

ABSTRACT
Epstein-Barr virus (EBV) infection of B cells leads to the sequential activation of two viral promoters, Wp and Cp, resulting in the expression of six EBV nuclear antigens (EBNAs) and the viral Bcl2 homologue BHRF1. The viral transactivator EBNA2 is required for this switch from Wp to Cp usage during the initial stages of infection. EBNA2-dependent Cp transcription is mediated by the EBNA2 response element (E2RE), a region that contains at least two binding sites for cellular factors; one of these sites, CBF1, interacts with RBP-JK, which then recruits EBNA2 to the transcription initiation complex. Here we demonstrate that the B cell-specific transcription factor BSAP/Pax5 binds to a second site, CBF2, in the E2RE. Deletion of the E2RE in the context of a recombinant virus greatly diminished levels of Cp-initiated transcripts during the initial stages of infection but did not affect the levels of Wp-initiated transcripts or EBNA mRNAs. Consistent with this finding, viruses deleted for the E2RE were not markedly impaired in their ability to induce B cell transformation in vitro. In contrast, a larger deletion of the entire Cp region did reduce EBNA mRNA levels early after infection and subsequently almost completely ablated lymphoblastoid cell line (LCL) outgrowth. Notably, however, rare LCLs could be established following infection with Cp-deleted viruses, and these were indistinguishable from wild-type-derived LCLs in terms of steady-state EBV gene transcription. These data indicate that, unlike Wp, Cp is dispensable for the virus' growth-transforming activity.
IMPORTANCE Epstein-Barr virus (EBV), a B lymphotropic herpesvirus etiologically linked to several B cell malignancies, efficiently induces B cell proliferation leading to the outgrowth of lymphoblastoid cell lines (LCLs). The initial stages of this growth-transforming infection are characterized by the sequential activation of two viral promoters, Wp and Cp, both of which appear to be preferentially active in target B cells. In this work, we have investigated the importance of Cp activity in initiating B cell proliferation and maintaining LCL growth. Using recombinant viruses, we demonstrate that while Cp is not essential for LCL outgrowth in vitro, it enhances transformation efficiency by >100-fold. We also show that Cp, like Wp, interacts with the B cell-specific activator protein BSAP/Pax5. We suggest that EBV has evolved this two-promoter system to ensure efficient colonization of the host B cell system in vivo.
INTRODUCTION
Epstein-Barr virus (EBV), a lymphotropic herpesvirus linked to a number of human lymphomas, efficiently transforms resting B cells in vitro into permanent lymphoblastoid cell lines (LCLs). Such LCLs are driven to continuously proliferate through the coordinated action of a limited set of viral genes; these include the six nuclear antigens (EBNA1, -2, -3A, -3B, -3C, and -LP), the viral Bcl2 homologue BHRF1, three latent membrane proteins (LMPs) (LMP1, LMP2A, and LMP2B), two small nonpolyadenylated EBV-encoded RNAs (EBERs), and series of microRNAs (miRNAs) (1). The early stages of this B cell transformation process are characterized by the sequential activation of two viral promoters (2). The initiating event is the activation of Wp, a viral promoter present in each of the 4 to 8 tandemly arranged BamHI W repeats, which is dependent on the B cell-specific transcription factor BSAP/Pax5 (3). At early time points, these Wp-initiated transcripts lead to the expression of BHRF1 and the two nuclear antigens EBNA2 and EBNA-LP. Subsequently, EBNA2 transactivates an alternative EBNA promoter, Cp, and this switch in promoter usage is accompanied by the expression of the remaining nuclear antigens EBNA1, EBNA3A, EBNA3B, and EBNA3C and the LMPs (1).
Much work has focused on the identification of sequences that govern Cp activity in infected B cells. Of these, the EBNA2 response element (E2RE) situated between positions −429 and −245 relative to the Cp transcription start site is the most critical (4–7). Genetic and biochemical studies have defined two binding sites within the E2RE, termed CBF1 and CBF2, which interact with cellular transcription factors (4, 8). Several groups demonstrated that the CBF1 site binds RBP-JK (9–12), a component of the Notch signaling pathway. RBP-JK subsequently recruits EBNA2 to the promoter, which simultaneously abrogates the RBP-JK-mediated repression of Cp while stimulating viral transcription through the EBNA2 transactivation domain (11, 13, 14). While the CBF2 site has been less well characterized, one study reported that this sequence interacts with the AU-rich element RNA binding protein 1 (AUF1), also known as heterogeneous nuclear ribonucleoprotein D (hnRNPD) (15). In addition, early DNase I footprinting studies revealed that the E2RE sequence from positions −362 to −327, which includes the CBF2 site, was specifically protected by an unidentified B lymphocyte-specific transcription factor (7). Taken together with the observation from in vitro reporter assays that Cp, like Wp, is preferentially active in B cells (16), these findings suggest that a B cell-specific factor may regulate Cp activity. However, both RBP-JK and AUF1 are ubiquitously expressed, and therefore, this B cell specificity remains unexplained. Other studies have also identified a number of promoter-proximal sequences critical for Cp activity. These include sites for NF-Y, Sp1, and C/EBP, which contribute to EBNA1-dependent activation of Cp (17, 18); E2F1, ARID3A/Bright, and Oct-2, which act as a bridging complex between Cp and oriP-bound EBNA1 (19); and the cell cycle regulatory proteins E2F1 and Rb, which interact with the histone H3K4 demethylase LSD1 (20).
While the above-described studies have been invaluable in identifying factors that regulate Cp activity, it remains unclear to what extent Cp is required for the virus' B cell growth-transforming activity. In this context, we previously used recombinant EBV technology to demonstrate that a functional Wp is absolutely required to initiate B cell growth transformation and for the outgrowth of virus-transformed LCLs. In contrast, previous studies with recombinant EBV genomes lacking either the entire Cp promoter or just the E2RE indicated that Cp is dispensable for LCL formation (21–24). However, many of the previous Cp studies used recombinant viruses generated by linked transformation marker rescue of the nontransforming P3HR1 virus, in which the EBNA2 deletion was repaired at the same time as the introduction of Cp mutations. Notably, this approach is unable to investigate the early events during the infection process or quantitatively measure the effects of Cp mutations on B cell transformation efficiency, because of the lack of productively infected cell lines carrying the recombinant EBV genomes.
Following our own finding that Wp activity is dependent on BSAP/Pax5 (3), the present work reexamines the B cell-specific nature of Cp and more accurately quantifies the role of Cp during B cell transformation. To this end, we used the B95.8 strain-derived 2089 bacterial artificial chromosome (BAC) system (25) to generate recombinant viruses with defined deletions within the Cp region. Our findings indicate that viral sequences in addition to the E2RE appear to contribute to Cp activation in the context of the virus genome and that while Cp is not absolutely essential for B cell transformation, Cp activity greatly enhances the efficient outgrowth of EBV-transformed LCLs.
MATERIALS AND METHODS
Ethics statement.
This research was approved by the Black Country Committee of the United Kingdom National Research Ethics Service (REC reference number 14/WM/0001).
Electrophoretic mobility shift assays (EMSAs).
The preparation of nuclear extracts and the in vitro binding assay conditions were described previously (26). The E2RE probe was a [γ-32P]ATP-end-labeled PCR-generated sequence corresponding to nucleotides −430 to −330 relative to the Cp transcription start site. The wild-type and mutant RBP-JK (CBF1) and CBF2 binding site sequences used as competitors were described previously (8, 27). Supershift reactions were carried out with a goat polyclonal BSAP (Pax5 [C-20]) antibody (Santa Cruz Biotechnology) (28). In vitro-translated (IVT) proteins were generated from plasmids pSG5-BSAP (29) and pu1093-5 (RBP-JK) (30) by using the TNT rabbit reticulocyte system (Promega).
Recombinant viruses.
Recombinant EBV genomes containing Cp deletions were created from the B95.8 strain-derived EBV BAC construct 2089 (25) and its 2W derivative (31) by homologous recombination. Shuttle vectors designed to delete the entire Cp promoter region from positions −429 to +846 (Cp knockout [CpKO]) or just the E2RE sequences from positions −429 to −245 (CpE2REKO) were created from a pBluescript vector, pBS-Cp, containing BamHI C sequences from positions 7315 to 14558 (32) and a tetracycline resistance cassette. pBS-CpKO was created by deleting the EagI-MscI fragment (positions10801 to 12190) (32), while pBS-CpE2REKO was made by deletion of the EagI-SacI fragment (positions 10801 to 11089). The flanking sequences acted as regions for homologous recombination with the relevant BAC following their introduction into recombinase-expressing Escherichia coli (33). As a control, a revertant virus containing a wild-type Cp sequence was made by homologous recombination between pBS-Cp and the 8W-CpKO BAC. BAC integrity and the number of W repeats were confirmed by restriction enzyme digestion and visualization of bands on ethidium bromide-stained 0.8% agarose gels following either standard or field inversion gel electrophoresis.
The recombinant BACs were transfected into HEK-293 cells by using Lipofectamine 2000 (Invitrogen), and clones were selected in the presence of 100 μg/ml hygromycin B. Individual clones were selected for their ability to produce high titers of virus following transfection and induction of the lytic cycle with BZLF1 and BALF4 expression plasmids. Virus supernatants were harvested and purified by density gradient centrifugation (Optiprep; Axis Shield), and virus titration was carried out by using quantitative PCR (QPCR) as previously described (34).
B cell infection.
B cells were positively selected from apheresis cones (National Health Service Blood and Transplant [NHSBT], Birmingham, United Kingdom) by using CD19 Dynabeads (Invitrogen), followed by detachment with CD19 Detachabead (Invitrogen), according to the manufacturer's protocols. Isolated B cells were incubated overnight with recombinant virus preparations at a known multiplicity of infection (MOI). The attachment of the recombinant virus to B cells was assessed by binding assays, as previously described (35).
Analysis of EBV gene transcription.
RNA was prepared from cells by using a Nucleospin II kit (Fisher). Residual genomic DNA was removed by using a DNA-free kit (Ambion), and cDNA was synthesized by using a QScript kit (VWR), according to the manufacturers' instructions. EBV transcripts were measured in aliquots of cDNA by using either a standard 96-well QPCR assay format (36) or a high-throughput 48:48 Dynamic Array Integrated Fluidics Circuit (Fluidigm) (37). For 48:48 arrays, cDNA samples were subjected to an initial 12 rounds of preamplification with primers directed against 45 EBV and 3 endogenous control human cellular sequences. The QPCR array was then performed on a 1-in-5 dilution of the preamplified product in a Biomark HD instrument (Fluidigm), using primers and probes directed toward the same 48 target sequences. The data were analyzed with Biomark real-time PCR analysis software, version 2.0 (Fluidigm). In both standard and 48:48 array QPCR experiments, the AQ plasmid (37), containing a single copy of each amplicon, was used to generate standard curves, permitting absolute quantification of transcript numbers.
Transformation assays.
B cell transformation assays were carried out in 96-well plates with fixed cell numbers and various virus doses, as previously described, and wells with growing colonies were scored after 6 weeks (3, 31).
RESULTS
The B cell-specific activator protein BSAP/Pax5 binds to the Cp promoter.
While a number of transcription factors have been shown to be involved in the regulation of Cp (Fig. 1A), the molecular basis of the preferential B cell-specific activity of Cp described in the literature (6, 16) and confirmed by our own unpublished Cp reporter assays is currently unclear. Since our previous studies showed that the B cell specificity of Wp was mediated by BSAP/Pax5 (3, 28), we considered whether the same factor also interacted with Cp sequences.
FIG 1.

BSAP/Pax5 interacts with the CBF2 binding site. (A) Schematic representation of the linear EBV genome indicating the multiple repeats of the BamHI W region, each containing a copy of Wp, and the second upstream EBNA promoter Cp. Also shown are the locations of the EBV latent genes, the latent origin of replication (oriP), and the terminal repeats (TR). The inset is a schematic illustration of the Cp/Wp region indicating the relative positions of known cellular transcription factors. (B) Alignment of the BSAP/Pax5 consensus binding sequence with BSAP/Pax5 binding sites from the H2B 2.1 promoter, low-affinity Wp site B, high-affinity EBV Wp site D, and the Cp CBF2 binding site. Shaded nucleotides indicate positions conserved in the BSAP/Pax5 consensus, while nucleotides in boldface type are conserved between Wp site D and CBF2. (C) Protein-DNA complexes formed by incubating the 100-bp radiolabeled E2RE probe with nuclear extracts from either the T cell line CEM or the B cell line Rael-BL, either alone (−) or in the presence of the indicated oligonucleotide competitors. Supershifts were performed by the addition of a Pax5 antibody. The last three tracks indicate protein-DNA complexes formed by incubating the E2RE probe with IVT RBP-JK and BSAP proteins. Ab, antibody.
The transcription factor BSAP/Pax5 plays an essential role in B cell development and is expressed from the pro-B cell stage through to the mature B cell stage but is absent in terminally differentiated plasma cells (38–41). Interestingly, a previously reported DNase I footprinting analysis suggested that a region of the E2RE spanning the CBF2 site interacted with a B cell-restricted factor. Notably, the CBF2 binding site in Cp bears striking sequence similarity to the high-affinity BSAP binding site D within Wp (Fig. 1B, boldface type) as well as homology to the H2B2.1 BSAP binding site, the low-affinity BSAP site B in Wp, and the published degenerate BSAP consensus sequence (Fig. 1B, gray shading) (42). We therefore carried out a series of EMSAs to determine whether BSAP/Pax5 bound to the Cp promoter (Fig. 1C). Incubation of a radiolabeled E2RE probe with a nuclear extract derived from a non-B cell line, CEM, led to the formation of a single protein-DNA complex, C1, whereas incubation with nuclear extract from a B cell line, Rael, yielded the same complex and two additional slower-migrating complexes, C2 and C3. This cell type-specific pattern of protein-DNA complexes was confirmed by using nuclear extracts from four other non-B cell lines and six additional B cell lines (data not shown). The formation of this C1 complex seen in both CEM and Rael nuclear extracts was totally blocked by preincubation with the RBP-JK/CBF1 oligonucleotide competitor but not the mutated RBP-JKmut or CBF2 competitor. Thus, the C1 complex likely corresponds to the ubiquitous factor RBP-JK bound at the CBF1 site. Regarding the two B cell-specific complexes, C2 formation was specifically inhibited by the CBF2 competitor, while C3 formation was abrogated by the RBP-JK and CBF2 competitors. These findings strongly suggest that the C3 complex contains RBP-JK bound to the CBF1 site and a second, B cell-restricted factor bound to the CBF2 site.
To investigate whether the B cell-specific complexes C2 and C3 contained BSAP/Pax5, the EMSAs were repeated in the presence of a Pax5 antibody. The addition of the anti-Pax5 antibody greatly reduced the formation of both complexes C2 and C3 (Fig. 1C), clearly indicating that these complexes contain BSAP/Pax5. Note that the anti-Pax5 antibody had no effect on complex C1 containing RBP-JK. BSAP/Pax5 binding was confirmed in subsequent experiments using in vitro-translated (IVT) RBP-JK and BSAP proteins. Thus, incubation of IVT RBP-JK and BSAP proteins, either singly or in combination, reproduced the same pattern of protein-DNA complexes as that observed in the presence of the Rael nuclear extract (Fig. 1C). Taken together, these data indicate a novel interaction between BSAP/Pax5 and the CBF2 site.
Generation of recombinant EBV genomes carrying Cp deletions.
In order to address the contribution of Cp sequences to the initiation and maintenance of B cell growth transformation, we constructed a series of recombinant viruses carrying either a 1.3-kb deletion spanning the entire Cp region from positions −429 to +846 (CpKO) or a deletion that removed just the E2RE sequences from positions −429 to −245, which included the newly identified BSAP/Pax5 site (CpE2REKO) (Fig. 2). These recombinant EBV genomes were created from B95.8-derived BAC construct 2089, which contains 11 copies of the major BamHI internal repeat, or a 2W derivative carrying just two BamHI W copies (3, 25, 31). As previously reported (31), we observed a tendency for the 2089-derived BACs to lose BamHI W repeats during the recombination process in bacteria (data not shown). Since our previous studies demonstrated that the number of BamHI W repeats markedly affects the efficiency of B cell outgrowth, with a minimum of five copies being necessary for optimal growth transformation (31), we therefore selected a series of 2089-derived recombinant BACs, each containing 8 copies of BamHI W (denoted 8W, 8WCpKO, and 8WE2REKO) for use in subsequent experiments (Fig. 2). As a control, we also created a Cp revertant in the context of an eight-BamHI W repeat-containing BAC (8WCp-rev) by homologous recombination of wild-type Cp sequences with the 8WCpKO BAC. In parallel, two corresponding series of CpKO and E2REKO recombinants were also created in the background of the 2W BAC (31).
FIG 2.

Construction of recombinant EBV genomes carrying Cp deletions. (A) Schematic illustration of the parental 8W recombinant BAC containing 8 copies of the BamHI W repeat region. HYG, hygromycin; TET, tetracycline; GFP, green fluorescent protein. (B) Linear representation of the Cp/Wp region indicating the size and position of the Cp deletions introduced into the 8W BAC and the location of known transcription factor binding sites. 8WE2REKO is deleted for sequences from positions −429 to −245 relative to the Cp transcription start (which includes the EBNA2 response element), while 8WCpKO is deleted for the entire Cp promoter region from positions −429 to +846.
BACs containing the desired mutations were introduced into HEK-293 cells, and the transfected cells were selected with hygromycin, before screening of individual clones for high levels of virus production. Density gradient-purified virus stocks from at least two different HEK-293 clones were made for each recombinant. No consistent difference was found in virus titers produced from the different clones, indicating that the introduced Cp mutations had no effect on virus production (data not shown). Likewise there were no consistent differences between the different virus preparations in virus binding assays (35).
EBV latent-cycle transcription following primary B cell infection.
We initially examined the early transcriptional events following B cell infection. CD19-positive B cells were infected at an MOI of 100 with the panel of wild-type and Cp recombinant EBVs, and aliquots of infected cells were harvested at intervals of 1, 2, 3, and 5 days for RNA analysis. In order to comprehensively screen viral transcripts in small numbers of infected cells, viral RNA was quantified by QPCR analysis using a 48:48 Dynamic Array (Fluidigm), which allows the simultaneous amplification of 48 samples with 48 different QPCR assays in individual nanoliter-scale reactions (37). In contrast to previous studies, absolute quantification of each transcript was achieved by using standard curves derived from preamplified dilutions of the AQ plasmid, which contained a single copy of each target amplicon.
The data in Fig. 3 show a subset of results from one representative experiment measuring EBV gene transcription in B cells infected with the 8W series of recombinant viruses. Wp-initiated transcripts were detectable with 8W, 8WE2REKO, and 8WCp-rev viruses at 1 day postinfection and reached similar levels by day 5, with up to 4,000 Wp transcripts per ng of RNA (Fig. 3A). However, the maximal levels of Wp transcripts were about 4-fold lower in 8WCpKO virus-infected cells. While we were unable to detect Cp-initiated transcription in cells infected with the 8WCpKO virus, Cp transcripts from the 8W and 8WCp-rev viruses were readily detectable at 2 days postinfection and steadily increased throughout the time course, eventually reaching 4,000 to 5,000 transcripts per ng RNA at day 5 (Fig. 3A); note that these values were similar to those seen for Wp transcripts. In sharp contrast, Cp-initiated transcription in cells infected with the 8WE2REKO virus followed a temporal pattern similar to that seen with the wild-type virus, but the absolute levels of transcription were >10-fold lower. Thus, the 8WE2REKO virus is clearly impaired in its ability to activate Cp during the early stages of the infection process. Notably, however, the levels of Wp-initiated transcripts with the 8WE2REKO virus were comparable to those in the wild-type 8W and revertant 8WCp-rev viruses, indicating that Wp activity had not increased to compensate for reduced levels of Cp activity.
FIG 3.

EBV latent gene transcription in primary B cells infected with recombinant viruses. Primary B cells were exposed to 8W wild-type, Cp-deleted, or revertant viruses at an MOI of 100, and EBV gene expression was quantified at 1, 2, 3, and 5 days postinfection by using a 48:48 Dynamic PCR array. (A) Absolute copy numbers of Wp- and Cp-initiated transcripts per ng total RNA. (B) Relative activity of Wp and Cp expressed as a percentage of the total number of Wp and Cp transcripts. (C) Absolute copy numbers of selected EBV latent gene transcripts expressed per ng total RNA. Histograms represent the means and standard errors of data from triplicate RNA samples independently preamplified and tested by QPCR.
Since our assays measure absolute copy numbers of viral transcripts, we were able to directly compare the levels of Cp- and Wp-initiated transcripts in infected cells. To allow a clearer comparison of promoter usage between different viruses, Wp and Cp levels were expressed as a percentage of the combined number of Wp and Cp transcripts (Fig. 3B). In the case of the 8W, 8WE2RE, and 8WCp-rev viruses, Wp activity accounted for 95 to 100% of the transcripts at day 1 postinfection. In the case of 8W and 8WCp-rev, the proportion of Wp-initiated transcripts fell substantially at day 2, which was accompanied by a concomitant increase in the level of Cp transcripts to ∼30 to 40% of the overall promoter usage, while at day 3 and day 5 postinfection, the proportion of Cp and Wp usage was approximately similar. In contrast, Wp remained the dominant promoter throughout the time course in cells infected with the 8WE2REKO virus, accounting for >90% of the transcripts.
Wp and Cp drive the transcription of a long primary transcript, which is subsequently processed to generate the individual EBNA mRNAs. At early times postinfection, the most abundant mRNA products in 8W (wild-type)-infected cells were EBNA2 and BHRF1 (Fig. 3C); transcripts encoding EBNA1 and EBNA3A (Fig. 3C) as well as those encoding EBNA3B and EBNA3C (data not shown) were also detectable but at much lower levels. Notably, despite the lack of efficient Cp activation, deletion of the E2RE did not reduce the overall levels of EBNA and LMP transcripts in infected cells. In contrast, all the latent genes were transcribed at lower levels in 8WCpKO-infected cells, consistent with the diminished levels of Wp activity and the complete absence of Cp activity. Note that while LMP1 and LMP2A transcript levels remained relatively low during the first 5 days postinfection, as these transcripts reach maximal expression levels only at later times, the lower level of LMP1 in the 8CpKO virus is consistent with reduced expression levels of EBNA2.
Efficiency of B cell transformation.
The early transcriptional events observed in the above-described experiments suggested that while the E2RE is necessary for efficient activation of Cp transcription, it did not impact latent gene expression during the early stages of EBV infection. We therefore investigated the importance of the E2RE for subsequent cell transformation and the establishment of LCLs. B cells were infected at a series of different MOIs before seeding of 104 cells into wells of a 96-well plate. Cultures were examined for the presence of transformed foci after 6 weeks. The results (Fig. 4A) are shown for two different clones of each of the 8W series of viruses. The 8W wild-type and 8WE2REKO viruses both transformed B cells very efficiently; the MOIs required for 50% transformation were <1 for both 8W wild-type clones and 0.5 and 4.0 for the two 8WE2REKO clones. In contrast, the 8WCpKO virus failed to yield any B cell transformants in this experiment (Fig. 4A).
FIG 4.
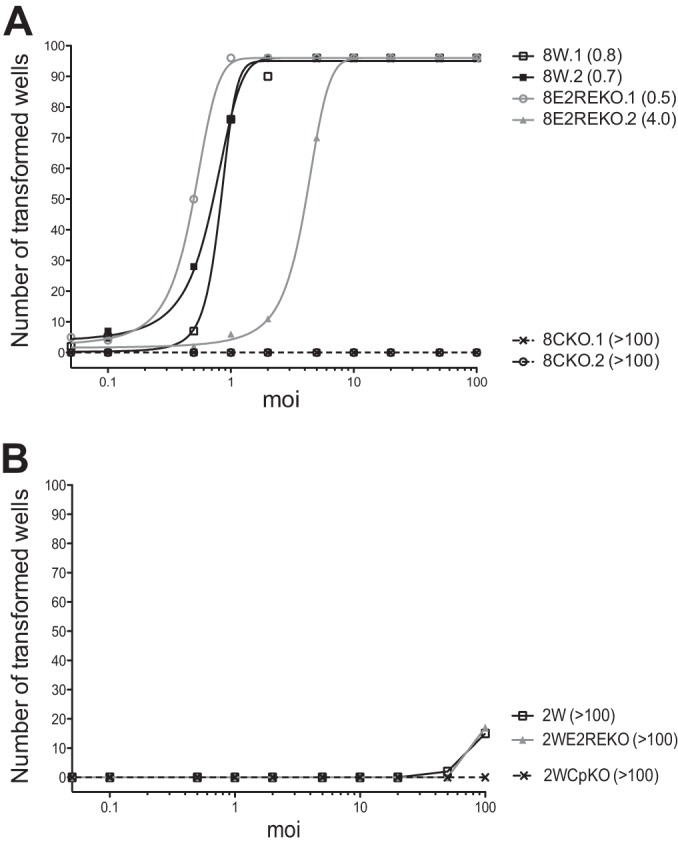
Transformation efficiencies of recombinant viruses. (A) Number of wells from a 96-well plate showing transformed foci within 6 weeks following exposure to the 8W wild-type or Cp-deleted virus at a range of MOIs. Virus preparations from two independent HEK-293 producer clones were assayed for each recombinant. The numbers in parentheses indicate the MOI required for transformation in 50% of wells. (B) Results of transformation assays using the 2W, 2WE2REKO, and 2WCpKO viruses.
In previous work, we showed that viruses with only two copies of the BamHI W repeat region were impaired for Wp/Cp activation and B cell transformation (31). We therefore examined whether deletion of the E2RE would have a more pronounced effect on B cell transformation in the 2W background. Note that these transformation assays were carried out at the same time and on the same batch of primary B cells as the above-described assays with the 8W series of viruses. As observed for the 8W recombinant viruses, the 2W wild-type and 2WE2REKO viruses had almost identical transformation efficiencies, although these efficiencies were much lower than those for the corresponding 8W viruses (Fig. 4B). In this particular experiment, there were insufficient wells with transformed foci to calculate the MOI required for 50% transformation. As expected from the 8WCpKO results, deletion of the entire Cp region in the context of this 2W virus also completely ablated the virus' growth-transforming capacity.
Although the 8WCpKO viruses failed to yield B cell transformants in the majority of the 96-well transformation assays described above, occasionally we were able to establish LCLs from rare wells showing transformed foci when B cells were infected at the highest virus doses. Furthermore, although the 2WCpKO virus never showed transformation in any of the 96-well assays, on some occasions we were able to establish LCLs from the 2WCpKO virus, as well as from the 8WCpKO virus, from bulk cultures of infected primary B cells. These cell lines took longer to establish than did the LCLs made with the other viruses, as, by microscopic examination, they represented the outgrowth of a very small number of transformed cells, unlike the situation with wild-type and E2REKO viruses, where the majority (8W) or many (2W) of the cells in each well were transformed. However, once the cultures were established and had been expanded, there was no obvious difference in the growth rates between the CpKO LCLs and the others. The absence of Cp transcripts from the 8WCpKO and 2WCpKO LCLs, together with hygromycin resistance, confirmed that these LCLs were genuine CpKO LCLs and not rare, spontaneously transformed LCLs derived from EBV-infected donor B cells.
EBV gene transcription in LCLs established with recombinant Cp viruses.
To investigate whether the Cp mutations affected the steady-state levels of Wp and Cp transcripts in established transformed lines, we screened an extensive panel of LCLs generated from different wild-type and mutant viruses; these LCLs included 12 8W wild-type LCLs, 4 8WE2REKO LCLs, 4 8WCpKO LCLs, 5 2W wild-type LCLs, 3 2WE2REKO LCLs, and 3 2WCpKO LCLs. Three observations can be made from these data (Fig. 5). First, there was a broad range in the absolute numbers of Wp- and Cp-initiated transcripts in wild-type 8W and 2W LCLs. These data emphasize the fact that Wp activity is readily detectable in established LCLs and contributes significantly to the overall levels of EBNA transcription even in the presence of a wild-type Cp sequence. Moreover, the relative levels of Cp and Wp usage are not significantly altered by the number of Wp copies present in the EBV genome. Second, deletion of the E2RE always resulted in Wp becoming the dominant promoter, with only very low levels of Cp transcripts being seen in LCLs derived from the 8WE2REKO and 2WE2REKO viruses. Thus, the pattern of promoter usage seen in the first few days after infection with E2RE-deleted viruses was maintained in the established LCLs. Third, in the absence of any detectable Cp transcription, Wp activity did not apparently increase to compensate.
FIG 5.

Wp and Cp activity in established LCLs infected with recombinant viruses. (A) Expression levels of Wp- and Cp-initiated transcripts in a range of established LCLs carrying the indicated EBV genomes determined by standard 96-well QPCR and normalized to the cellular glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression level. (B) Relative activity of Wp and Cp expressed as a percentage of the total number of Wp and Cp transcripts in the same LCLs as described above for panel A.
We then extended the analysis to a subset of these LCLs, two of each type, using a high-throughput QPCR array to screen a broader range of EBV latent- and lytic-cycle transcripts. These data (Fig. 6) are expressed as EBV transcript copy numbers normalized to the cellular PGK1 expression level. As noted above, there was considerable variation in the individual transcript levels between different LCLs, but the values were within the ranges previously reported for a panel of LCLs analyzed by using this technology (37). Importantly, with the exception of Cp activity, there were no consistent differences in the levels of any latent or lytic transcripts that could be attributed to the Cp mutations. In conclusion, while deletion of the entire Cp region or just the E2RE greatly reduces Cp activity during the initial stages of B cell infection and dramatically decreases transformation efficiency, rare LCLs carrying these mutant viruses transcribe the full spectrum of growth-transforming genes at levels comparable to those in LCLs derived from a wild-type EBV strain.
FIG 6.

EBV gene transcription in established LCLs infected with recombinant viruses. EBV transcripts from a subset of the wild-type (W), E2REKO (E), and CpKO (C) 8W and 2W LCLs were quantified by using a 48:48 Dynamic PCR array, and the data were normalized to the cellular PGK1 expression level. Data are shown for Wp- and Cp-initiated transcripts; the latent transcripts encoding BHRF1 (containing the Y2-HF splice), EBNA2, EBNA3A, EBNA1 (U-K splice), LMP1, and LMP2A; and lytic transcripts encoding BZLF1 and BHRF1 (containing the H2-HF splice). Histograms represent the means and standard errors of data from triplicate RNA samples independently preamplified and tested by QPCR.
DISCUSSION
EBV-induced B cell transformation is characterized by the sequential activation of two viral promoters, which drive the expression of six EBV nuclear antigens and the viral Bcl2 homologue BHRF1 in latently infected cells. The first promoter to be activated immediately after infection is Wp, present in each of the tandemly arranged BamHI W repeat sequences. Several cellular factors regulate transcription initiation from Wp, including the B cell transcription factor BSAP/Pax5, which is absolutely essential for both Wp activation and B cell transformation in vitro (2, 3, 31). These Wp-initiated transcripts lead to the expression of EBNA2 and the subsequent activation of an alternative EBNA promoter, Cp, located in the upstream BamHI C region. This raises an important question, namely, to what extent EBV-induced growth transformation depends on the activity of Cp, both in the early stages of the infection process and in established LCLs.
The starting point for this work was a reexamination of a previously reported observation that Cp reporter constructs are preferentially active in B cell lines (16, 34). This B cell specificity is achieved, at least in part, by interactions of available cellular factors with the E2RE (6). In this study, we identified sequence homology between the BSAP/Pax5 consensus site and sequences within the Cp E2RE and subsequently demonstrated that BSAP/Pax5 binds to the E2RE in vitro (Fig. 1). This finding supports the results of previous DNase I footprinting studies, which revealed the binding of a lymphocyte-specific factor to this region (7), and is also consistent with the results of a recent chromatin immunoprecipitation sequencing (ChIP-Seq) analysis of BSAP/Pax5 binding across the entire EBV genome (43). This newly identified BSAP binding site overlaps the CBF2 site, a region previously shown to be critical for Cp activity (27). Notably, a mutation within the CBF2 site (27), previously shown to diminish Cp activity, disrupted BSAP/Pax5 binding (Fig. 1C). Intriguingly, a previous study reported that the CBF2 site interacted with the cellular factor AUF1/hnRNPD (15), a result at odds with the results of the present work, since AUF1 is ubiquitously expressed. Although we were unable to reproduce the AUF1 interaction using EMSAs (data not shown), we cannot exclude the possibility that under certain conditions, AUF1 is recruited to Cp as part of a larger transcription factor complex that includes BSAP/Pax5. One such multiprotein complex, containing the B cell transcription factor Oct2, ARID3A, and E2F1, has been shown to bind to Cp sequences from positions −149 to −109 and mediates EBNA1-dependent activation of Cp (19). Importantly, our data demonstrate a novel interaction between BSAP/Pax5 and the E2RE, which provides a mechanistic basis for the apparent B cell specificity of Cp.
In the next series of experiments, we sought to examine the effects of specifically deleting the E2RE sequences in the context of a recombinant virus. Note that it was previously reported that E2RE-deleted viruses are still capable of inducing B cell transformation (23, 24). However, those previous studies relied upon marker rescue experiments using P3HR1-derived recombinants and were limited in their quantitative analyses of early events during the infection process and the efficiency of LCL outgrowth. In the present work, we have used purified recombinant viruses of known titers generated by using the EBV BAC system, quantified promoter usage and gene expression from early times post-B cell infection through to the establishment of LCLs using QPCR, and rigorously monitored LCL outgrowth at a range of MOIs (3, 31, 35). Our data confirm that the E2RE plays an important role in efficiently activating Cp during the early stages of infection (Fig. 3) but also show that the E2RE-deleted mutant virus was still capable of transcribing latent transcripts at levels similar to those of the wild-type virus. Consistent with these observations, E2RE-deleleted recombinants were not markedly impaired in B cell transformation assays in either the 8W or 2W virus background (Fig. 4).
Notably, the absolute levels of Wp-initiated transcripts induced by our E2REKO viruses, either at early stages postinfection or in established LCLs, were not elevated compared to those induced by wild-type viruses (Fig. 3A and 5A), suggesting that Wp activity does not simply compensate for reduced levels of Cp transcription. This result contradicts previously reported findings that Wp transcription is increased in viruses lacking the E2RE (24, 44) but is consistent with results from another report, which analyzed the effect of a point mutation in the CBF1/RBP-JK binding site (23). Interestingly, LCLs derived from E2RE-deleted viruses still retained low levels of Cp activity, indicating that additional viral sequences, such as the interaction of oriP with EBNA1, can also contribute to Cp transcription in the context of the viral genome. Indeed, a previous study using a recombinant EBV deleted for amino acids 65 to 89 of EBNA1 demonstrated the importance of EBNA1 in the activation and maintenance of Cp transcription (45). Overall, our data argue that while the binding of RBP-JK and BSAP/Pax5 to Cp is not essential for EBV-induced B cell transformation, these proteins may cooperate with factors bound to other regions of Cp (19, 46), ensuring optimal Cp activity and the expression of the latency III program of viral gene expression in infected B cells.
In contrast to the situation with the E2RE-deleted viruses, recombinants lacking the entire Cp region transformed B cells at least 100-fold less efficiently than did the wild-type virus (Fig. 4). Given that E2RE- and Cp-deleted viruses are both highly defective for Cp activity, the dramatic difference in the transforming capacities of these two mutants is surprising. However, this disparity may reflect the fact that Cp-deleted viruses expressed 3- to 4-fold-lower numbers of Wp-initiated transcripts during the early stages of infection, leading to correspondingly reduced levels of EBNA and BHRF1 transcripts at days 1 to 5 (Fig. 3). Note that there is no evidence for other unintentional mutations being present in these Cp-deleted viruses, since Wp levels were restored in a CpKO revertant (Fig. 3A). We speculate that the large 1.4-kb genomic deletion potentially disrupted the nucleosomal architecture of the oriP/Cp/Wp region or altered the position of enhancer sequences, with a concomitant reduction in Wp activity (47).
Despite being greatly impaired for B cell transformation, rare LCLs carrying Cp-deleted viruses were established, and following expansion, they proliferated in culture at rates similar to those of wild-type LCLs. These LCLs provide important clues regarding the relative contribution of Wp and Cp during B cell transformation. First, our data support previous studies demonstrating that Cp is dispensable for LCL outgrowth while also clearly showing that the presence of a functional Cp promoter greatly increases the transformation efficiency. Second, Cp-deleted LCLs exclusively used Wp to drive EBNA expression yet expressed steady-state levels of latent transcripts similar to those seen in wild-type LCLs and E2RE-deleted LCLs (Fig. 5B and 6). In a recent study (37), we described a method for estimating the absolute number of EBV transcripts on a per-cell basis. Using the same approach, the most abundant transcripts in this LCL panel were EBNA2 (median, 87 transcripts per cell), LMP1 (median, 98 transcripts), and latent BHRF1 (median, 46 transcripts), while EBNA1, EBNA3A, and LMP2A transcripts were present at much lower levels; importantly, these values are comparable to those seen previously for a larger panel of LCLs (37). These findings argue that during clonal selection following infection with a Cp-deleted virus, there is strong pressure to maintain sufficient levels of Wp-driven latent gene expression to support LCL growth. Third, it has been suggested that the switch from Wp to Cp usage is required to ensure efficient transcriptional elongation of the primary EBV transcript harboring the EBNA3 and EBNA1 coding sequences located at least 70 kb downstream of the promoter (48). Our data suggest that under certain conditions, Wp-initiated transcripts are capable of efficiently driving the transcription of the downstream EBNA genes.
The present findings differ from those of a previous study that reported that Cp deletion did not affect B cell transformation efficiency (21, 22). That previous study used a virus carrying a larger 3.3-kb deletion, spanning from upstream of the glucocorticoid response element (GRE) to beyond the Cp transcription start site (Fig. 1A), and relied on the rescue of transformation-competent recombinants derived from P3HR1. The recovery of LCLs carrying these Cp-deleted viruses was apparently no less efficient than that with a control wild-type virus (22). In addition, a cell-free supernatant containing Cp-deleted recombinant virus produced from such LCLs infected and transformed fresh B cells as efficiently as the wild-type virus (21, 22). These discrepancies may reflect technical differences between the two studies. While the previous study used crude virus preparations, our virus stocks were density gradient purified and titers were accurately determined by QPCR to ensure that equivalent MOIs were used in all experiments, while our virus transformation assays were done with a range of MOIs. The P3HR1-rescued virus may also have contained second-site mutations, which could have been selected during the initial rescue by B cell transformation. Additionally, the BACs used here are based on the B95.8 EBV strain, which has a deletion that removes ∼12 kb of sequence compared to wild-type viruses and the recombinants produced in the P3HR1 system. Importantly, however, both studies concurred that Cp activity is dispensable for B cell transformation and demonstrated that established LCLs carrying wild-type or Cp-deleted viruses had similar levels of latent gene expression (21, 22).
EBV efficiently colonizes the B cell system in vivo through the transient expression of a series of growth-transforming genes, which together induce B cell proliferation and ultimately drive the expansion of a pool of latently infected cells. This growth-transforming program involves the sequential activation of two viral promoters, Wp and Cp, which are highly conserved among primate lymphocryptoviruses (49–51), suggesting that these promoters have evolved as part of the virus' strategy to establish lifelong persistence. By recruiting a key B cell transcription factor, BSAP/Pax5, both of these promoters may be optimized to ensure high levels of viral gene transcription in the infected target cell. While the present work confirms that Wp activity is sufficient for LCL outgrowth, the efficiency of B cell transformation is greatly enhanced by the presence of functional Cp sequences.
ACKNOWLEDGMENTS
We thank Debbie Croom-Carter for technical assistance.
This work was funded by a Cancer Research UK Programme Grant award (C5575/A15032) to M.R., A.I.B., and A.B.R.
REFERENCES
- 1.Longnecker R, Kieff E, Cohen J. 2013. Epstein-Barr virus, p 1898–1959 InKnipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Woisetschlaeger M, Yandava CN, Furmanski LA, Strominger JL, Speck SH. 1990. Promoter switching in Epstein-Barr virus during the initial stages of infection of B lymphocytes. Proc Natl Acad Sci U S A 87:1725–1729. doi: 10.1073/pnas.87.5.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tierney R, Nagra J, Hutchings I, Shannon-Lowe C, Altmann M, Hammerschmidt W, Rickinson A, Bell A. 2007. Epstein-Barr virus exploits BSAP/Pax5 to achieve the B-cell specificity of its growth-transforming program. J Virol 81:10092–10100. doi: 10.1128/JVI.00358-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin XW, Speck SH. 1992. Identification of critical cis elements involved in mediating Epstein-Barr virus nuclear antigen 2-dependent activity of an enhancer located upstream of the viral BamHI C promoter. J Virol 66:2846–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rooney CM, Brimmell M, Buschle M, Allan G, Farrell PJ, Kolman JL. 1992. Host cell and EBNA-2 regulation of Epstein-Barr virus latent-cycle promoter activity in B lymphocytes. J Virol 66:496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sung NS, Kenney S, Gutsch D, Pagano JS. 1991. EBNA-2 transactivates a lymphoid-specific enhancer in the BamHI C promoter of Epstein-Barr virus. J Virol 65:2164–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woisetschlaeger M, Jin XW, Yandava CN, Furmanski LA, Strominger JL, Speck SH. 1991. Role for the Epstein-Barr virus nuclear antigen 2 in viral promoter switching during initial stages of infection. Proc Natl Acad Sci U S A 88:3942–3946. doi: 10.1073/pnas.88.9.3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling PD, Rawlins DR, Hayward SD. 1993. The Epstein-Barr virus immortalizing protein EBNA-2 is targeted to DNA by a cellular enhancer-binding protein. Proc Natl Acad Sci U S A 90:9237–9241. doi: 10.1073/pnas.90.20.9237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grossman SR, Johannsen E, Tong X, Yalamanchili R, Kieff E. 1994. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc Natl Acad Sci U S A 91:7568–7572. doi: 10.1073/pnas.91.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henkel T, Ling PD, Hayward SD, Peterson MG. 1994. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science 265:92–95. doi: 10.1126/science.8016657. [DOI] [PubMed] [Google Scholar]
- 11.Hsieh JJ, Hayward SD. 1995. Masking of the CBF1/RBPJ kappa transcriptional repression domain by Epstein-Barr virus EBNA2. Science 268:560–563. doi: 10.1126/science.7725102. [DOI] [PubMed] [Google Scholar]
- 12.Waltzer L, Logeat F, Brou C, Israel A, Sergeant A, Manet E. 1994. The human J kappa recombination signal sequence binding protein (RBP-J kappa) targets the Epstein-Barr virus EBNA2 protein to its DNA responsive elements. EMBO J 13:5633–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen JI, Kieff E. 1991. An Epstein-Barr virus nuclear protein 2 domain essential for transformation is a direct transcriptional activator. J Virol 65:5880–5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sjoblom A, Nerstedt A, Jansson A, Rymo L. 1995. Domains of the Epstein-Barr virus nuclear antigen 2 (EBNA2) involved in the transactivation of the latent membrane protein 1 and the EBNA Cp promoters. J Gen Virol 76(Part 11):2669–2678. [DOI] [PubMed] [Google Scholar]
- 15.Fuentes-Panana EM, Peng R, Brewer G, Tan J, Ling PD. 2000. Regulation of the Epstein-Barr virus C promoter by AUF1 and the cyclic AMP/protein kinase A signaling pathway. J Virol 74:8166–8175. doi: 10.1128/JVI.74.17.8166-8175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Contreras-Brodin B, Karlsson A, Nilsson T, Rymo L, Klein G. 1996. B cell-specific activation of the Epstein-Barr virus-encoded C promoter compared with the wide-range activation of the W promoter. J Gen Virol 77:1159–1162. doi: 10.1099/0022-1317-77-6-1159. [DOI] [PubMed] [Google Scholar]
- 17.Nilsson T, Zetterberg H, Wang YC, Rymo L. 2001. Promoter-proximal regulatory elements involved in oriP-EBNA1-independent and -dependent activation of the Epstein-Barr virus C promoter in B-lymphoid cell lines. J Virol 75:5796–5811. doi: 10.1128/JVI.75.13.5796-5811.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borestrom C, Zetterberg H, Liff K, Rymo L. 2003. Functional interaction of nuclear factor y and sp1 is required for activation of the Epstein-Barr virus C promoter. J Virol 77:821–829. doi: 10.1128/JVI.77.2.821-829.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borestrom C, Forsman A, Ruetschi U, Rymo L. 2012. E2F1, ARID3A/Bright and Oct-2 factors bind to the Epstein-Barr virus C promoter, EBNA1 and oriP, participating in long-distance promoter-enhancer interactions. J Gen Virol 93:1065–1075. doi: 10.1099/vir.0.038752-0. [DOI] [PubMed] [Google Scholar]
- 20.Chau CM, Deng Z, Kang H, Lieberman PM. 2008. Cell cycle association of the retinoblastoma protein Rb and the histone demethylase LSD1 with the Epstein-Barr virus latency promoter Cp. J Virol 82:3428–3437. doi: 10.1128/JVI.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swaminathan S. 1996. Characterization of Epstein-Barr virus recombinants with deletions of the BamHI C promoter. Virology 217:532–541. doi: 10.1006/viro.1996.0148. [DOI] [PubMed] [Google Scholar]
- 22.Swaminathan S, Hesselton R, Sullivan J, Kieff E. 1993. Epstein-Barr virus recombinants with specifically mutated BCRF1 genes. J Virol 67:7406–7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans TJ, Farrell PJ, Swaminathan S. 1996. Molecular genetic analysis of Epstein-Barr virus Cp promoter function. J Virol 70:1695–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoo L, Speck SH. 2000. Determining the role of the Epstein-Barr virus Cp EBNA2-dependent enhancer during the establishment of latency by using mutant and wild-type viruses recovered from cottontop marmoset lymphoblastoid cell lines. J Virol 74:11115–11120. doi: 10.1128/JVI.74.23.11115-11120.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delecluse HJ, Hammerschmidt W. 2000. The genetic approach to the Epstein-Barr virus: from basic virology to gene therapy. Mol Pathol 53:270–279. doi: 10.1136/mp.53.5.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bell A, Skinner J, Kirby H, Rickinson A. 1998. Characterisation of regulatory sequences at the Epstein-Barr virus BamHI W promoter. Virology 252:149–161. doi: 10.1006/viro.1998.9440. [DOI] [PubMed] [Google Scholar]
- 27.Fuentes-Panana EM, Ling PD. 1998. Characterization of the CBF2 binding site within the Epstein-Barr virus latency C promoter and its role in modulating EBNA2-mediated transactivation. J Virol 72:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tierney R, Kirby H, Nagra J, Rickinson A, Bell A. 2000. The Epstein-Barr virus promoter initiating B-cell transformation is activated by RFX proteins and the B-cell-specific activator protein BSAP/Pax5. J Virol 74:10458–10467. doi: 10.1128/JVI.74.22.10458-10467.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reimold AM, Ponath PD, Li YS, Hardy RR, David CS, Strominger JL, Glimcher LH. 1996. Transcription factor B cell lineage-specific activator protein regulates the gene for human X-box binding protein 1. J Exp Med 183:393–401. doi: 10.1084/jem.183.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zimber-Strobl U, Kremmer E, Grasser F, Marschall G, Laux G, Bornkamm GW. 1993. The Epstein-Barr virus nuclear antigen 2 interacts with an EBNA2 responsive cis-element of the terminal protein 1 gene promoter. EMBO J 12:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tierney RJ, Kao KY, Nagra JK, Rickinson AB. 2011. Epstein-Barr virus BamHI W repeat number limits EBNA2/EBNA-LP coexpression in newly infected B cells and the efficiency of B-cell transformation: a rationale for the multiple W repeats in wild-type virus strains. J Virol 85:12362–12375. doi: 10.1128/JVI.06059-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Jesus O, Smith PR, Spender LC, Elgueta Karstegl C, Niller HH, Huang D, Farrell PJ. 2003. Updated Epstein-Barr virus (EBV) DNA sequence and analysis of a promoter for the BART (CST, BARF0) RNAs of EBV. J Gen Virol 84:1443–1450. doi: 10.1099/vir.0.19054-0. [DOI] [PubMed] [Google Scholar]
- 33.Neuhierl B, Delecluse HJ. 2005. Molecular genetics of DNA viruses: recombinant virus technology. Methods Mol Biol 292:353–370. [DOI] [PubMed] [Google Scholar]
- 34.Shannon-Lowe C, Adland E, Bell AI, Delecluse HJ, Rickinson AB, Rowe M. 2009. Features distinguishing Epstein-Barr virus infections of epithelial cells and B cells: viral genome expression, genome maintenance, and genome amplification. J Virol 83:7749–7760. doi: 10.1128/JVI.00108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shannon-Lowe C, Baldwin G, Feederle R, Bell A, Rickinson A, Delecluse HJ. 2005. Epstein-Barr virus-induced B-cell transformation: quantitating events from virus binding to cell outgrowth. J Gen Virol 86:3009–3019. doi: 10.1099/vir.0.81153-0. [DOI] [PubMed] [Google Scholar]
- 36.Bell AI, Groves K, Kelly GL, Croom-Carter D, Hui E, Chan AT, Rickinson AB. 2006. Analysis of Epstein-Barr virus latent gene expression in endemic Burkitt's lymphoma and nasopharyngeal carcinoma tumour cells by using quantitative real-time PCR assays. J Gen Virol 87:2885–2890. doi: 10.1099/vir.0.81906-0. [DOI] [PubMed] [Google Scholar]
- 37.Tierney RJ, Shannon-Lowe CD, Fitzsimmons L, Bell AI, Rowe M. 2015. Unexpected patterns of Epstein-Barr virus transcription revealed by a high throughput PCR array for absolute quantification of viral mRNA. Virology 474:117–130. doi: 10.1016/j.virol.2014.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adams B, Dorfler P, Aguzzi A, Kozmik Z, Urbanek P, Maurer-Fogy I, Busslinger M. 1992. Pax-5 encodes the transcription factor BSAP and is expressed in B lymphocytes, the developing CNS, and adult testis. Genes Dev 6:1589–1607. doi: 10.1101/gad.6.9.1589. [DOI] [PubMed] [Google Scholar]
- 39.Barberis A, Widenhorn K, Vitelli L, Busslinger M. 1990. A novel B-cell lineage-specific transcription factor present at early but not late stages of differentiation. Genes Dev 4:849–859. doi: 10.1101/gad.4.5.849. [DOI] [PubMed] [Google Scholar]
- 40.Nutt SL, Heavey B, Rolink AG, Busslinger M. 1999. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 401:556–562. doi: 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- 41.Urbanek P, Wang ZQ, Fetka I, Wagner EF, Busslinger M. 1994. Complete block of early B cell differentiation and altered patterning of the posterior midbrain in mice lacking Pax5/BSAP. Cell 79:901–912. doi: 10.1016/0092-8674(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Z, Espinoza CR, Yu Z, Stephan R, He T, Williams GS, Burrows PD, Hagman J, Feeney AJ, Cooper MD. 2006. Transcription factor Pax5 (BSAP) transactivates the RAG-mediated V(H)-to-DJ(H) rearrangement of immunoglobulin genes. Nat Immunol 7:616–624. doi: 10.1038/ni1339. [DOI] [PubMed] [Google Scholar]
- 43.Arvey A, Tempera I, Lieberman PM. 2013. Interpreting the Epstein-Barr virus (EBV) epigenome using high-throughput data. Viruses 5:1042–1054. doi: 10.3390/v5041042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoo LI, Mooney M, Puglielli MT, Speck SH. 1997. B-cell lines immortalized with an Epstein-Barr virus mutant lacking the Cp EBNA2 enhancer are biased toward utilization of the oriP-proximal EBNA gene promoter Wp1. J Virol 71:9134–9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Altmann M, Pich D, Ruiss R, Wang J, Sugden B, Hammerschmidt W. 2006. Transcriptional activation by EBV nuclear antigen 1 is essential for the expression of EBV's transforming genes. Proc Natl Acad Sci U S A 103:14188–14193. doi: 10.1073/pnas.0605985103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Almqvist J, Zou J, Linderson Y, Borestrom C, Altiok E, Zetterberg H, Rymo L, Pettersson S, Ernberg I. 2005. Functional interaction of Oct transcription factors with the family of repeats in Epstein-Barr virus oriP. J Gen Virol 86:1261–1267. doi: 10.1099/vir.0.80620-0. [DOI] [PubMed] [Google Scholar]
- 47.Nilsson T, Sjoblom A, Masucci MG, Rymo L. 1993. Viral and cellular factors influence the activity of the Epstein-Barr virus BCR2 and BWR1 promoters in cells of different phenotype. Virology 193:774–785. doi: 10.1006/viro.1993.1186. [DOI] [PubMed] [Google Scholar]
- 48.Palermo RD, Webb HM, West MJ. 2011. RNA polymerase II stalling promotes nucleosome occlusion and pTEFb recruitment to drive immortalization by Epstein-Barr virus. PLoS Pathog 7:e1002334. doi: 10.1371/journal.ppat.1002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fuentes-Panana EM, Swaminathan S, Ling PD. 1999. Transcriptional activation signals found in the Epstein-Barr virus (EBV) latency C promoter are conserved in the latency C promoter sequences from baboon and rhesus monkey EBV-like lymphocryptoviruses (cercopithicine herpesviruses 12 and 15). J Virol 73:826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rivailler P, Cho YG, Wang F. 2002. Complete genomic sequence of an Epstein-Barr virus-related herpesvirus naturally infecting a New World primate: a defining point in the evolution of oncogenic lymphocryptoviruses. J Virol 76:12055–12068. doi: 10.1128/JVI.76.23.12055-12068.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rivailler P, Jiang H, Cho YG, Quink C, Wang F. 2002. Complete nucleotide sequence of the rhesus lymphocryptovirus: genetic validation for an Epstein-Barr virus animal model. J Virol 76:421–426. doi: 10.1128/JVI.76.1.421-426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]