFP. (F) PLA was conducted on endogenous G3BP1 and Tia1, and foci per cell were quantified under each condition. (A to E) Red, PLA signal; blue, DAPI. (A to D and F) The graphs depict quantification of PLA as described in Materials and Methods. The error bars represent standard errors. *, P ≤ 0.05; **, P < 0.01; ***P < 0.001; Student's t test.
Since we did not observe robust PLA signal under any conditions, we sought to confirm that our results are within the linear range of the assay and report close proximity interactions relevant to SG biology. Therefore, PLA was conducted on HeLa cells expressing G3BP1-GFP using antibodies directed against G3BP1 and GFP. We rationalized that most PLA foci result from recognition on two epitopes of the same molecule (G3BP1 and GFP within the G3BP1-GFP protein) and should therefore establish an upper limit of PLA foci per cell under the conditions of our assay. Indeed, we saw on average only 10 foci per cell under these conditions, suggesting our PLA findings with G3BP1 and PKR are within the linear range and consistent with the expected number of foci if two distinct proteins are interacting within a cell (Fig. 6E). Notably, the red PLA foci are not enriched within SGs, where G3BP1-GFP is enriched, suggesting that aggregation of proteins in SGs may hinder epitope recognition of proteins despite their close proximity (Fig. 5B, D, and E). To further confirm that changes in G3BP1-PKR PLA foci are consistent with recognition of two distinct proteins in SGs, we performed PLA in HeLa cells for G3BP1 and Tia1. Tia1 is another canonical stress granule protein that is recruited to SGs from the nucleus during stress. We observed a 2-fold increase in PLA foci during arsenite treatment that generates SGs (Fig. 6F), which is similar to that observed for G3BP1-PKR PLA and supports our data indicating that G3BP1 interacts with PKR in an SG.
PKR determinants for localization in SGS.
The picture compiled from these results indicated that a subset of PKR is recruited to G3BP1-induced SGs. Since there were variations in colocalization of PKR with G3BP1 stress granules depending on the epitope being examined, we considered whether one determinant of localization is the activation state of PKR, which is accompanied by conformational changes. Using an antibody that recognizes active PKR marked by T446 phosphorylation, we used IFA to investigate the localization of active PKR. We found increased levels of active PKR in cells containing large G3BP1-induced SGs but not in adjacent untransfected cells, consistent with our previous work (15). The phosphorylated PKR (P-PKR) detected with this antibody presented as numerous discrete punctae rather than the much more diffuse staining pattern of total PKR (compare Fig. 7A with Fig. 4 and 5). However, active PKR was not enriched in, or detectably recruited to, G3BP1-induced stress granules (Fig. 7A). Pearson's correlation coefficient, calculated from several fields, was 0.04 with a high P value, further supporting the conclusion that active PKR does not colocalize with G3BP1-induced SGs.
FIG 7.

Determinants of PKR recruitment to SGs. (A) Immunofluorescence microscopy with PKR phospho-T446-specific antibody. Cells were transfected with GFP or G3BP1-GFP, as indicated, and GFP localization was detected directly by fluorescence. T, transfected cells; U, untransfected cell. Pearson's correlation coefficient (PCC) for colocalization of P-PKR (red) with G3BP1-GFP (green), along with the relevant P value, are indicated on the right. (B) Domain map of human PKR with mutated amino acids indicated. RBD, RNA-binding domain. K60/64 and K150/154 are residues implicated in RNA binding; K296 is a key residue within the catalytic site of PKR. (C) G3BP1 expressed from pcDNA 3.1 HisC-G3BP1 (green) and hPKR-mCherry (red) were coexpressed in HeLa cells to investigate colocalization. Colocalization of the wild type, a K60/64/150/154A mutant deficient in RNA binding, and the K296W catalytically inactive PKR mutant was examined, as indicated. G3BP1 transgene expression was detected with anti-T7 antibodies. The PCC and P value for colocalization of each PKR mutant with G3BP1 are indicated.
We also tested colocalization of PKR-mCherry fusion proteins containing mutations in PKR. We expressed a mutant with four point mutations that knock out RNA-binding domain function (K60/64 and K150/154) (19) or a catalytically dead mutant of PKR-mCherry (Fig. 7B). Both of these PKR-mCherry mutants displayed partial localization to G3BP1-induced SGs that was indistinguishable from that of wt PKR-mCherry (Fig. 7C). These findings are supported by Pearson's correlation coefficients of 0.51 and 0.74 for the RNA-binding and catalytically dead mutants, respectively, indicating colocalization. We confirmed that wt PKR-mCherry could be activated and was functional with the tag (data not shown). Taken together, since mutant PKR can localize to SGs normally but activated PKR does not, the data are most consistent with localization of a subset of PKR into granules that occurs prior to activation.
The G3BP1 PXXP domain is required for the recruitment of PKR to SGs and activation.
To probe G3BP1 determinants for PKR recruitment, we investigated a mutant of G3BP1 lacking the internal flexible domain containing a PXXP motif (designated the PXXP domain) (Fig. 1A). When G3BP1ΔPXXP was expressed, it could still nucleate granules in HeLa and U2OS cells and G3BP1 KO MEFs, but unlike wild-type G3BP1, G3BP1ΔPXXP-induced granules did not contain detectable PKR (Fig. 8A). Consistent with the microscopy, Pearson's correlation coefficients go from high positive values for wild-type G3BP1 expression, indicative of strong colocalization, to near zero for G3BP1ΔPXXP, thus confirming our data. Since PKR is the only eIF2α kinase induced by G3BP1 expression (15), eIF2α phosphorylation serves as an effective indicator of PKR activation in this system. Cells with G3BP1ΔPXXP-induced granules also failed to produce high levels of eIF2α phosphorylation (Fig. 8B), in contrast to expression of wt G3BP1 in both HeLa and U2OS cells (40- and 7-fold reduction, respectively). A similar but less pronounced effect on eIF2α phosphorylation was observed in G3BP1 KO MEFs (72 versus 49% of cells for wild-type G3BP1 versus G3BP1ΔPXXP granules, respectively) (Fig. 8B). Taken together, these data suggest that recruitment of PKR to SGs and its activation require the G3BP1 PXXP domain.
FIG 8.

G3BP1ΔPXXP-induced granules uncouple G3BP1 aggregation from eIF2α phosphorylation. (A) Stress granules were induced with either wild-type G3BP1 or G3BP1ΔPXXP (green) expression in HeLa and U2OS cells and G3BP1 KO MEFs, as indicated. The cells were fixed and stained for PKR (red). The PCC and P value are indicated for each condition. The insets represent the boxed areas. (B) HeLa, U2OS, and G3BP1 KO cells were transfected with wild-type G3BP1 or G3BP1ΔPXXP and stained for phospho-eIF2α as previously described (15). As a control, untransfected cells were treated with sodium arsenite at 500 μM for 30 min. eIF2α phosphorylation was quantified as described in Materials and Methods. Standard errors were calculated for HeLa cells in three experiments, and representative experiments with U2OS cells and MEFs are shown. The y axis represents the percentage of cells with large G3BP1-induced SGs that scored positive for eIF2α phosphorylation, as previously described (15).
The PXXP domain within G3BP1 is essential for nucleating SGs of normal composition.
The morphology of G3BP1ΔPXXP granules differed from that of wild-type G3BP1 granules because G3BP1ΔPXXP granules could grow larger and often partly surrounded the nucleus (Fig. 9B shows examples of large granules). Thus, we probed whether other canonical SG markers are recruited to G3BP1ΔPXXP-induced granules in these cell lines. Indeed, we found that several translation initiation factors were not efficiently recruited to G3BP1ΔPXXP-induced granules in HeLa and U2OS cells, unlike G3BP1-induced granules. They included the key factors eIF4G1 and poly(A)-binding protein (PABP), as shown in Fig. 9A and B, and eIF3a (data not shown). In G3BP1 KO MEFs, partial recruitment of eIF4G1 and eIF3a to G3BP1ΔPXXP-induced granules was observed. Recruitment of factors in G3BP1 KO MEFs was restricted to smaller, and not larger, granules for reasons that are not yet understood. Overall these results indicate that granule assembly mediated by G3BP1ΔPXXP differs from wild-type G3BP1-mediated SG assembly and that the PXXP domain and/or another missing factor(s) is required for PKR recruitment and activation in response to assembly of G3BP1-induced SGs. Differences in expression levels of this unknown factor(s) between the cell lines examined may alter PKR activation.
FIG 9.

Enteroviruses replicate in cells with G3BP1ΔPXXP granules. (A) HeLa, U2OS, and G3BP1 KO cells were transfected with wild-type G3BP1-GFP or G3BP1ΔPXXP (green) and counterstained using antibodies against eIF4G (red). (B) HeLa and U2OS cells were transfected with wild-type G3BP1 or G3BP1ΔPXXP (green) and stained with antibodies against PABP (red). The insets represent the boxed areas. (C) HeLa cells were transfected with the indicated transgene and infected 16 h posttransfection with CVB5 at an MOI of 0.1, and the kinetics of virus in the supernatant were measured with TCID50 assays. Transfection efficiency approached 90% in the experiment. (D) At each time point in panel C, HeLa cells were harvested and IFN-β mRNA was measured using qPCR. IFN-β mRNA levels are expressed relative to a β-actin internal-control mRNA. (E) HeLa cells expressing the indicated GFP-tagged transgenes (green; G3BP1ΔNTF2-GFP [G3BP1ΔNTF2] and G3BP1ΔPXXP-GFP [G3BP1ΔPXXP]) were infected at an MOI of 5 for 7.5 h with CVB3-dsRed (red). Cells were harvested and imaged by epifluorescence microscopy. Mock-infected cells showed no red signal and are therefore not shown. Cells expressing transgenes are marked with a T. All other cells were untransfected. (F) Transfected HeLa cells from panel E were quantified as described in Materials and Methods for the presence of red signal indicative of CVB3 infection. The values represent the percentages of transgene-expressing cells that were red. (D and E) The error bars represent standard errors. *, P < 0.05; **, P < 0.01; ***P < 0.001; Student's t test.
Since G3BP1ΔPXXP does not recruit PKR to SGs and PKR is not activated in cells with G3BP1ΔPXXP-induced stress granules, we considered whether G3BP1ΔPXXP may exhibit impaired antiviral activity. Therefore, we expressed either GFP, G3BP1-GFP, G3BP1ΔNTF2, or G3BP1ΔPXXP in HeLa cells, followed by infection with CVB5 at an MOI of 0.1. Supernatant was collected at several time points, and virus titers were measured to obtain kinetic information about CVB5 replication in the presence of G3BP1 mutants. Consistent with the results shown in Fig. 1 and previous findings for enteroviruses (10, 37), G3BP1 expression restricted CVB5 replication by approximately 11-fold. In contrast, expression of either the G3BP1ΔNTF2 or the G3BP1ΔPXXP mutant devoid of functional SGs did not inhibit CVB5 replication significantly (Fig. 9C). We analyzed IFN-β mRNA expression during the course of CVB5 infection because NF-κB participates in IFN-β transcription, NF-κB is activated during G3BP1 expression (Fig. 2), and NF-κB has been reported to act downstream of PKR (18, 38). Indeed, we observed statistically significant, elevated IFN-β mRNA levels by qPCR at 12 and 18 h p.i. only during expression of wild-type G3BP1 but not that of either the G3BP1ΔNTF2 or the G3BP1ΔPXXP mutant (Fig. 9D).
To confirm that the G3BP1ΔPXXP mutant does not contain the wt G3BP1 antiviral activity, presumably because it cannot recruit PKR to SGs, we transfected HeLa cells with either GFP alone, wild-type G3BP1, G3BP1ΔNTF2, or G3BP1ΔPXXP. The transfected cells were then infected with CVB3 expressing dsRed at an MOI of 3. IFA was used to monitor infection of individual cells expressing transgenes. As expected, cells expressing GFP, G3BP1ΔNTF2, or G3BP1ΔPXXP were infected at similar levels, while expression of wild-type G3BP1 showed reduced infection by CVB3-dsRed (Fig. 9E). Even cells with large G3BP1ΔPXXP stress granules were infected as often as cells expressing either GFP or G3BP1ΔNTF2, unlike the expression of wild-type G3BP1. Quantification of transfected cells with red signal indicative of CVB3 infection shows that expression of wild-type G3BP1 protected the cells from infection by 2.5- to 3-fold over GFP alone, G3BP1ΔNTF2, or G3BP1ΔPXXP (Fig. 9F). These findings support our findings that assembly of stress granules is important for PKR activation and that PKR recruitment to SGs and innate immune activation are dependent on the PXXP domain of G3BP1.
DISCUSSION
Many virus systems have been found to resist SG formation and to target key SG nucleating proteins, particularly G3BP1 and Tia1, to inhibit SG assembly. We previously found that poliovirus 3Cpro cleaves G3BP1 to accomplish this task, but why these functions have evolved is unknown. Since SGs are an extension of the translational control pathway, one reason could be to block sequestration of translation factors and machinery within SGs to enable their use for virus protein translation. However, a more compelling reason might be to block innate immune activation and signaling. An attractive emerging hypothesis is that stress granules can link cell stress responses to innate immune activation by serving as platforms to initiate or amplify innate immune signaling (7, 12). Most viruses induce cell stress at multiple levels as replication cycles initiate. A stress-activated virus surveillance system could play an ancillary role to the well-evolved pathogen-associated molecular pattern (PAMP) and pattern recognition receptor (PRR) systems. PKR is a key PRR in this system, and thus, the G3BP1-PKR axis represents a logical linkage.
Here, we have built on previous findings that G3BP1 expression induces SG formation that somehow results in PKR activation (15) and have uncovered additional determinants for G3BP1 and PKR functions in this activation pathway. First, we established that G3BP1 possesses antiviral activities against several enteroviruses using overexpression in cells or rescue of G3BP1 in G3BP1 KO MEFs. We show that G3BP1 can activate effectors of the innate immune transcriptional program, culminating in enhanced expression of a set of cytokines. We demonstrate that a subset of PKR is recruited to SGs, that close-proximity interactions between G3BP1 and PKR complexes increase in response to stress and PKR activation, that once activated PKR no longer associates with SGs, and that the PXXP domain of G3BP1 is essential for PKR recruitment to SGs and PKR activation in cells. Together, these findings suggest that G3BP1 plays an important role in the recruitment of PKR to SGs and suggest that activation of PKR can take place at the SG. Finally, we show that innate immune activation and antiviral functions of G3BP1 are dependent on both the NTF2 and PXXP domains of G3BP1, likely because the domains participate in assembly of normal stress granules. Thus, G3BP1 provides a mechanistic link for cross talk between stress-responsive and innate immune pathways and acts as an antiviral protein.
G3BP1 activation of NF-κB and JNK.
In this work, we showed that G3BP1 activates innate immunity, including NF-κB and JNK transcriptional responses, but only weakly activates STAT1 and, curiously, does not activate IRF3 transcription. However, during CVB5 infection, a transient elevation of IFN-β mRNA was observed that was dependent on functional G3BP1 and may be linked to NF-κB. Preliminary data indicate G3BP1-induced NF-κB transcriptional activation may not be stress granule dependent, whereas AP1 (JNK) transcriptional control does appear to require G3BP1-induced SGs (data not shown). PKR has been shown to modulate NF-κB activity by interacting with the IκK complex (38, 39). Our finding that G3BP1 can recruit PKR suggests G3BP1 may act as a signaling scaffold that regulates the downstream innate immune response. In this role, G3BP1 could bridge signaling components like PKR to transduce a signal to the transcription factor NF-κB, thereby mediating an innate immune transcriptional response. Work is under way to determine if G3BP1 can activate NF-κB directly.
AP1 is a transcription factor complex activated in response to activation of Jun N-terminal protein kinase (JNK). It is interesting that the NF-κB and JNK pathways have previously been reported to antagonize each other by modulating expression of GADD45 subunits and communicating with the programmed cell death machinery (40, 41). These studies provide a plausible mechanism by which G3BP1 and/or SGs may link NF-κB and JNK signaling, thereby controlling cell fate. SGs themselves may also play a role in the regulation of apoptosis induction, considering that they recruit Rack1 during some stresses, which prevents activation of MAPK signaling that is important for activation of the cell death program (42).
Determinants for PKR recruitment to SGs.
We show that G3BP1 is responsible for PKR recruitment to SGs, but whether G3BP1 directly or indirectly participates in activation of PKR is still unclear. The G3BP1-PKR proximity foci we observed suggest G3BP1 and PKR assemble in complexes as SGs form in response to oxidative stress or poly(I · C) treatment, with either indirect or direct interactions. The increase in G3BP1-PKR complexes under the two distinct conditions may simply represent their coordinate recruitment to stress granules. However, some reports indicate that PKR is activated in arsenite-treated cells (43, 44), so increased PLA signal under arsenite-treated conditions may increase similarly to poly(I·C)-treated cells as a result of a PKR activation complex containing G3BP1 and PKR. Since there is very little PKR present under conditions devoid of IFN and PKR is a catalytic protein, few G3BP1-PKR complexes would theoretically be needed to mount a significant response to a viral infection.
It is clear that PKR activation is not required for recruitment to SGs, since PKR point mutants that are catalytically dead or abrogated for RNA binding are recruited to SGs to the same extent as wild-type PKR in HeLa cells. These results suggest PKR recruitment depends on other, larger regions or conformation for stress granule localization. On the other hand, localization data must be interpreted with caution, since PKR can dimerize and endogenous, functional PKR protein was present in these experiments. However, the current paradigm for PKR activation indicates that these mutants should not be dimerized with wild-type, untagged, endogenous PKR (45). It is likely that PKR, like G3BP1, is rapidly exchanged in and out of SGs. In addition, the finding that active P-PKR is not found in SGs supports our working model, where inactive PKR is recruited to SGs and undergoes activation at the granule and active kinase is released back into the surrounding cytoplasm to engage with its substrates and activate the innate immune response.
Previous work has employed G3BP1 and PKR knockdown experiments to identify a role for SGs as a scaffolding platform to orchestrate the innate immune response, culminating in induction of IFN-β (7, 12). Our findings using G3BP1-induced SGs where no additional stressor is added to cells support the role of properly assembled stress granules in mediating innate immune activation. Indeed, we observed a spike in IFN-β mRNA following infection with CVB5 only when proper stress granules could be assembled by wild-type G3BP1 expression. In this system, the peak in IFN-β mRNA may be transient, since CVB5 can disassemble stress granules similarly to CVB3 and poliovirus (10, 37), and the small magnitude of IFN-β synthesis is a result of the rapid infection kinetics of CVB5 that result in cleavage of multiple upstream IFN activators.
The data support a role for the PXXP domain in PKR recruitment and activation, since eIF2α phosphorylation was deficient in cells with G3BP1ΔPXXP granules and, critically, these granules did not contain PKR. G3BP1ΔPXXP-induced aggregates are not normal SGs but rather appear to be defective granules lacking key translation factors characteristic of bona fide SGs. The lack of PKR recruitment could reveal a dependence on a third factor(s) missing from the defective granules or, more simply, be a consequence of G3BP1 interacting with PKR through the PXXP domain. Still, deletion of the PXXP domain uncouples G3BP1 aggregation from PKR recruitment, activation, and PKR-mediated translational repression. Previous results in picornavirus systems where G3BP1 is cleaved have suggested that the importance of G3BP1 in SG biology originates from the ability of the N-terminal protein-interacting domains to be physically linked with the C-terminal RNA interaction domains (7, 10, 37). Here, we show that the PXXP domain also regulates proper aggregation, recruitment of SG factors, and translational repression. Consistent with an important role for the PXXP domain of G3BP1 in innate immunity, deletion of this domain renders cells sensitive to enterovirus infection.
We performed extensive characterizations of biochemical interactions of G3BP1 and G3BP1 truncations with PKR. We determined that G3BP1 and inactive, but not active, PKR directly interact as purified proteins, dependent on both the NTF2-like and PXXP domains (L. C. Reineke, N. Kedersha, M. A. Langereis, F. J. M. van Kuppeveld, and R. E. Lloyd, submitted for publication). Further, in cells, an intricate binding relationship exists between inactive PKR and G3BP1, which is aided by Caprin 1 and retains binding to activated PKR after its release from G3BP1 (Reineke et al., submitted). Since only inactive PKR enters SGs, such a scenario highlights an important function of SGs in the PKR activation process.
Roles of G3BP1 and other factors in PKR activation.
This work shows a role for G3BP1 in PKR recruitment to SGs and indicates that PKR activation takes place at the granule. More work is required to determine if G3BP1 plays only scaffolding/recruitment roles, can activate PKR alone, or requires a third factor or several other cofactors to activate PKR. Other PKR-interacting proteins have been described that either activate PKR (PACT, IPS1, and NFAR1/2) or inhibit PKR (MDA7, hDUS2, and p58IPK) (46–52). It would be of interest to investigate whether these proteins also interact with G3BP1-induced SGs and modulate SG assembly. NF45 has been shown to antagonize SG assembly, in contrast to the NF45-interacting proteins NFAR1/2 (47). NFAR1 (NF90) has been shown to promote PKR activation and SG assembly (46). As such, these proteins are attractive candidates for future studies delineating proteins involved in the SG-dependent mechanism for PKR activation.
In conclusion, this study is the first to show that a stress granule structural protein can facilitate the innate immune response by interacting with the innate immune machinery. On one hand, G3BP1 and SGs can signal through PKR to the translation apparatus to regulate translation of viral genomes. On the other hand, G3BP1 can regulate an innate immune transcriptional response to promote expression of other proteins that dampen the severity of viral infection. Since these responses are triggered by a stress granule protein in conjunction with nucleation of stress granules, this establishes a mechanistic linkage between cellular stress responses and innate immune activation that can help control enterovirus infection.
ACKNOWLEDGMENTS
We thank James White for scientific contributions that set up this work.
This work was funded by NIH Public Health Service grant AI50237 and NCI Cancer Center Support grant (P30CA125123). Additional support was provided by the Integrated Microscopy Core at Baylor College of Medicine with funding from the NIH (HD007495, DK56338, and CA125123), the Dan L. Duncan Cancer Center, and the John S. Dunn Gulf Coast Consortium for Chemical Genomics.
REFERENCES
- 1.Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, Anderson P. 2002. Evidence that ternary complex (eIF2-GTP-tRNA (i) (Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell 13:195–210. doi: 10.1091/mbc.01-05-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. 2005. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 169:871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ariumi Y, Kuroki M, Kushima Y, Osugi K, Hijikata M, Maki M, Ikeda M, Kato N. 2011. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J Virol 85:6882–6892. doi: 10.1128/JVI.02418-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cristea IM, Rozjabek H, Molloy KR, Karki S, White LL, Rice CM, Rout MP, Chait BT, MacDonald MR. 2010. Host factors associated with the Sindbis virus RNA-dependent RNA polymerase: role for G3BP1 and G3BP2 in virus replication. J Virol 84:6720–6732. doi: 10.1128/JVI.01983-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pager CT, Schütz S, Abraham TM, Luo G, Sarnow P. 2013. Modulation of hepatitis C virus RNA abundance and virus release by dispersion of processing bodies and enrichment of stress granules. Virology 435:472–484. doi: 10.1016/j.virol.2012.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reineke LC, Lloyd RE. 2013. Diversion of stress granules and P-bodies during viral infection. Virology 436:255–267. doi: 10.1016/j.virol.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng CS, Jogi M, Yoo JS, Onomoto K, Koike S, Iwasaki T, Yoneyama M, Kato H, Fujita T. 2013. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J Virol 87:9511–9522. doi: 10.1128/JVI.03248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parker F, Maurier F, Delumeau I, Duchesne M, Faucher D, Debussche L, Dugue A, Schweighoffer F, Tocque B. 1996. A Ras-GTPase-activating protein SH3-domain-binding protein. Mol Cell Biol 16:2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panas MD, Varjak M, Lulla A, Eng Merits KEA, Karlsson Hedestam GB, McInerney GM. 2012. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol Biol Cell 23:4701–4712. doi: 10.1091/mbc.E12-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White JP, Cardenas AM, Marissen WE, Lloyd RE. 2007. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2:295–305. doi: 10.1016/j.chom.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Bidet K, Dadlani D, Garcia-Blanco MA. 2014. G3BP1, G3BP2 and CAPRIN1 are required for translation of interferon stimulated mRNAs and are targeted by a Dengue virus non-coding RNA. PLoS Pathog 10:e1004242. doi: 10.1371/journal.ppat.1004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Onomoto K, Jogi M, Yoo J-S, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, Matsumiya T, Namiki H, Yoneyama M, Fujita T. 2012. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7:e43031. doi: 10.1371/journal.pone.0043031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langereis MA, Feng Q, van Kuppeveld FJ. 2013. MDA5 localizes to stress granules, but this localization is not required for the induction of type I interferon. J Virol 87:6314–6325. doi: 10.1128/JVI.03213-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takahashi M, Higuchi M, Matsuki H, Yoshita M, Ohsawa T, Oie M, Fujii M. 2013. Stress granules inhibit apoptosis by reducing reactive oxygen species production. Mol Cell Biol 33:815–829. doi: 10.1128/MCB.00763-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reineke LC, Dougherty JD, Pierre P, Lloyd RE. 2012. Large G3BP-induced granules trigger eIF2α phosphorylation. Mol Biol Cell 23:3499–3510. doi: 10.1091/mbc.E12-05-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tourriere H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Tazi J. 2003. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J Cell Biol 160:823–831. doi: 10.1083/jcb.200212128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Varshavsky A. 2011. The N-end rule pathway and regulation by proteolysis. Protein Sci 20:1298–1345. doi: 10.1002/pro.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taghavi N, Samuel CE. 2012. Protein kinase PKR catalytic activity is required for the PKR-dependent activation of mitogen-activated protein kinases and amplification of interferon beta induction following virus infection. Virology 427:208–216. doi: 10.1016/j.virol.2012.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel RC, Stanton P, Sen GC. 1996. Specific mutations near the amino terminus of double-stranded RNA-dependent protein kinase (PKR) differentially affect its double-stranded RNA binding and dimerization properties. J Biol Chem 271:25657–25663. doi: 10.1074/jbc.271.41.25657. [DOI] [PubMed] [Google Scholar]
- 20.Wasserman T, Katsenelson K, Daniliuc S, Hasin T, Choder M, Aronheim A. 2010. A novel c-Jun N-terminal kinase (JNK)-binding protein WDR62 is recruited to stress granules and mediates a nonclassical JNK activation. Mol Biol Cell 21:117–130. doi: 10.1091/mbc.E09-06-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee SH, Wang X, DeJong J. 2000. Functional interactions between an atypical NF-kappaB site from the rat CYP2B1 promoter and the transcriptional repressor RBP-Jkappa/CBF1. Nucleic Acids Res 28:2091–2098. doi: 10.1093/nar/28.10.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tabor-Godwin JM, Ruller CM, Bagalso N, An N, Pagarigan RR, Harkins S, Gilbert PE, Kiosses WB, Gude NA, Cornell CT, Doran KS, Sussman MA, Whitton JL, Feuer R. 2010. A novel population of myeloid cells responding to coxsackievirus infection assists in the dissemination of virus within the neonatal CNS. J Neurosci 30:8676–8691. doi: 10.1523/JNEUROSCI.1860-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zekri L, Chebli K, Tourriere H, Nielsen FC, Hansen TVO, Rami A, Tazi J. 2005. Control of fetal growth and neonatal survival by the RasGAP-associated endoribonuclease G3BP. Mol Cell Biol 25:8703–8716. doi: 10.1128/MCB.25.19.8703-8716.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kedersha N, Tisdale S, Hickman T, Anderson P. 2008. Real-time and quantitative imaging of mammalian stress granules and processing bodies RNA turnover in eukaryotes: nucleases, pathways and anaylsis of mRNA decay, 1st ed Elsevier Inc., New York, NY. [DOI] [PubMed] [Google Scholar]
- 25.Byrd MP, Zamora M, Lloyd RE. 2005. Translation of eukaryotic translation initiation factor 4GI (eIF4GI) proceeds from multiple mRNAs containing a novel cap-dependent internal ribosome entry site (IRES) that is active during poliovirus infection. J Biol Chem 280:18610–18622. doi: 10.1074/jbc.M414014200. [DOI] [PubMed] [Google Scholar]
- 26.White JP, Reineke LC, Lloyd RE. 2011. Poliovirus switches to an eIF2-independent mode of translation during infection. J Virol 85:8884–8893. doi: 10.1128/JVI.00792-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vandermark ER, Deluca KA, Gardner CR, Marker DF, Schreiner CN, Strickland DA, Wilton KM, Mondal S, Woodworth CD. 2012. Human papillomavirus type 16 E6 and E7 proteins alter NF-kB in cultured cervical epithelial cells and inhibition of NF-kB promotes cell growth and immortalization. Virology 425:53–60. doi: 10.1016/j.virol.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nees M, Geoghegan JM, Hyman T, Frank S, Miller L, Woodworth CD. 2001. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J Virol 75:4283–4296. doi: 10.1128/JVI.75.9.4283-4296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ronco LV, Karpova AY, Vidal M, Howley PM. 1998. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Gene Dev 12:2061–2072. doi: 10.1101/gad.12.13.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hebner CM, Wilson R, Rader J, Bidder M, Laimins LA. 2006. Human papillomaviruses target the double-stranded RNA protein kinase pathway. J Gen Virol 87:3183–3193. doi: 10.1099/vir.0.82098-0. [DOI] [PubMed] [Google Scholar]
- 31.Martin S, Zekri L, Metz A, Maurice T, Chebli K, Vignes M, Tazi J. 2013. Deficiency of G3BP1, the stress granules assembly factor, results in abnormal synaptic plasticity and calcium homeostasis in neurons. J Neurochem 125:175–184. doi: 10.1111/jnc.12189. [DOI] [PubMed] [Google Scholar]
- 32.Piotrowska J, Hansen SJ, Park N, Jamka K, Sarnow P, Gustin KE. 2010. Stable formation of compositionally unique stress granules in virus-infected cells. J Virol 84:3654–3665. doi: 10.1128/JVI.01320-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoo J-S, Takahasi K, Ng CS, Ouda R, Onomoto K, Yoneyama M, Lai JC, Lattmann S, Nagamine Y, Matsui T, Iwabuchi K, Kato H, Fujita T. 2014. DHX36 Enhances RIG-I signaling by facilitating PKR-mediated antiviral stress granule formation. PLoS Pathog 10:e1004012. doi: 10.1371/journal.ppat.1004012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang P, Langland JO, Jacobs BL, Samuel CE. 2009. Protein kinase PKR-dependent activation of mitogen-activated protein kinases occurs through mitochondrial adapter IPS-1 and is antagonized by vaccinia virus E3L. J Virol 83:5718–5725. doi: 10.1128/JVI.00224-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius K-J, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson L-G, Landegren U. 2006. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 36.Söderberg O, Leuchowius K-J, Gullberg M, Jarvius M, Weibrecht I, Larsson L-G, Landegren U. 2008. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45:227–232. doi: 10.1016/j.ymeth.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 37.Fung G, Ng CS, Zhang J, Shi J, Wong J, Piesik P, Han L, Chu F, Jagdeo J, Jan E, Fujita T, Luo H. 2013. Production of a dominant-negative fragment Due to G3BP1 cleavage contributes to the disruption of mitochondria-associated protective stress granules during CVB3 infection. PLoS One 8:e79546. doi: 10.1371/journal.pone.0079546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonnet MC, Weil R, Dam E, Hovanessian AG, Meurs EF. 2000. PKR stimulates NF-kappa B irrespective of its kinase function by interacting with the Ikappa B kinase complex. Mol Cell Biol 20:4532–4542. doi: 10.1128/MCB.20.13.4532-4542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zerbini LF, Wang Y, Czibere A, Correa RG, Cho J-Y, Ijiri K, Wei W, Joseph M, Gu X, Grall F, Goldring MB, Zhou J-R, Libermann TA, Zhou J-R. 2004. NF-kappa B-mediated repression of growth arrest- and DNA-damage-inducible proteins 45alpha and gamma is essential for cancer cell survival. Proc Natl Acad Sci U S A 101:13618–13623. doi: 10.1073/pnas.0402069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G. 2001. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature 414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- 42.Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M. 2008. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat Cell Biol 10:1324–1332. doi: 10.1038/ncb1791. [DOI] [PubMed] [Google Scholar]
- 43.Daher A, Laraki G, Singh M, Melendez-Peña CE, Bannwarth S, Peters AHFM, Meurs EF, Braun RE, Patel RC, Gatignol A. 2009. TRBP control of PACT-induced phosphorylation of protein kinase R is reversed by stress. Mol Cell Biol 29:254–265. doi: 10.1128/MCB.01030-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patel CV, Handy I, Goldsmith T, Patel RC. 2000. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem 275:37993–37998. doi: 10.1074/jbc.M004762200. [DOI] [PubMed] [Google Scholar]
- 45.Lemaire PA, Anderson E, Lary J, Cole JL. 2008. Mechanism of PKR activation by dsRNA. J Mol Biol 381:351–360. doi: 10.1016/j.jmb.2008.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wen X, Huang X, Mok BW-Y, Chen Y, Zheng M, Lau S-Y, Wang P, Song W, Jin D-Y, Yuen K-Y, Chen H. 2014. NF90 exerts antiviral activity through regulation of PKR phosphorylation and stress granules in infected cells. J Immunol 192:3753–3764. doi: 10.4049/jimmunol.1302813. [DOI] [PubMed] [Google Scholar]
- 47.Shiina N, Nakayama K. 2014. RNA granule assembly and disassembly modulated by nuclear factor associated with double-stranded RNA 2 and nuclear factor 45. J Biol Chem 289:21163–21180. doi: 10.1074/jbc.M114.556365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee TG, Tomita J, Hovanessian AG, Katze MG. 1990. Purification and partial characterization of a cellular inhibitor of the interferon-induced protein kinase of Mr 68,000 from influenza virus-infected cells. Proc Natl Acad Sci U S A 87:6208–6212. doi: 10.1073/pnas.87.16.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel RC, Sen GC. 1998. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J 17:4379–4390. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang P, Li Y, Xia J, He J, Pu J, Xie J, Wu S, Feng L, Huang X, Zhang P. 2014. IPS-1 plays an essential role in dsRNA-induced stress granule formation by interacting with PKR and promoting its activation. J Cell Sci 127:2471–2482. doi: 10.1242/jcs.139626. [DOI] [PubMed] [Google Scholar]
- 51.Pataer A, Vorburger S, Chada S, Balachandran S, Barber G, Roth J, Hunt K, Swisher S. 2005. Melanoma differentiation-associated gene-7 protein physically associates with the double-stranded RNA-activated protein kinase PKR. Mol Ther 11:717–723. doi: 10.1016/j.ymthe.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 52.Mittelstadt M, Frump A, Khuu T, Fowlkes V, Handy I, Patel CV, Patel RC. 2008. Interaction of human tRNA-dihydrouridine synthase-2 with interferon-induced protein kinase PKR. Nucleic Acids Res 36:998–1008. doi: 10.1093/nar/gkm1129. [DOI] [PMC free article] [PubMed] [Google Scholar]