

ABSTRACT
Ebola virus (EBOV) causes hemorrhagic fevers with high mortality rates. During cellular entry, the virus is internalized by macropinocytosis and trafficked through endosomes until fusion between the viral and an endosomal membrane is triggered, releasing the RNA genome into the cytoplasm. We found that while macropinocytotic uptake of filamentous EBOV viruslike particles (VLPs) expressing the EBOV glycoprotein (GP) occurs relatively quickly, VLPs only begin to enter the cytoplasm after a 30-min lag, considerably later than particles bearing the influenza hemagglutinin or GP from lymphocytic choriomeningitis virus, which enter through late endosomes (LE). For EBOV, the long lag is not due to the large size or unusual shape of EBOV filaments, the need to prime EBOV GP to the 19-kDa receptor-binding species, or a need for unusually low endosomal pH. In contrast, since we observed that EBOV entry occurs upon arrival in Niemann-Pick C1 (NPC1)-positive endolysosomes (LE/Lys), we propose that trafficking to LE/Lys is a key rate-defining step. Additional experiments revealed, unexpectedly, that severe acute respiratory syndrome (SARS) S-mediated entry also begins only after a 30-min lag. Furthermore, although SARS does not require NPC1 for entry, SARS entry also begins after colocalization with NPC1. Since the only endosomal requirement for SARS entry is cathepsin L activity, we tested and provide evidence that NPC1+ LE/Lys have higher cathepsin L activity than LE, with no detectable activity in earlier endosomes. Our findings suggest that both EBOV and SARS traffic deep into the endocytic pathway for entry and that they do so to access higher cathepsin activity.
IMPORTANCE Ebola virus is a hemorrhagic fever virus that causes high fatality rates when it spreads from zoonotic vectors into the human population. Infection by severe acute respiratory syndrome coronavirus (SARS-CoV) causes severe respiratory distress in infected patients. A devastating outbreak of EBOV occurred in West Africa in 2014, and there was a significant outbreak of SARS in 2003. No effective vaccine or treatment has yet been approved for either virus. We present evidence that both viruses traffic late into the endocytic pathway, to NPC1+ LE/Lys, in order to enter host cells, and that they do so to access high levels of cathepsin activity, which both viruses use in their fusion-triggering mechanisms. This unexpected similarity suggests an unexplored vulnerability, trafficking to NPC1+ LE/Lys, as a therapeutic target for SARS and EBOV.
INTRODUCTION
Filoviruses are large filamentous viruses that cause deadly hemorrhagic fevers (1–3). Recently, much has been learned about how these viruses enter cells to initiate replication (for reviews, see references 4–7). After engaging host cell surface proteins, including C-type lectins and T-cell immunoglobulin and mucin domain proteins and Tyro3/Axl/Mer family members, Ebola virus (EBOV) particles are internalized by macropinocytosis and traffic through endosomes. En route, the receptor binding subunit of the EBOV glycoprotein (GP) is cleaved by cysteine proteases to generate a key 19-kDa form. The 19-kDa GP binds to its intracellular receptor, Niemann-Pick C1 (NPC1) (8–10), a multipass membrane protein that facilitates cholesterol egress from late endosomes (LEs) (11, 12). Yet, NPC1 binding appears to be insufficient to trigger the 19-kDa EBOV GP, and the final fusion-inducing events remain to be elucidated (4, 7, 10).
In the present study, we first compared the entry kinetics of EBOV and other viral particles. We found that, in the cells studied, EBOV begins to enter the cytoplasm only after a 30-min lag, considerably later than particles bearing the GP of lymphocytic choriomeningitis virus (LCMV) or influenza virus, which enter the cytoplasm through LEs (13–15). We further showed that the late EBOV entry profile is not governed by the unusually large size or shape of EBOV filaments, by their macropinocytotic internalization kinetics, by processing of EBOV GP to the 19-kDa species, or by a need for unusually low endosomal pH. Rather, a key rate-defining step is arrival in an NPC1+ late endosome-lysosome (LE/Lys), a hybrid organelle also referred to as an endolysosome. We also found, unexpectedly, that endosomal entry of severe acute respiratory virus coronavirus (SARS-CoV) also displays a late entry profile, commencing only after colocalization with NPC1. Since the only factor required for SARS endosomal entry is cathepsin (cat) L, we tested and provide evidence that NPC1+ LE/Lys contain higher levels of cat L activity than earlier endosomes. The implications of our findings for the sites of EBOV and SARS entry are discussed.
MATERIALS AND METHODS
Cells and viruses.
HEK293T cells (ATCC CRL-11268) and BSC-1 cells (grivet kidney; gift of Xiaowei Zhuang, Harvard University) were maintained in high-glucose Dulbecco's modified Eagle medium, 1% l-glutamine, 1% antibiotic/antimycotic, and 1% sodium pyruvate (Gibco Life Technologies). Media were supplemented with 10% supplemented calf serum (SCS) (HyClone) or 10% fetal bovine serum (Atlanta Biologicals) for HEK293T and BSC-1 cells, respectively. These media (with 10% serum) are referred to, respectively, as HEK293T medium and BSC-1 medium.
Plasmids.
Plasmids encoding VP40, mCherry-VP40, βlam-VP40, vesicular stomatitis virus (VSV) G, Zaire EBOV GPΔ, and LCMV GP were described previously (16). The plasmid encoding Zaire EBOV GP tagged (C-terminal end) with a V5 epitope was a gift of Paul Bates (University of Pennsylvania). The plasmid encoding SARS SΔ19 (SARS S containing a truncation of 19 C-terminal amino acids) was a gift of Shutoku Matsuyama and Fumihiro Taguchi (NIID, Tokyo, Japan). Green fluorescent protein (GFP)-Rab5 and GFP-Rab7 were gifts of Marino Zerial (Max Planck Institute of Molecular Cell Biology, Dresden, Germany), and NPC1-GFP was a gift of Matthew Scott (Stanford University).
Production of VLPs and HIV pseudovirions.
EBOV VP40-based filamentous VLPs were prepared as described previously (16) with two minor modifications: the ratio of plasmids (glycoprotein/VP40/βlam-VP40/mCherry-VP40) was 2:1:2:2. VLPs were pelleted through 20% sucrose-HM (20 mM HEPES, 20 mM morpholineethanesulfonic acid [MES], 130 mM NaCl, pH 7.4) and resuspended in 10% sucrose-HM. VLPs were snap-frozen in liquid N2 and stored at −80°C for long-term storage (in single-use aliquots). To generate the 19-kDa form of EBOV GP, EBOV GP-V5 VLPs were washed by pelleting through 20% sucrose as described above but resuspended in HM buffer overnight, followed by repelleting (124,214 × g for 2 h at 4°C) in an SW55 rotor. Washed EBOV GP-V5 VLPs were then resuspended in 10% sucrose-HM (1:100 starting volume of medium), and their protein concentration was determined by bicinchoninic acid (BCA). A total of 25 μg washed VLPs bearing EBOV GP-V5 (in 2 mM CaCl2, 10% sucrose, 20 mM HEPES, 20 mM MES, 150 mM NaCl, pH 7.4) was treated with 0.25 mg/ml thermolysin (VitaCyte) containing 0.5 mM CaCl2 at 37°C for 30 min. The reaction was quenched with 500 μM phosphoramidon (Sigma-Aldrich). The resultant 19-kDa EBOV GP VLPs were kept on ice until use. Cleavage of GP to 19 kDa was confirmed by Western blotting with mouse monoclonal antibody (MAb) H3C8 (against GP1 peptide 72 to 109; gift of Carolyn Wilson, FDA, Bethesda, MD).
HIV pseudovirions bearing EBOV GP or SARS S and Vpr-βlam were produced in HEK 293T cells as described previously (17) with minor modifications and clarifications: 10 μg instead of 6 μg of glycoprotein cDNA was used, the medium was changed at 4 h posttransfection to HEK293T medium (with 5% SCS), and the cells were not treated with sodium butyrate. Total media were collected at 48 h posttransfection and cleared twice of cell debris by centrifugation at 1,070 × g for 10 min at 4°C. Pseudovirions were then pelleted through 20% sucrose-HM for 2 h at 112,398 × g in an SW28 rotor at 4°C. Pseudovirions were resuspended overnight in 1:100 starting medium volume in 10% sucrose-HM at 4°C and then snap-frozen in liquid N2 and stored at −80°C for long-term storage (in single-use aliquots). Pseudovirions bearing SARS S were produced in HEK293T cells that were continually passaged with a nonenzymatic cell disassociation reagent (Sigma-Aldrich) to prevent S protein cleavage during pseudovirus production.
EBOV VLP internalization and EBOV VLP, HIV pseudovirion, and influenza entry assays.
EBOV VLP internalization assays were conducted as described previously (16). For EBOV VLP entry assays, 40 to 50,000 target cells were seeded into each well of a 96-well microtiter plate. After 18 to 24 h, when the cells were 90 to 100% confluent, VLPs (5 to 10 μl) diluted in chilled Opti-MEM I (OMEM; Gibco Life Technologies) were bound to cells by centrifugation at 250 × g for 60 min at 4°C, washed with OMEM, and then placed in a 37°C, 5% CO2 incubator. At the indicated times, the plates were placed on ice, and the medium was immediately replaced with OMEM containing 200 nM bafilomycin A1 (Sigma-Aldrich) to prevent further entry. After 10 min, the cells were moved to room temperature (RT), and at the end of the time course, all cells were loaded with the βlam substrate CCF2-AM (Invitrogen) and processed and analyzed as described previously (16). For experiments testing inhibitors, cells were incubated with the indicated inhibitors (indicated concentrations) for 30 to 60 min prior to the experiment, and inhibitors were present at all steps until substrate (CCF2-AM) loading. The titer was determined for each VLP prep for entry efficiency, and for experiments we used volumes of VLPs aimed to achieve a final extent of 40 to 50% entry. Across all experiments, the average final percentages of entry for VLPs with EBOV GP, VSV G, and LCMV GP were 43%, 56%, and 52%, respectively. Bald VLPs bearing no glycoprotein showed no cytoplasmic entry (not shown). HIV pseudovirion entry assays were conducted in the same manner, using 5 to 10 μl pseudovirions, with the addition of 5 μM E64d (EMD Millipore) to the bafilomycin quench medium and substrate-loading buffer. For experiments involving pseudovirions bearing SARS S, the target cells were BSC-1 cells continually passaged with a nonenzymatic cell disassociation reagent (Sigma-Aldrich). To assay influenza entry, 125,000 BSC-1 cells were seeded into each well of 24-well microtiter plates. After 18 to 24 h (cells at ∼90% confluence), 655 hemagglutinin (HA) units of X:31 influenza virus (Charles River) were diluted into 250 μl chilled OMEM and added to each well. After centrifuging (250 × g) for 30 min at 4°C, the plate was warmed in a 37°C, 5% CO2 incubator. At the indicated times, entry was quenched with 200 nM bafilomycin A1 diluted in OMEM. After a total of 6 h at 37°C (to allow for new HA expression), cells were lifted with Accumax (Innovative Cell Technologies, Inc.), pelleted at 800 × g (4°C), blocked with 3% bovine serum albumin (BSA) in phosphate-buffered saline (PBS++) for 15 min on ice, and incubated with the site A (HC3) anti-HA MAb (diluted 1:500 in 3% BSA-PBS++) for 45 min on ice. Cells were washed twice with 1 ml chilled PBS++, pelleted as described above, and stained with Alexa 488-labeled anti-mouse IgG (Molecular Probes; diluted 1:1,000 in 3% BSA-PBS++) for 30 min on ice. After being washed, as described above, cells were fixed with 4% paraformaldehyde (PFAM; Boston Bioproducts) for 15 min at RT. HA expression was then determined by flow cytometry.
EBOV GP VLP and SARS S HIV pseudovirion colocalization with NPC1.
For EBOV VLP colocalization, BSC-1 cells (125,000 cells) were seeded on glass coverslips in 24-well plates. After 18 to 24 h, the cells (∼90% confluent) were washed once with 4°C OMEM, EBOV GP VLPs (5 to 10 μl diluted in 250 μl chilled OMEM) were added, and the plates were centrifuged for 1 h at 250 × g (4°C). After replacing the medium with fresh OMEM, the cells were incubated at 37°C in a 5% CO2 incubator. At the indicated times, the coverslips were removed, fixed in 4% PFAM for 15 min, and then permeabilized and blocked in saponin solution (0.05% saponin, 10% SCS, 10 mM glycine, and 10 mM HEPES in PBS, pH 7.4) for 30 min. Rabbit anti-NPC1 MAb (Abcam) in saponin solution (1:500 dilution) was then added to each coverslip for 45 min. After the coverslips were washed three times with PBS (∼5 min per wash), Alexa 488-tagged anti-rabbit antibody IgG (Molecular Probes) in saponin solution (1:1,500 dilution) was added and the coverslips were incubated for 30 min and washed as described above. Coverslips were then mounted with Fluoromount G (Southern Biotech) and imaged with a Nikon C1 laser scanning confocal unit attached to a Nikon Eclipse TE2000-E microscope with a 100×, 1.45-numerical-aperature (NA) Plan Apochromat objective. A total of 20 to 30 images of cell fields with internalized VLPs were then acquired (per sample) and analyzed using the ImageJ software with the JACoP plugin to obtain Manders colocalization coefficients. Colocalization of SARS S pseudovirions with NPC1 was analyzed as described above, except that pseudovirion location was detected with an anti-HIV p24 MAb (183-H12-5C; NIH AIDS Reagent Program).
Characterization of NPC1+ endosomes.
BSC-1 cells (125,000) were seeded on glass coverslips in 24-well plates. After 18 to 24 h, the cells (∼90% confluent) were washed with PBS−, fixed for 15 min with 4% PFAM, permeabilized with 0.05% saponin for 15 min, and then blocked for 15 min with 3% BSA-PBS. Cells were then stained with the rabbit anti-NPC1 MAb described above and mouse MAbs specific for CD63 (1:500; Developmental Studies Hybridoma Bank [DSHB]), Lamp-1 (1:500; DSHB), LBPA (1:500; gift of Jean Gruenberg, University of Geneva, Switzerland), M6PR (1:100; Thermo-Scientific; mouse MAb), or EEA1 (1:500; BD Biosciences) for 45 min at RT. After cells were washed, primary antibodies were detected with (1:1,500) anti-rabbit Alexa Fluor 647 and (1:1,500) anti-mouse Alexa Fluor 546 (Invitrogen) for 45 min at RT. All antibodies were diluted in 3% BSA-PBS. After being washed with PBS and distilled and deionized H2O, coverslips were mounted using Fluoromount G (Southern Biotech). Images were then collected with a Nikon C1 laser scanning confocal microscope with a 100× objective as described above. Ten random fields were collected for each sample, and images were quantified for overlap using the JACoP plugin in ImageJ. Automatic thresholds were applied for both the green and red channels (uniformly to all images). The degree of colocalization is reported as an averaged Manders correlation coefficient.
Transient transfection of GFP-Rab5, GFP-Rab7, and NPC1-GFP.
BSC-1 cells (600,000) were seeded in 60-mm Falcon tissue culture dishes in BSC-1 medium (without antibiotic/antimycotic) and grown at 37°C with 5% CO2 for 14 to 18 h, at which time they were transfected as follows: 2.5 μg of DNA encoding GFP-Rab5, GFP-Rab7, or NPC1-GFP was diluted in phenol red-free DMEM without serum or other additives to a final volume of 150 μl. A total of 15 μl of PolyFect (Qiagen) transfection reagent was mixed into the diluted DNA and allowed to incubate for 10 min to allow complex formation. Meanwhile, cells were washed with PBS++ and replenished with 3 ml of fresh BSC-1 medium. After 10 min, 1 ml BSC-1 medium was added to the DNA-PolyFect solution, mixed up and down twice, and immediately added to the BSC-1 cells. A total of 5 to 7 h posttransfection, cells were washed twice with PBS++, lifted with 0.05% trypsin-EDTA, and quenched with BSC-1 medium. A total of 200,000 to 300,000 of the transfected cells were then seeded into 35-mm glass-bottom tissue culture dishes (MatTek), which had been coated with fibronectin (20 μg/ml in PBS++; Sigma-Aldrich) for 60 min prior to plating. Live-cell imaging experiments were conducted within 24 to 48 h posttransfection.
Live-cell imaging of cathepsin L (cat L) Magic Red activity probe and data analysis.
BSC-1 cells, transfected as described above, were pretreated with 10 μM CA-074-Me (Sigma-Aldrich), to inhibit cathepsin B (cat B) activity, in BSC-1 medium for 2 h. The cells were then washed and covered with 500 μl of imaging medium (10% FBS-FluoroBrite-DMEM, 10 mM HEPES, 10 μM CA-074-Me). Cells were then imaged every 5 s for 5 to 10 min on a motorized stage maintained at 37°C using a 100×/1.49 Nikon objective on a Nikon Eclipse TE2000-E microscope equipped with a Yokogawa CSU 10 spinning-disk confocal unit and a 512-by-512 Hamamatsu 9100c-13 EM-BT camera. A total of 10 to 25 s after commencing imaging, 500 μl of 37°C imaging medium containing the Magic Red cat L activity probe (ImmunoChemistry Technologies) as per the manufacturer's instructions was added without interrupting imaging. Image analysis was performed on images that were taken 2 to 3 min after addition of the probe. Individual endosomes (based on GFP fluorescence) were selected as regions of interest (ROI) using NIS-Elements (Nikon). Pearson's correlation coefficients between each ROI and the cat L probe fluorescence (red) were calculated using the same software. A total of 1,332 GFP-Rab5 endosomes, 1,238 GFP-Rab7 endosomes, and 1,314 NPC1-GFP endosomes were analyzed. Box plots of the Pearson's coefficients were generated using BoxPlotR (18). Statistical analysis was performed using GraphPad Prism 6 (GraphPad Software). To test for normality, a D'Agostino-Pearson omnibus normality test was performed. A Kruskal-Wallis test followed by a Dunn's posttest was completed to test for significance.
RESULTS
EBOV GP VLPs enter the cytoplasm later than particles bearing VSV G, LCMV GP, or influenza HA.
We first compared the internalization and entry kinetics of filamentous EBOV VLPs bearing either VSV G or EBOV GP. On authentic bullet-shaped VSV particles, the G protein directs virus internalization by clathrin-mediated endocytosis (CME) and entry primarily through early endosomes (19, 20). The VLPs we employed contain mCherry-VP40 and β-lactamase-VP40 (βlam-VP40) in addition to native VP40, which drives assembly of long filamentous particles. mCherry-VP40 is used to monitor VLP internalization from the cell surface and trafficking within the cell, and βlam-VP40 is used to monitor cytoplasmic entry using a well-characterized virus entry assay (16). Like standard EBOV VLPs (21, 22), these entry reporter VLPs exhibit the same morphology and entry properties of infectious EBOV, and they can be engineered to incorporate foreign viral GPs (16, 23, 24).
Filamentous VP40-based VLPs expressing VSV G were internalized quickly from the cell surface and entered the cytoplasm soon thereafter (Fig. 1A), recapitulating internalization and entry kinetics of authentic (smaller) bullet-shaped VSV particles (19). In contrast, although internalized relatively quickly, VLPs bearing EBOV GP began to enter the cytoplasm only after a 30-min lag (Fig. 1B) and therefore with considerably slower overall kinetics than similarly sized VLPs expressing VSV-G. After internalization by macropinocytosis (25, 26), LCMV and other arenaviruses enter cells through LEs (13). Nonetheless, filamentous VLPs bearing LCMV GP initiated cytoplasmic entry after a significantly shorter lag (15 min) than ones bearing either full-length EBOV GP or EBOV GPΔ, which lacks the mucin-like domain (Fig. 2A). VP40-driven VLPs expressing VSV G and LCMV GP were shown, by negative-stain electron microscopy, to be similarly shaped filaments as those bearing EBOV GP (data not shown). As for filamentous LCMV GP VLPs, spherical and relatively small X:31 influenza virus particles (∼100-nm diameter), which are known to enter the cytoplasm through LEs (14), also commenced cytoplasmic entry sooner (15-min lag) than VLPs bearing EBOV GP (Fig. 2B). The average half-times for entry mediated by VSV G, influenza HA, LCMV GP, and EBOV GP were 22.5 (±3.5), 40.0 (±7.1), 55.0 (±8.7), and 73.3 (±12.9) min, respectively. Collectively, the results in Fig. 1 and 2 indicate that while VLPs bearing EBOV GP are internalized from the cell surface relatively rapidly, they enter the cytoplasm slowly, even slower than influenza and LCMV, late-penetrating viruses that enter the cytoplasm through LE (15, 27). Furthermore, the results with VSV G and LCMV GP on filamentous VLPs (Fig. 1A and 2A) suggest that the unusual size and shape of the VLPs employed do not affect their entry kinetics.
FIG 1.
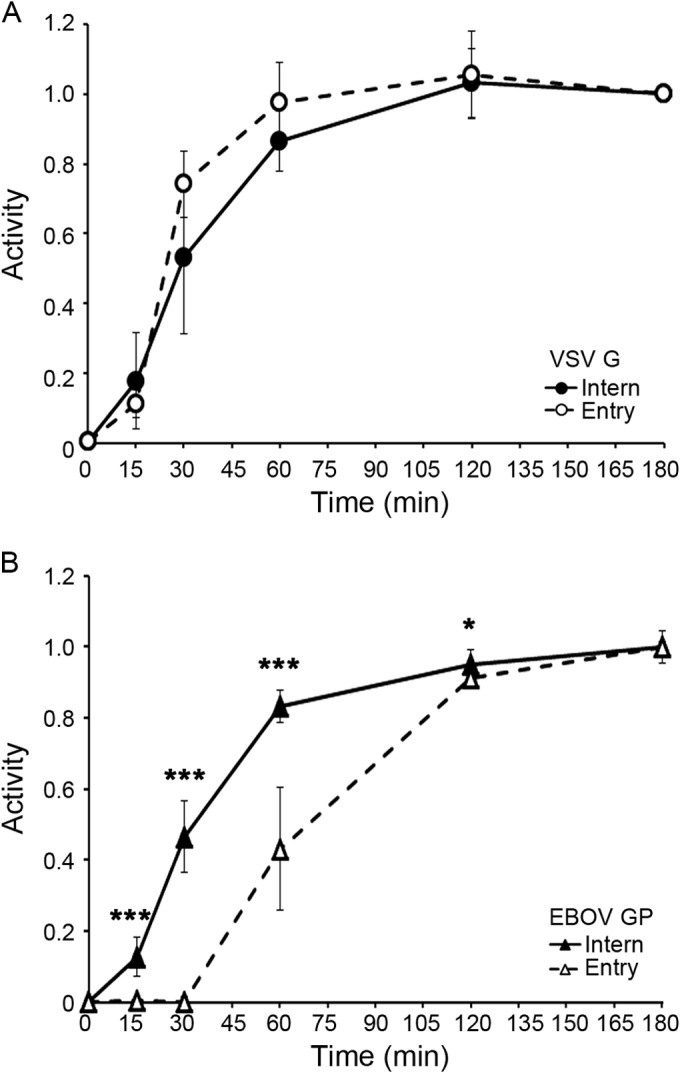
EBOV VLPs are internalized quickly but begin to enter the cytoplasm only after a 30-min lag. VLPs bearing either VSV G (A) or EBOV GP (B) were bound to the surface of BSC-1 cells at 4°C. After being washed, the cells were warmed to 37°C and assayed at the indicated times for internalization from the cell surface (closed symbols, solid lines) and cytoplasmic entry (open symbols, dashed lines) as described in Materials and Methods. Data points (normalized to values at 180 min) are averages from five samples from two experiments (A) and eight samples from three experiments (B); error bars indicate standard deviations (SD). Asterisks in panel B indicate statistical differences between values for EBOV internalization and entry (*, P < 0.05; *** P < 0.0005). Similar results were obtained in 293AD and SNB19 cells (not shown).
FIG 2.
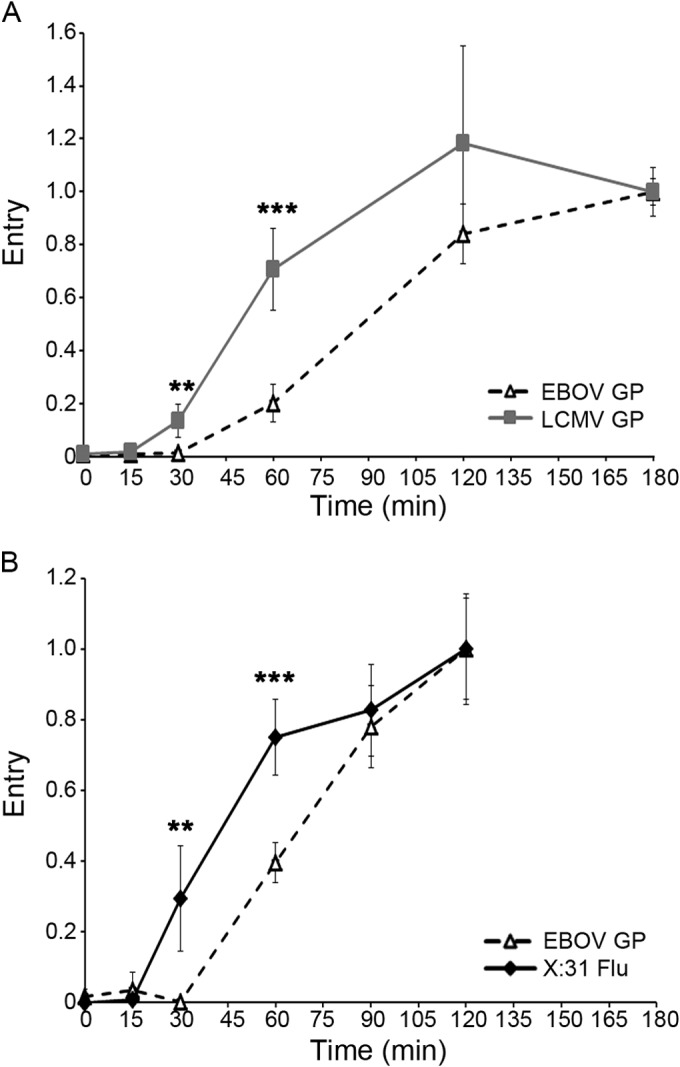
EBOV GP VLPs begin to enter the cytoplasm later than LCMV and influenza particles. (A) VLPs bearing either LCMV GP (squares, gray line) or EBOV GP (open triangles, dashed line) were bound to BSC-1 cells and assayed for cytoplasmic entry as described for Fig. 1. Each data point is the average from six samples (from three experiments performed in duplicate). Similar results were observed in experiments comparing LCMV GP and EBOV GPΔ-mediated entry into BSC-1 cells and LCMV GP and EBOV GP-mediated entry into SNB19 cells (not shown). (B) X:31 influenza virus particles (diamonds, solid line) or EBOV GP VLPs (triangles, dashed line) were bound to BSC-1 cells at 4°C. Cells were then warmed and processed and analyzed for cytoplasmic entry of EBOV GP VLPs as described for panel A. Entry of X:31 influenza virus was monitored as described in Materials and Methods. Each data point is the average from six samples (normalized to the values at 120 min) (from two experiments performed in triplicate). Error bars indicate SD. Asterisks indicate statistical significance between the extent of entry for EBOV GP versus LCMV GP (A) or EBOV GP versus influenza (B) particles: **, P < 0.005; ***, P < 0.0005. Both the lag before the onset of and the half-life for entry of EBOV GP VLPs are independent of the final extent of entry observed in individual experiments (not shown).
The lag before EBOV entry is not governed by internalization kinetics, particle size or shape, or need for EBOV GP priming or unusually low pH.
Figure 1 demonstrates that the long lag preceding EBOV cytoplasmic entry is not due to slow internalization kinetics. Here, we tested other properties that might influence the late EBOV GP entry kinetics. We first confirmed that late EBOV entry is not due to the large size (average length = 1 μm) or unusual filamentous shape of EBOV particles, as suggested previously (28) and above. Spherical, ∼100-nm-diameter HIV-βlam pseudovirions bearing EBOV GP entered the cytoplasm with the same kinetics as the large filamentous VLPs bearing EBOV GP (Fig. 3). In addition, VLPs bearing EBOV GP preprimed to 19-kDa GP (the form that binds to NPC1) entered the cytoplasm with the same kinetics as ones bearing either full-length GP or GPΔ (Fig. 4). Moreover, EBOV GP does not require exposure to unusually low pH to induce entry; VLPs bearing EBOV GP were not more sensitive to either ammonium chloride (Fig. 5A) or bafilomycin (Fig. 5B) than VLPs bearing VSV G or LCMV GP. In addition, VLPs bearing the 19-kDa GP showed the same sensitivity to bafilomycin as ones bearing the full-length GP (Fig. 5C). Hence, the ∼30-min lag before initiation of EBOV GP-mediated entry (Fig. 1 and 2) is not rate-limited by internalization kinetics (Fig. 1), particle size or shape (Fig. 3), priming to the 19-kDa GP1 (Fig. 4), or a need for unusually low endosomal pH (Fig. 5).
FIG 3.
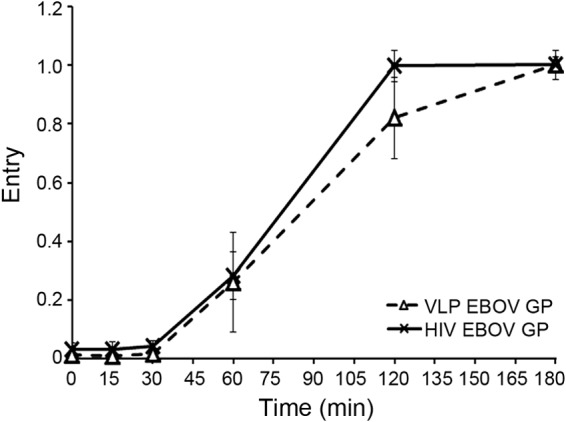
EBOV GP begins to mediate cytoplasmic entry after a 30-min lag whether on a filamentous VLP or a spherical retroviral pseudovirion. VLPs (triangles, dashed line) or HIV pseudovirions (x, solid line) bearing EBOV GP were bound to BSC-1 cells at 4°C, warmed to 37°C, and analyzed for cytoplasmic entry as described for Fig. 1. Each data point is the average from four samples (from two experiments performed in duplicate). Error bars indicate SD. None of the data points are statistically different at P values of <0.05.
FIG 4.
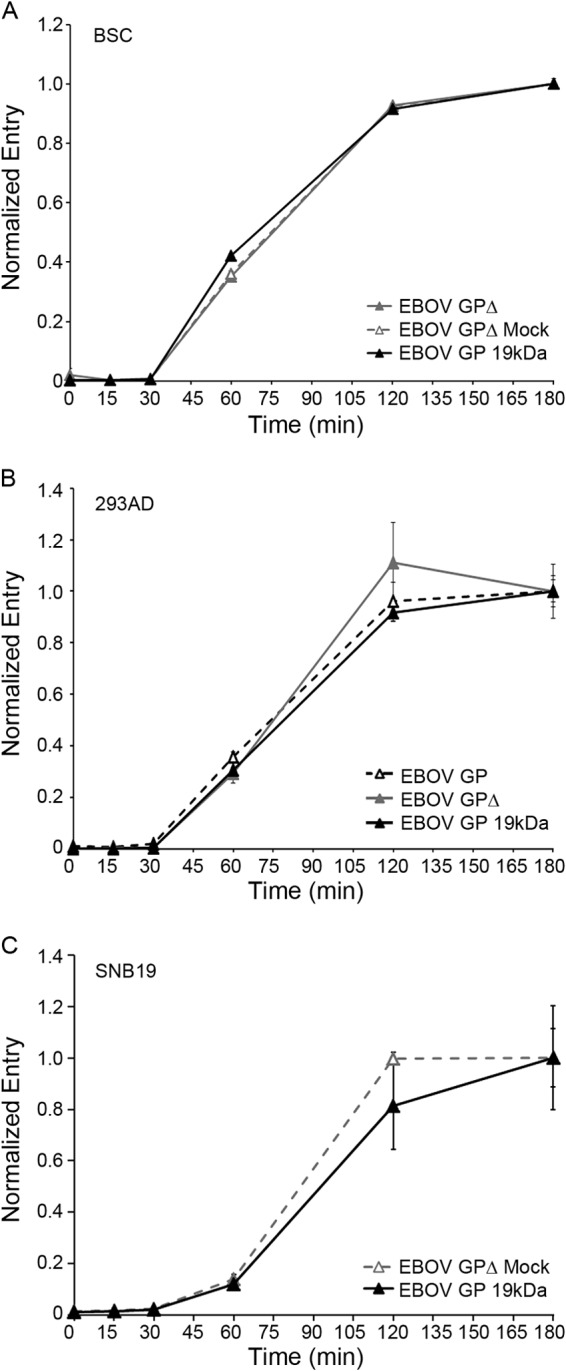
EBOV GP VLPs begin to enter the cytoplasm after a 30-min lag whether they bear full-length, Δmucin, or 19-kDa EBOV GP. VLPs bearing the indicated form of EBOV GP (full length, open black triangles, dashed black line; GPΔ, solid gray triangles, solid gray line; mock-cleaved GPΔ, open gray triangles, dashed gray line; 19-kDa GP, solid black triangle, solid black line) were bound to the surface of BSC (A), 293AD (B), or SNB19 (C) cells at 4°C, warmed, and assayed for cytoplasmic entry as described for Fig. 1. Data points in each panel are the averages from duplicate samples, and error bars indicate SD. Data for panel 4C are from Fig. 2.5B from a Ph.D. thesis (24). None of the comparative data points (for different forms of GP) are statistically different at P values of <0.05.
FIG 5.
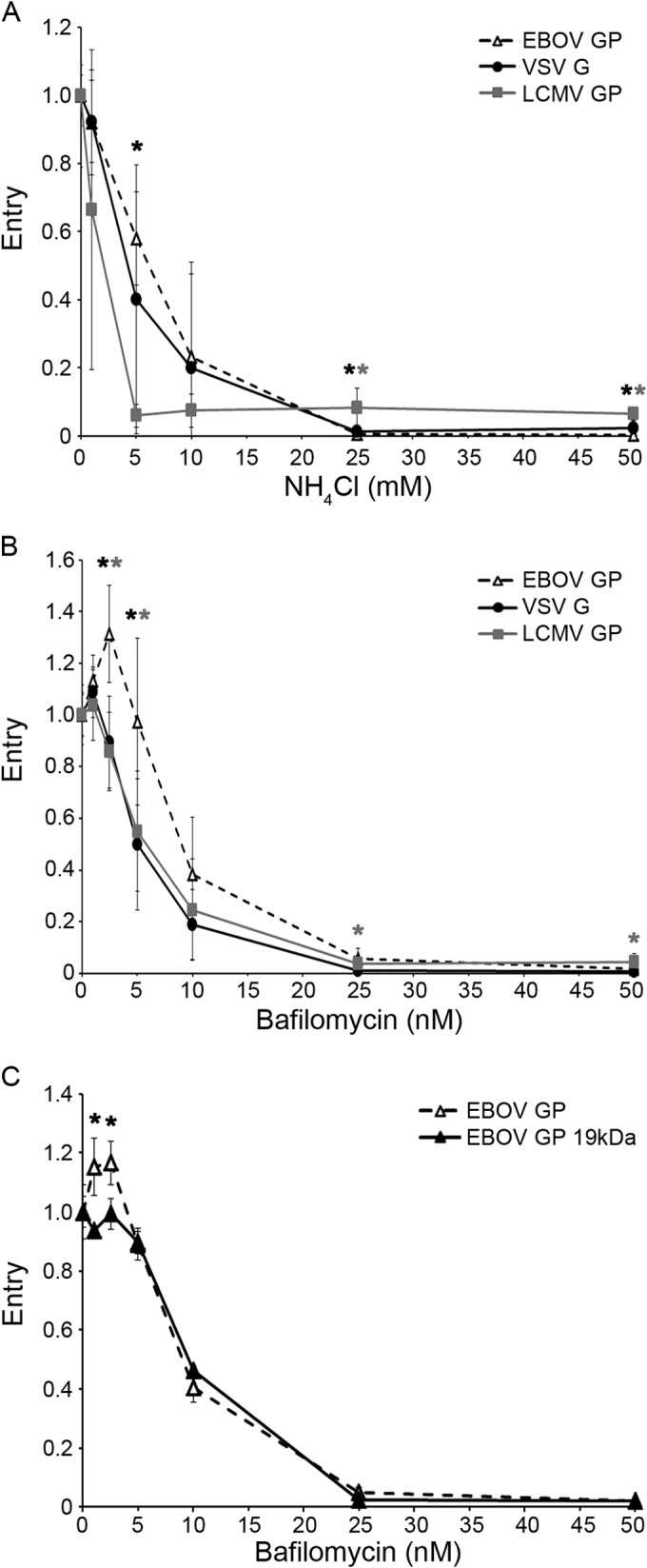
EBOV GP does not require unusually low pH to mediate cytoplasmic entry. BSC-1 cells were pretreated for 15 min with the indicated concentration of either NH4Cl (A) or bafilomycin (B, C). VLPs bearing EBOV GP (open triangles, dashed line), LCMV GP (squares, gray line), VSV G (circles, solid black line), or 19-kDa EBOV GP (black triangles, black line) were then bound, and the cells were processed and analyzed for cytoplasmic entry as described for Fig. 1, except that the indicated concentration of the indicated inhibitor was present for all steps. Each data point in panels A and B is the average from six samples (from three experiments performed in duplicate). Each data point in panel C is the average from three samples from one experiment. In all panels, error bars indicate SD. Asterisks in panels A and B indicate that the value for EBOV GP is higher than that for LCMV GP (black) or VSV G (gray) at P values of <0.05 (*) or <0.005 (**). Note that a higher value indicates, if anything, lower sensitivity to the lysosomotropic agent. Asterisks in panel C indicate a higher value for EBOV GP than for the 19-kDa GP (*, P < 0.05).
Evidence that EBOV enters the cytoplasm soon after arrival in NPC1+ endolysosomes (LE/Lys), where it remains sensitive to the cysteine protease inhibitor E64d.
We next asked if arrival in NPC1+ LE/Lys is a rate-defining step for EBOV entry. We observed that VLPs bearing EBOV GP entered the cytoplasm with kinetics indistinguishable from their colocalization with NPC1+ LE/Lys (Fig. 6). In the cells studied, NPC1+ LE/Lys contain relatively high levels of CD63, Lamp1, and Lamp2, which are reported to be present in LEs and Lys. Conversely, these NPC1+ LE/Lys contain only modest levels of lysobisphosphatidic acid and only low levels of the early endosome marker EEA1 and the LE marker mannose-6-phosphate receptor (M6PR) (Fig. 7). A major suggestion of Fig. 6 is that arrival in NPC1+ LE/Lys is a rate-defining step for EBOV GP-mediated entry. The data further suggest that once within NPC1+ LE/Lys, the conditions are suitable for relatively rapid fusion triggering.
FIG 6.

EBOV VLPs begin to enter the cytoplasm soon after arrival in an NPC1+ endosome. (A) EBOV GP VLPs were bound to parallel sets of BSC-1 cells at 4°C and, after being washed, incubated at 37°C. At the indicated time, cells were analyzed for cytoplasmic entry (open triangles, dashed line) as described for Fig. 1 and for colocalization with NPC1 (solid triangles, solid line) as described in Materials and Methods. Data are the averages from six experiments. In each experiment, entry was monitored in duplicate samples and NPC1 colocalization was analyzed from 20 to 30 microscope fields. Colocalization is presented as Manders coefficients normalized to the average Manders coefficient at the 120-min time point (0.45 ± 0.12). We did not extend the analysis to 180 min, as across all experiments entry at 120 min was 94.5% of that seen at 180 min (i.e., virtually maximal). Error bars indicate SD. There was no statistically significant difference between the extent of colocalization and the extent of entry at any of the time points. (B) Representative micrographs from 15-, 60-, and 120-min time points. Red, VLPs (Cherry-VP40); green, NPC1 (antibody staining). Yellow arrows indicate areas of overlap.
FIG 7.

Characterization of NPC1+ endosomes in BSC-1 cells. BSC-1 cells were fixed, permeabilized, and stained with a rabbit monoclonal antibody against NPC1 as well as mouse monoclonal antibodies versus CD63, Lamp1, LBPA, M6PR, or EEA1, as indicated. They were then stained with corresponding Alexa Fluor 647 anti-rabbit or anti-mouse Alexa Fluor 546 secondary antibodies, as appropriate, and observed by confocal microscopy. For each sample, 10 random fields were analyzed for overlap between NPC1 and the indicated marker. (A, C) Manders colocalization coefficients. Error bars represent SD. Representative images (B, D) are shown below each graph. The data are from two separate experiments shown on the left (A, B) and right (C, D). In another experiment, high colocalization was also seen between Lamp2 and NPC1 (not shown).
Processing of EBOV GP to 19 kDa fosters binding to NPC1 (10) and potentiates GP for in vitro conformational changes (29). We previously showed that entry mediated by the 19-kDa EBOV GP remains sensitive to the cysteine protease inhibitor E64d and to a cathepsin L (cat L) inhibitor (30), but it has remained unclear when, where, and why this is so. Here, we found that EBOV GP remains sensitive to E64d over the same time course as it remains sensitive to the fusion-inhibiting agent bafilomycin (Fig. 8A). Moreover, E64d neither blocked nor delayed trafficking of EBOV GP VLPs to NPC1+ LE/Lys (Fig. 8B and C). These findings suggest that the post-19-kDa E64d-sensitive step (30, 31) occurs within the lumen of NPC1+ LE/Lys. This further suggests that the final E64d-sensitive factor acts at, or temporally just upstream of, virus-endosome fusion, perhaps by cleaving 19-kDa GP to a final fusion-ready state (30, 32).
FIG 8.
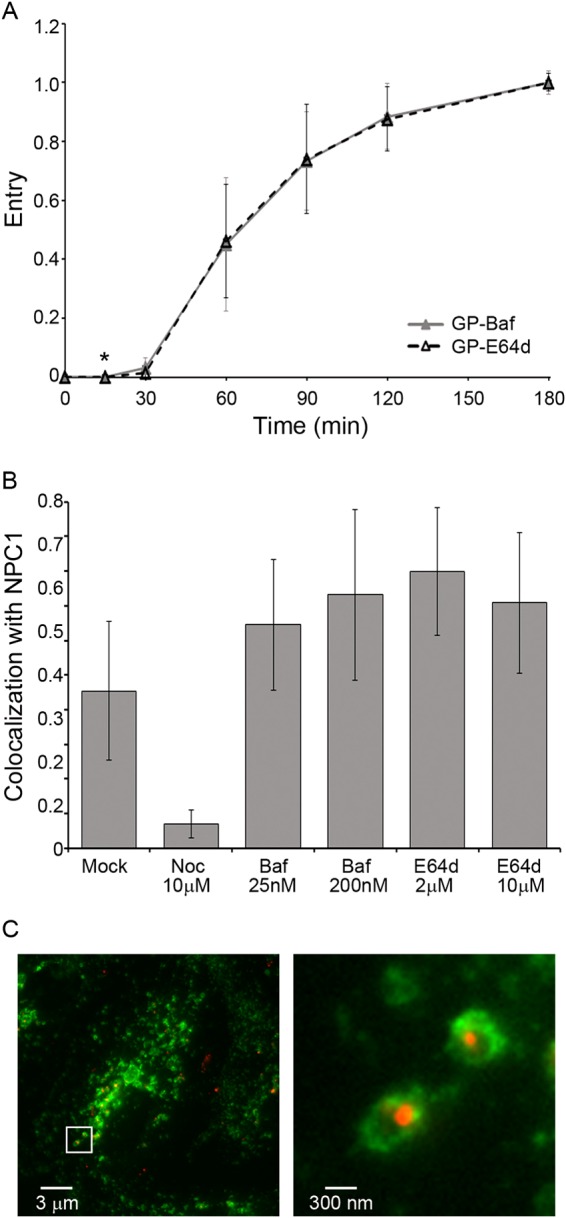
EBOV GP VLPs remain sensitive to E64d within NPC1+ endosomes. (A) EBOV GP VLPs overcome their sensitivity to E64d and bafilomycin with the same kinetics. EBOV GP VLPs were bound to BSC-1 cells at 4°C and then warmed to 37°C. At the indicated times, cells were treated with bafilomycin (200 nM) or E64d (2 μM). Cells were then processed for EBOV GP VLP cytoplasmic entry as described for Fig. 1. Each data point is the average from eight samples (from three experiments; two performed in triplicate and one performed in duplicate). Only one point showed a statistical difference: *, P < 0.05. The same result was obtained in 293AD cells (not shown). (B) Neither E64d nor bafilomycin prevents the arrival of EBOV GP VLP in NPC1+ endosomes: BSC-1 cells were pretreated for 15 min with dimethyl sulfoxide (DMSO; mock) or the indicated concentration of inhibitor (Noc, nocodazole; Baf, bafilomycin; E64d). Samples taken 120 min post-warm-up to 37°C were then analyzed for VLP colocalization with NPC1 as described for Fig. 6 (20 fields/sample; error bars represent SD). (C) EBOV GP VLPs are found within the lumen of NPC1+ endosomes in the presence of E64d. Samples from the 120-min time point (2 μM E64d) were processed as described for panel B and observed with a Plan Apo A/1.40 60× oil objective on a Nikon Eclipse TE2000-E fluorescence microscope. Left, representative image; right, ×10 zoom of white box. Red, EBOV GP VLP (mCherry-VP40); green, NPC1.
SARS CoV entry also begins after a 30-min lag and after arrival in NPC1+ LE/Lys.
SARS engages its receptor, ACE2, on the cell surface. In cells that do not express trypsin-like proteases on their surface, SARS CoV enters the cytoplasm through endosomes, where fusion is triggered by cat L cleavage of the SARS S (fusion) protein. This endosomal entry route has been substantiated by showing that E64d and cat L inhibitors block SARS S-mediated endosomal entry and that cat L can induce fusion of SARS particles bound to ACE2 (33–36). However, the class of (cat L-containing) endosomes that serve as portals for SARS entry has not been determined. Since LEs are said to contain cathepsins (37), we predicted that SARS S-mediated entry would occur with a time course similar to that seen for LCMV and influenza virus (Fig. 2; i.e., with an ∼15-min lag), in other words, earlier than entry mediated by EBOV GP, which first begins after a 30-min lag. To test our prediction, we used HIV-βlam pseudovirions expressing either EBOV GP or SARS S. To prevent SARS entry through the cell surface, the cells used to produce pseudovirions, as well as the target cells, were passaged without trypsin (see Materials and Methods). In sharp contrast to our expectation, pseudovirions bearing SARS S exhibited the same ∼30-min lag before initiating entry as pseudovirions bearing EBOV GP (Fig. 9). Moreover, as for EBOV (Fig. 6), SARS entry was first seen after detecting colocalization of SARS S pseudovirions with NPC1+ LE/Lys, which also occurred after an ∼30-min lag (Fig. 10). After the 30-min lag, the rate of SARS entry appeared somewhat slower than the rate of EBOV GP-mediated entry. In sum, the results in Fig. 9 and 10 uncover unexpected similarities in the timing and location of entry of SARS, a CoV, and EBOV, a filovirus.
FIG 9.
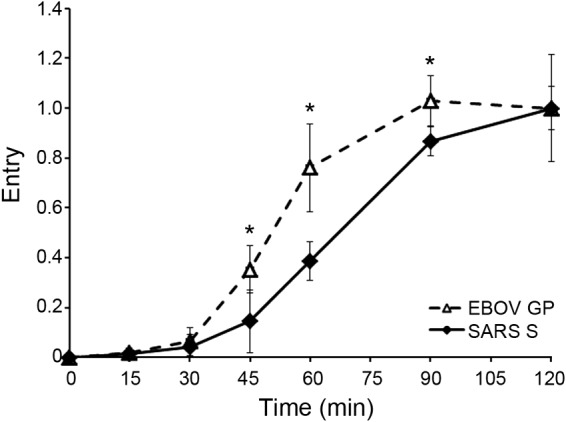
Entry mediated by both SARS CoV S and EBOV GP begins after a 30-min lag. HIV pseudovirions bearing either SARS S (solid diamonds, solid line) or EBOV GP (open triangles, dashed line) were processed and analyzed for cytoplasmic entry (quenched with 5 μM E64d) as described for Fig. 1. Each data point is the average from four samples (from two experiments performed in duplicate). Error bars indicate SD, and asterisks indicate statistically significant differences (*, P < 0.05) between EBOV GP and SARS S at the indicated time point.
FIG 10.

SARS S pseudovirions begin to enter the cytoplasm after colocalization with NPC1. (A) SARS S pseudovirions were bound to parallel sets of BSC-1 cells at 4°C and then incubated at 37°C to allow internalization and entry. At the indicated times, cells were analyzed for cytoplasmic entry (open diamonds, dashed line) as described for Fig. 3 or colocalization with NPC1 (solid diamonds, solid line) as described in Materials and Methods. Entry data are the averages from five samples (from two experiments). NPC1 colocalization was analyzed from 55 microscope fields (from two experiments) and presented as Manders coefficients normalized to the average Manders coefficient at 120 min (0.43 ± 0.11). Error bars indicate SD. ***, P < 0.0005, for the 60-min time point. (B) Representative images from the 15-min, 60-min, and 120-min time points are shown. Red, staining for HIV p24; green, staining for NPC1. Yellow arrows indicate areas of overlap.
Evidence that NPC1+ LE/Lys have higher cat L activity than earlier endosomes.
The late entry kinetics of EBOV, and especially of SARS, suggested that NPC1+ LE/Lys have higher cathepsin activity than earlier endosomes, including LEs through which influenza and LCMV enter. To test this idea, BSC-1 cells were transfected to express GFP-Rab5, GFP-Rab7, or NPC1-GFP, and cat L activity was detected using a fluorescent cat L probe in conjunction with live-cell microscopy, as described in Materials and Methods. We first analyzed cat L activity in endosomes marked with GFP-Rab5 (Fig. 11A), which comprise both early endosomes (Rab5+/Rab7−) as well as transitional (Rab5+/Rab7+) endosomes (Fig. 12) (38–41). As seen in Movie S1 in the supplemental material and Fig. 11A (asterisks), we did not detect cat L activity in GFP-Rab5+ endosomes. Therefore, neither early endosomes nor transitional endosomes have detectable cat L activity. We next analyzed cat L activity in endosomes marked with GFP-Rab7. In addition to being in transitional endosomes, Rab7 is found in LE and LE/Lys (Fig. 12). We detected two populations of endosomes marked with GFP-Rab7: one without cat L activity (Fig. 11B, asterisks; see also Movie S2 in the supplemental material) and one with cat L activity (Fig. 11B, hatch marks; see also Movie S2). In this analysis, we also detected a population of vesicles with strong cat L activity but with no or minimal GFP-Rab7 (Fig. 11B, arrows). The latter population may represent lysosomes. We next analyzed cat L activity in endosomes marked with NPC1-GFP. Most of these endosomes contained cat L activity (Fig. 11C, hatch marks; see also Movie S3).
FIG 11.

Colocalization of cat L activity with Rab5+, Rab7+, and NPC1+ endosomes. BSC-1 cells were transfected to express GFP-Rab5, GFP-Rab7, or NPC1-GFP, incubated with a cat L activity probe, and analyzed by live-cell microscopy on a spinning disk confocal microscope as described in Materials and Methods. Shown are images from the respective movies at 2 to 3 min after the addition of substrate for cat L activity (red) and endosomes (green) marked by GFP-Rab5 (A), GFP-Rab7 (B), or NPC1-GFP (C). (D) Box plots showing the Pearson correlation coefficients (calculated as described in Materials and Methods) between GFP-Rab5 (n = 1,332), GFP-Rab7 (n = 1,238), or NPC1-GFP (n = 1,314) and cat L activity. Asterisk denotes statistical significance (P ≤ 0.0001). Medians are represented by center lines and the 25th and 75th percentiles by upper and lower box boundaries. Whiskers cover the 5th and 95th percentiles, and circles show outliers. (E) Manders values representing the percent overlap of GFP-Rab7 with endogenous NPC1 and endogenous NPC1 and GFP-Rab7. Averaged percent colocalization was calculated after analyzing 33 cells. Error bars represent SD.
FIG 12.
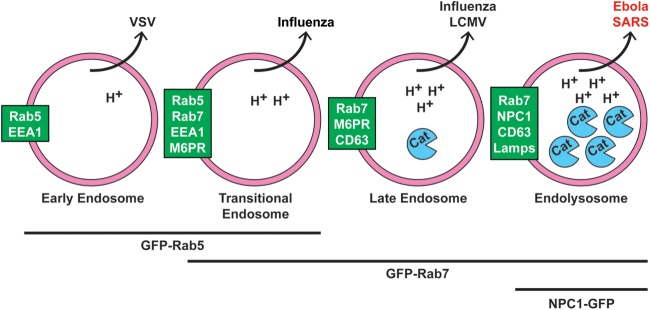
Working model for cellular sites of EBOV and SARS CoV entry. We propose that Ebola and SARS enter the cytoplasm later than other “late-penetrating viruses” (e.g., influenza and LCMV), through NPC1+ endolysosomes (LE/Lys). We further propose that they do so to access high levels of endosomal cathepsins (and, for EBOV, to bind to NPC1). Text in green boxes indicates markers of the respective organelles (pink circles). Viruses known to exit each organelle are indicated above. While one study showed that influenza enters through transitional endosomes (40), others refer to influenza and LCMV as entering through LE (15, 26). H+ indicates protons, and Cat indicates cathepsins. Lines and text below the organelles indicate GFP-tagged markers used in Fig. 11 and Movies S1 to S3 in the supplemental material.
As a measure of colocalization, the Pearson's correlation coefficients (r) between the GFP marker and the cat L probe are plotted in Fig. 11D. For Rab5+ endosomes, the coefficient was 0.02, supporting a lack of cat L activity in early endosomes or transitional endosomes (Fig. 12). The r value for colocalization of GFP-Rab7 and cat L activity was 0.28, while that for NPC1-GFP and cat L activity was statistically higher (P < 0.0001), with a value of 0.35. A high proportion of cat L activity recorded in Rab7+ endosomes likely stems from LE/Lys (Rab7+/NPC1+), due to the high degree of colocalization of Rab7 and NPC1 (Fig. 11E) (42). Therefore, LE (Rab7+/NPC1−) likely have even lower levels of cat L activity than LE/Lys (Rab7+/NPC1+), as might be inferred from Fig. 11D. Collectively, the results in Fig. 11 (and Movies S1 to S3 in the supplemental material) provide strong evidence that along the endocytic pathway, cat L activity is, indeed, highest in NPC1+ LE/Lys (Fig. 12).
DISCUSSION
In the present study, we provide new insights into the entry processes of the filovirus EBOV and SARS CoV. We found that (in the cells studied) EBOV enters the cytoplasm later than LCMV and influenza virus, viruses that enter the cytoplasm through LE. We further showed that late EBOV entry is not due to the filamentous morphology of EBOV particles, their internalization kinetics, their need for proteolytic priming of GP to the 19-kDa species, or a need for unusually low endosomal pH. Rather, a key rate-defining step for EBOV entry appears to be arrival in NPC1+ LE/Lys. Moreover, contrary to our expectation, we found that SARS CoV displays similarly late endosomal entry kinetics, also commencing after colocalization with NPC1. And lastly, we found that NPC1+ LE/Lys have higher levels of cat L activity than LE, with no detectable cat L activity in early or transitional endosomes. We discuss our findings in terms of the sites of entry, mechanisms of fusion activation, and therapeutic opportunities for SARS and EBOV.
Sites of EBOV and SARS endosomal entry.
Our findings indicate that both SARS and EBOV enter the cytoplasm after a noticeably longer lag, and with considerably slower overall kinetics, than either influenza virus or particles bearing the GP of LCMV, viruses that enter the cytoplasm through LEs (15). Additionally, endosomal entry of both SARS and EBOV first begins after detectable colocalization with NPC1. Given the closeness in timing between colocalization of EBOV GP VLPs with NPC1, its obligate endosomal receptor (8, 9), and entry (Fig. 6A), we propose (Fig. 12) that entry of EBOV occurs through NPC1+ LE/Lys. Based on similar findings in Fig. 10, we propose that SARS also enters through NPC1+ LE/Lys. Interestingly, two other shared features have recently been revealed for SARS and EBOV: entry of both viruses (from disparate families) is restricted by (i) similar interferon-inducible transmembrane proteins (43) as well as by (ii) some of the same FDA-approved drugs (16, 23, 44). Our findings suggesting a late, perhaps common, entry portal for EBOV and SARS may help explain the latter two observations. Moreover, recent evidence suggests that two other CoVs, mouse hepatitis virus and feline CoV, enter the cell through LE/Lys (45).
Why might SARS and EBOV need to traffic late in the endocytic pathway for entry? (i) SARS.
The current model for SARS entry posits that binding to its cell surface receptor, ACE2, induces conformational changes that potentiate the S glycoprotein for a fusion-inducing proteolytic cleavage event (33, 34, 36). In many cells, including cells of the respiratory tract, it is thought that cell surface trypsin-like proteases enact this cleavage (35, 46). However, in cells that lack such proteases, the fusion-activating cleavage is carried out by endosomal cathepsins. Evidence supporting cat L-dependent SARS endosomal entry stems from the observation that cat L treatment is sufficient to induce fusion of SARS (bound to ACE2) (47, 48). This suggests that cat L activity may be the only endosomal factor needed to trigger SARS S. While there is a consensus that cat L cleavage of SARS S occurs in endosomes (33, 34, 36), the specific class of endosomes where cat L cleavage, and hence SARS entry, occurs has not been established. Our observations suggest that although SARS does not need the NPC1 protein for entry (49), it traffics deep and late into the endocytic pathway, to or through NPC1+ LE/Lys, to fuse and gain access to the cytoplasm. Since the only post-ACE2 binding requirement for SARS endosomal entry is cat L activity, we propose that SARS travels deep down the endocytic pathway to access sufficiently high levels of cat L to trigger fusion. Supporting this proposal, we provide evidence that NPC1+ LE/Lys do, indeed, contain higher levels of cat L activity than LE and that early and transitional endosomes lack detectable cat L activity.
(ii) EBOV.
NPC1, which is essential for EBOV entry (8, 9, 49), serves as a high-affinity intracellular receptor for a proteolytically primed form of EBOV GP (4–7). Whereas full-length EBOV GP and EBOV GPΔ cannot bind to NPC1, primed 19-kDa EBOV GP can (8, 10, 16). The 20-kDa EBOV GP, which can be produced by in vitro treatment of EBOV GP with cat L (29, 30, 50) or chymotrypsin (31), can also bind to NPC1 (51) (data not shown). As discussed for SARS, it has remained unclear where along the endocytic pathway cathepsin cleavage of EBOV GP, to an NPC1 binding form (∼20 or ∼19 kDa), occurs (4). Furthermore, EBOV particles remain sensitive to E64d and a cat L inhibitor even after cleavage of GP to the 19-kDa species (30), and E64d sensitivity appears to be maintained up until, or shortly before, the moment of fusion (Fig. 8). In addition to providing a docking protein (NPC1) on the limiting endosomal membrane, NPC1+ LE/Lys may also provide needed levels of a cathepsin or another cysteine protease to cleave the 19/20-kDa GP (30, 32, 52), a cleavage event that might occur after binding to NPC1 (which may first induce a conformational change in 19-kDa GP). By analogy with SARS, a post-receptor (NPC1) binding cleavage event in 19-kDa GP might trigger EBOV fusion. A major difference would be that whereas SARS S engages its receptor, ACE2, at the cell surface, primed (19/20-kDa) EBOV GP first binds its receptor (NPC1) in LE/Lys. In addition to optimal cathepsin levels and low pH (29, 53, 54), NPC1+ LE/Lys may possess a lipid composition and/or other factors required for optimal EBOV fusion (16).
Implications for therapeutics and other late-penetrating viruses.
As discussed above, there appear to be unexpected similarities in the location, timing, requirements for, and restrictions of entry of SARS and EBOV. This suggests that it may be possible to identify small molecules that thwart both EBOV and SARS. Indeed, several small molecules (in addition to cathepsin inhibitors [55–57]) have been found that block both viruses (16, 23, 44), although it is not yet clear if they are working through common mechanisms against both EBOV and SARS. The latter might include drugs that block virus trafficking to, or maturation of, NPC1+ LE/Lys. Reoviruses require cathepsin processing for entry (58), and cathepsins also appear to enhance rotavirus entry (59). While it has generally been thought that these viruses enter through LEs, it is possible that they too enter deeper in the endocytic pathway, and that some drugs that block SARS and EBOV may block entry of other late-penetrating viruses that also require cathepsins for cell entry.
Supplementary Material
ACKNOWLEDGMENTS
We thank Shutoku Matsuyama for advice on preparing and examining endosomal entry of pseudovirions bearing SARS S, David Castle for many helpful discussions, and other colleagues for reagents (as indicated in Materials and Methods).
The work was supported by AI103601 (to J.M.W.) and DK58536 and AI093708 (to J.E.C.). C.J.S. and J.A.S. were supported, in part, by training grants T32AI055432 and T32AI007046, respectively.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03398-14.
REFERENCES
- 1.Feldmann H, Geisbert TW. 2011. Ebola haemorrhagic fever. Lancet 377:849–862. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartman AL, Towner JS, Nichol ST. 2010. Ebola and Marburg hemorrhagic fever. Clin Lab Med 30:161–177. doi: 10.1016/j.cll.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Gire SK, Goba A, Andersen KG, Sealfon RSG, Park DJ, Kanneh L, Jalloh S, Momoh M, Fullah M, Dudas G, Wohl S, Moses LM, Yozwiak NL, Winnicki S, Matranga CB, Malboeuf CM, Qu J, Gladden AD, Schaffner SF, Yang X, Jiang PP, Nekoui M, Colubri A, Coomber MR, Fonnie M, Moigboi A, Gbakie M, Kamara FK, Tucker V, Konuwa E, Saffa S, Sellu J, Jalloh AA, Kovoma A, Koninga J, Mustapha I, Kargbo K, Foday M, Yillah M, Kanneh F, Robert W, Massally JLB, Chapman SB, Bochicchio J, Murphy C, Nusbaum C, Young S, Birren BW, Grant DS, Scheiffelin JS, Lander ES, Happi C, Gevao SM, Gnirke A, Rambaut A, Garry RF, Khan SH, Sabeti PC. 2014. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345:1369–1372. doi: 10.1126/science.1259657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chandran K. 2012. Filovirus entry into cells—new insights. Curr Opin Virol 2:206–214. doi: 10.1016/j.coviro.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hofmann-Winkler H, Kaup F, Pöhlmann S. 2012. Host cell factors in filovirus entry: novel players, new insights. Viruses 4:3336–3362. doi: 10.3390/v4123336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunt CL, Lennemann NJ, Maury W. 2012. Filovirus entry: a novelty in the viral fusion world. Viruses 4:258–275. doi: 10.3390/v4020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White JM, Schornberg KL. 2012. A new player in the puzzle of filovirus entry. Nat Rev Microbiol 10:317–322. doi: 10.1038/nrmicro2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. 2011. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. 2011. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, Ruthel G, Pfeffer SR, Dye JM, Whelan SP, Brummelkamp TR, Chandran K. 2012. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J 31:1947–1960. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ko DC, Gordon MD, Jin JY, Scott MP. 2001. Dynamic movements of organelles containing Niemann-Pick C1 protein: NPC1 involvement in late endocytic events. Mol Biol Cell 12:601–614. doi: 10.1091/mbc.12.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cruz JC, Sugii S, Yu C, Chang T-Y. 2000. Role of Niemann-Pick type C1 protein in intracellular trafficking of low density lipoprotein-derived cholesterol. J Biol Chem 275:4013–4021. doi: 10.1074/jbc.275.6.4013. [DOI] [PubMed] [Google Scholar]
- 13.Nunberg JH, York J. 2012. The curious case of arenavirus entry, and its inhibition. Viruses 4:83–101. doi: 10.3390/v4010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lakadamyali M, Rust MJ, Babcock HP, Zhuang X. 2003. Visualizing infection of individual influenza viruses. Proc Natl Acad Sci U S A 100:9280–9285. doi: 10.1073/pnas.0832269100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lozach P-Y, Huotari J, Helenius A. 2011. Late-penetrating viruses. Curr Opin Virol 1:35–43. doi: 10.1016/j.coviro.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Shoemaker CJ, Schornberg KL, Delos SE, Scully C, Pajouhesh H, Olinger GG, Johansen LM, White JM. 2013. Multiple cationic amphiphiles induce a Niemann-Pick C phenotype and inhibit Ebola virus entry and infection. PLoS One 8:e56265. doi: 10.1371/journal.pone.0056265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delos SE, La B, Gilmartin A, White JM. 2010. Studies of the “chain reversal regions” of the avian sarcoma/leukosis virus (ASLV) and Ebolavirus fusion proteins: analogous residues are important, and a His residue unique to EnvA affects the pH dependence of ASLV entry. J Virol 84:5687–5694. doi: 10.1128/JVI.02583-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spitzer M, Wildenhain J, Rappsilber J, Tyers M. 2014. BoxPlotR: a Web tool for generation of box plots. Nat Meth 11:121–122. doi: 10.1038/nmeth.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johannsdottir HK, Mancini R, Kartenbeck J, Amato L, Helenius A. 2009. Host cell factors and functions involved in vesicular stomatitis virus entry. J Virol 83:440–453. doi: 10.1128/JVI.01864-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mire CE, White JM, Whitt MA. 2010. A spatio-temporal analysis of matrix protein and nucleocapsid trafficking during vesicular stomatitis virus uncoating. PLoS Pathog 6:e1000994. doi: 10.1371/journal.ppat.1000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Licata JM, Johnson RF, Han Z, Harty RN. 2004. Contribution of Ebola virus glycoprotein, nucleoprotein, and VP24 to budding of VP40 virus-like particles. J Virol 78:7344–7351. doi: 10.1128/JVI.78.14.7344-7351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noda T, Sagara H, Suzuki E, Takada A, Kida H, Kawaoka Y. 2002. Ebola virus VP40 drives the formation of virus-like filamentous particles along with GP. J Virol 76:4855–4865. doi: 10.1128/JVI.76.10.4855-4865.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johansen LM, Brannan JM, Delos SE, Shoemaker CJ, Stossel A, Lear C, Hoffstrom BG, Dewald LE, Schornberg KL, Scully C, Lehár J, Hensley LE, White JM, Olinger GG. 2013. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci Transl Med 5:190ra79. doi: 10.1126/scitranslmed.3005471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shoemaker CJ. 2012. Mechanisms of Ebolavirus entry. Ph.D. thesis University of Virginia, Charlottesville, VA. [Google Scholar]
- 25.Iwasaki M, Ngo N, la Torre de JC. 2013. Sodium hydrogen exchangers contribute to arenavirus cell entry. J Virol 88:643–654. doi: 10.1128/JVI.02110-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quirin K, Eschli B, Scheu I, Poort L, Kartenbeck J, Helenius A. 2008. Lymphocytic choriomeningitis virus uses a novel endocytic pathway for infectious entry via late endosomes. Virology 378:21–33. doi: 10.1016/j.virol.2008.04.046. [DOI] [PubMed] [Google Scholar]
- 27.Lozach P-Y, Mancini R, Bitto D, Meier R, Oestereich L, Overby AK, Pettersson RF, Helenius A. 2010. Entry of bunyaviruses into mammalian cells. Cell Host Microbe 7:488–499. doi: 10.1016/j.chom.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saeed MF, Kolokoltsov AA, Freiberg AN, Holbrook MR, Davey RA. 2008. Phosphoinositide-3 kinase-Akt pathway controls cellular entry of Ebola virus. PLoS Pathog 4:e1000141. doi: 10.1371/journal.ppat.1000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brecher M, Schornberg KL, Delos SE, Fusco ML, Saphire EO, White JM. 2012. Cathepsin cleavage potentiates the Ebola virus glycoprotein to undergo a subsequent fusion-relevant conformational change. J Virol 86:364–372. doi: 10.1128/JVI.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J. 2006. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J Virol 80:4147. doi: 10.1128/JVI.80.8.4147-4156.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong AC, Sandesara RG, Mulherkar N, Whelan SP, Chandran K. 2010. A forward genetic strategy reveals destabilizing mutations in the Ebolavirus glycoprotein that alter its protease dependence during cell entry. J Virol 84:163–175. doi: 10.1128/JVI.01832-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. 2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belouzard S, Millet JK, Licitra BN, Whittaker GR. 2012. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4:1011–1033. doi: 10.3390/v4061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heald-Sargent T, Gallagher T. 2012. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 4:557–580. doi: 10.3390/v4040557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S. 2012. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol 86:6537–6545. doi: 10.1128/JVI.00094-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simmons G, Zmora P, Gierer S, Heurich A, Pöhlmann S. 2013. Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antiviral Res 100:605–614. doi: 10.1016/j.antiviral.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guha S, Padh H. 2008. Cathepsins: fundamental effectors of endolysosomal proteolysis. Indian J Biochem Biophys 45:75–90. [PubMed] [Google Scholar]
- 38.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. 2005. Rab conversion as a mechanism of progression from early to late endosomes. Cell 122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 39.Vonderheit A, Helenius A. 2005. Rab7 associates with early endosomes to mediate sorting and transport of Semliki forest virus to late endosomes. PLoS Biol 3:e233. doi: 10.1371/journal.pbio.0030233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lakadamyali M, Rust MJ, Zhuang X. 2006. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell 124:997–1009. doi: 10.1016/j.cell.2005.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Weering JRT, Verkade P, Cullen PJ. 2012. SNX-BAR-mediated endosome tubulation is co-ordinated with endosome maturation. Traffic 13:94–107. doi: 10.1111/j.1600-0854.2011.01297.x. [DOI] [PubMed] [Google Scholar]
- 42.Blom TS. 2003. Defective endocytic trafficking of NPC1 and NPC2 underlying infantile Niemann-Pick type C disease. Hum Mol Genet 12:257–272. doi: 10.1093/hmg/ddg025. [DOI] [PubMed] [Google Scholar]
- 43.Huang I-C, Bailey CC, Weyer JL, Radoshitzky SR, Becker MM, Chiang JJ, Brass AL, Ahmed AA, Chi X, Dong L, Longobardi LE, Boltz D, Kuhn JH, Elledge SJ, Bavari S, Denison MR, Choe H, Farzan M. 2011. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog 7:e1001258. doi: 10.1371/journal.ppat.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dyall J, Coleman CM, Hart BJ, Venkataraman T, Holbrook MR, Kindrachuk J, Johnson RF, Olinger GG, Jahrling PB, Laidlaw M, Johansen LM, Lear-Rooney CM, Glass PJ, Hensley LE, Frieman MB. 2014. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob Agents Chemother 58:4885–4893. doi: 10.1128/AAC.03036-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burkard C, Verheije MH, Wicht O, van Kasteren SI, van Kuppeveld FJ, Haagmans BL, Pelkmans L, Rottier PJM, Bosch BJ, de Haan CAM. 2014. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog 10:e1004502. doi: 10.1371/journal.ppat.1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bertram S, Glowacka I, Müller MA, Lavender H, Gnirss K, Nehlmeier I, Niemeyer D, He Y, Simmons G, Drosten C, Soilleux EJ, Jahn O, Steffen I, Pöhlmann S. 2011. Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin-like protease. J Virol 85:13363–13372. doi: 10.1128/JVI.05300-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simmons G. 2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A 102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsuyama S, Ujike M, Morikawa S, Tashiro M, Taguchi F. 2005. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc Natl Acad Sci U S A 102:12543–12547. doi: 10.1073/pnas.0503203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haines KM, Vande Burgt NH, Francica JR, Kaletsky RL, Bates P. 2012. Chinese hamster ovary cell lines selected for resistance to Ebolavirus glycoprotein mediated infection are defective for NPC1 expression. Virology 432:20–28. doi: 10.1016/j.virol.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hood CL, Abraham J, Boyington JC, Leung K, Kwong PD, Nabel GJ. 2010. Biochemical and structural characterization of cathepsin L-processed Ebola virus glycoprotein: implications for viral entry and immunogenicity. J Virol 84:2972–2982. doi: 10.1128/JVI.02151-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Misasi J, Chandran K, Yang JY, Considine B, Filone CM, Cote M, Sullivan N, Fabozzi G, Hensley L, Cunningham J. 2012. Filoviruses require endosomal cysteine proteases for entry but exhibit distinct protease preferences. J Virol 86:3284–3292. doi: 10.1128/JVI.06346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marzi A, Reinheckel T, Feldmann H. 2012. Cathepsin B & L are not required for Ebola virus replication. PLoS Negl Trop Dis 6:e1923. doi: 10.1371/journal.pntd.0001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gregory SM, Harada E, Liang B, Delos SE, White JM, Tamm LK. 2011. Structure and function of the complete internal fusion loop from Ebolavirus glycoprotein 2. Proc Natl Acad Sci U S A 108:11211–11216. doi: 10.1073/pnas.1104760108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harrison JS, Koellhoffer JF, Chandran K, Lai JR. 2012. Marburg virus glycoprotein GP2: pH-dependent stability of the ectodomain α-helical bundle. Biochemistry 51:2515–2525. doi: 10.1021/bi3000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shah PP, Wang T, Kaletsky RL, Myers MC, Purvis JE, Jing H, Huryn DM, Greenbaum DC, Smith AB, Bates P, Diamond SL. 2010. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and Ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol Pharmacol 78:319–324. doi: 10.1124/mol.110.064261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adedeji AO, Severson W, Jonsson C, Singh K, Weiss SR, Sarafianos SG. 2013. Novel inhibitors of severe acute respiratory syndrome coronavirus entry that act by three distinct mechanisms. J Virol 87:8017–8028. doi: 10.1128/JVI.00998-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elshabrawy HA, Fan J, Haddad CS, Ratia K, Broder CC, Caffrey M, Prabhakar BS. 2014. Identification of a broad-spectrum antiviral small molecule against SARS-CoV, Ebola, Hendra, and Nipah viruses using a novel high throughput screening assay. J Virol 88:4353–4365. doi: 10.1128/JVI.03050-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mainou BA, Dermody TS. 2012. Transport to late endosomes is required for efficient reovirus infection. J Virol 86:8346–8358. doi: 10.1128/JVI.00100-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Díaz-Salinas MA, Silva-Ayala D, López S, Arias CF. 2014. Rotaviruses reach late endosomes and require the cation-dependent mannose-6-phosphate receptor and the activity of cathepsin proteases to enter the cell. J Virol 88:4389–4402. doi: 10.1128/JVI.03457-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.