

Abstract
In this study, we show that replication-competent subgenomic hepatitis C virus (HCV) RNA can be transferred to permissive Huh7 cells, leading to the establishment of viral RNA replication. Further, we show that these events are mediated by exosomes rather than infectious virus particles. If similar events occur in vivo, this could represent a novel, albeit inefficient, mechanism of viral spread and immune escape.
TEXT
Hepatitis C virus is a positive-sense RNA virus that causes acute and chronic hepatitis, cirrhosis, and hepatocellular carcinoma. Infectious virions contain full-length HCV RNA encapsidated by the viral core protein and enveloped by the E1 and E2 viral glycoproteins. It was recently reported that HCV RNA can be transferred from HCV subgenomic replicon (SGR) cells (that lack the viral structural proteins and cannot produce virus particles) to plasmacytoid dendritic cells (pDCs) in a virion-independent, cell contact-dependent manner, triggering the secretion of type 1 interferon (1), and that SGR-to-pDC cell-cell transfer is mediated by HCV RNA-containing exosomes (2) produced by the SGR cells (3), in which HCV RNA is highly enriched relative to cellular (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]) mRNA (3).
Extending these findings, Ramakrishnaiah and colleagues recently reported that exosome preparations from infected cells can “infect” naive Huh7.5.1 cells (4). However, because exosomes display the same size, density, and sedimentation characteristics as infectious HCV particles (5–9), exosome preparations unavoidably contain infectious virions, making it difficult to separate canonical virus infection from exosome-mediated HCV RNA transfer in those studies. To avoid that ambiguity, here and previously (1, 3) we used HCV SGR cells to investigate the role of exosome-mediated HCV RNA transfer under conditions in which the contribution of virions can be excluded.
Replication-competent subgenomic HCV RNA is transferred from SGR cells to cocultured Huh7 cells.
In these studies, Huh7 cells that contain (i) an HCV SGR that encodes the viral nonstructural proteins and an antibiotic resistance protein (either hygromycin [hygro] or neomycin [G418] [neo] resistance) (10, 11) and (ii) retroviruses (12) that encode the reciprocal antibiotic resistance genes (neomycin/G418 or hygromycin resistance) served as “donor” cell lines (Huh7-SGRneoR-RVhygroR and Huh7-SGRhygroR-RVneoR) in which neomycin/G418 and hygromycin resistance were encoded either by cytosolic HCV RNA or chromosomal DNA (via retroviral insertion). We also engineered an Huh7-derived “recipient” cell line (Huh7-RVblastR-YFP) by retrovirally inserting a blasticidin (blast) resistance gene and a yellow fluorescent protein (YFP) cassette. By applying dual-antibiotic selection with either neo and blast or hygro and blast (Fig. 1A and B), we could distinguish between cell-cell transfer of HCV SGR RNA to HCV-negative recipient cells by an RNA transfer mechanism and a DNA transfer mechanism mediated by cell-cell fusion which would also produce HCV RNA-positive daughter cells via cytoplasmic mixing. For example, neo and blast double-resistant cell colonies derived after 19 days of antibiotic selection of cocultured Huh7-SGRneoR-RVhygroR donor cells and Huh7-RVblastR-YFP recipient cells would reflect HCV RNA transfer from donor to recipient cells, whereas hygro and blast double resistance in a parallel culture would reflect DNA transfer presumably due to cell-cell fusion. The reverse would be expected for Huh7-SGRhygroR-RVneoR donor cells cocultured with the Huh7-RVblastR-YFP recipient cells.
FIG 1.

Virion-independent RNA transfer between Huh7 cells. A total of 1 × 105 SGR cells were cocultured with 1 × 105 Huh7-RVblastR-YFP cells for 2 days in the absence of selection. On day 2, cells were split 1:2 and cultured in the presence of antibiotics, as indicated using blasticidin S (5 μg/ml; InvivoGen), hygromycin B (75 μg/ml; InvivoGen), and neomycin/G418 (250 μg/ml; InvivoGen). Selection medium was replaced twice a week. After 19 days of selection, the cells were fixed and stained with crystal violet. Resistant colonies were counted blindly by three people, and cell mass was subsequently measured. Both donor cell lines, Huh7-SGRneoR-RVhygroR (A) and Huh7-SGRhygroR-RVneoR (B), were able to transfer RNA-encoded antibiotic resistance to the recipient cells. DNA transfer was less efficient. Shown are representative images of resistant crystal violet-stained colonies after 19 days of selection and quantification of the means from 3 independent experiments done in duplicate. P values were calculated by a two-tailed, paired Student t test. Error bars indicate standard deviations.
As shown in Fig. 1A and B, approximately 55 resistant colonies per well survived after a coculture containing 1E5 donor cells and 1E5 recipient cells was subjected to double selection for RNA transfer, suggesting that replication-competent HCV RNA can indeed be transmitted in a virion-independent manner to HCV-permissive uninfected cells. In contrast, fewer and smaller colonies were observed after double selection for DNA transfer, suggesting that cell fusion cannot account for more than a fraction of the HCV RNA-containing colonies after selection for RNA transfer. Cell mass measured by crystal violet staining confirmed the colony counts. Consistent with the need for direct SGR cell-pDC contact for exosome-mediated pDC activation by HCV SGR cells in previous experiments (3), no RNA or DNA transfer was observed when donor and recipient cells were separated in a Corning Transwell plate and subjected to double-resistant colony formation (not shown).
HCV RNA transfer leads to HCV replication in recipient cells.
To verify that drug resistance after double selection reflected HCV RNA transfer and subsequent RNA replication, we performed fluorescence in situ hybridization (FISH) analysis to visualize HCV RNA (Fig. 2A) as well as real-time reverse transcription-quantitative PCR (RT-qPCR) to measure HCV RNA levels in the resistant colonies (Fig. 2B) (3, 13). Indeed, HCV RNA was easily detected in recipient cells selected for RNA transfer (Fig. 2A, lower left), although it replicates less efficiently in the resistant colonies than in the donor cells (Fig. 2B). Cells selected for DNA transfer may contain less HCV RNA than cells selected for RNA transfer, suggesting that cytosolic HCV RNA is transferred during fusion but may not be able to replicate in all fused cells (Fig. 2A, lower right, and B).
FIG 2.

Visualization of replication-competent HCV RNA transfer to recipient cells. (A) After coculture as described for Fig. 1A, donor SGR, recipient, and resistant colony cells were plated at equal densities and fixed 24 h later, and HCV RNA was labeled using FISH. HCV plus-strand RNA (HCV RNA+; red) and HCV minus-strand RNA (HCV RNA−; yellow) were detected using probe sets that target a region comprised within NS3 and NS4 proteins (reference no. VF4-11069; Panomics/Affymetrix, Santa Clara, CA) according to the manufacturer's instructions and as described previously (12). Nuclei were stained by Hoechst33342 dye (blue; Life Technologies product no. H1399). The picture on the bottom right shows the overlay. Images were acquired with a Zeiss LSM 710 laser scanning confocal microscope. Both positive- and negative-strand HCV RNA are readily detected in cells selected for RNA transfer but are found rarely in cells selected for DNA transfer. (B) Real-time RT-qPCR of the same cells as in panel A, detecting HCV RNA. Shown are the averages from 3 independent experiments. Error bars indicate standard deviations. GE, genome equivalents; YFP, yellow fluorescent protein (green). P values were calculated using a two-tailed, unpaired Student t test with GraphPad Prism software.
To asses HCV RNA transfer at earlier time points, we stained cocultured cells by FISH for HCV plus-strand RNA at 0, 3, and 10 days after coculture (Fig. 3A) and detected HCV RNA (red) in recipient cells (green) over time at low frequency, consistent with the low rate of RNA transfer observed in Fig. 1.
FIG 3.

Chromosomal DNA transfer leads to polyploidy, whereas RNA transfer does not. (A) Donor and recipient cells were cocultured as described for Fig. 1, and cocultured cells were labeled for plus-strand HCV RNA using FISH as described for Fig. 2 after 0, 3, and 10 days of coculture. In rare instances, HCV RNA can be detected over time in YFP-positive (green) recipient cells (B). After coculture as described for Fig. 1A, donor SGR, recipient, and resistant colony live cells were labeled with Hoechst33342 dye (Life Technologies product no. H1399) to detect DNA levels and analyzed by flow cytometry.
We hypothesized that DNA transfer likely occurs via cell fusion, which should yield cells with double the normal chromosome count. To test this hypothesis, we examined the DNA profile of cells in double-resistant colonies by flow cytometry using the DNA marker Hoechst (Fig. 3B). Whereas cells selected for RNA transfer exhibit a normal DNA profile similar to the donor and recipient cell lines, cells selected for DNA transfer show a shift in Hoechst staining, suggesting they have double the amount of chromosomes as would be expected after cell-cell fusion.
Exosome release inhibitors strongly reduce the rate of HCV RNA transfer.
Since HCV RNA is known to be transferred from SGR cells to pDCs via exosomes (3), we hypothesized that exosomes may be responsible for HCV RNA transfer from donor to recipient Huh7 cells. To test this hypothesis, we performed the coculture experiments described in Fig. 1 in the presence of two different neutral sphingomyelinase inhibitors (GW4869 and spiroepoxide), known to block exosome secretion (14–19). Both inhibitors severely reduced the level of RNA transfer (Fig. 4A) without affecting cell viability or HCV RNA replication in the donor cells (not shown). This suggests that exosomes are likely the dominant mode of transmission of HCV RNA between hepatocytes. In support of this, we also found that HCV RNA extracted from exosomes released by our donor cell line is replication competent when transfected into Huh7 cells (Fig. 4B).
FIG 4.
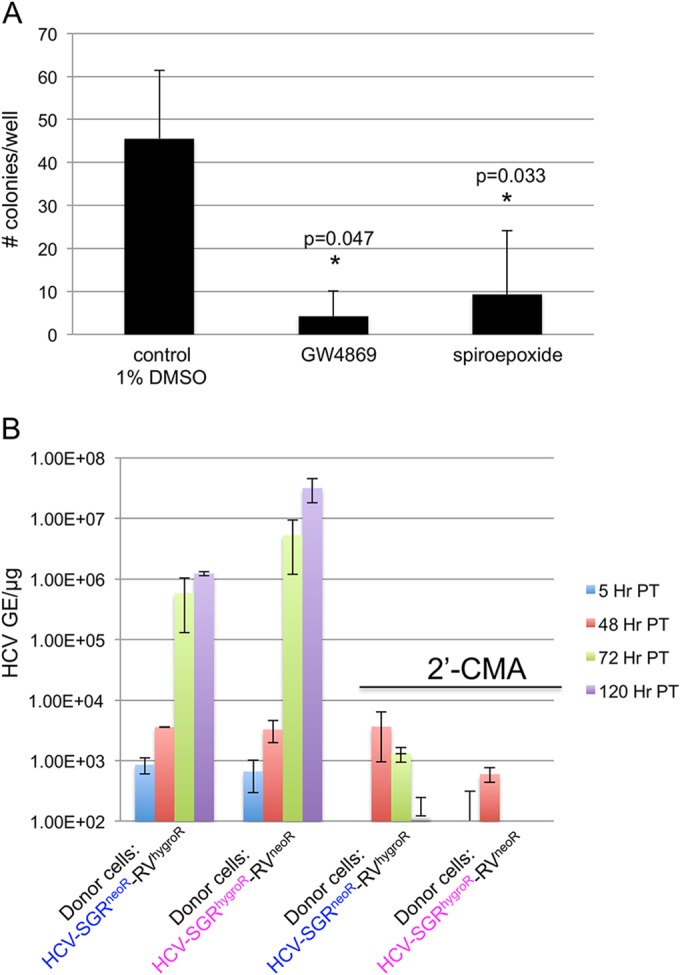
Exosome release inhibitors GW4869 and spiroepoxide inhibit RNA transfer. (A) Cocultures as described for Fig. 1B were performed in the presence of 1% dimethyl sulfoxide (DMSO), GW4869 (10 μM; Sigma), or spiroepoxide (5 μM; Santa Cruz). Colonies were counted, and shown are the means from 3 independent experiments done in duplicate. (B) Exosomes were isolated and RNA was extracted as described previously (13, 21). Purified RNA was transfected into uninfected Huh7 cells using Mirus TransIT mRNA transfection reagent (Mirus Bio, Madison, WI) according to the manufacturer's instructions. 2′-C-methyladenosine (2′-CMA; kindly provided by W. Zhong, Gilead Sciences) was added at the time of transfection. P values were calculated by a two-tailed, paired Student t test. Error bars indicate standard deviation.
We are intrigued by the fact that although HCV RNA transfer is exosome mediated, it appears to require cell-cell contact since coculture experiments done in Transwell plates did not lead to exosomal HCV RNA transfer, and exosomes concentrated from SGR cells were not able to transmit HCV RNA to target cells directly (data not shown). This suggests that, similar to exosome-mediated transfer of HCV RNA from SGR cells to plasmacytoid dendritic cells (3), SGR cells appear to secrete exosomes into the culture supernatant at concentrations that are below a functional threshold that is easily reached in the intercellular space during cell-cell contact. Furthermore, in pilot studies (not shown), we found that only 0.1% of all secreted exosomes contain HCV RNA, and we were unable to isolate sufficient numbers of exosomes from SGR cell supernatants to achieve a high enough HCV RNA/cell ratio to initiate RNA replication. These results contrast with previous reports (4, 20) in which the authors conclude that exosomes prepared from HCV-infected cell supernatants efficiently transfer infection to recipient cells. The interpretation of those results is complicated by the difficulty inherent in separating exosomes from infectious HCV particles because of their similar size and density characteristics, raising the possibility that infection was mediated by virus particles in those experiments.
In summary, these data support the notion that replication-competent HCV RNA can be transferred in a virion-independent manner between permissive Huh7 cells that support genome expansion and expression. These events do not require infectious virus particles, because subgenomic replicon cells lacking viral structural genes were used in all experiments, and they appear to be dependent on an exosome-mediated, cell-cell transfer mechanism. If these events occur in vivo, exosomes containing replication-competent HCV RNA may be able to “infect” replication-permissive hepatocytes and contribute to viral spread in a manner that would avoid immune inhibition, although our results indicate that this process is much less efficient than infection by authentic virus particles.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01-AI079043 and U19-AI088778 and the Swiss National Science Foundation, Swiss Foundation for Grants in Biology and Medicine SFGBM, fellowship number PASMP3_145759.
We thank Takaji Wakita (National Institute of Infectious Diseases, Tokyo, Japan) for kindly providing the infectious JFH-1 molecular clone and replicon constructs, Francois-Loïc Cosset (Ecole Normale Superieure de Lyon, INSERM U758, Lyon, France) for retrovirus-based vectors, and Weidong Zhong (Gilead Sciences) for the inhibitor 2′-C-methyladenosine (2′-CMA). We are grateful to Stefan Wieland, Urtzi Garaigorta, Cesar Virgen, Pablo Gastaminza, and Marlene Dreux for constructive comments and suggestions and our Scripps laboratory colleagues for encouragement and help.
REFERENCES
- 1.Takahashi K, Asabe S, Wieland S, Garaigorta U, Gastaminza P, Isogawa M, Chisari FV. 2010. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci U S A 107:7431–7436. doi: 10.1073/pnas.1002301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bobrie A, Colombo M, Raposo G, Thery C. 2011. Exosome secretion: molecular mechanisms and roles in immune responses. Traffic 12:1659–1668. doi: 10.1111/j.1600-0854.2011.01225.x. [DOI] [PubMed] [Google Scholar]
- 3.Dreux M, Garaigorta U, Boyd B, Decembre E, Chung J, Whitten-Bauer C, Wieland S, Chisari FV. 2012. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 12:558–570. doi: 10.1016/j.chom.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramakrishnaiah V, Thumann C, Fofana I, Habersetzer F, Pan Q, de Ruiter PE, Willemsen R, Demmers JA, Stalin Raj V, Jenster G, Kwekkeboom J, Tilanus HW, Haagmans BL, Baumert TF, van der Laan LJ. 2013. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc Natl Acad Sci U S A 110:13109–13113. doi: 10.1073/pnas.1221899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradley D, McCaustland K, Krawczynski K, Spelbring J, Humphrey C, Cook EH. 1991. Hepatitis C virus: buoyant density of the factor VIII-derived isolate in sucrose. J Med Virol 34:206–208. doi: 10.1002/jmv.1890340315. [DOI] [PubMed] [Google Scholar]
- 6.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol 82:2120–2129. doi: 10.1128/JVI.02053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gastaminza P, Kapadia SB, Chisari FV. 2006. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J Virol 80:11074–11081. doi: 10.1128/JVI.01150-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meckes DG Jr, Raab-Traub N. 2011. Microvesicles and viral infection. J Virol 85:12844–12854. doi: 10.1128/JVI.05853-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pumeechockchai W, Bevitt D, Agarwal K, Petropoulou T, Langer BC, Belohradsky B, Bassendine MF, Toms GL. 2002. Hepatitis C virus particles of different density in the blood of chronically infected immunocompetent and immunodeficient patients: implications for virus clearance by antibody. J Med Virol 68:335–342. doi: 10.1002/jmv.10208. [DOI] [PubMed] [Google Scholar]
- 10.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. doi: 10.1053/j.gastro.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 11.Pietschmann T, Lohmann V, Kaul A, Krieger N, Rinck G, Rutter G, Strand D, Bartenschlager R. 2002. Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J Virol 76:4008–4021. doi: 10.1128/JVI.76.8.4008-4021.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dreux M, Dao Thi VL, Fresquet J, Guerin M, Julia Z, Verney G, Durantel D, Zoulim F, Lavillette D, Cosset FL, Bartosch B. 2009. Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains. PLoS Pathog 5:e1000310. doi: 10.1371/journal.ppat.1000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chairoungdua A, Smith DL, Pochard P, Hull M, Caplan MJ. 2010. Exosome release of beta-catenin: a novel mechanism that antagonizes Wnt signaling. J Cell Biol 190:1079–1091. doi: 10.1083/jcb.201002049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kogure T, Lin WL, Yan IK, Braconi C, Patel T. 2011. Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology 54:1237–1248. doi: 10.1002/hep.24504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kosaka N, Iguchi H, Yoshioka Y, Hagiwara K, Takeshita F, Ochiya T. 2012. Competitive interactions of cancer cells and normal cells via secretory microRNAs. J Biol Chem 287:1397–1405. doi: 10.1074/jbc.M111.288662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. 2010. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem 285:17442–17452. doi: 10.1074/jbc.M110.107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, Simons M. 2008. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319:1244–1247. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- 19.Yuyama K, Sun H, Mitsutake S, Igarashi Y. 2012. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. J Biol Chem 287:10977–10989. doi: 10.1074/jbc.M111.324616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bukong TN, Momen-Heravi F, Kodys K, Bala S, Szabo G. 2014. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog 10:e1004424. doi: 10.1371/journal.ppat.1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thery C, Amigorena S, Raposo G, Clayton A. 2006. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol Chapter 3:Unit 3.22. doi: 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]