

ABSTRACT
Human cytomegalovirus (HCMV) transmission within the host is important for the pathogenesis of HCMV diseases. Natural killer (NK) cells are well known to provide a first line of host defense against virus infections. However, the role of NK cells in the control of HCMV transmission is still unknown. Here, we provide the first experimental evidence that NK cells can efficiently control HCMV transmission in different cell types. NK cells engage different mechanisms to control the HCMV transmission both via soluble factors and by cell contact. NK cell-produced interferon gamma (IFN-γ) suppresses HCMV production and induces resistance of bystander cells to HCMV infection. The UL16 viral gene contributes to an immune evasion from the NK cell-mediated control of HCMV transmission. Furthermore, the efficacy of the antibody-dependent NK cell-mediated control of HCMV transmission is dependent on a CD16-158V/F polymorphism. Our findings indicate that NK cells may have a clinical relevance in HCMV infection and highlight the need to consider potential therapeutic strategies based on the manipulation of NK cells.
IMPORTANCE Human cytomegalovirus (HCMV) infects 40% to 100% of the human population worldwide. After primary infection, mainly in childhood, the virus establishes a lifelong persistence with possible reactivations. Most infections remain asymptomatic; however, HCMV represents a major health problem since it is the most frequent cause of infection-induced birth defects and is responsible for high morbidity and mortality in immunocompromised patients. The immune system normally controls the infection by antibodies and immune effector cells. One type of effector cells are the natural killer (NK) cells, which provide a rapid response to virus-infected cells. NK cells participate in viral clearance by inducing the death of infected cells. NK cells also secrete antiviral cytokines as a consequence of the interaction with an infected cell. In this study, we investigated the mechanisms by which NK cells control HCMV transmission, from the perspectives of immune surveillance and immune evasion.
INTRODUCTION
Human cytomegalovirus (HCMV) is an enveloped virus that belongs to the family Herpesviridae. HCMV remains the most important viral pathogen in severely immunocompromised individuals, with substantial morbidity and mortality. It is also the major cause of congenital infections that lead to developmental abnormalities and fetal death (1).
Following initial infection, HCMV transmission can occur in cell cultures either by cell-free virus through the supernatant or by cell-to-cell transmission involving direct cell contact (2). In clinical practice, HCMV detection by isolation in cell cultures, pp65 antigenemia, and detection of viral DNA from blood (DNAemia) are used to diagnose an active systemic HCMV infection. From in vitro data, it can be concluded that antigenemia requires cell-to-cell contact between infected cells and polymorphonuclear leukocytes (PMN), which allows PMNs to load with viral antigens, mainly pp65 (3). Although the mechanisms of HCMV cell-to-cell transmission are not fully clear, many authors hypothesized that this mode is more important in vivo. This is also supported by the fact that most clinical HCMV isolates spread strictly from cell to cell in fibroblast cultures at low passage numbers (4).
During infection, HCMV encounters the host immune defense mechanisms, including intrinsic, innate, and adaptive immunity. The clinical outcome of HCMV infection mainly depends on the antiviral immunity. As effectors of the innate immunity, NK cells provide a rapid response to virus-infected cells (5). The most prominent functions of NK cells are their ability to produce antiviral cytokines and lyse virus-infected cells. NK cells can be activated by HCMV-infected cells (6, 7), and they are supposed to be important for the protection against HCMV infections in vivo. A case report indicated that NK cell deficiency was associated with severe herpesvirus infections, including active HCMV infection confirmed by virus isolation (8). Another case report showed that NK cell expansion was correlated with the control of an HCMV infection in the absence of T cells in a T− B− NK+ SCID patient (9). Furthermore, HCMV is the only virus known that can shape the human NK cell's receptor repertoire (10, 11). These clues highly suggest a role of NK cells in the defense against HCMV infection; however, direct proof of NK cells in controlling HCMV infection is absent.
By using an improved focus expansion assay (4, 12), we used a systematic approach to investigate the contribution of NK cells to HCMV transmission. We characterized HCMV transmission in various cell types and the NK cell contribution in controlling HCMV transmission. The assay we developed not only provides a clinically relevant readout for determination of the immune cell function in HCMV research, but will also allow other researchers to study the role of immune cells in other viral infections.
MATERIALS AND METHODS
Ethics statement.
All buffy coat samples were purchased from the Transfusion Center of the Ulm University Hospital (Institut für Klinische Transfusionsmedizin und Immungenetik Ulm GmbH, Ulm, Germany) and were randomly obtained from HCMV-seronegative healthy donors. All donors gave written informed consent to approve and authorize the use of their blood for medical, pharmaceutical, and research purposes.
Cells.
Peripheral blood mononuclear cells (PBMCs) were isolated and cryopreserved as previously described (13). Thawed PBMCs were preactivated overnight before NK cell isolation with or without 500 IU/ml interleukin-2 (IL-2) (Proleukin). Next, NK cells were enriched by negative selection from PBMCs using the human NK cell enrichment kit (Miltenyi). Parental NK-92 cells, lacking endogenous CD16, were stably retrovirally transduced to express the 176F variant (GFP [green fluorescent protein]-CD16 176F NK-92 [lower affinity]) and 176V variant (GFP-CD16 176V NK-92 [higher affinity]), as described previously (14). All NK-92 cell lines were obtained from Conkwest, San Diego, CA. NK-92 cells were maintained in minimum essential medium α (MEM α) (GIBCO/Invitrogen) supplemented with 10% fetal bovine serum (FBS), 5% human plasma (HCMV negative, 56°C inactivated), and 500 IU/ml IL-2 (Proleukin). Human foreskin fibroblasts (HFFs) and retinal pigment epithelial cells (ARPE19) were cultured in MEM (GIBCO/Invitrogen) containing 10% FBS. Human umbilical vein endothelial cells (HUVECs) were cultured in RPMI 1640 (GIBCO/Invitrogen) with 50 μg/ml endothelial cell growth supplement (Becton Dickinson), 10% human serum (HCMV negative, 56°C inactivated), and 5 IU/ml heparin. For the coculture of NK cells and HFFs or NK cells and ARPE19 cells, MEM α containing 10% FBS with or without 500 IU/ml IL-2 was used. For the coculture of NK cells and HUVECs, HUVEC medium with 500 IU/ml IL-2 was used.
Preparation of viral stocks.
Clinical isolates 1 (E30546) and 4 (E68240) originated from blood of two kidney transplant patients. Clinical isolates 2 (E52812) and 5 (E56647) originated from throat swabs of two children with acute infection. Clinical isolate 3 (E57300) originated from blood of one bone marrow transplant patient. The cell-associated HCMV clinical isolates were demonstrated by the lack of detectable infectivity in cell culture supernatants. For preparation of virus-infected HFF stocks, clinical isolates were initially propagated on HFFs until about 60% of cells showed cytopathic effect (CPE) and were used before passage 6. Infected cultures were subsequently trypsinized, the infection rate was determined, and the viruses were stored for the experiments.
HCMV strain TB40/E was derived from throat wash of a bone marrow transplant recipient by propagation for 5 passages in fibroblasts and 22 passages in endothelial cells (15). Mutant bacterial artificial chromosomes (BACs) were generated by markerless mutagenesis (16). HCMV strain TB40/E and viruses reconstituted from TB40/E BACs (BAC4 and BAC4ΔUL16) were propagated in HFFs. HCMV strain AD169 was obtained from ATCC, and AD169ΔUL16GFP was generated as described previously (17). For preparation of virus-infected HFF stocks, HFFs were infected with cell-free virus and frozen at 3 days postinfection.
Focal expansion assay.
Infected fibroblasts were cocultured with a 2,000-fold excess of uninfected HFF in 96-well plates. To analyze the HCMV transmission on other cell types, infected fibroblasts were cocultured with a 2,000-fold excess of uninfected endothelial or epithelial cells. NK cells were added in the cultures at the beginning at the effector-to-target (E:T) cell ratios indicated. After cocultivation, supernatants were carefully discarded without affecting cell monolayers. Next, the cultures were fixed with 80% acetone, and immediate early antigens (IEAs) were detected. Transwell inserts (1-μm pore; Greiner Bio-One) in 24-well plates were used for Transwell experiments.
Immunofluorescence.
To detect viral antigens, cells were fixed with 80% acetone and incubated with IEA antibodies (Argene-Biosoft) or the late major capsid protein (MCP) (pUL86, MAb 28-4, kindly provided by W. Britt), followed by staining with AF555- or AF488-conjugated goat anti-mouse immunoglobulins (Molecular Probes/Invitrogen). Nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole).
To determine HCMV antibody binding, HFFs infected at a low multiplicity of infection (MOI) were fixed 96 h postinfection with precooled methanol for 10 min at 4°C, incubated for 20 min at 4°C with FcR blocking reagent (Miltenyi), and incubated for 120 min at 4°C with the indicated dilutions of HCMV antibodies. During the last 30 min of incubation, rabbit serum (1:10; Sigma) was added to block unspecific binding of secondary antibodies. After washing, cells were incubated for 1 h at 4°C with fluorescein isothiocyanate (FITC)-conjugated rabbit anti-human IgG (Dako). Infected and noninfected cells were stained as described above.
HCMV antibody preparation and neutralization assay.
Pooled immunoglobulin for intravenous use (Gamunex; Talecris Biotherapeutics) was purchased commercially and used at different dilutions. Aliquots of HCMV IgG-negative and -positive individual serum samples were collected from whole blood by centrifugation at 4,000 rpm for 20 min. All sera were heat inactivated at 56°C for 30 min and stored at −20°C. HCMV IgG serology was determined with an enzyme-linked immunofluorescence assay (VIDAS CMV IgG; bioMérieux). For neutralization studies, media containing TB40/E (MOI, 1) were incubated with the indicated HCMV antibodies for 30 min at 37°C, and then the indicated mixtures were added to the fibroblasts.
Antiviral treatments and assays.
The antiviral drug ganciclovir (GCV) (Cymeven; Syntex, Germany) was added to cocultures from the beginning at concentrations of 7.5, 15, and 30 μM. Pooled immunoglobulin was added to cocultures from the beginning at dilutions of 1:200, 1:40, and 1:20. After 4 days of coculture, HFF monolayer cultures were fixed with 80% acetone, and viral IEA was determined.
IFN detection and neutralization.
Production of interferons (IFNs) was assessed in coculture supernatants by commercial enzyme-linked immunosorbent assay (ELISA) kits for IFN-α (41100; R&D Systems), IFN-β (41410; R&D Systems), and IFN-γ (430104; Biolegend). For blocking experiments, cocultures were kept in the presence of the following neutralization antibodies from the beginning of the experiments: IFN-α (10 μg/ml), clone MMHA-13 (R&D Systems); IFN-β (5 μg/ml), clone IFNb/A1 (Biolegend); IFN-γ (10 μg/ml), clone B27 (Biolegend); and mouse IgG1 isotype control (10 μg/ml), clone MOPC-21 (Biolegend).
Flow cytometry.
The following monoclonal antibodies (MAbs) were used: peridinin chlorophyll protein (PerCP)–Cy5.5–anti-CD3 (UCHT1), allophycocyanin (APC)–anti-CD56 (B159), and phycoerythrin (PE)–anti-NKG2D (1D11) from BD Biosciences; PE–anti-TRAIL (RIK-2) from Biolegend; and anti-DNAM-1 antibody (clone 4), a gift from Stipan Jonjic, Department of Histology and Embryology, Medical Faculty, University of Rijeka, Croatia. Cells were analyzed using FACSCalibur (BD Biosciences).
Statistical analysis.
One-way analysis of variance (ANOVA) (followed by multiple comparisons tests with least significant difference [LSD]) was used to compare data. Results were considered to be significant at a P value of 0.05.
RESULTS
Establishment of cell culture models to investigate cell-to-cell and cell-free HCMV transmission.
The cell-free HCMV infection starts with binding of free virions to permissive target cells, followed by entry and replication. Once the initial infection has occurred, HCMV may further be transmitted through cell-to-cell contact or cell-free virus for subsequent rounds of infection. Epithelial cells, endothelial cells, fibroblasts, and smooth muscle cells are major targets for HCMV infection in vivo (18).
To establish the experimental setting for studying the transmission of HCMV in fibroblasts, endothelial cells, and epithelial cells, we included 5 low-passage-number (less than passage 6) clinical HCMV isolates and the HCMV laboratory strain TB40/E. We mixed infected HFFs with a 2,000-fold excess of uninfected HCMV permissive cells and cocultured them for 2 to 5 days, which allowed HCMV to spread to adjacent uninfected cells. Newly infected cells could be identified as infectious foci in different cell types by HCMV immediate early antigen (EIA) staining. To further quantitatively analyze HCMV transmission in various cell types, we counted the number of infected cells of all the newly formed infectious foci. Infectious foci were defined as clusters of at least three infected cells. In this assay, depending on the experimental setting, 5 to 15 foci could be identified per well in 96-well plates. The kinetics of focus growth could be clearly identified from day 2 to day 5 in the three cell types, except for clinical isolate 5, which was unable to infect endothelial and epithelial cells (Fig. 1A). This might be explained by a lack of the protein complex formed by gH/gL and the pUL128-131A gene products in clinical isolate 5, which is required for endothelial and epithelial cell tropism. The sequence of clinical isolate 5 is still under investigation. The cell-free transmission was indicated by foci with isolated infected cells in the periphery of a larger focus, which were obviously infected by cell-free virus (4). Clinical isolates 1, 2, and 3 strictly spread through cell-to-cell transmission in fibroblasts. Clinical isolates 4 and 5 and laboratory strain TB40/E spread through both cell-to-cell and cell-free transmission in fibroblasts. After 5 days of coculture, most fibroblasts were infected in cultures with clinical isolates 4 and 5 and strain TB40/E: thus infectious foci could no longer be identified. There was no difference in the transmission patterns in endothelial cells and epithelial cells with the different virus strains. The representative kinetics of focus formation of clinical isolate 1 and TB40/E are shown in Fig. 1B. In general, the foci grew slower in endothelial and epithelial cells than in fibroblasts. Furthermore, when endothelial cells were used, the infected cells tended to remain less concentrated in the foci, and it could be shown by live microscopy that infected endothelial cells migrated during coculture (Kerstin Laib Sampaio, personal communication).
FIG 1.

Kinetics of HCMV transmission in different cell types. (A) Fibroblasts infected by clinical isolates and laboratory strain TB40/E were cocultured with a 2,000-fold excess of uninfected fibroblasts, endothelial cells, or epithelial cells for 2, 3, 4, and 5 days. Monolayers were fixed at the indicated times, and newly infected cells were monitored by HCMV IEA staining. The numbers of infected cells per focus were counted. One dot represents the number of infected cells of an individual focus. Bars indicate mean values of all foci. (B) Representative infectious foci of clinical isolate 1 and TB40/E in fibroblasts, endothelial cells, and epithelial cells are shown. The presence of HCMV IEA (green fluorescence) indicates infected cells, and cell nuclei are stained in blue (DAPI). *, the foci could not be counted due to the high infection rate in fibroblasts; **, days of coculture.
NK cells can control HCMV transmission in different cell types.
To investigate the NK cell contribution in the control of HCMV transmission in various cell types, NK-92 cells were used for the initial experiments. The NK-92 cell line displays a surface marker phenotype and functional profile of NK cells, and their growth is dependent on IL-2 (19). NK-92 cells were added to the indicated cell types at various effector-to-target (E:T) ratios ranging from 0.0625 to 0.5. With E:T ratios greater than 1, the NK-92 cells induced frank cytolysis of not infected cells of different types. Infectious foci were evaluated on day 4 post-coculture. NK-92 cells efficiently controlled the transmission of all HCMV strains tested in fibroblasts and endothelial and epithelial cells at different E:T ratios (Fig. 2A). NK-92 cells controlled HCMV transmission in fibroblasts at an E:T ratio of 0.125, in endothelial cells at an E:T ratio of 0.25, and in epithelial cells at an E:T ratio of 0.5. The reason for the different efficacies of NK cells in these three cell types is unknown but needs further investigation.
FIG 2.

NK cells efficiently inhibit HCMV transmission in fibroblasts and endothelial and epithelial cells. (A) Fibroblasts infected by the clinical isolates and laboratory strain TB40/E were cocultured with a 2,000-fold excess of uninfected fibroblasts, endothelial cells, or epithelial cells for 4 days. NK-92 cells were added to the cocultures immediately at different E:T ratios. Monolayers were fixed at the indicated times, and newly infected cells were identified by HCMV IEA staining. The numbers of infected cells per focus were counted. One dot represents the number of infected cells of an individual focus. Bars indicate mean values of all foci. (B) Thawed PBMCs were cultured using NK cell medium with IL-2 for 20 h, and then NK cells were negatively purified from PBMCs. Fibroblasts infected by the clinical isolate 1 and TB40/E were cocultured with a 2,000-fold excess of uninfected fibroblasts, endothelial cells, or epithelial cells for 3 days. Primary NK cells were added to the focus expansion assay immediately at different E:T ratios with IL-2-containing medium. Infectious foci were monitored by HCMV IEA staining.
To test whether primary NK cells could similarly control HCMV transmission, we purified NK cells from HCMV-seronegative donors. Primary NK cells with IL-2 treatment also efficiently controlled the transmission of clinical isolate and laboratory-adapted HCMV strains in fibroblasts and endothelial and epithelial cells at different E:T ratios (Fig. 2B). NK cells controlled HCMV transmission in fibroblasts at an E:T ratio of 0.0625, in endothelial cells at an E:T ratio of 0.25, and in epithelial cells at an E:T ratio of 0.5. The efficacy of primary NK cells in these three cell types is very similar to that of NK-92 cells.
To assess our focus expansion assay for the evaluation of antiviral drugs and HCMV antibodies on the inhibition of HCMV transmission in human fibroblasts (which also served as positive controls), cultures were incubated with different concentrations of ganciclovir (GCV) or a commercially available Ig preparation containing neutralizing antibodies against HCMV from the beginning. Ganciclovir inhibited the transmission of all HCMV strains in fibroblasts at a concentration of 7.5 μM, which was the lowest concentration tested. However, HCMV antibodies exhibited different effects when different viruses were used. It should be noted that the neutralizing activity of HCMV antibody could only be observed with the clinical isolates 4 and 5 and laboratory strain TB40/E, which had exhibited a cell-free transmission mode in fibroblasts. These findings demonstrate that our focus expansion assay can also be used to study the effects of antiviral drugs and HCMV antibodies on HCMV transmission.
IL-2 enhances primary NK cells in the control of HCMV transmission.
From initial experiments, we found that NK cells that were purified from PBMCs without any treatment also inhibited the HCMV transmission at low E:T ratios, but they were not as effective as NK-92 cells at the same E:T ratios. However, the inhibitory capacity of primary NK cells was strongly enhanced with IL-2 treatment when the same concentration as that for NK-92 cells was used (Fig. 3A). Ex vivo NK cells without IL-2 treatment mediated frank cytolysis of noninfected fibroblasts at E:T ratios of greater than 4, whereas IL-2-treated NK cells induced a marked cytolysis of fibroblasts at E:T ratios greater than 0.5. The frank cytolysis means that the intact monolayers of fibroblasts were affected by NK cells. This could be observed by light microscopy and staining of nuclei. This proves that IL-2 treatment enhances cytotoxicity of primary NK cells. We also found that primary NK cells alone produced a small amount of IFN-γ with IL-2 treatment. They produced more IFN-γ when kept in contact with uninfected fibroblasts. Furthermore, they produced significantly more IFN-γ when cocultured with infected HFFs at the same E:T ratio (Fig. 3B). Primary NK cells produce only small amounts of IFN-γ without IL-2 treatment, even in the presence of infected HFFs.
FIG 3.
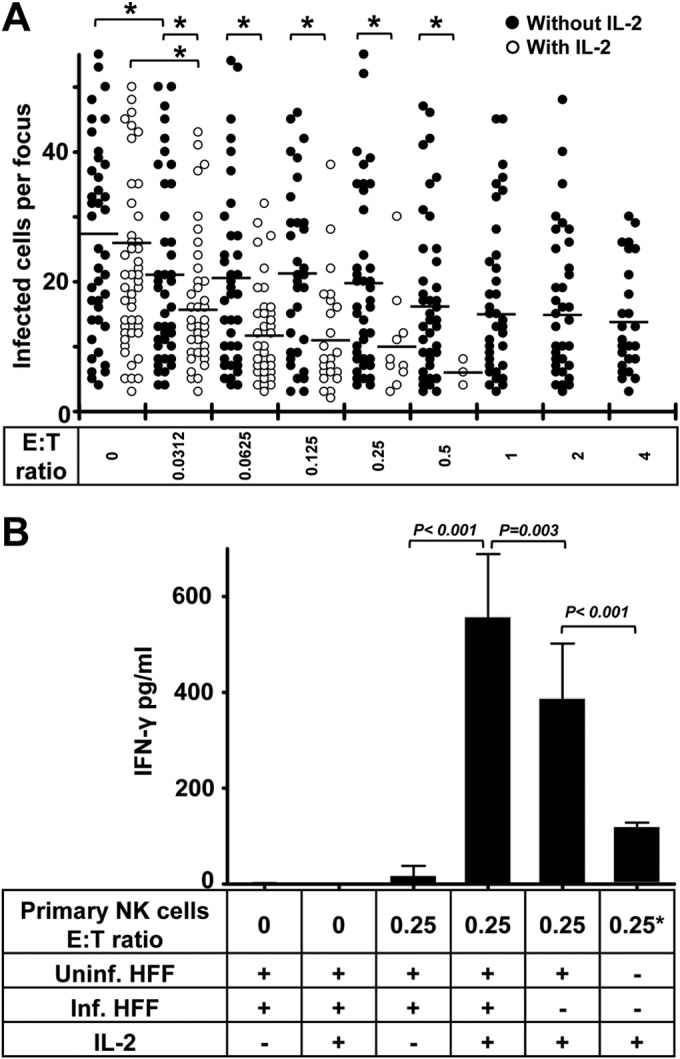
IL-2 enhances the inhibition of HCMV transmission by primary NK cells. (A) Thawed PBMCs were cultured using NK cell medium with or without IL-2 for 20 h. Next, NK cells were negatively selected from PBMCs. TB40/E-infected fibroblasts were cocultured with a 2,000-fold excess of uninfected fibroblasts for 3 days. Purified NK cells were added to the focus expansion assay immediately at different E:T ratios with or without IL-2-containing medium. Monolayers were fixed at the indicated times, and newly infected cells were monitored by HCMV IEA staining. The numbers of infected cells per focus were counted. One dot represents the number of infected cells of an individual focus. Bars represent mean values of all foci from three donors tested. *, P < 0.05. (B) The concentrations of IFN-γ in supernatants from indicated cultures were tested by ELISA. Uninf., uninfected; Inf., infected. “0.25*” indicates that the same amount of NK cells was used.
NK cells control HCMV transmission via soluble factors and by cell contact.
NK cells contribute to the immune defense against viruses by a cytotoxic activity against infected cells, as well as by producing antiviral cytokines. Activated NK cells are the main source of IFN-γ, which plays a pivotal role in the antiviral response (5) (Fig. 3B). Exogenous addition of IFN-α, IFN-β, and IFN-γ potently inhibits the replication of HCMV in fibroblasts (20). Cell contact between effectors and target cells is required for cytolysis medicated by NK cells, including (i) the perforin-granzyme pathway induced by ligation of activating NK receptors, (ii) death receptor ligand-mediated induction of apoptosis, and (iii) antibody-dependent cellular cytotoxicity (ADCC).
To investigate the mechanisms of the inhibition of HCMV transmission by NK cells, primary NK cells and fibroblasts were separated in a Transwell system when the focus expansion assay was performed. As shown in Fig. 4, the separation of NK cells and fibroblasts partially reduced the capacity of the NK cells to control HCMV transmission, indicating that cell-to-cell contact contributes to the effect. Despite the reduced capacity to control HCMV transmission in the Transwell experiments, NK cells separated from fibroblasts still inhibited the transmission to some extent, proving also the contribution of soluble factors. The same experiments were also performed with NK-92 cells with the same results (data not shown).
FIG 4.
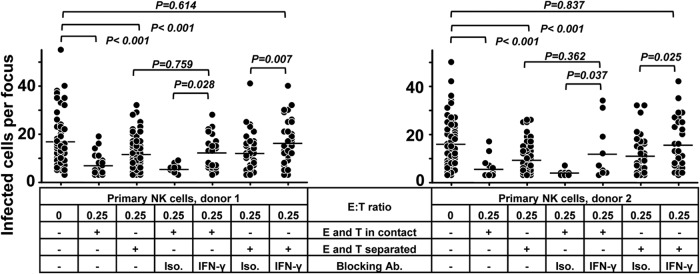
Cell contact and soluble factors both contribute to the control of HCMV transmission by NK cells. Thawed PBMCs were cultured using NK cell medium with IL-2 for 20 h, and then NK cells were negatively selected from PBMCs. TB40/E-infected fibroblasts were cocultured with a 2,000-fold excess of uninfected fibroblasts for 3 days. Purified NK cells were added to the focus expansion assay from the beginning at an E:T ratio of 0.25 with IL-2-containing medium. Primary NK cells were either added to the cocultures directly or kept separated from fibroblasts using a Transwell filter system. Specific blocking MAb against IFN-γ or an isotype antibody as a control was added at the beginning of cocultures as indicated. Monolayers were fixed, and newly infected cells were monitored by HCMV IEA staining. The numbers of infected cells per focus were counted. One dot represents the number of infected cells of an individual focus. Bars indicate mean values of all foci.
Interferons have been known to suppress virus replication and make uninfected cells resistant to infection (20, 21). To determine the possible soluble antiviral factors, we measured the levels of interferon-α, -β, and -γ in the supernatants. Indeed, IFN-γ could be detected only in supernatants containing NK cells. Furthermore, primary NK cells or NK-92 cells cocultured with HCMV-infected cells contained significantly more IFN-γ (Fig. 3B) (data not shown). However, IFN-α (detected with a high-sensitive-range detection kit) and IFN-β could not be detected in the same supernatants. In subsequent blocking experiments, we also tested the effect of interferon-neutralizing antibodies on the inhibitory effect described. Only neutralizing MAbs against IFN-γ but not against IFN-α or IFN-β partially inhibited the suppressive effect of NK-92 cells on HCMV transmission (data not shown). Using primary NK cells, the Transwell system and/or neutralizing MAbs against IFN-γ were also applied in focus expansion assays. As shown in Fig. 4, neutralizing MAbs against IFN-γ also partially blocked the suppressive effect of primary NK cells on HCMV transmission. However, the combination of Transwell separation and neutralizing MAbs against IFN-γ completely eliminated the capacity of the NK cells to control HCMV transmission. In summary, these experiments indicate that IFN-γ alone as well as cell contact contributes to the NK cells mediated inhibition of HCMV transmission, although we could not define the exact quantitative contributions of these two mechanisms.
To further characterize the mechanisms of soluble factors involved in the control of HCMV transmission, supernatants were collected from cocultures with or without NK cells, and cell-free virus was removed with a 0.1-μm-pore filter (Fig. 5A). First, we tested whether these supernatants could suppress HCMV viral antigen expression and cell-free virus production. We prepared infected fibroblasts by infection with TB40/E at an MOI of 7.5. After 30 min of incubation, cells were washed with phosphate-buffered saline (PBS) and supplemented with fresh medium. Thereafter, the indicated supernatants were added to infected fibroblasts at different times postinfection and were kept on the cocultures for different times. As shown in Fig. 5B, the initial phase of infection, monitored by IEA staining, was not affected by any of the supernatants when added to infected fibroblasts from 0.5 h until 24 h postinfection. We further kept supernatants on infected cells from 24 h to 96 h postinfection and compared the differences in late antigen expression and cell-free virus production (Fig. 5B and C). HCMV major capsid protein (MCP [encoded by the UL86 gene]) can be detected after viral DNA replication and corresponds to the release of progeny virus. Although nearly all fibroblasts were infected (98% of cells IEA positive in all groups), the expression of the MCP was strongly inhibited in the presence of supernatants from NK cells. The expression pattern of MCP (speckled) was also different from that in untreated infected cells (whole nuclei). Thus, we quantified the MCP expression by MCP-to-DAPI surface area ratios. At 96 h postinfection, cell-free virus production was also reduced significantly in the presence of supernatants from NK cells (Fig. 5C). Both inhibitory effects could be partially inhibited by IFN-γ-neutralizing antibodies. This indicates that IFN-γ produced by NK cells plays an important role in these inhibitory effects. However, supernatants from cultures containing NK cells also were ineffective when applied at very late times (96 to 120 h postinfection), when MCP was already fully expressed (Fig. 5B and C).
FIG 5.

NK cell-containing supernatants reduce HCMV production. (A) Supernatants were collected from the indicated cocultures and filtered through a 0.1-μm-pore filter to remove cell-free virus. Uninf., uninfected; Inf., infected; Ab, antibody; Iso., isotype antibody control. (B) Fibroblasts were infected by TB40/E (MOI, 7.5) for 30 min and washed with PBS. Thereafter, supernatants were added to infected fibroblasts from 30 min postinfection (p.i.) until 24 h p.i., from 24 to 96 h p.i., or from 96 to 120 h p.i., and then infection rates were determined by HCMV IEA staining. IEA staining is shown by green fluorescence and MCP staining is red at late times infection, and cell nuclei are stained in blue (DAPI). The infection rates (IEA/DAPI) are indicated by the numbers. The MCP-to-DAPI surface area ratios are also indicated by the numbers. (C) Fibroblasts were infected by TB40/E (MOI, 7.5) for 30 min and washed with PBS. Thereafter, supernatants were added to the infected fibroblasts from 24 to 96 h p.i. or from 96 to 120 h p.i. Then 100 μl of 8-fold-diluted supernatants containing cell-free virus was transferred to uninfected fibroblasts. After 30 min of incubation, cells were washed with PBS, and the infection rates (IEA/DAPI) were determined after 24 h.
Furthermore, we investigated whether the supernatants from cultures containing primary NK cells had an effect on the resistance of bystander cells against HCMV infection. Uninfected fibroblasts were pretreated with indicated supernatants I to VIII for 24 h (Fig. 5A) and then infected with TB40/E at an MOI of 0.05. We found that uninfected fibroblasts were resistant to HCMV infection if they were exposed to supernatants from cocultures containing primary NK cells compared to NK cell-free supernatants (Table 1). The induced resistance against HCMV infection seems to be correlated to the level of IFN-γ (Fig. 3B). In addition, neutralizing MAbs against IFN-γ also partially inhibited this effect (Table 1). This indicates that IFN-γ plays an important role in this induced resistance of HCMV infection. The same experiments were also performed with NK-92 cells (data not shown).
TABLE 1.
Determination of IEA-positive HFFs
| Donor | % of IEA+ HFFs after pretreatment with coculture supernatanta: |
|||||||
|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | VII | VIII | |
| 1 | 1.225 | 1.1 | 0.23 | 0.85 | 0.085 | 0.275 | 0.075 | 0.365 |
| 2 | 1.145 | 1.105 | 0.115 | 0.945 | 0.045 | 0.285 | 0.095 | 0.335 |
HFFs were pretreated with the supernatants shown for 24 h before infection with TB40/E at an MOI of 0.05. Coculture supernatants from Fig. 5A were used.
HCMV genes modulate the NK cell-mediated control of HCMV transmission: implications for immune evasion.
NK cell activity is regulated by activating and inhibitory receptors, and death receptors are important for NK cell-mediated cytolysis (22). Studies have shown that HCMV expresses multiple gene products that downregulate the ligands of activating receptors and death receptors, thereby rendering infected cells resistant to NK cell lysis (23, 24).
We tested whether HCMV genes can induce an evasion from the NK cell-mediated control of HCMV transmission. HCMV encodes glycoprotein UL16 to prevent cell surface expression of MICB, ULBP1, and ULBP2. Thus, UL16 suppresses NK cell recognition by impeding cell surface expression of these ligands of NKG2D (23). NK-92 cells express NKG2D at low levels (19; data not shown), whereas primary NK cells express high levels of NKG2D. Thus, we applied primary NK cells in our study. We first applied the HCMV TB40/E-derived BAC4 as well as a mutant of BAC4 lacking the UL16 viral gene (BAC4ΔUL16) in our study. BAC4- or BAC4ΔUL16-infected fibroblasts were applied in the focus expansion assay, and IL-2-treated primary NK cells were used at an E:T ratio of 0.25. As shown in Fig. 6A, BAC4ΔUL16-infected fibroblasts were nearly completely eliminated by NK cells, whereas infectious foci still could be identified in BAC4-infected cells. This indicates that HCMV UL16 contributes to the immune evasion of the NK cell-mediated control of HCMV transmission. To better characterize the immune evasion mechanisms, we applied neutralizing MAbs against IFN-γ in the same experimental setting. The neutralizing MAbs against IFN-γ slightly compromised the effect of NK cells without statistical significance (Fig. 6A). Furthermore, the levels of IFN-γ between the two groups showed no difference (Fig. 6B). This indicates that UL16 modulates the NK cell killing of the infected cells. To further confirm this, we also included a UL16 deletion mutant on the background of AD169 marked with GFP (AD169ΔUL16GFP) in our assay, providing the possibility of live imaging. The GFP expression is under the control of the UL16 promoter, which can be detected 6 h postinfection and applied as a marker of HCMV infection (17). Again, AD169ΔUL16GFP-infected fibroblasts were efficiently eliminated by primary NK cells compared to AD169-infected cells (Fig. 6C). During the live imaging, we also observed that NK cells mainly aggregated surrounding infected cells.
FIG 6.
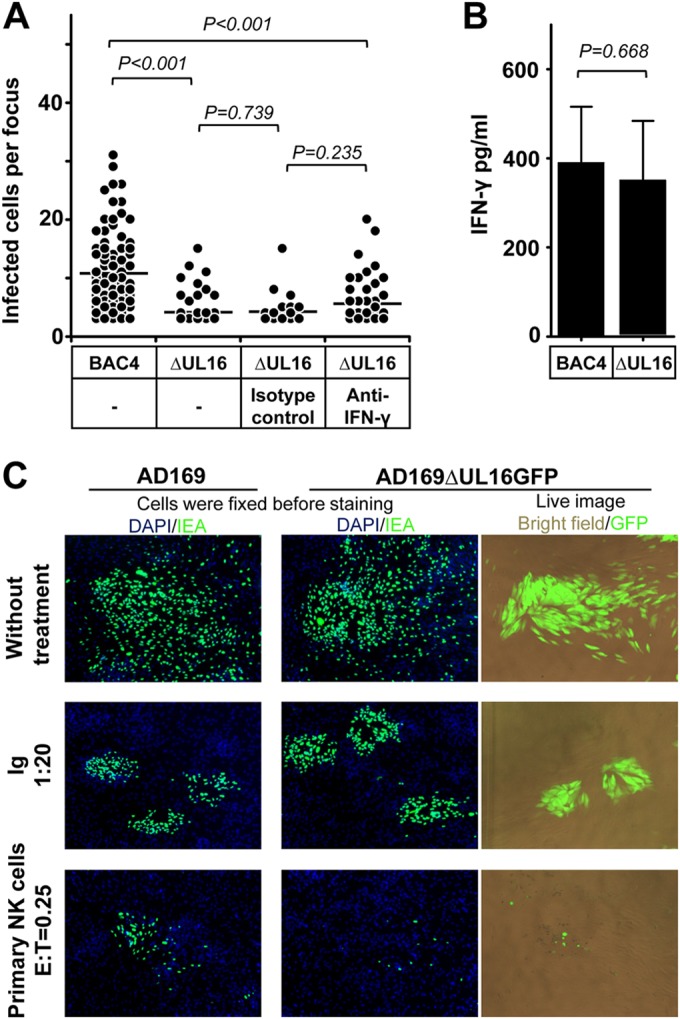
HCMV gene UL16 modulates the control of HCMV transmission by NK cells. (A) Thawed PBMCs were cultured using NK cell medium with IL-2 for 20 h, and then NK cells were negatively selected from PBMCs. Fibroblasts infected with BAC4 or BAC4ΔUL16 were cocultured with a 2,000-fold excess of uninfected fibroblasts for 3 days. Purified NK cells were added to the focus expansion assay from the beginning at an E:T ratio of 0.25 with IL-2-containing medium. Specific blocking MAb against IFN-γ or an isotype antibody as a control was added immediately as indicated. Monolayers were fixed, and newly infected cells were monitored by HCMV IEA staining. The numbers of infected cells per focus were counted. One dot represents the number of infected cells of an individual focus. Bars represent mean values of all foci from four donors tested. (B) The concentration of IFN-γ in supernatants from indicated cultures were tested by ELISA. (C) Fibroblasts infected with AD169 or AD169ΔUL16GFP were used in the focus expansion assay. Infected cells were monitored by live imaging of GFP or IEA staining after fixation. HCMV antibodies from a commercial Ig preparation (Ig) at a 1:20 dilution were used as a positive control.
FcγRIIIa-158F/V polymorphism contributes to the NK cell-mediated control of HCMV transmission: implications for immune surveillance.
The control of HCMV requires a continuous immune surveillance in vivo involving both innate and adaptive immunity. NK cells are main effectors of antibody-dependent cell-mediated cytotoxicity (ADCC), which provides a known bridge between innate and adaptive immunity. NK cell-mediated ADCC depends on the surface expression of FcγRIIIa (CD16) (25). The human FcγRIIIa gene displays a dimorphism in the position coding for amino acid residue 158; with one allele encoding a higher-Fc-affinity receptor variant having a valine at amino acid residue 158 (158V) and another, lower-affinity receptor variant carrying phenylalanine (158F) (26). NK cells are the principal effectors of ADCC mediated by therapeutic monoclonal antibodies (biological). It has been shown that lymphoma patients homozygous for the high-affinity allele (158VV) of FcγRIIIa have a survival advantage upon rituximab monotherapy (27). We previously demonstrated that the activity of NK cells against HCMV-infected cells can be enhanced by HCMV antibodies (6). Thus, we also investigated the role of ADCC in the control of HCMV transmission using the focus expansion assay.
We applied three essential components in our experiments. First, we used three NK-92 cell lines expressing FcγRIIIa with different affinities. Cells from the parental NK-92 cell line do not express FcγRIIIa and therefore cannot lyse target cells via ADCC. However, NK-92 cells stably transfected with FcγRIIIa were fully capable of mediating ADCC (14). We applied FcγRIIIa-transfected NK cell lines in our study expressing either the low-affinity (158F) or high-affinity (158V) variant of FcγRIIIa (Fig. 7A). We were able to obtain clear results irrespective of the considerable interdonor variation in ADCC assays. Second, we applied HCMV antibodies that were able to neutralize cell-free virus and to bind to infected cells. We demonstrated that serum from an HCMV-seropositive donor and HCMV antibodies from a commercial Ig preparation using pooled sera could efficiently neutralize HCMV cell-free virus infection of fibroblasts (Fig. 7B). Furthermore, infected fibroblasts (red fluorescence) showed a clear binding of HCMV antibodies (green fluorescence) (Fig. 7C), but no binding of IgG from HCMV antibody-negative serum could be observed. This indicates the HCMV-infected fibroblasts did express HCMV antigens on their surface that could be recognized by HCMV antibodies. Third, we applied one clinical HCMV isolate that strictly spread through cell-to-cell transmission and was fully resistant to HCMV antibody neutralization. This clinical isolate thus provides the possibility to exclude the influence of antibody neutralization.
FIG 7.

CD16 receptor-158V/F polymorphism contributes to the efficacy of HCMV antibody-dependent NK cell inhibition of cell-to-cell virus transmission. (A) The levels of CD16 expression on three NK-92 cell lines are shown. (B) One-hundred-microliter cell-free suspensions of TB40/E with or without (w/o) HCMV antibodies (Abs) were used in neutralization experiments on fibroblasts. Virus suspensions and indicated HCMV antibodies (HCMV-positive serum used at a 1:10 final dilution, HCMV antibodies from a commercial Ig preparation [Ig] used at a 1:200 final dilution) were incubated for 30 min and then used to inoculated the cells. The infection rates were assessed after 24 h of incubation. The presence of HCMV IEA (red fluorescence) indicates infected fibroblasts, and cell nuclei are stained in blue (DAPI). (C) Fibroblasts were infected after 3 days and incubated with the Ig preparation followed by FITC–anti-human IgG staining. The presence of HCMV IEA (red fluorescence) indicates infected cells, human IgG binding on fibroblasts is shown by green fluorescence, and nuclei are stained in blue (DAPI). (D) Fibroblasts infected by clinical isolate 1 were cocultured with a 2,000-fold excess of uninfected fibroblasts for 3 days. HCMV antibody preparations (HCMV-positive [Pos.] and -negative [Neg.] sera used at a 1:10 final dilution, Ig preparation used at a 1:200 final dilution) were added at the beginning of cocultures in the presence or absence of the different NK-92 cell lines at an E:T ratio of 0.25. Monolayers were fixed at the indicated times, and newly infected cells were monitored by HCMV IEA staining. The numbers of infected cells per focus were counted. One dot represents the number of infected cells of an individual focus. Bars indicate mean values of all foci.
As shown in Fig. 7D, HCMV antibodies have no effect on the transmission of the clinical isolate, and all three NK-92 cell lines equally did control HCMV transmission in the absence of HCMV antibodies. Importantly, only NK92-CD16-158V cells showed an enhanced effect on the control of the HCMV transmission in the presence of HCMV antibodies. This indicates that expression of high-affinity FcγRIIIa-158V plays a role in the antibody-dependent NK cell-mediated control of cell-to-cell HCMV transmission.
DISCUSSION
In this study, we evaluated the contribution of NK cells to the control of HCMV transmission by mechanisms depending on innate and adaptive immunity. The experimental system we have established provides a new and very sensitive method to directly study the control of HCMV transmission by immune cells in vitro. We thereby also provide a methodology to evaluate viral immune evasion and immune surveillance using patient materials. We are confident that our work will open new perspectives to quantitatively study human immunity in viral infections.
We applied 5 preselected clinical HCMV isolates and the laboratory strain TB40/E in our study. These virus strains can be classified into two groups based on their transmission pattern in fibroblasts. The clinical isolates 1, 2, and 3 expanded strictly by cell-to-cell transmission. Clinical isolates 4 and 5 and laboratory strain TB40/E are transmitted both via cell-free virus and cell-to-cell contact (Fig. 1). The reasons for this difference are not fully understood so far but might at least partially be related to the viral RL13 and UL128 genes as reported previously (28, 29). The mechanisms of HCMV cell-to-cell transmission are also not fully clear (17), but the assumption is that HCMV disseminates predominantly by cell-to-cell transmission in vivo, where many cell types can be infected by HCMV (18). Most but not all clinical HCMV isolates grow strictly by cell-to-cell transmission in vitro after primary isolation (17). So far, we have applied fibroblasts to study the underlying mechanisms. Later on, it would be advisable to confirm the results in other cell types. Our data show that NK cells can efficiently control the transmission of HCMV clinical isolates and the laboratory strain TB40/E in fibroblasts, epithelial cells, and endothelial cells (Fig. 2). These results highly suggest a general importance of NK cells in anti-HCMV immunity.
Our Transwell study clearly indicates that both direct cell contact between effector and target cells and also soluble factors are involved in the control of HCMV transmission by NK cells. When NK cells were kept in direct contact with fibroblasts, the infectious foci were smaller than those resulting from Transwell experiments without direct contact between target end effector cells. This indicates that an NK cell-mediated lysis of infected fibroblasts is involved in the reduction of infectious focus expansion. In the focus expansion assay, we observed that NK cells preferred to aggregate around fibroblasts showing infection by GFP-labeled virus. This suggests that NK cells could somehow recognize, migrate to, and interact with HCMV-infected cells.
We demonstrated that supernatants from cocultures containing NK cells can inhibit HCMV production but can also induce resistance of bystander fibroblasts against HCMV infection. Type I IFNs (IFN-α and IFN-β) and type II IFN (IFN-γ) are important components of the host immune response to viral infections. IFN-α and IFN-β are produced by most cells as a direct response to viral infection, while IFN-γ is synthesized almost exclusively by activated NK cells and activated T cells in response to virus-infected cells (30). It has been shown that exogenous addition of IFN-α/β and IFN-γ synergistically inhibits HCMV replication in fibroblasts (20). In our experiments, we could not detect any IFN-α and IFN-β production by our cocultures but could detect IFN-γ, which was obviously produced by the NK cells. We confirmed that only IFN-γ contributes to the inhibition of HCMV transmission in antibody blocking experiments. It has been shown that HCMV infection might interfere with IFN-α production (31). Other studies showed by looking at IFN-β mRNA that lymphotoxins and cytomegalovirus cooperatively induce IFN-β production. IFN-β was suggested to be an anti-HCMV cytokine by antibody blocking experiments without having shown IFN-β concentrations (32, 33). So, the discrepancy from our results might be explained by different experimental settings or different blocking antibodies.
NK cell activation can be mediated through cytokine stimulation (34). Our results demonstrated that IL-2 treatment highly enhanced the capacity of primary NK cells in the control of HCMV transmission. This is consistent with the fact that IL-2 treatment enhances NK cell-mediated inhibition of HCMV replication (33). We could show that primary NK cells produced more IFN-γ with IL-2 treatment. Furthermore, IL-2 treatment highly enhances the TRAIL expression on the primary NK cells we used (data not shown). Another study also had shown that primary NK cells expressed more lymphotoxin α (LTα), LTβ, and tumor necrosis factor (TNF) with IL-2 treatment, which in turn could enhance the IFN-β production by fibroblasts (33). Yet again, we could not detect IFN-β production in our experiments using primary NK cells. IL-2 can be produced by CD4+ T cells during the early stage of reexposure to HCMV, and NK cell activity can be enhanced by IL-2 in response to HCMV-infected cells (unpublished data). Thus, IL-2 may play an important role in the NK cell-mediated innate immunity against HCMV.
It is known that NK cell activity is regulated by activating and inhibitory receptors. Five viral genes (UL16, UL83, miR-UL112, UL141, and UL142) have been identified as being capable of suppressing NK cell recognition of activating receptors (23). UL16, miR-UL112, and UL142 suppress the presentation of ligands on the cell surface for the NK cell-activating receptor NKG2D. UL141 modulates the immune response by downregulating the surface expression of DNAM-1-activating ligands CD155 and CD112. Additionally, a study also had shown that UL141 downregulates TRAIL DR surface expression on fibroblasts, thus thwarting the NK cells' activity (24). UL83 encoding tegument protein pp65 directly binds to the NK cells activating receptor NKp30. The binding leads to the dissociation of the linked CD3ζ from NKp30 and thus impedes NK cell activation (35). By using mutants derived from TB40/E-BAC4, we tested the contribution of all above-mentioned viral genes in the control of HCMV by NK cells. We demonstrated that UL16 contributes to NK cell-mediated suppression of HCMV transmission. In addition, we also found that UL141 was also involved in the modulation of NK cell-mediated suppression of HCMV transmission (data not shown). However UL83, miR-UL112, and UL142 did not contribute to NK cell-mediated virus control. This discrepancy might be explained by different experimental settings or by the fact that the previous study did not use an assay to assess the role of these genes in the HCMV control. Two viral genes (UL18 and UL40) have been identified as capable of enhancing NK cell inhibitory receptor recognition of NK cells (23). These inhibitory receptors are expressed only by certain subsets of NK cells. The contributions of UL18 and UL40 are still under investigation with purified subsets of NK cells.
HCMV antibodies show a very strong antiviral effect in various experimental settings in vitro. HCMV antibodies can efficiently neutralize cell-free virus infectivity in different cell types. They have much higher cell-free virus neutralization potency on epithelial cells and endothelial cells than on fibroblasts (36). Although HCMV antibodies have no effect on the cell-to-cell transmission in fibroblasts, they can fully block cell-to-cell transmission in epithelial cells and endothelial cells (37; unpublished data). The discrepancy in neutralizing potency may be due to the fact that HCMV utilizes different modes of entry into epithelial cells and endothelial cells versus fibroblasts. We have previously shown that HCMV antibodies highly enhance the activity of NK cells in the response to HCMV-infected macrophages (6). However, the exact role of HCMV antibodies in the control of infection in vivo is still unclear. The reported results of HCMV antibody treatment in patients differ very much between different studies. This might be explained by an inefficiency of HCMV antibody to block cell-to-cell virus transmission in fibroblasts, different IgG preparations (38), and by differences between NK cells from different donors to mediate an ADCC effect (39). We demonstrated that NK92-CD16-158V cells had an enhanced effect on the control of HCMV transmission through ADCC. This might provide an explanation for differences in the efficiency of antibody treatment in patients. From an application perspective, a characterization of the CD16 phenotype and the ability of antibody preparations to control HCMV transmission might define the individuals who could benefit from antibody treatment. To this end, the assay presented here could be very helpful.
The reduction of infected cells in the focus expansion assay by NK cells is correlated to the results of the IFN-γ secretion assay. In the same experimental setting, NK cells exposed to infected fibroblasts secreted more IFN-γ. By using this assay, we identified UL16 and UL141 as being involved in NK cell-mediated inhibition of HCMV transmission. The same genes were identified to be involved in the direct killing of infected fibroblasts by using a chromium 51 release assay with high E:T ratios (data not shown). In the first experiments, we could show that the killing of cells infected by the HCMV UL16 deletion mutant was due to the induction of apoptosis, as determined by morphological changes, and furthermore, death receptor ligand blocking antibodies could partially inhibit NK cell-induced apoptosis and NK cell-mediated control of virus transmission in different cell types (unpublished data).
Our focus expansion assay to study the contribution of NK cells in the control of virus transmission offers important possibilities for a quantitative analysis of HCMV transmission, such as the following. (i) The assay provides a practical method to directly study the immune surveillance and evasion in viral infections. The readout of the focus expansion assay is the control of infection instead of just measuring the activation of immune cells. (ii) The direct contribution of immune cells in infection can be quantitatively evaluated, and thus also its clinical relevance is high. (iii) Another advantage of the assay is that we can dissect different mechanisms in the same experimental setting. (iv) The assay requires very small amounts of NK cells (5,000 NK cells per well) and low E:T ratios, which makes functional analysis of rare immune cell populations possible. (v) The infectious foci consist of infected cells at different infectious stages (data not shown), which might represent the situation in vivo.
Our systematic investigations of NK cells in HCMV transmission may be translated to manage HCMV-associated diseases by manipulating NK cells. NK cells execute the anti-HCMV activity by their unique mechanisms, which are different from those of antiviral drugs and HCMV immunoglobulin. It is reasonable to evaluate the effect of NK cell adoptive therapy in HCMV diseases. Our study also highlights two possible clinical available interventions to enhance the activity of NK cells: IL-2 and HCMV antibody. We hypothesize that these two potential interventions might be very efficient in certain groups of patients. Thus, whether these in vitro data could be translated into management of HCMV-associated diseases by enhancing NK cell activity using HCMV antibody and/or IL-2, potentially in combination with antiviral therapy, should seriously be considered.
ACKNOWLEDGMENTS
We thank Ingrid Bennett for help in preparing the manuscript and Kerstin Laib Sampaio for helpful discussions.
This work was supported by grants from the DFG-Program “International Graduate School in Molecular Medicine, Ulm University.”
Z.W. designed and performed research, analyzed data, and wrote the paper. C.S. and J.J.R. provided reagents and HCMV laboratory strains. M.J. performed clinical isolate cultures. T.M. designed research, analyzed data, and wrote the paper.
The authors declare they have no financial or commercial conflicts of interest.
REFERENCES
- 1.Pass RF. 2001. Cytomegalovirus, p 2675–2705 InFields BN, Knipe DM, Howley PM, Griffin DE (ed), Fields virology, 4th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Sattentau Q. 2008. Avoiding the void: cell-to-cell spread of human viruses. Nat Rev Microbiol 6:815–826. doi: 10.1038/nrmicro1972. [DOI] [PubMed] [Google Scholar]
- 3.Revello MG, Percivalle E, Arbustini E, Pardi R, Sozzani S, Gerna G. 1998. In vitro generation of human cytomegalovirus pp65 antigenemia, viremia, and leukoDNAemia. J Clin Invest 101:2686–2692. doi: 10.1172/JCI1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sinzger C, Knapp J, Plachter B, Schmidt K, Jahn G. 1997. Quantification of replication of clinical cytomegalovirus isolates in cultured endothelial cells and fibroblasts by a focus expansion assay. J Virol Methods 63:103–112. doi: 10.1016/S0166-0934(97)02082-X. [DOI] [PubMed] [Google Scholar]
- 5.Jost S, Altfeld M. 2013. Control of human viral infections by natural killer cells. Annu Rev Immunol 31:163–194. doi: 10.1146/annurev-immunol-032712-100001. [DOI] [PubMed] [Google Scholar]
- 6.Wu Z, Sinzger C, Frascaroli G, Reichel J, Bayer C, Wang L, Schirmbeck R, Mertens T. 2013. Human cytomegalovirus-induced NKG2Chi CD57hi natural killer cells are effectors dependent on humoral antiviral immunity. J Virol 87:7717–7725. doi: 10.1128/JVI.01096-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magri G, Muntasell A, Romo N, Saez-Borderias A, Pende D, Geraghty DE, Hengel H, Angulo A, Moretta A, Lopez-Botet M. 2011. NKp46 and DNAM-1 NK-cell receptors drive the response to human cytomegalovirus-infected myeloid dendritic cells overcoming viral immune evasion strategies. Blood 117:848–856. doi: 10.1182/blood-2010-08-301374. [DOI] [PubMed] [Google Scholar]
- 8.Biron CA, Byron KS, Sullivan JL. 1989. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med 320:1731–1735. doi: 10.1056/NEJM198906293202605. [DOI] [PubMed] [Google Scholar]
- 9.Kuijpers TW, Baars PA, Dantin C, van den Burg M, van Lier RA, Roosnek E. 2008. Human NK cells can control CMV infection in the absence of T cells. Blood 112:914–915. doi: 10.1182/blood-2008-05-157354. [DOI] [PubMed] [Google Scholar]
- 10.Beziat V, Liu LL, Malmberg JA, Ivarsson MA, Sohlberg E, Bjorklund AT, Retiere C, Sverremark-Ekstrom E, Traherne J, Ljungman P, Schaffer M, Price DA, Trowsdale J, Michaelsson J, Ljunggren HG, Malmberg KJ. 2013. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 121:2678–2688. doi: 10.1182/blood-2012-10-459545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guma M, Angulo A, Vilches C, Gomez-Lozano N, Malats N, Lopez-Botet M. 2004. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 104:3664–3671. doi: 10.1182/blood-2004-05-2058. [DOI] [PubMed] [Google Scholar]
- 12.Sinzger C, Mangin M, Weinstock C, Topp MS, Hebart H, Einsele H, Jahn G. 2007. Effect of serum and CTL on focal growth of human cytomegalovirus. J Clin Virol 38:112–119. doi: 10.1016/j.jcv.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 13.Wu Z, Frascaroli G, Mertens T. 2013. Assessment of natural killer cell responses to human cytomegalovirus-infected macrophages. Methods Mol Biol 1064:289–298. doi: 10.1007/978-1-62703-601-6_21. [DOI] [PubMed] [Google Scholar]
- 14.Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. 2008. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol 180:6392–6401. doi: 10.4049/jimmunol.180.9.6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J Gen Virol 89:359–368. doi: 10.1099/vir.0.83286-0. [DOI] [PubMed] [Google Scholar]
- 16.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 17.Digel M, Sampaio KL, Jahn G, Sinzger C. 2006. Evidence for direct transfer of cytoplasmic material from infected to uninfected cells during cell-associated spread of human cytomegalovirus. J Clin Virol 37:10–20. doi: 10.1016/j.jcv.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Sinzger C, Digel M, Jahn G. 2008. Cytomegalovirus cell tropism. Curr Top Microbiol Immunol 325:63–83. doi: 10.1007/978-3-540-77349-8_4. [DOI] [PubMed] [Google Scholar]
- 19.Grier JT, Forbes LR, Monaco-Shawver L, Oshinsky J, Atkinson TP, Moody C, Pandey R, Campbell KS, Orange JS. 2012. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J Clin Invest 122:3769–3780. doi: 10.1172/JCI64837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sainz B Jr, LaMarca HL, Garry RF, Morris CA. 2005. Synergistic inhibition of human cytomegalovirus replication by interferon-alpha/beta and interferon-gamma. Virol J 2:14. doi: 10.1186/1743-422X-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan N, Chen ZJ. 2012. Intrinsic antiviral immunity. Nat Immunol 13:214–222. doi: 10.1038/ni.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. 2011. Innate or adaptive immunity? The example of natural killer cells. Science 331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilkinson GW, Tomasec P, Stanton RJ, Armstrong M, Prod'homme V, Aicheler R, McSharry BP, Rickards CR, Cochrane D, Llewellyn-Lacey S, Wang EC, Griffin CA, Davison AJ. 2008. Modulation of natural killer cells by human cytomegalovirus. J Clin Virol 41:206–212. doi: 10.1016/j.jcv.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith W, Tomasec P, Aicheler R, Loewendorf A, Nemcovicova I, Wang EC, Stanton RJ, Macauley M, Norris P, Willen L, Ruckova E, Nomoto A, Schneider P, Hahn G, Zajonc DM, Ware CF, Wilkinson GW, Benedict CA. 2013. Human cytomegalovirus glycoprotein UL141 targets the TRAIL death receptors to thwart host innate antiviral defenses. Cell Host Microbe 13:324–335. doi: 10.1016/j.chom.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daeron M. 1997. Fc receptor biology. Annu Rev Immunol 15:203–234. doi: 10.1146/annurev.immunol.15.1.203. [DOI] [PubMed] [Google Scholar]
- 26.Ravetch JV, Perussia B. 1989. Alternative membrane forms of Fc gamma RIII(CD16) on human natural killer cells and neutrophils. Cell type-specific expression of two genes that differ in single nucleotide substitutions. J Exp Med 170:481–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. 2002. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 99:754–758. doi: 10.1182/blood.V99.3.754. [DOI] [PubMed] [Google Scholar]
- 28.Dargan DJ, Douglas E, Cunningham C, Jamieson F, Stanton RJ, Baluchova K, McSharry BP, Tomasec P, Emery VC, Percivalle E, Sarasini A, Gerna G, Wilkinson GW, Davison AJ. 2010. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J Gen Virol 91:1535–1546. doi: 10.1099/vir.0.018994-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stanton RJ, Baluchova K, Dargan DJ, Cunningham C, Sheehy O, Seirafian S, McSharry BP, Neale ML, Davies JA, Tomasec P, Davison AJ, Wilkinson GW. 2010. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J Clin Invest 120:3191–3208. doi: 10.1172/JCI42955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katze MG, He Y, Gale M Jr. 2002. Viruses and interferon: a fight for supremacy. Nat Rev Immunol 2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 31.Miller DM, Zhang Y, Rahill BM, Waldman WJ, Sedmak DD. 1999. Human cytomegalovirus inhibits IFN-alpha-stimulated antiviral and immunoregulatory responses by blocking multiple levels of IFN-alpha signal transduction. J Immunol 162:6107–6113. [PubMed] [Google Scholar]
- 32.Benedict CA, Banks TA, Senderowicz L, Ko M, Britt WJ, Angulo A, Ghazal P, Ware CF. 2001. Lymphotoxins and cytomegalovirus cooperatively induce interferon-beta, establishing host-virus detente. Immunity 15:617–626. doi: 10.1016/S1074-7613(01)00222-9. [DOI] [PubMed] [Google Scholar]
- 33.Iversen AC, Norris PS, Ware CF, Benedict CA. 2005. Human NK cells inhibit cytomegalovirus replication through a noncytolytic mechanism involving lymphotoxin-dependent induction of IFN-beta. J Immunol 175:7568–7574. doi: 10.4049/jimmunol.175.11.7568. [DOI] [PubMed] [Google Scholar]
- 34.Pak-Wittel MA, Yang L, Sojka DK, Rivenbark JG, Yokoyama WM. 2013. Interferon-gamma mediates chemokine-dependent recruitment of natural killer cells during viral infection. Proc Natl Acad Sci U S A 110:E50–E59. doi: 10.1073/pnas.1220456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnon TI, Achdout H, Levi O, Markel G, Saleh N, Katz G, Gazit R, Gonen-Gross T, Hanna J, Nahari E, Porgador A, Honigman A, Plachter B, Mevorach D, Wolf DG, Mandelboim O. 2005. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat Immunol 6:515–523. doi: 10.1038/ni1190. [DOI] [PubMed] [Google Scholar]
- 36.Fouts AE, Chan P, Stephan JP, Vandlen R, Feierbach B. 2012. Antibodies against the gH/gL/UL128/UL130/UL131 complex comprise the majority of the anti-cytomegalovirus (anti-CMV) neutralizing antibody response in CMV hyperimmune globulin. J Virol 86:7444–7447. doi: 10.1128/JVI.00467-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerna G, Sarasini A, Patrone M, Percivalle E, Fiorina L, Campanini G, Gallina A, Baldanti F, Revello MG. 2008. Human cytomegalovirus serum neutralizing antibodies block virus infection of endothelial/epithelial cells, but not fibroblasts, early during primary infection. J Gen Virol 89:853–865. doi: 10.1099/vir.0.83523-0. [DOI] [PubMed] [Google Scholar]
- 38.Frenzel K, Ganepola S, Michel D, Thiel E, Kruger DH, Uharek L, Hofmann J. 2012. Antiviral function and efficacy of polyvalent immunoglobulin products against CMV isolates in different human cell lines. Med Microbiol Immunol 201:277–286. doi: 10.1007/s00430-012-0229-2. [DOI] [PubMed] [Google Scholar]
- 39.Schnueriger A, Grau R, Sondermann P, Schreitmueller T, Marti S, Zocher M. 2011. Development of a quantitative, cell-line based assay to measure ADCC activity mediated by therapeutic antibodies. Mol Immunol 48:1512–1517. doi: 10.1016/j.molimm.2011.04.010. [DOI] [PubMed] [Google Scholar]