

ABSTRACT
Bioengineering of viruses and virus-like particles (VLPs) is a well-established approach in the development of new and improved vaccines against viral and bacterial pathogens. We report here that the capsid of a major avian pathogen, infectious bursal disease virus (IBDV), can accommodate heterologous proteins to induce protective immunity. The structural units of the ∼70-nm-diameter T=13 IBDV capsid are trimers of VP2, which is made as a precursor (pVP2). The pVP2 C-terminal domain has an amphipathic α helix that controls VP2 polymorphism. In the absence of the VP3 scaffolding protein, 466-residue pVP2 intermediates bearing this α helix assemble into genuine VLPs only when expressed with an N-terminal His6 tag (the HT-VP2-466 protein). HT-VP2-466 capsids are optimal for protein insertion, as they are large enough (cargo space, ∼78,000 nm3) and are assembled from a single protein. We explored HT-VP2-466-based chimeric capsids initially using enhanced green fluorescent protein (EGFP). The VLP assembly yield was efficient when we coexpressed EGFP-HT-VP2-466 and HT-VP2-466 from two recombinant baculoviruses. The native EGFP structure (∼240 copies/virion) was successfully inserted in a functional form, as VLPs were fluorescent, and three-dimensional cryo-electron microscopy showed that the EGFP molecules incorporated at the inner capsid surface. Immunization of mice with purified EGFP-VLPs elicited anti-EGFP antibodies. We also inserted hemagglutinin (HA) and matrix (M2) protein epitopes derived from the mouse-adapted A/PR/8/34 influenza virus and engineered several HA- and M2-derived chimeric capsids. Mice immunized with VLPs containing the HA stalk, an M2 fragment, or both antigens developed full protection against viral challenge.
IMPORTANCE Virus-like particles (VLPs) are multimeric protein cages that mimic the infectious virus capsid and are potential candidates as nonliving vaccines that induce long-lasting protection. Chimeric VLPs can display or include foreign antigens, which could be a conserved epitope to elicit broadly neutralizing antibodies or several variable epitopes effective against a large number of viral strains. We report the biochemical, structural, and immunological characterization of chimeric VLPs derived from infectious bursal disease virus (IBDV), an important poultry pathogen. To test the potential of IBDV VLPs as a vaccine vehicle, we used the enhanced green fluorescent protein and two fragments derived from the hemagglutinin and the M2 matrix protein of the human murine-adapted influenza virus. The IBDV capsid protein fused to influenza virus peptides formed assemblies able to protect mice against viral challenge. Our studies establish the basis for a new generation of multivalent IBDV-based vaccines.
INTRODUCTION
Virus capsids are used as protein cages or platforms to incorporate various types of materials at inner and/or outer capsid surfaces or as nanocontainers to encapsulate proteins or other biomolecules with potential application in nanomedicine and nanobiotechnology (1, 2). The use of virus-like particles (VLPs) is a promising strategy for vaccine development (3–5). VLPs generally stimulate strong B and T cell immune responses and, in the absence of adjuvants, target dendritic cells to promote their maturation and migration, a step essential for activating the innate and adaptive immune responses. These features, which led to the description of VLPs as self-adjuvanting immunogen delivery systems (6, 7), make VLPs attractive stand-alone vaccine candidates for many diseases (8–11). In addition, VLPs can also be used as platforms for the multimeric display of foreign antigens (12–15). Here we introduce a strategy for engineering chimeric VLPs for presentation of heterologous proteins to the immune system, using the capsid of the infectious bursal disease virus (IBDV).
IBDV, a major pathogen in the poultry industry worldwide, is a double-stranded RNA virus with an ∼70-nm-diameter T=13 icosahedral capsid (16). The capsid protein VP2 is synthesized as a precursor, pVP2, which is part of the polyprotein NH2-pVP2-VP4-VP3-COOH. The pVP2 C terminus, which is processed by several proteases, bears the molecular switch (the amphipathic α5 helix) that controls VP2 structural plasticity (17). VP3 participates during capsid assembly as a canonical scaffolding protein (18, 19). Expression of VP2 alone results in the assembly of ∼23-nm-diameter T=1 subviral particles (SVPs) (20–22). VP2 is folded into three domains, termed the projection (P), shell (S), and base (B) domains. Domains S and P are β barrels, whereas the B domain is formed by N- and C-terminal α helices facing the shell interior.
In addition to attenuated or inactivated IBDV-based vaccines (23), VP2 expression provides complete protection against IBDV (24–26). T=1 SVPs have also been used as vaccine carriers to target diseases such as cancer, after incorporation of a 54-residue E7 oncoprotein fragment of human papillomavirus 16 to the VP2 C terminus (27). The T=1 SVP platform nevertheless entails considerable space limitations, as its cargo space is only 380 nm3. Exposed loops at the tip of the trimeric VP2 spikes could be alternative targets for heterologous peptide insertion (28), such as the PBC loop (between β strands B and C of the P domain) that incorporates foot-and-mouth disease virus (FMDV) epitopes in T=1 SVPs (29).
In the absence of VP3, 466-residue pVP2 intermediates with helix α5 (VP2-466) assemble into VLPs only when expressed with an N-terminal His6 tag (the HT-VP2-466 protein), which plays a role that emulates the scaffolding role of VP3 (17). This HT-VP2-466 protein assembles into genuine T=13 capsids and related assemblies that lack the other four viral proteins and are >200 times larger than the T=1 SVPs, making them optimal for insertion of much larger peptides or proteins.
We have established the structural basis for the development of new HT-VP2-466-based chimeric capsids as a vaccine carrier, initially using enhanced green fluorescent protein (EGFP) fused at the HT-VP2-466 N-terminal end. As these chimeric VLPs proved to be excellent containers for carrying macromolecules to living cells, we tested two fragments derived from the human murine-adapted influenza virus A/PR/8/34 (H1N1), the hemagglutinin (HA) and matrix (M2) proteins, both of which elicit protective antibodies with broad activity. Our approach allowed the generation of a bivalent vaccine that included epitopes for VP2 as well as influenza virus HA and M2 and shows the potential for further use in the development of other multivalent vaccines.
MATERIALS AND METHODS
Cells and viruses.
For IBDV infections, we used the Soroa isolate (30), adapted to growth in QM7 quail muscle cells. Influenza virus A/PR/8/34 (H1N1), adapted to the mouse and grown in MDCK canine kidney cells, was used in challenge experiments. Recombinant baculovirus (rBV) FB/CAP (expressing a 466-residue capsid [CAP] protein with an N-terminal His6 tag) and recombinant vaccinia virus (rVV) vT7lacOI-POLY have been described previously (17, 31). Expression experiments were carried out in BSC-40 cells for rVV infections and Trichoplusia ni (H5) insect cells (Invitrogen) for rBV infections. QM7, BSC-40, and MDCK cells were cultured in Dulbecco modified Eagle medium (DMEM) with 10% fetal calf serum. H5 cells were cultured as described previously (19). rBVs were grown and titrated as reported elsewhere (30, 32).
Construction of recombinant baculoviruses.
rBVs were prepared from pFastBac (pFB)-derived vectors as described previously (17). Plasmids pFB/His-VP2-466 and pEGFP-C1 (Clontech) were used as the templates to generate both the pFB/EGFP-CAP and pFB/CAP-EGFP fusions (referred to as the N-terminal [Nt] and C-terminal [Ct] fusions, respectively) by generating two overlapping PCR fragments. First, the His-VP2-466 fragment was amplified using primers 5′-GGAGGTGGAGGTGGAGGTCATCACCATCACCATCAC and 3′-GCGCAAGCTTAGGCAGGTGGGAACAATGTGG for the Nt fusion and primers 5′-GCGCAGATCTATGACAAACCTGTCAGATCAAACCC and 3′-ACCTCCACCTCCACCTCCGGCAGGTGGGAAC for the Ct fusion. The corresponding EGFP fragments were amplified using primers 5′-GCGCAGATCTATGGTGAGCAAGGGC and 3′-ACCTCCACCTCCACCTCCCTTGTACAGCTC (for the Nt fusion) or primers 5′-GGAGGTGGAGGTGGAGGTATGGTGAGCAAGGGC and 3′-GCGCAAGCTTTTACTTGTACAGCTCGTC (for the Ct fusion). The overlapping fragments where then used as a template with primers 5′-GCGCAGATCTATGGTGAGCAAGGGC and 3′-GCGCAAGCTTAGGCAGGTGGGAACAATGTGG (for the Nt fusion) or primers 5′-GCGCAGATCTATGACAAACCTGTCAGATCAAACCC and 3′-GCGCAAGCTTTTACTTGTACAGCTCGTC (for the Ct fusion). The resulting DNA fragment was BglII/HindIII digested and cloned into plasmid pFB/HTa (Invitrogen).
For construction of the pFB-HT-EGFP vector, the EGFP sequence was extracted from pEGFPC1 by use of NcoI/HindIII and cloned into the pFB-HTa vector, which had previously been digested with the same enzymes. Using PCR overlap extension, two versions of the CAP gene containing a NotI site at the N-terminal end or at the PHI loop, which connects β strands H and I in the VP2 P domain, were generated (22). For PCRs, we used pFB/CAP as the template and a set of appropriate primers (available on request). PCR overlap extension was carried out with the corresponding flanking primers. SnaBI/PshAI (containing a NotI site at the N terminus) and SacI/BamHI (containing a NotI site in the PHI loop) fragments were cloned into pFB/CAP, which had previously been digested with the same enzymes. Both versions were used to construct fusion proteins with the HA and M2 proteins of influenza virus A/PR/8/34. Distinct HA fragment-coding sequences including the complete HA, the stalk domain, or HA2 and the long α helix of HA2 (LAH) were amplified using pFB/HT-HAPR8 as the template, primer 3′-HA NotI, and different 5′ end primers (available on request). The LAH-coding sequence was amplified with primers specific for the 3′ and 5′ ends. The resulting PCR fragments and the pFB/CAP plasmids with the NotI site at the N terminus or in the PHI loop were NotI digested and ligated. To generate fusion proteins with the M2 fragment, two synthetic complementary oligonucleotides encoding the M2 fragment flanked by NotI protruding ends (Biomers) were hybridized and ligated with NotI-digested plasmids.
Bacmids derived from the DH10Bac Escherichia coli strain were selected and prepared for Lipofectin transfection according to the manufacturer's protocols (Invitrogen). The constructs were expressed in H5 insect cells (33).
Purification of chimeric VLPs and IBDV capsids.
H5 cells (2 × 108 to 5 × 108 cells) were infected with the corresponding rBV at a multiplicity of infection (MOI) of 1 to 5 PFU/cell. We coinfected the cells with rBV EGFP-CAP and CAP at different MOI ratios ranging from 1:1 (EGFP-CAP/CAP) to 10:1. Cells were harvested at 48 h postinfection (hpi) and lysed in PES buffer (25 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], pH 6.2, 150 mM NaCl, 20 mM CaCl2) plus 1% IGEPAL CA-630 (Sigma) and 1% protease inhibitors (Complete Mini; Roche) on ice (30 min). The lysate was clarified by centrifugation (1,000 × g, 10 min) and processed on a 25% sucrose cushion (170,000 × g, 150 min, 4°C). The pellet was resuspended in PES buffer and processed in a linear 25 to 50% sucrose gradient (200,000 × g, 45 min, 4°C). Gradients were collected in 12 fractions, concentrated 10-fold by ultracentrifugation, and subjected to SDS-PAGE, Western blotting, and electron microscopy (EM) analysis or used for mouse immunization experiments. The IBDV Soroa strain and IBDV polyprotein-derived VLPs from rVV vT7lacOI-POLY were purified from QM7 cells as described previously (19).
Purification of HT-EGFP.
H5 cells infected with rBV FB/HT-EGFP (MOI, 2 to 5 PFU/cell) were harvested at 72 hpi and lysed in 50 mM Tris-HCl, pH 8.0, 500 mM NaCl, and 0.1% IGEPAL CA-630 (Sigma) with 1% protease inhibitors (Complete Mini; Roche) on ice. After sonication and clarification, the supernatant was mixed with TALON metal affinity resin (Clontech), equilibrated in lysis buffer, and incubated for 2 h. The resin was washed three times with washing buffer (50 mM Tris-HCl, pH 8, 500 mM NaCl, 0.1% IGEPAL CA-630 5 mM imidazole), and bound protein was eluted with the same buffer containing 200 mM imidazole. The sample was dialyzed against phosphate-buffered saline (PBS) buffer.
SDS-PAGE and Western blot analysis.
Extracts of infected cells (10 to 15 μl) or concentrated sucrose gradient fractions (2 to 5 μl) were added to Laemmli sample buffer, boiled (100°C, 3 min), and resolved in 11% SDS-polyacrylamide gels. Western blot analyses were carried out using anti-VP2 (30), anti-His, or anti-GFP antibodies (both from Sigma).
Quantification of protein incorporation into VLPs.
Stoichiometric analysis of chimeric VLPs to quantitate the EGFP-CAP fusion protein was performed on Coomassie blue-stained gels with bovine serum albumin (BSA) standards. The gels were scanned and analyzed with Quantity One software (Bio-Rad). The copy numbers of the EGFP-CAP and CAP molecules were normalized to the internal standard, derived from 780 copies of VP2, as established by X-ray crystallography and three-dimensional (3D) cryo-electron microscopy (cryo-EM) of virions (17, 20).
Electron microscopy analysis and three-dimensional image processing.
For conventional EM, 5-μl samples were applied to glow-discharged carbon-coated grids and negatively stained with 2% aqueous uranyl acetate. Micrographs were recorded with a JEOL 1200 EXII electron microscope operating at 100 kV and a nominal magnification of ×40,000.
For cryo-EM, 5 μl of each sample was incubated on Quantifoil R2/2 grids and vitrified in liquid ethane following established procedures (31, 34). The samples were observed in a Tecnai G2 electron microscope operating at 200 kV. Images were recorded at a nominal magnification of ×50,000 under minimal-dose conditions (∼10 e−/Å2) on Kodak SO-163 film. Micrographs were digitized in a Photoscan TD scanner (Zeiss) at a 14-μm/pixel sampling rate (2.4 Å/pixel for the specimen).
Image processing was performed using Xmipp software (http://xmipp.cnb.csic.es/) (35). Particles were manually selected using the X3d tool (36) and extracted and normalized with Xmipp. Contrast transfer function (CTF) correction was performed using the Bshow application (http://lsbr.niams.nih.gov/bsoft/bshow/bshow.html) (37). For initial determination of the center and orientation of particles, a previously obtained model of the HT-VP2-466 (CAP) VLP (17) filtered at 30 Å was used. A new density map was generated and used for subsequent iterative refinement. Resolution was assessed by use of the Fourier shell correlation (FSC) criterion between independent half-data-set maps, applying a correlation limit of 0.5. After independent refinements, 1,267 and 600 particles were included in the CAP and EGFP-CAP 3D reconstructions (3DRs), respectively, and the estimated resolutions were 20.8Å and 22.7 Å, respectively.
For difference map calculations, spherically averaged radial density profiles of CAP and EGFP-CAP maps computed at a 22.7-Å resolution were calculated, normalized, and scaled with Xmipp to match the fit between the cryo-EM map profiles. The EGFP-CAP map was scaled such that, at the radii corresponding to the outer capsid surface, its densities matched those of the CAP map. A difference map was obtained by the arithmetic subtraction of the CAP map from the EGFP-CAP map. Graphics were produced by the UCSF Chimera program.
Immunization and viral challenge of mice.
Female BALB/c mice (6 to 8 weeks old, six animals per group) were immunized subcutaneously three times with 150 μl PBS containing 30 to 40 μg EGFP-CAP VLPs, CAP VLPs, soluble EGFP, or a mixture of CAP and soluble EGFP. A control group received buffer only. The first dose was mixed with an equal volume of complete Freund's adjuvant, and subsequent doses were mixed with incomplete Freund's adjuvant. Immunizations were also performed in the absence of adjuvants, using antigen diluted in 300 μl PBS administered by intraperitoneal injection. Mice were immunized at 4-week intervals. For enzyme-linked immunosorbent assay (ELISA) analysis of sera, ∼200 μl of blood was extracted from the submandibular vein 10 days after the last immunization.
Immunizations with CAP VLPs with fused influenza virus proteins were similarly performed at 21-day intervals. At 14 days after the last immunization, mice received a lethal viral challenge. Mice were anesthetized with a parenteral mixture of 10% (vol/vol) ketamine (Imalgene 500; Merial) and 10% (vol/vol) xylazine (Xilagesic; Carlier) in PBS, followed by administration of 500 PFU (∼5 times the calculated 50% lethal dose [LD50]) of influenza virus A/PR/8/34 in 40 μl PBS via the intranasal route, allowing intervals for recovery of the respiratory rate. Body weight was monitored daily, and mice that lost >25% of their initial body weight were sacrificed.
ELISA.
Antibody levels in serum were evaluated by an indirect ELISA (38). To evaluate the levels of antibodies specific to CAP and EGFP, we coated 96-well MaxiSorp plates (Nunc) with 1 μg/ml IBDV-derived VLPs from rVV vT7lacOI-POLY or purified bacterial HT-EGFP in PBS (4°C, 16 h). The plates were blocked with 0.5% BSA, washed, and incubated with serial dilutions of mouse sera (1 h, 20°C). Rabbit anti-VP2 and mouse anti-GFP sera were included as positive controls. Bound antibody was detected using horseradish peroxidase (HRP)-conjugated goat antimouse (or antirabbit) antibody (Dako) and developed with o-phenylenediamine, and the absorbance at 492 nm was measured.
To analyze specific anti-M2 antibodies, EIA/RIA high-bind plates (Corning) were coated with 50 μl/well of synthetic M2 ectodomain (M2e) peptide (SLLTEVETPIRNEWGCR; 20 μg/μl; Sigma) in 50 mM bicarbonate buffer, pH 9.6 (overnight, 4°C), blocked, and incubated as described above. The signal was developed with tetramethylbenzidine (TMB; Invitrogen), terminated with 0.2 M H2SO4, and read at 450 nm.
Dot blot assay.
Purified, UV-inactivated influenza virus (4 μl, ∼105 PFU) and 4-μl BSA (0.1-μg/μl) droplets were dotted on a nitrocellulose membrane (Protran; Schleicher & Schuell), blocked with PBS containing 5% nonfat dry milk, and incubated with sera (1:300, 2 h, 20°C). After three washes, the blots were incubated with HRP-conjugated goat anti-mouse IgG (Roche) and developed using a commercial ECL kit (GE Healthcare). The signal was registered on X-ray films, digitized, and quantified with ImageJ software (NIH, Bethesda, MD). Relative intensities were calculated as the percentages of the maximum signal recorded in each experiment.
Serum antibody binding to native M2 and HA expressed on influenza virus-infected MDCK cells.
Immune sera were assessed for specific binding to native tetrameric M2 protein expressed on influenza virus-infected MDCK cells as described previously (39). MDCK cells were cultured to near confluence in DMEM with 10% FBS in 96-well plates. Cells were infected with 105 PFU in DMEM or incubated with DMEM alone (uninfected control) (1 h, 37°C) and then in serum-free DMEM (12 h, 37°C). Cells were washed in DMEM, fixed with 10% formalin (10 min, 20°C), washed with PBS, and blocked with 1% BSA in PBS (1 h). Mouse serum samples diluted 1:200 in PBS–0.1% BSA were added and incubated in triplicate (90 min, 20°C), washed, and incubated with HRP-conjugated goat anti-mouse IgG (1 h, 20°C), followed by detection with TMB. Uninfected MDCK cells were used to control background immune reactivity. The reaction was terminated, and the absorbance at 492 nm was measured. Specific anti-M2 antibody signals were calculated as the difference in absorbance between specific cells (influenza virus-infected MDCK cells) and nonspecific cells (uninfected MDCK cells).
Neutralization assay.
The neutralizing activity of VLP-immunized mouse sera was measured by plaque reduction assay using MDCK cells, as described previously (40). Serum diluted 1:13 was mixed with 100 PFU of influenza virus A/PR/8/34, and the mixture was incubated (1 h, 37°C). Confluent MDCK cell monolayers were inoculated with the mixture and adsorbed (1 h, 21°C), and the mixture was removed. Cells were washed with PBS and overlaid with DMEM containing 1 μg/ml trypsin, 0.1 mg/ml DEAE-dextran, and 0.5% Noble agar. After 72 h of incubation, cells were formaldehyde fixed and stained with 2% crystal violet and the plaques were counted.
Ethics statement.
Mice were maintained according to European Union and national guidelines for animal experimentation, and treatment protocols were approved by the CNB/CSIC Ethics Committee for Animal Research. Experiments were performed at the CNB/CSIC animal facility, Madrid, Spain (permit no. 28079-29-A), and were approved by the Ethical Review Committee (CEEA-CNB).
Accession numbers.
The 3DRs for the CAP and EGFP-CAP capsids are deposited in the Electron Microscopy Data Bank (EMDB; http://www.ebi.ac.uk/pdbe/emdb) under accession no. EMD-5997 and EMD-5996, respectively.
RESULTS
Design and characterization of HT-VP2-466-based fusion proteins with EGFP.
Previous structural studies of the VP2 structural polymorphism using a simple recombinant baculovirus (rBV) that expressed HT-VP2-466 allowed us to establish the minimal morphogenetic elements required for efficient VLP assembly, which are similar to those required for efficient assembly of IBDV virions (17). Here we developed chimeric VLPs using HT-VP2-466 (CAP) as a platform to incorporate heterologous proteins. We used enhanced green fluorescent protein (EGFP), which consists of a β barrel of ∼25 by ∼45 Å with 11 β chains, on the basis of its relatively large size (Fig. 1A). Correct EGFP folding can be confirmed, as it is necessary for fluorescent activity. As the VP2 N and C termini face the capsid interior in close proximity (Fig. 1A), we generated two CAP-based chimeric genes that we expressed in insect cells. EGFP-CAP has EGFP fused at the VP2 N terminus preceding the His6 tag, and CAP-EGFP has EGFP at the VP2 C terminus downstream of the α5 helix. To facilitate protein folding and interactions between morphogenetic peptides, a 6-glycine linker was included between CAP and EGFP for both chimeras. H5 insect cells were infected with rBV/EGFP-CAP or rBV/CAP-EGFP, and cultures were harvested at 48 hpi and analyzed by SDS-PAGE and Western blotting; anti-GFP (Fig. 1B, bottom left) and anti-VP2 (Fig. 1B, bottom right) antibodies detected a band with a molecular mass of ∼75 kDa, which coincides with the theoretical molecular mass of the EGFP-CAP chimera (80.3 kDa). For reasons that we were unable to determine, we detected no expression for the C-terminal version of CAP-EGFP.
FIG 1.
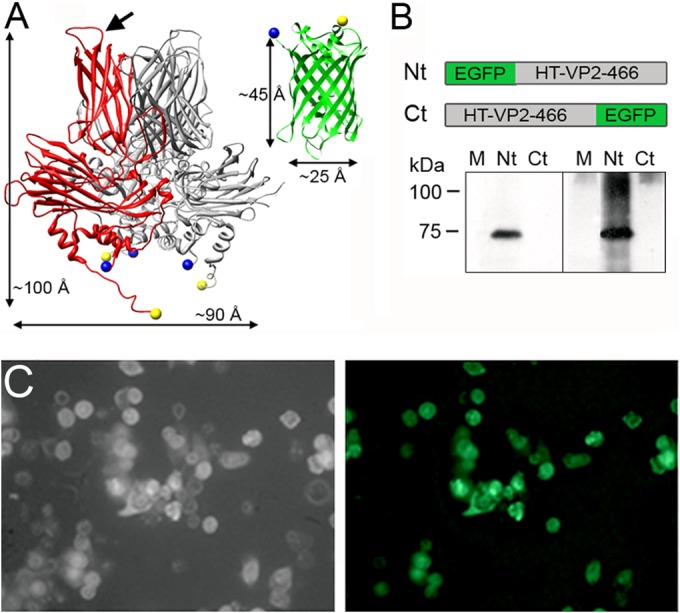
VP2 and EGFP structure and expression of EGFP-fused HT-VP2-466-based chimeric proteins. (A) (Left) VP2 trimer X-ray model (Protein Data Bank [PDB] accession number 2GSY) with a VP2 monomer (red). The VP2 chain (452 residues) lacks 7 N-terminal and 11 C-terminal residues; the first visible N-terminal amino acids and the last C-terminal amino acids are represented as blue and yellow circles, respectively (note that the N and C termini are close to each other). Arrow, PHI loop (located between the H and I β strands of domain P). (Right) EGFP X-ray model (green; PDB accession number 1GFL), with the N and C termini colored as described above. Dimensions are shown. (B) (Top) Scheme for HT-VP2-466-based chimeric proteins with EGFP fused to the N terminus (Nt; EGFP-HT-VP2-466) or to the C terminus (Ct; HT-VP2-466-EGFP). (Bottom) Extracts of H5 cells infected with rBV expressing both chimeras (Nt or Ct) were analyzed by SDS-PAGE and Western blotting using anti-GFP (left) or anti-VP2 (right) antibodies. Lanes M, molecular size markers (kDa). (C) Phase-contrast (left) and fluorescence (right) images of H5 cells expressing EGFP-HT-VP2-466. The fluorescent image was acquired with a Leica DMI6000B inverted microscope with the filter set for GFP.
In parallel experiments, we visualized a strong fluorescent signal in rBV/EGFP-CAP-infected H5 cells at 48 hpi (Fig. 1C), suggesting a natively folded EGFP structure. No signal was detected in rBV/CAP-EGFP-infected insect cells or in the rBV/CAP control.
Optimization of chimeric CAP-EGFP capsid assembly.
We studied the capacity of EGFP-CAP to assemble into particulate material, such as VLPs and/or tubular structures. H5 cells were infected with rBV/EGFP-CAP (48 h), and extracts were ultracentrifuged on a sucrose cushion, followed by a linear sucrose gradient. The resulting fractions were concentrated and analyzed by SDS-PAGE, Western blotting, and negative-staining electron microscopy (EM) (Fig. 2A). The biochemical profile of Coomassie blue-stained gels (Fig. 2A, gel) and Western blots developed with anti-VP2 (Fig. 2Ai) and anti-GFP (Fig. 2Aiii) antibodies indicated that most fusion protein migrated as small aggregates and soluble proteins in the top fractions (fractions 11 and 12). A weak specific signal found in the middle fractions (fractions 5 and 6) corresponded to assemblies with a morphology and size resembling those of viral capsids (Fig. 2Aiv). These results indicate that capsid assembly is very inefficient compared to the efficiency of CAP expression, in which tubes and virion-like capsids migrated from fraction 7 to the top of the gradient (Fig. 2B).
FIG 2.

Expression of the HT-VP2-466-based chimeric VLP and optimization of its assembly. (A) EGFP-HT-VP2-466 chimeric protein was expressed in insect cells, and the assemblies were purified on sucrose gradients; 12 fractions (indicated by the numbers over the lanes) were collected, concentrated, and analyzed by SDS-PAGE and Coomassie staining (i) and by Western blotting using anti-VP2 (ii) and anti-GFP (iii) antibodies. The direction of sedimentation was from right to left, with fraction 12 being at the gradient top. (iv) The image shows a representative electron micrograph (negative staining) of particulate material in fraction 6. Bar = 100 nm. (B) Wild-type HT-VP2-466 (control) was analyzed as described for panel A in a Coomassie-stained gel (i) or by Western blotting for anti-VP2 antibodies (ii). (iii) The image shows HT-VP2-466 assemblies from fraction 6 that correspond to tubes and isometric capsids with a morphology and size similar to those of IBDV capsids (inset). Bar = 100 nm. (Ci to Ciii) Assemblies from cells coexpressing EGFP-HT-VP2-466 and HT-VP2-466 at a 5:1 ratio were analyzed as described for panel Ai to Aiii, respectively. (iv) The electron micrograph shows fraction 6. Bar = 100 nm. (A to C) Arrows labeled 1, HT-VP2-466 (∼50-kDa) bands; arrows labeled 2, EGFP-HT-VP2-466 (∼75-kDa) bands. (D) Native agarose gel electrophoresis after Coomassie staining (left) or UV illumination (right) of EGFP (lanes 1), HT-VP2-466 VLPs (lanes 2), and EGFP-HT-VP2-466 VLPs (lanes 3). Images of the same gel are shown.
We hypothesized that steric hindrance limited assembly. To improve assembly yield, we coinfected rBV/EGFP-CAP and rBV/CAP at distinct ratios. H5 cells were initially coinfected at a 1:1 MOI ratio, which was progressively increased for rBV/EGFP-CAP to 3:1, 5:1, and 10:1 (Table 1). IBDV-related assemblies were purified from cell extracts, and two major proteins (∼75 and ∼50 kDa) were identified in the gradient fractions that corresponded to EGFP-CAP and CAP (Fig. 2Ci to iii; 5:1 MOI ratio). Fractions were analyzed by negative-staining EM. VLPs similar to those produced by wild-type CAP were abundant in middle fractions (T=13 capsids band in this region) (Fig. 2C).
TABLE 1.
Incorporation of EGFP-HT-VP2-466 into chimeric capsidsa
| Coinfection ratiob | % EGFP-HT-VP2-466 incorporated into capsids | Mean copy no. of EGFP-HT-VP2-466b |
|---|---|---|
| 1:1 | 18 | 148 |
| 3:1 | 27 ± 4 | 210 ± 31 |
| 5:1 | 31 ± 2 | 240 ± 16 |
| 10:1 | 25 | 195 |
The percentage of capsids incorporating EGFP-HT-VP2-466 and the mean copy number of EGFP-HT-VP2-466 were estimated from Coomassie blue-stained gels of fractions 5 to 7, assuming that all assemblies are T=13 capsids.
Ratio of rBV/EGFP-HT-VP2-466 to rBV/HT-VP2-466.
Chimeric VLPs were analyzed by agarose gel electrophoresis and illuminated with a UV lamp, which showed fluorescence and mobility (Fig. 2D, lanes 3) similar to those of wild-type VLPs (Fig. 2D, left, lane 2). Stoichiometric analysis of Coomassie blue-stained gels of fractions 5 to 7 showed the highest EGFP incorporation efficiency at a 5:1 MOI ratio (Table 1), in which ∼240 copies of EGFP were incorporated per capsid (∼31% of capsid proteins had EGFP).
Three-dimensional structure of EGFP-CAP icosahedral capsids.
Cryo-EM of VLP-enriched fractions showed that, like CAP particles (17), EGFP-CAP capsids are a complex mixture of distinct assemblies that range from ∼50 to ∼80 nm in diameter (Fig. 3A and D). Capsids of both VLPs are similar to T=13 IBDV virions (diameter, ∼65 nm) and were selected to calculate a three-dimensional reconstruction at an ∼21-Å resolution. Although their outer surfaces were nearly superimposable (Fig. 3B and E), the EGFP-CAP protein shell was thicker (Fig. 3C and F), presumably due to EGFP fusion to the inner capsid surface. Comparison of radial density profiles showed a peak of extra density for EGFP-CAP located in a 280-Å radius (Fig. 3G, arrow). To define these differences more precisely, we calculated a difference map by arithmetic subtraction of CAP from EGFP-CAP (Fig. 3H, in green on the inner VLP surface). These difference density islands are located mainly at the local 6-fold and 5-fold axes. They represented 90% of the EGFP molecule volume, probably due to low EGFP occupancy (31%; Table 1) and EGFP-CAP linker flexibility.
FIG 3.

Three-dimensional cryo-EM reconstructions of chimeric EGFP-HT-VP2-466 and wild-type HT-VP2-466 capsids. (A, D) Cryo-electron micrographs of purified EGFP-HT-VP2-466 (A) and HT-VP2-466 capsids (D). Bar = 50 nm. (B, E) Surface-shaded representations of the outer surface, viewed along an icosahedral 2-fold axis, of the T=13 capsids of EGFP-HT-VP2-466 (B) and HT-VP2-466 (E). Bar = 20 nm. (C, F) Transverse central sections from the 3DR of EGFP-HT-VP2-466 (C) and HT-VP2-466 (F) T=13 VLPs. Lighter shading indicates a higher density. (G) Radial density profiles from 3D maps of EGFP-HT-VP2-466 (green) and HT-VP2-466 (blue) T=13 VLPs computed at a 23-Å resolution. Arrow, extra peak of density on the inner surface of the EGFP-HT-VP2-466 capsid (radius = ∼280 Å). (H) Difference map calculated by arithmetic subtraction of the map for the HT-VP2-466 capsid from the map for the EGFP-HT-VP2-466 capsid. The map on the inner surface of a HT-VP2-466 capsid viewed along an icosahedral 2-fold axis is shown in green. The GFP atomic structure is shown at the same scale (top left). Bar = 20 nm.
Immune response to chimeric EGFP-CAP particles.
BALB/c mice (six per group) were immunized with purified EGFP-CAP VLP, CAP VLP, soluble EGFP, or a mixture of CAP and soluble EGFP (Fig. 4A). The amount of EGFP and CAP injected was constant for each group; mice were primed with antigen in complete Freund's adjuvant and received two boosts with antigen in incomplete Freund's adjuvant. Blood was collected 10 days after the last boost, and sera were tested by ELISA for antibodies to VP2 and EGFP. Serum titers were expressed as the inverse of the serum dilution that yielded a 50% signal intensity at saturation (plateau) levels (or the maximum signal). All immunized mice developed VP2-specific antibodies with high titers (2 × 104 to 6 × 104; Fig. 4B), whereas anti-EGFP antibodies had lower titers (∼2 × 103; Fig. 4C), which indicated that the VP2 response did not mask that of EGFP. Anti-EGFP antibody production was independent of whether the EGFP administered with CAP VLPs was fused (located inside the VLP), mixed (located outside the VLP), or soluble (administered in the absence of CAP VLP).
FIG 4.
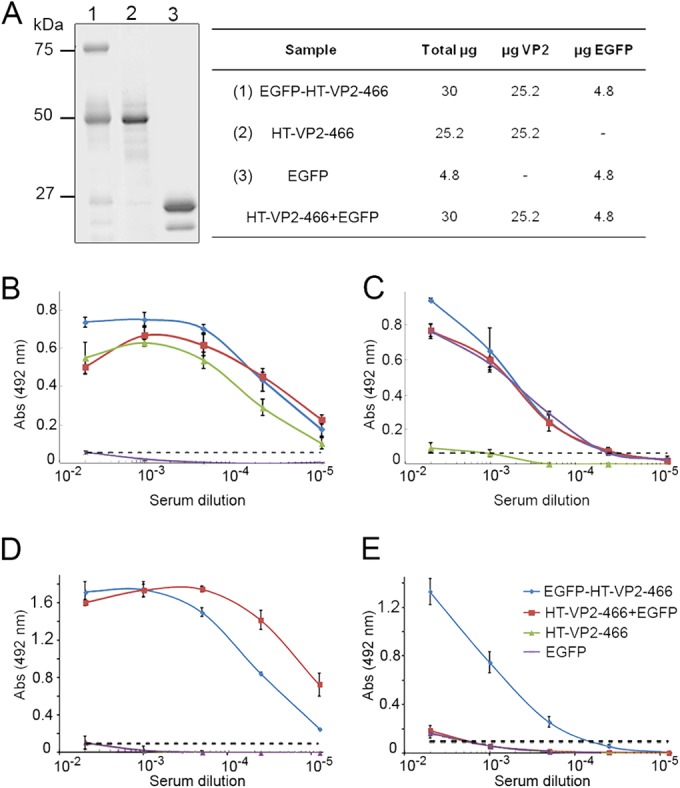
ELISA titration of mouse antisera after immunization with EGFP-derived chimeric assemblies. (A) SDS-PAGE of antigens used for mouse immunization: EGFP-HT-VP2-466 VLP (lane 1), HT-VP2-466 VLP (lane 2), and bacterial GFP (lane 3). The table shows the antigen dose administered per mouse (μg/dose; three injections). (B, C) ELISA titration of mouse antisera in plates coated with IBDV-derived VLPs from rVV vT7lacOI-POLY (B) or purified bacterial HT-EGFP (C). Mice were immunized with GFP-CAP (blue), CAP (green), CAP-GFP (red), and GFP (purple), all with adjuvant. (D, E) ELISA titration of mouse antisera after immunization with the same antigens described for panels B and C without adjuvant; plates were coated as described for panels B and C. Dashed lines, background level measured for an unrelated mouse serum sample.
Given the VLP adjuvant effect on the fused protein, immunization experiments were repeated without adjuvants. Three groups of mice (four mice per group) were immunized with EGFP-CAP VLPs, a mixture of CAP and soluble EGF, or soluble EGFP alone. The serum titers of anti-VP2-specific antibodies were ∼3 × 104 (Fig. 4D); EGFP-specific antibodies had a titer of ∼103, which is similar to the titer obtained with Freund's adjuvant, only when EGFP was fused to CAP (Fig. 4E). The response to soluble EGFP, alone or with CAP VLPs, was negligible. The IBDV capsid thus showed a major adjuvant effect on the heterologous protein.
Design and analysis of chimeric HT-VP2-466 capsids with influenza virus proteins.
The results presented above showed that HT-VP2-466 capsids can incorporate ∼240 copies of a foreign protein without altering the structural integrity of the T=13 capsid. Following the same strategy used for EGFP, we generated several chimeric proteins bearing influenza virus A/PR/8/34 (H1N1) proteins and fragments. These chimeras included HA, the membrane-proximal stalk of HA (the so-called HA2 domain, involved in virus and cell membrane fusion), the HA2 long α helix (LAH) (Fig. 5A), and the ectodomain of the channel protein M2 (Fig. 5B), which comprises the 23 N-terminal amino acid residues of M2 (LAH and the M2 ectodomain are highly conserved among different strains). rBV-based expression of M2-CAP or LAH-CAP alone was efficient and resulted in the assembly of T=13 capsid-like particles and tubular structures (Fig. 5C) and irregular structures (Fig. 5D), respectively. HA2-CAP and HA-CAP formed VP2-related assemblies only when coexpressed with wild-type CAP; the highest levels of chimeric protein were incorporated at coinfection ratios of 3:1 and 10:1, respectively (Fig. 5E and F). Since M2-CAP assembled well into capsids and tubes, we coinfected M2-CAP and HA2-CAP. At a 1:1 ratio, they yielded T=13 capsids and related assemblies of different sizes (Fig. 5G). In addition to the HT-VP2-466 N terminus, the VP2 PHI loop on the IBDV particle surface was also used as an insertion site for M2 and HA2. M2 is a relatively small, unstructured peptide, and the HA2 N and C termini are proximal in its tertiary structure. Insertion of M2 or HA2 in the VP2 PHI loop, alone or coexpressed with wild-type CAP or M2-CAP at various ratios, did not result in the incorporation of these chimeric proteins.
FIG 5.

Expression and purification of influenza virus-derived HT-VP2-466-based chimeric assemblies. (A) Structure of the influenza virus hemagglutinin. Ribbon diagram of an HA trimer (PDB accession number 1RU7), with one monomer colored for clarity (orange and blue, HA1 and HA2 chains, respectively; light blue, LAH). (B) Structure of the influenza virus M2 protein. Tetrameric M2 (PDB accession number 2KWX) is shown as a ribbon diagram, with one monomer shown in violet. The unstructured N-terminal M2 ectodomains are represented as dashed lines. (C to G) Assemblies were purified as described in the legend to Fig. 2A and then analyzed by SDS-PAGE, Coomassie staining, and negative-staining EM. Cells were infected with rBV/M2-CAP (C), rBV/LAH-CAP (D), rBV/HA2-CAP+rBV/CAP (E), rBV/HA-CAP+rBV/CAP (F), or rBV/HA2-CAP+rBV/M2-CAP (G). Electron micrographs (right) in panel C are for fractions 6 (top) and 2 (bottom), and micrographs to the right of panels D to G are for fraction 6. Bars = 100 nm.
CAP with influenza virus proteins induces HA- and M2-specific antibody responses.
BALB/c mice were immunized with 30 to 40 μg (of which 4 to 5 μg corresponds to influenza virus peptides) of purified VLP of M2-CAP, LAH-CAP, HA2-CAP, HA-CAP, and HA2- and M2-CAP (Fig. 6A). As controls, we immunized one group with GFP-CAP VLPs and one group with PBS alone. Serum titers of antibodies against VP2 for all six groups, calculated by ELISA, ranged from 3.5 × 104 to 12.5 × 104. Serum antibodies specific for influenza virus proteins were evaluated by quantitative dot blot analysis using inactivated influenza virus particles adhered to nitrocellulose. This assay essentially measured IgG antibodies to the most abundant viral protein, HA, as protein M2 expression is low in the viral particle. The HA2-CAP–M2-CAP VLP-immunized group showed high levels of HA-specific antibodies (four serum samples showed ≥80% of the maximum signal, and the remaining two showed >50%; Fig. 6B, yellow). Two serum samples from the HA2-CAP VLP-immunized group also showed strong signals (>80%), with weaker signals being seen for the remaining sera (<50%; Fig. 6B, blue). The LAH-, HA-, and M2-CAP-immunized groups showed weak specific antibodies (Fig. 6B). Sera were tested by ELISA for antibodies to M2 synthetic peptide; the M2-CAP VLP-immunized group had a homogeneous response, with higher levels of M2-specific antibodies being seen in that group than in the M2-CAP–HA2-CAP VLP-immunized group (Fig. 6C). Immune sera were assessed by ELISA for specific binding to native tetrameric M2 expressed on the surface of influenza virus-infected MDCK cells, which was efficiently recognized by sera from M2-CAP VLP-immunized mice (Fig. 6D).
FIG 6.
Antibody response after immunizations with influenza virus-derived chimeric assemblies. (A) SDS-PAGE of antigens (Ag) used for immunizations: Lanes: 1, HA-CAP–CAP; 2, HA2-CAP–CAP; 3, HA2-CAP–M2-CAP; 4, LAH-CAP; 5, M2-CAP. Molecular size markers (kDa) are indicated on the left. The table indicates the amounts of antigen per dose and mouse (μg). (B) Results of a dot blot assay of mouse antisera (1:300 dilution) after immunization with the indicated chimeric assembly to test for IgG antibody to inactivated influenza virus. Relative intensity was calculated as a percentage of the maximum signal recorded; each bar represents serum from a single mouse. (C) Results of an ELISA of M2-CAP and HA2-CAP–M2-CAP antisera (1:200 dilution) on plates coated with a synthetic M2 ectodomain peptide (SLLTEVETPIRNEWGCR). (D) Results for sera (1:200 dilution) from mice immunized with M2-CAP, HA2-CAP, or HA2-CAP–M2-CAP tested by ELISA on influenza virus-infected MDCK cells. Monoclonal anti-M2 antibody 14C2 (1 mg/ml) was diluted 1/2,000 as a control for panels B and C (optical density [OD] at 492 nm, 1.57 ± 0.26). GFP-CAP serum and PBS were used as negative controls (optical densities at 492 nm, 0.08 and 0.05, respectively).
Vaccination with CAP containing HA and M2 protects against influenza virus challenge.
Groups of mice immunized with HA-CAP, HA2-CAP, HA2-CAP–M2-CAP, LAH-CAP, and M2-CAP VLP and two control groups immunized with GFP-CAP VLP and PBS alone were challenged intranasally with 500 PFU/mouse (5 times the LD50) of influenza virus A/PR/8/34 (H1N1). Body weight and survival rates were monitored for 14 days after virus challenge (Fig. 7). Control groups showed a progressive loss of weight from day 3, and 83% had died by days 7 to 9 postchallenge. The response of mice immunized with HA-CAP was similar to that of the controls, with the mice showing severe weight loss and high rates of mortality. Mice immunized with LAH-CAP VLPs had a heterogeneous response; 83% suffered considerable weight loss, but only 17% died. In contrast, HA2-CAP-, M2-CAP-, and HA2-CAP–M2-CAP-immunized mice were 100% protected against lethal challenge, although the reductions in morbidity varied. In the HA2-CAP group, 50% showed a transient loss of ∼20% of their body weight, which returned to normal values; weight loss was negligible in the other 50%. In the M2-CAP group, the initial body weight that was lost (with ≤15% loss) was recovered in 84% of the individuals, indicative of a high degree of protection; 16% (1 mouse) lost ∼20% of their body weight, but they also recovered. The HA2-CAP–M2-CAP-immunized mice were protected. Initial losses of up to 15% of body weight were rapidly recovered in all individuals, indicating mild disease.
FIG 7.

Protection against lethal challenge. (A) Groups of vaccinated mice and a mock-vaccinated control were challenged intranasally with a lethal dose of influenza virus A/PR/8/34 (5× LD50). Mice were immunized with HA-CAP, HA2-CAP, HA2-CAP–M2-CAP, LAH-CAP, and M2-CAP VLPs and with GFP-CAP VLPs and PBS (mock) alone. Survival was monitored for 14 days and is expressed as the percentage of surviving mice. (B) The body weights of the infected mouse groups (n = 6) were monitored for 14 days. Values represent the weights of individual mice expressed as a percentage of the initial weight on the day of inoculation (100%).
DISCUSSION
Viral vaccine research is an active field. Whereas attenuation of virulence and chemical inactivation are the oldest (and still-used) approaches to classical vaccine production, new strategies for human and veterinary medicine are constantly emerging due to improvements in safety and efficiency, among other factors (41). Approaches that use recombinant viral vector- and subunit (VLP)-based vaccines have been demonstrated to offer effective protection against diseases (42–45). Our model for vaccine development is the IBDV VP2 capsid protein, which we tested as a platform to anchor foreign antigens. Based on the VP2 atomic structure resolved by X-ray crystallography (20–22), we evaluated its capacity to incorporate fused foreign peptides or proteins to specific sites, without interfering with its natural tendency to assemble into icosahedral capsids or helical tubes.
Molecular studies of the basis of VP2 structural polymorphism allowed us to establish that although VP2 is the only component of the icosahedral capsid (17, 20), it requires other scaffolding (as VP3) and proteolytic proteins to assemble into T=13 capsids (19). In a simple recombinant baculovirus-based expression system, assembly of ∼70-nm-diameter T=13 VLPs similar to virion capsids is nevertheless efficient with HT-VP2-466, a 466-residue capsid protein intermediate with an N-terminus-bound His tag. Here we show proof of concept for HT-VP2-466 assemblies to be carriers of heterologous protein in their interior cargo space.
Our nonexhaustive analysis of VP2 insertion sites for foreign peptides confirms that some regions are inappropriate, including the VP2 P-domain loops as well as the VP2 C terminus. Structural constraints are unclear and must be established empirically; in our system, VLP assembly is impeded in VP2 chimeras with M2 or HA2 inserted into the PHI loop (which does not contribute to intratrimeric interactions), whereas the PBC loop incorporated a 12-amino-acid FMDV epitope in T=1 SVPs (29).
When expressed alone, HT-VP2-466 with EGFP fused to the N terminus (EGFP-HT-VP2-466) was almost unable to assemble into particulate material, such as VLPs or tubular structures, probably due to the proximity of N-terminal ends at the internal hexameric and pentameric surfaces. To minimize steric restrictions, EGFP-HT-VP2-466 was coexpressed with the wild-type HT-VP2-466 to promote VLP assembly. The maximum level of EGFP-HT-VP2-466 incorporation into VLPs (assuming an icosahedral T=13 capsid) was ∼250 copies/capsid; this value implies that a single molecule of each VP2 trimer can contain a large foreign protein and that the antibody response to foreign protein is adequate at this ratio. Chimeras with the influenza virus M2 ectodomain (23 residues, 2.7 kDa) or the HA long α helix (LAH; 54 residues, 6.7 kDa) can self-assemble in the absence of the wild-type subunit, and the HA2 chimeric capsid can incorporate ∼45% of its structural subunits with the fused HA2 domain (672 residues, 73.5 kDa). These results show that the HT-VP2-466-based system is sufficiently robust for assembly into particulate material (necessary for self-adjuvanting ability). Its flexibility allows inclusion of large antigens, as steric constraints are counteracted by incorporation of a smaller number of foreign protein copies. HT-VP2-466 capsids could plausibly bear two fused antigens in the interior surface, as is likely for the capsids with HA2 and M2 inserts.
The size of the antigens that can be incorporated into VLPs is an important limitation of many reported VLP platforms for foreign antigen display. Chimeric peptide insertions longer than 20 to 30 amino acids often fail to assemble into VLPs (46). These limitations restrict the number of epitopes that can be targeted with an individual chimeric VLP and make the targeting of complex conformational B cell epitopes difficult, if not impossible. There are nonetheless marked differences in the versatility of distinct VLPs as antigen display platforms in terms of the size of the insertions that they tolerate. Chimeric VLPs with large protein inserts have been designed from many viral systems, as is the case for rotavirus double-layered particles composed of VP2 and VP6 proteins (DLP2/6s) harboring green fluorescent protein (GFP) (47). In an IBDV-based system, full proteins were encapsulated by fusion to the VP3 C terminus of the polyprotein gene, but fluorescent VLPs are complex, as they contain three viral proteins (VP2, VP3, and VP4) (48). GFP insertion into the HT-VP2-466-based capsid is an important step forward. This new generation of VLP-based vaccines offers several advantages, as it allows discrimination between vaccinated and pathogen-infected animals. As we demonstrate in this study, it also allows the construction of multivalent chimeric VLPs.
Another important factor in constructing a functional chimeric capsid protein is retention of antigen secondary structural elements in the chimeric VLP. LAH-CAP-immunized mice were less protected than those immunized with complete HA2 in chimeric capsids. The conformation of LAH fused to the capsid protein might differ from its native structure, with the consequent distortion of conformational epitopes. HA-immunized mouse mortality was as high as that of the control groups, possibly because HA did not acquire its native structure. The so-called foldon element from the T4 bacteriophage is absent in our constructions but is necessary for correct HA folding and trimer assembly (49).
Current influenza vaccines are mainly based on the antibody response to HA derived from three influenza virus strains (A/H1N1, A/H3N2, and B). Their effectiveness is limited by virus mutation (antigenic drift) and by segment or gene reassortment (antigenic shift) (50). Vaccines against influenza virus are reevaluated annually (51), and new vaccine research focuses on conserved antigenic motifs that elicit broadly protective antibodies. Promising cross-protective or universal vaccines use the nucleoprotein, the HA2 stalk domain of HA (which lacks the immunodominant head variable domain), and the M2 ectodomain (which has low immunogenicity) (52). The 16 influenza virus HA subtypes are further divided into two major phylogenetic groups, differing in the structure of the LAH region. LAH-directed antibodies could provide cross-protection among viruses that belong to one of these groups but limited intergroup cross-protection (53). The M2e sequence is highly conserved in most influenza virus A strains, although the pandemic California strain has four mutations in this region (54, 55). Vaccine approaches that combine the presentation of both epitopes might thus elicit a broader, more protective immune response. Here we tested HT-VP2-466 VLPs as an immunogenic carrier for HA2 and M2 influenza virus antigens. When administered subcutaneously to mice, these VLPs elicited a strong antibody response to HA2 and M2 and efficiently protected against mortality and weight loss following influenza virus A challenge.
The IBDV-based chimeric VLPs incorporate foreign epitopes in their interior. Although it is assumed that foreign target antigens must be displayed on the chimeric VLP exterior to induce an efficient humoral response, there are several reports of antibody responses to antigens incorporated inside VLPs. For instance, immunization of mice with rotavirus-derived double-layered VLPs composed of VP2 and VP6 proteins (DLP2/6s) induces antibodies to VP2, which forms the innermost layer (56, 57). Chimeric DLP2/6s with GFP fused to the VP2 N terminus (inside the VLPs) elicit anti-GFP antibodies (47). Although vaccination with CAP containing HA and M2 protects against influenza virus challenge, a standard plaque reduction assay showed no neutralization of the inserted influenza virus antigens in sera (not shown). This lack of observable neutralization was not unanticipated, given previous results involving these influenza virus antigens.
The mechanisms by which M2-based immunogens provide protection against influenza virus infection have been analyzed in detail; M2 immunization of mice induces a protective humoral response but not neutralization of virus entry (54, 55, 58). HA2 or LAH immunization-induced immune responses are more complex. The conserved HA stalk domain and the LAH peptide are targeted by several cross-reactive, broadly neutralizing antibodies (bnAbs) (53, 59, 60). HA stalk-directed bnAbs mediate neutralization by inhibiting virus-host membrane fusion during influenza virus entry, whereas HA globular head-directed neutralizing antibodies inhibit viral binding to host cell receptors. Vaccination of mice with HA stalk-derived immunogens gives rise to broadly cross-reactive antisera and provides protection against lethal virus challenge (61–65). In several reports of conventional in vitro neutralization assays, no neutralization activity was detected in the protective sera (61, 62, 64), as we also observed. The collection of conclusive evidence for the presence of HA stalk-directed neutralizing antibodies in serum has proven to be technically challenging, given the limited sensitivity and high detection limit of standard assays. Antibody-dependent effector functions, such as antibody-dependent cell-mediated cytotoxicity (ADCC), contribute markedly to the protection afforded by M2 and HA stalk-derived immunogens (55, 61).
The HT-VP2-466-based system could be used to construct a multivalent platform with antigens from different viruses to formulate a vaccine against animal or human diseases, thus reducing production costs. It would be interesting to construct an HT-VP2-466-based capsid with HA2 and M2 epitopes from the highly pathogenic avian influenza virus H5N1 for evaluation as a vaccine against both avian viruses. The PBC loop on the VLP exterior surface, into which an FMDV-derived 12-residue epitope was successfully inserted, could be an additional site for construction of multivalent vaccines. Our VP2-based system would allow the detection of inefficient chimeric VLPs, for example, when preservation of neutralizing epitopes is too poor to elicit an optimal antibody response (as is the case for LAH and complete HA). We are currently pursuing the use of HT-VP2-466 as an assembly/disassembly system for heterologous nucleic acid packaging and encapsulation of drugs and other molecules.
ACKNOWLEDGMENTS
We thank José F. Rodríguez for technical and intellectual support and stimulating discussion, Mercedes Llorente (CNB-CSIC Protein Tools Unit) for the design and development of immune response analyses, and Catherine Mark for editorial assistance.
E.P. and N.M. were recipients of a Ph.D. fellowship from the Spanish Ministry of Science and Innovation (JAE and FPI grants, respectively), and C.P.M. is a Ph.D. fellow of the La Caixa Foundation International Fellowship Program (La Caixa/CNB). This work was supported by Spanish Ministry of Economy and Competitivity grants AGL-2010-22200-C02-02 (to J.B.), BFU 2011-29038 (to J.L.C.), and BFU 2011-25902 (to J.R.C.).
REFERENCES
- 1.Bárcena J, Blanco E. 2013. Design of novel vaccines based on virus-like particles or chimeric virions. Subcell Biochem 68:631–665. doi: 10.1007/978-94-007-6552-8_21. [DOI] [PubMed] [Google Scholar]
- 2.Yildiz I, Shukla S, Steinmetz NF. 2011. Applications of viral nanoparticles in medicine. Curr Opin Biotechnol 22:901–908. doi: 10.1016/j.copbio.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bachmann MF, Jennings GT. 2010. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol 10:787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 4.Spohn G, Bachmann MF. 2008. Exploiting viral properties for the rational design of modern vaccines. Expert Rev Vaccines 7:43–54. doi: 10.1586/14760584.7.1.43. [DOI] [PubMed] [Google Scholar]
- 5.Jennings GT, Bachmann MF. 2008. The coming of age of virus-like particle vaccines. Biol Chem 389:521–536. doi: 10.1515/BC.2008.064. [DOI] [PubMed] [Google Scholar]
- 6.Grgacic EV, Anderson DA. 2006. Virus-like particles: passport to immune recognition. Methods 40:60–65. doi: 10.1016/j.ymeth.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ludwig C, Wagner R. 2007. Virus-like particles—universal molecular toolboxes. Curr Opin Biotechnol 18:537–545. doi: 10.1016/j.copbio.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vacher G, Kaeser MD, Moser C, Gurny R, Borchard G. 2013. Recent advances in mucosal immunization using virus-like particles. Mol Pharm 10:1596–1609. doi: 10.1021/mp300597g. [DOI] [PubMed] [Google Scholar]
- 9.Noad R, Roy P. 2013. Virus-like particles, p 167–186 InRehm BHA. (ed), Bionanotechnology: biological self-assembly and its applications. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 10.Kushnir N, Streatfield SJ, Yusibov V. 2012. Virus-like particles as a highly efficient vaccine platform: diversity of targets and production systems and advances in clinical development. Vaccine 31:58–83. doi: 10.1016/j.vaccine.2012.10.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roldao A, Mellado MC, Castilho LR, Carrondo MJ, Alves PM. 2010. Virus-like particles in vaccine development. Expert Rev Vaccines 9:1149–1176. doi: 10.1586/erv.10.115. [DOI] [PubMed] [Google Scholar]
- 12.Plummer EM, Manchester M. 2011. Viral nanoparticles and virus-like particles: platforms for contemporary vaccine design. Wiley Interdiscip Rev Nanomed Nanobiotechnol 3:174–196. doi: 10.1002/wnan.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawano M, Morikawa K, Suda T, Ohno N, Matsushita S, Akatsuka T, Handa H, Matsui M. 2014. Chimeric SV40 virus-like particles induce specific cytotoxicity and protective immunity against influenza A virus without the need of adjuvants. Virology 448:159–167. doi: 10.1016/j.virol.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 14.Ye X, Ku Z, Liu Q, Wang X, Shi J, Zhang Y, Kong L, Cong Y, Huang Z. 2014. Chimeric virus-like particle vaccines displaying conserved enterovirus 71 epitopes elicit protective neutralizing antibodies in mice through divergent mechanisms. J Virol 88:72–81. doi: 10.1128/JVI.01848-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crisci E, Fraile L, Moreno N, Blanco E, Cabezon R, Costa C, Mussa T, Baratelli M, Martinez-Orellana P, Ganges L, Martínez J, Bárcena J, Montoya M. 2012. Chimeric calicivirus-like particles elicit specific immune responses in pigs. Vaccine 30:2427–2439. doi: 10.1016/j.vaccine.2012.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luque D, Rivas G, Alfonso C, Carrascosa JL, Rodríguez JF, Castón JR. 2009. Infectious bursal disease virus is an icosahedral polyploid dsRNA virus. Proc Natl Acad Sci U S A 106:2148–2152. doi: 10.1073/pnas.0808498106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saugar I, Luque D, Ona A, Rodríguez JF, Carrascosa JL, Trus BL, Castón JR. 2005. Structural polymorphism of the major capsid protein of a double-stranded RNA virus: an amphipathic alpha helix as a molecular switch. Structure 13:1007–1017. doi: 10.1016/j.str.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 18.Oña A, Luque D, Abaitua F, Maraver A, Castón JR, Rodríguez JF. 2004. The C-terminal domain of the pVP2 precursor is essential for the interaction between VP2 and VP3, the capsid polypeptides of infectious bursal disease virus. Virology 322:135–142. doi: 10.1016/j.virol.2004.01.025. [DOI] [PubMed] [Google Scholar]
- 19.Saugar I, Irigoyen N, Luque D, Carrascosa JL, Rodríguez JF, Castón JR. 2010. Electrostatic interactions between capsid and scaffolding proteins mediate the structural polymorphism of a double-stranded RNA virus. J Biol Chem 285:3643–3650. doi: 10.1074/jbc.M109.075994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coulibaly F, Chevalier C, Gutsche I, Pous J, Navaza J, Bressanelli S, Delmas B, Rey FA. 2005. The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell 120:761–772. doi: 10.1016/j.cell.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 21.Lee CC, Ko TP, Chou CC, Yoshimura M, Doong SR, Wang MY, Wang AH. 2006. Crystal structure of infectious bursal disease virus VP2 subviral particle at 2.6AÅ resolution: implications in virion assembly and immunogenicity. J Struct Biol 155:74–86. doi: 10.1016/j.jsb.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 22.Garriga D, Querol-Audi J, Abaitua F, Saugar I, Pous J, Verdaguer N, Castón JR, Rodríguez JF. 2006. The 2.6-angstrom structure of infectious bursal disease virus-derived T=1 particles reveals new stabilizing elements of the virus capsid. J Virol 80:6895–6905. doi: 10.1128/JVI.00368-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller H, Islam MR, Raue R. 2003. Research on infectious bursal disease—the past, the present and the future. Vet Microbiol 97:153–165. doi: 10.1016/j.vetmic.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Huang Z, Elankumaran S, Yunus AS, Samal SK. 2004. A recombinant Newcastle disease virus (NDV) expressing VP2 protein of infectious bursal disease virus (IBDV) protects against NDV and IBDV. J Virol 78:10054–10063. doi: 10.1128/JVI.78.18.10054-10063.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Francois A, Chevalier C, Delmas B, Eterradossi N, Toquin D, Rivallan G, Langlois P. 2004. Avian adenovirus CELO recombinants expressing VP2 of infectious bursal disease virus induce protection against bursal disease in chickens. Vaccine 22:2351–2360. doi: 10.1016/j.vaccine.2003.10.039. [DOI] [PubMed] [Google Scholar]
- 26.Taghavian O, Spiegel H, Hauck R, Hafez HM, Fischer R, Schillberg S. 2013. Protective oral vaccination against infectious bursal disease virus using the major viral antigenic protein VP2 produced in Pichia pastoris. PLoS One 8:e83210. doi: 10.1371/journal.pone.0083210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martín Caballero J, Garzón A, González-Cintado L, Kowalczyk W, Jiménez Torres I, Calderita G, Rodríguez M, Gondar V, Bernal JJ, Ardavin C, Andreu D, Zurcher T, von Kobbe C. 2012. Chimeric infectious bursal disease virus-like particles as potent vaccines for eradication of established HPV-16 E7-dependent tumors. PLoS One 7:e52976. doi: 10.1371/journal.pone.0052976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Letzel T, Coulibaly F, Rey FA, Delmas B, Jagt E, van Loon AA, Mundt E. 2007. Molecular and structural bases for the antigenicity of VP2 of infectious bursal disease virus. J Virol 81:12827–12835. doi: 10.1128/JVI.01501-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Remond M, Da Costa B, Riffault S, Parida S, Breard E, Lebreton F, Zientara S, Delmas B. 2009. Infectious bursal disease subviral particles displaying the foot-and-mouth disease virus major antigenic site. Vaccine 27:93–98. doi: 10.1016/j.vaccine.2008.10.036. [DOI] [PubMed] [Google Scholar]
- 30.Lombardo E, Maraver A, Castón JR, Rivera J, Fernández-Arias A, Serrano A, Carrascosa JL, Rodríguez JF. 1999. VP1, the putative RNA-dependent RNA polymerase of infectious bursal disease virus, forms complexes with the capsid protein VP3, leading to efficient encapsidation into virus-like particles. J Virol 73:6973–6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castón JR, Martínez-Torrecuadrada JL, Maraver A, Lombardo E, Rodríguez JF, Casal JI, Carrascosa JL. 2001. C terminus of infectious bursal disease virus major capsid protein VP2 is involved in definition of the T number for capsid assembly. J Virol 75:10815–10828. doi: 10.1128/JVI.75.22.10815-10828.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martínez-Torrecuadra JL, Castón JR, Castro M, Carrascosa JL, Rodríguez JF, Casal JI. 2000. Different architectures in the assembly of infectious bursal disease virus capsid proteins expressed in insect cells. Virology 278:322–331. doi: 10.1006/viro.2000.0559. [DOI] [PubMed] [Google Scholar]
- 33.Maraver A, Oña A, Abaitua F, Gonzalez D, Clemente R, Ruiz-Díaz JA, Castón JR, Pazos F, Rodríguez JF. 2003. The oligomerization domain of VP3, the scaffolding protein of infectious bursal disease virus, plays a critical role in capsid assembly. J Virol 77:6438–6449. doi: 10.1128/JVI.77.11.6438-6449.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P. 1988. Cryo-electron microscopy of vitrified specimens. Q Rev Biophys 21:129–228. doi: 10.1017/S0033583500004297. [DOI] [PubMed] [Google Scholar]
- 35.Marabini R, Masegosa IM, San Martín MC, Marco S, Fernández JJ, de la Fraga LG, Vaquerizo C, Carazo JM. 1996. Xmipp: an image processing package for electron microscopy. J Struct Biol 116:237–240. doi: 10.1006/jsbi.1996.0036. [DOI] [PubMed] [Google Scholar]
- 36.Conway JF, Trus BL, Booy FP, Newcomb WW, Brown JC, Steven AC. 1993. The effects of radiation damage on the structure of frozen hydrated HSV-1 capsids. J Struct Biol 111:222–233. doi: 10.1006/jsbi.1993.1052. [DOI] [PubMed] [Google Scholar]
- 37.Heymann JB, Belnap DM. 2007. Bsoft: image processing and molecular modeling for electron microscopy. J Struct Biol 157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 38.Engvall E, Perlmann P. 1971. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry 8:871–874. [DOI] [PubMed] [Google Scholar]
- 39.Hashemi H, Pouyanfard S, Bandehpour M, Noroozbabaei Z, Kazemi B, Saelens X, Mokhtari-Azad T. 2012. Immunization with M2e-displaying T7 bacteriophage nanoparticles protects against influenza A virus challenge. PLoS One 7:e45765. doi: 10.1371/journal.pone.0045765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshida R, Igarashi M, Ozaki H, Kishida N, Tomabechi D, Kida H, Ito K, Takada A. 2009. Cross-protective potential of a novel monoclonal antibody directed against antigenic site B of the hemagglutinin of influenza A viruses. PLoS Pathog 5:e1000350. doi: 10.1371/journal.ppat.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plotkin S. 2011. History of vaccine development. Springer, New York, NY. [Google Scholar]
- 42.Brun A, Albina E, Barret T, Chapman DA, Czub M, Dixon LK, Keil GM, Klonjkowski B, Le Potier MF, Libeau G, Ortego J, Richardson J, Takamatsu HH. 2008. Antigen delivery systems for veterinary vaccine development. Viral-vector based delivery systems. Vaccine 26:6508–6528. doi: 10.1016/j.vaccine.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brun A, Barcena J, Blanco E, Borrego B, Dory D, Escribano JM, Le Gall-Recule G, Ortego J, Dixon LK. 2011. Current strategies for subunit and genetic viral veterinary vaccine development. Virus Res 157:1–12. doi: 10.1016/j.virusres.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 44.Crisci E, Bárcena J, Montoya M. 2012. Virus-like particles: the new frontier of vaccines for animal viral infections. Vet Immunol Immunopathol 148:211–225. doi: 10.1016/j.vetimm.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patterson DP, Rynda-Apple A, Harmsen AL, Harmsen AG, Douglas T. 2013. Biomimetic antigenic nanoparticles elicit controlled protective immune response to influenza. ACS Nano 7:3036–3044. doi: 10.1021/nn4006544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chackerian B. 2007. Virus-like particles: flexible platforms for vaccine development. Expert Rev Vaccines 6:381–390. doi: 10.1586/14760584.6.3.381. [DOI] [PubMed] [Google Scholar]
- 47.Charpilienne A, Nejmeddine M, Berois M, Parez N, Neumann E, Hewat E, Trugnan G, Cohen J. 2001. Individual rotavirus-like particles containing 120 molecules of fluorescent protein are visible in living cells. J Biol Chem 276:29361–29367. doi: 10.1074/jbc.M101935200. [DOI] [PubMed] [Google Scholar]
- 48.Chevalier C, Lepault J, Da Costa B, Delmas B. 2004. The last C-terminal residue of VP3, glutamic acid 257, controls capsid assembly of infectious bursal disease virus. J Virol 78:3296–3303. doi: 10.1128/JVI.78.7.3296-3303.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu Y, Welsh JP, Swartz JR. 2014. Production and stabilization of the trimeric influenza hemagglutinin stem domain for potentially broadly protective influenza vaccines. Proc Natl Acad Sci U S A 111:125–130. doi: 10.1073/pnas.1308701110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elderfield R, Barclay W. 2011. Influenza pandemics. Adv Exp Med Biol 719:81–103. doi: 10.1007/978-1-4614-0204-6_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boni MF. 2008. Vaccination and antigenic drift in influenza. Vaccine 26(Suppl 3):C8–C14. doi: 10.1016/j.vaccine.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stanekova Z, Vareckova E. 2010. Conserved epitopes of influenza A virus inducing protective immunity and their prospects for universal vaccine development. Virol J 7:351. doi: 10.1186/1743-422X-7-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang TT, Tan GS, Hai R, Pica N, Petersen E, Moran TM, Palese P. 2010. Broadly protective monoclonal antibodies against H3 influenza viruses following sequential immunization with different hemagglutinins. PLoS Pathog 6:e1000796. doi: 10.1371/journal.ppat.1000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El Bakkouri K, Descamps F, De Filette M, Smet A, Festjens E, Birkett A, Van Rooijen N, Verbeek S, Fiers W, Saelens X. 2011. Universal vaccine based on ectodomain of matrix protein 2 of influenza A: Fc receptors and alveolar macrophages mediate protection. J Immunol 186:1022–1031. doi: 10.4049/jimmunol.0902147. [DOI] [PubMed] [Google Scholar]
- 55.Jegerlehner A, Schmitz N, Storni T, Bachmann MF. 2004. Influenza A vaccine based on the extracellular domain of M2: weak protection mediated via antibody-dependent NK cell activity. J Immunol 172:5598–5605. doi: 10.4049/jimmunol.172.9.5598. [DOI] [PubMed] [Google Scholar]
- 56.Lawton JA, Zeng CQ, Mukherjee SK, Cohen J, Estes MK, Prasad BV. 1997. Three-dimensional structural analysis of recombinant rotavirus-like particles with intact and amino-terminal-deleted VP2: implications for the architecture of the VP2 capsid layer. J Virol 71:7353–7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O'Neal CM, Crawford SE, Estes MK, Conner ME. 1997. Rotavirus virus-like particles administered mucosally induce protective immunity. J Virol 71:8707–8717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neirynck S, Deroo T, Saelens X, Vanlandschoot P, Jou WM, Fiers W. 1999. A universal influenza A vaccine based on the extracellular domain of the M2 protein. Nat Med 5:1157–1163. doi: 10.1038/13484. [DOI] [PubMed] [Google Scholar]
- 59.Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA. 2009. Antibody recognition of a highly conserved influenza virus epitope. Science 324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ekiert DC, Friesen RH, Bhabha G, Kwaks T, Jongeneelen M, Yu W, Ophorst C, Cox F, Korse HJ, Brandenburg B, Vogels R, Brakenhoff JP, Kompier R, Koldijk MH, Cornelissen LA, Poon LL, Peiris M, Koudstaal W, Wilson IA, Goudsmit J. 2011. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 333:843–850. doi: 10.1126/science.1204839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bommakanti G, Citron MP, Hepler RW, Callahan C, Heidecker GJ, Najar TA, Lu X, Joyce JG, Shiver JW, Casimiro DR, ter Meulen J, Liang X, Varadarajan R. 2010. Design of an HA2-based Escherichia coli expressed influenza immunogen that protects mice from pathogenic challenge. Proc Natl Acad Sci U S A 107:13701–13706. doi: 10.1073/pnas.1007465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Steel J, Lowen AC, Wang TT, Yondola M, Gao Q, Haye K, Garcia-Sastre A, Palese P. 2010. Influenza virus vaccine based on the conserved hemagglutinin stalk domain mBio 1(1):e00018-10. doi: 10.1128/mBio.00018-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang TT, Tan GS, Hai R, Pica N, Ngai L, Ekiert DC, Wilson IA, Garcia-Sastre A, Moran TM, Palese P. 2010. Vaccination with a synthetic peptide from the influenza virus hemagglutinin provides protection against distinct viral subtypes. Proc Natl Acad Sci U S A 107:18979–18984. doi: 10.1073/pnas.1013387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bommakanti G, Lu X, Citron MP, Najar TA, Heidecker GJ, ter Meulen J, Varadarajan R, Liang X. 2012. Design of Escherichia coli-expressed stalk domain immunogens of H1N1 hemagglutinin that protect mice from lethal challenge. J Virol 86:13434–13444. doi: 10.1128/JVI.01429-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mallajosyula VV, Citron M, Ferrara F, Lu X, Callahan C, Heidecker GJ, Sarma SP, Flynn JA, Temperton NJ, Liang X, Varadarajan R. 2014. Influenza hemagglutinin stem-fragment immunogen elicits broadly neutralizing antibodies and confers heterologous protection. Proc Natl Acad Sci U S A 111:E2514–E2523. doi: 10.1073/pnas.1402766111. [DOI] [PMC free article] [PubMed] [Google Scholar]