

ABSTRACT
Subacute sclerosing panencephalitis (SSPE) is caused by persistent measles virus (MV) infection in the central nervous system (CNS). Since human neurons, its main target cells, do not express known MV receptors (signaling lymphocyte activation molecule [SLAM] and nectin 4), it remains to be understood how MV infects and spreads in them. We have recently reported that fusion-enhancing substitutions in the extracellular domain of the MV fusion (F) protein (T461I and S103I/N462S/N465S), which are found in multiple SSPE virus isolates, promote MV spread in human neuroblastoma cell lines and brains of suckling hamsters. In this study, we show that hyperfusogenic viruses with these substitutions also spread efficiently in human primary neuron cultures without inducing syncytia. These substitutions were found to destabilize the prefusion conformation of the F protein trimer, thereby enhancing fusion activity. However, these hyperfusogenic viruses exhibited stronger cytopathology and produced lower titers at later time points in SLAM- or nectin 4-expressing cells compared to the wild-type MV. Although these viruses spread efficiently in the brains of SLAM knock-in mice, they did not in the spleens. Taken together, the results suggest that enhanced fusion activity is beneficial for MV to spread in neuronal cells where no cytopathology occurs, but detrimental to other types of cells due to strong cytopathology. Acquisition of enhanced fusion activity through substitutions in the extracellular domain of the F protein may be crucial for MV's extensive spread in the CNS and development of SSPE.
IMPORTANCE Subacute sclerosing panencephalitis (SSPE) is a fatal disease caused by persistent measles virus (MV) infection in the central nervous system (CNS). Its cause is not well understood, and no effective therapy is currently available. Recently, we have reported that enhanced fusion activity of MV through the mutations in its fusion protein is a major determinant of efficient virus spread in human neuronal cells and brains of suckling hamsters. In this study, we show that those mutations render the conformation of the fusion protein less stable, thereby making it hyperfusogenic. Our results also show that enhanced fusion activity is beneficial for MV to spread in the CNS but detrimental to other types of cells in peripheral tissues, which are strongly damaged by the virus. Our findings provide important insight into the mechanism for the development of SSPE after MV infection.
INTRODUCTION
Measles is a highly contagious disease characterized by high fever, cough, and maculopapular rash (1). The causative agent, measles virus (MV), is thought to first infect alveolar macrophages and dendritic cells of the respiratory tract, which transport the virus to local lymph nodes, where it replicates efficiently (2, 3). This allows MV to spread to other lymphoid organs throughout the body via the bloodstream, followed by infection of respiratory epithelia at later stages. Progeny viral particles are then shed from the epithelial cells through coughing and sneezing and transmitted to new hosts (4–8).
The lymphotropism and immunosuppressive nature of MV are explained by its use of signaling lymphocyte activation molecule (SLAM; also called CD150) expressed on immune cells as a cellular receptor (9–12). On the other hand, another receptor nectin 4 accounts for infection of epithelial cells (6, 7). In addition to these authentic receptors, vaccine and laboratory-adapted strains of MV can use CD46, a complement regulatory molecule, as an alternative receptor (13–15), through specific amino acid changes in its receptor-binding protein hemagglutinin (H) (16, 17). Furthermore, MV sometimes persists in the central nervous system (CNS) and causes a fatal degenerative disease, subacute sclerosing panencephalitis (SSPE), several years after acute measles (1, 18–20). In SSPE, MV appears to infect neuronal cells independently of SLAM and nectin 4, since neither SLAM nor nectin 4 is expressed at significant levels in the human CNS (21–23). Thus, how MV infects neuronal cells and causes the disease has not been well defined.
MV is an enveloped virus with a nonsegmented, negative-strand RNA genome and belongs to the genus Morbillivirus of the family Paramyxoviridae. The MV genome has six genes that encode the nucleocapsid, phospho- (P), matrix (M), fusion (F), H, and large proteins (1). The P gene encodes two additional proteins V and C. There are two envelope glycoproteins, the H and F proteins, which are responsible for receptor-binding and membrane fusion, respectively (1). The F protein forms homotrimer, and each monomer contains a hydrophobic fusion peptide (FP), two heptad repeat (HR) domains (HR-A and HR-B), a transmembrane domain, and a cytosolic tail. During virus entry into cells, binding of the H protein to a cellular receptor likely triggers a series of conformational changes of the F protein trimer. It was proposed that the MV F protein trimer in the prefusion conformation comprises large globular head domains supported by a short three-helical bundle (3-HB) stalk, based on the homology modeling with an X-ray crystal structure of the parainfluenza virus type 5 (PIV5) F trimer (24, 25). Upon triggering, the packed HR-A segment may refold to an extended structure after the 3-HB F stalk dissociates, leading to the exposure of the FP domain to the target cell membrane. As the two HRs (HR-A and HR-B) within a monomer interact with each other, the six-helix band (6-HB) structure is formed, leading to virus-to-cell fusion (24, 26). By the same mechanism, syncytium formation (cell-to-cell fusion) is induced in MV-infected cells expressing the H and F proteins (18). In fact, multinucleated giant cells are found pathologically in human cases with measles and in experimentally infected rhesus monkeys (18).
Amino acid sequences of the F protein are highly conserved among MV clinical isolates and vaccine strains (27, 28). However, we found that most SSPE strains possess amino acid substitutions in the extracellular domain of the F protein (29). Our study revealed that the substitutions found in multiple SSPE strains, T461I and S103I/N462S/N465S, confer enhanced fusion activity on the F protein. Furthermore, recombinant viruses with these fusion-enhancing mutations induced syncytium formation even in SLAM- and nectin 4-negative cells, including human neuroblastoma cell lines and Vero cells, and exhibited neurovirulence in suckling hamsters, unlike the parental wild-type MV (29).
In the present study, we examined the growth and spread of these hyperfusogenic recombinant MVs in neuronal and non-neuronal cells, using human cell lines and primary neuron cultures, as well as SLAM knock-in mice (30) inoculated intracerebrally or intraperitoneally. Our results revealed that the fusion-enhancing mutations in the F protein confer on MV growth advantage in neuronal cells but not in other types of cells. To understand why the growth of these viruses is reduced in non-neuronal cells, their cytopathogenicity was also examined.
MATERIALS AND METHODS
Cells.
Vero/hSLAM (10) cells were maintained in Dulbecco minimum essential medium (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS). NCI-H358 cells were maintained in RPMI 1640 medium (Wako Pure Chemical Industry, Osaka, Japan) supplemented with 10% FBS.
Plasmids.
The plasmids pCA7-ICH and pCA7-ICF that encode the H and F proteins of the pathogenic IC-B strain of MV, respectively, were established previously (17), using the eukaryotic expression vector pCA7 (31), a derivative of pCAGGS (32). The expression plasmids which express mutant F protein, pCA7-ICF(T461I) and pCA7-ICF(S103I/N462S/N465S), were described previously (29). The full-length genome plasmids pHHRz(+)MV323-Luci and pHHRz(+)MV323-F(T461I)-Luci were, respectively, generated from pHHRz(+)MV323-EGFP and pHHRz(+)MV323-F(T461I)-EGFP (29, 33), which encode the antigenomic full-length cDNA of the IC-B strain with the wild-type or mutant F protein (T461I), together with the enhanced green fluorescent protein (EGFP). The Renilla luciferase gene was amplified from pRL-TK by the PCR using the primer pair 5′-AGGCGCGCCATGACTTCGAAAGTTTATGATCCAG-3′ and 5′-ATGACGTCTTATTGTTCATTTTTGAGAACTCGC-3′. After digestion with AscI and AatII followed by ligation, the EGFP gene of pHHRz(+)MV323-EGFP or pHHRz(+)MV323-F(T461I)-EGFP was replaced with the amplified PCR product, producing pHHRz(+)MV323-Luci or pHHRz(+)MV323-F(T461I)-Luci, respectively.
Viruses.
The recombinant viruses IC323-F(T461I)-EGFP and IC323-F(S103I/N462S/N465S)-EGFP had been generated based on the wild-type MV expressing EGFP, IC323-EGFP, as reported previously (29, 33, 34). The recombinant MV expressing the Renilla luciferase, IC323-Luci or IC323-F(T461I)-Luci, was generated with plasmid pHHRz(+)MV323-Luci or pHHRz(+)MV323-F(T461I)-Luci, respectively, using reverse genetics as reported previously (33). The generated MVs were propagated in Vero/hSLAM cells. Titers of each recombinant virus were determined as PFU in Vero/hSLAM cells.
Infection of human primary neurons with recombinant viruses.
Human primary neurons prepared from human brains were obtained from Sciencell Research Laboratories (ScienCell, San Diego, CA) and seeded on the 24-well cluster plates pretreated with 0.2% gelatin (ScienCell). Primary neurons were maintained in neuronal medium (ScienCell) supplemented with neuronal growth supplement (ScienCell) according to the manufacturer's instructions. The majority of the cells were determined to be positive for the neurofilament, microtubule-associated protein 2, and β-tubulin III by immunofluorescence staining. Primary neurons were infected with recombinant MVs at an multiplicity of infection (MOI) of 0.1.
LDH release assay.
A lactate dehydrogenase (LDH) release assay was performed using CytoTox 96 nonradioactive cytotoxicity assay kit (Promega) in accordance with the manufacturer's instructions. Fifty microliters of the culture supernatant from virus-infected Vero/hSLAM cells was used for the assay. The data were expressed as percentages of the maximum LDH release, and the maximum LDH release was determined with the culture supernatant from mock-infected cells after freezing and thawing.
Plasmid-mediated fusion assay in Vero/hSLAM cells.
Vero/hSLAM cells cultured in 12-well cluster plate were transfected with pCA7-ICH (0.2 μg) plus pCA7 encoding wild-type or each mutant F protein (1.5 μg), using Polyethylenimine-Max (Polysciences, Inc., Warrington, PA). One or two days after transfection, the cells were observed under a light microscope after Giemsa staining.
Infection of the mouse model with recombinant viruses.
The type I IFN receptor subunit 1 (IFNAR)−/− SLAM knock-in mice were established previously (30). All mice used for the experiment were 6 to 10 weeks of age and kept under specific-pathogen-free conditions. For intracerebral infection, four mice were anesthetized using a combination anesthetic (0.3 mg/kg of medetomidine, 4.0 mg/kg of midazolam, and 5.0 mg/kg of butorphanol), and 104 PFU of IC323-F(T461I)-EGFP, IC323-F(S103I/N462S/N465S)-EGFP, or IC323-EGFP was inoculated into the right or left hemisphere of their brains. At 6 days postinfection (p.i.), all mice were euthanized under anesthesia, and EGFP autofluorescence of the collected brains was observed under a fluorescent stereomicroscope. For intraperitoneal infection, eight mice were anesthetized with sevoflurane and infected with 1.5 × 105 PFU of the recombinant MVs [IC323-Luci or IC323-F(T461I)-Luci]. At 7 days p.i., Renilla luciferase activities in spleens of infected mice were measured using the Renilla luciferase assay system (Promega). All animal experiments were reviewed by the Institutional Committee of Ethics on Animal Experiments and carried out according to the Guidelines for Animal Experiments of the Faculty of Medicine, Kyushu University, Japan.
RESULTS
Spread of hyperfusogenic viruses in human primary neuron cultures.
The EGFP-expressing recombinant MVs with the T461I or S103I/N462S/N465S substitution in the F protein [IC323-F(T461I)-EGFP and IC323-F(S103I/N462S/N465S)-EGFP] had been established previously, which were based on the wild-type IC-B strain (29, 34). Since these recombinant viruses with enhanced fusion activity were found to spread efficiently in human neuroblastoma cell lines lacking SLAM and nectin 4 and in the brains of suckling hamsters, unlike the parental virus IC323-EGFP (29), we next examined whether they also spread in human primary neuron cultures. Primary neurons were infected at an MOI of 0.1 with these hyperfusogenic recombinant viruses or IC323-EGFP, and EGFP fluorescence in infected cells was observed daily for a month. At 24 h p.i., small numbers of singly infected cells were observed after infection with any of the three viruses (Fig. 1). Whereas IC323-EGFP never spread beyond originally infected cells, both hyperfusogenic viruses spread directly to surrounding cells or to remote cells via extended axon-like structures up to 4 days p.i. (Fig. 1). Although cell-to-cell spread was evident, no apparent syncytium formation was observed in primary neurons infected with these hyperfusogenic viruses. Infected EGFP-positive cells survived for a month without apparent further spread of EGFP after 4 days p.i., while solitary EGFP-positive cells infected with IC323-EGFP also remained alive and continued to express EGFP for a month (data not shown).
FIG 1.

Spread of the recombinant viruses in human primary neuron culture. Human primary neurons were infected with the parental recombinant MV expressing EGFP (IC323-EGFP) or its mutants expressing the mutant F protein containing the indicated substitutions at an MOI of 0.1. Primary neurons tended to grow in clusters, and these clusters were connected by extended axon-like structures. Panels show representative images obtained with a phase-contrast and a fluorescence microscope 1 day (upper) or 4 days (lower) after infection. White arrowheads show singly infected cells. Scale bar, 200 μm.
Stability of the prefusion F protein trimer.
Why do these viruses that exhibit enhanced fusion activity in SLAM- or nectin 4-expressing cells, cause fusion in cells lacking either receptor molecule (29) and spread in human primary neuron cultures? In our previous study, we excluded the increase in the surface expression of the F protein as the cause of enhanced fusion activity (29). Then, we reasoned that the mutant F proteins are more easily triggered by the H protein upon receptor-binding, compared to the wild-type F protein. To examine the conformational stability of the MV prefusion F trimer, a plasmid-mediated fusion assay was performed in Vero/hSLAM cells at different temperatures (Fig. 2). At 37°C the F proteins with the T461I or S103I/N462S/N465S substitution, in combination with the wild-type H protein, induced larger syncytia than the wild-type F protein, a finding consistent with our previous study (29). Notably, the wild-type F protein did not induce syncytium formation at 25°C, presumably because the full conformational changes of the F protein require higher temperatures to overcome the activation energy barrier (35). In contrast, the mutant F proteins formed distinct syncytia even at 25°C. The results suggest that the substitutions T461I and S103I/N462S/N465S destabilize the prefusion F trimer, thereby allowing its triggering at the lower temperature and promoting its fusion activity.
FIG 2.
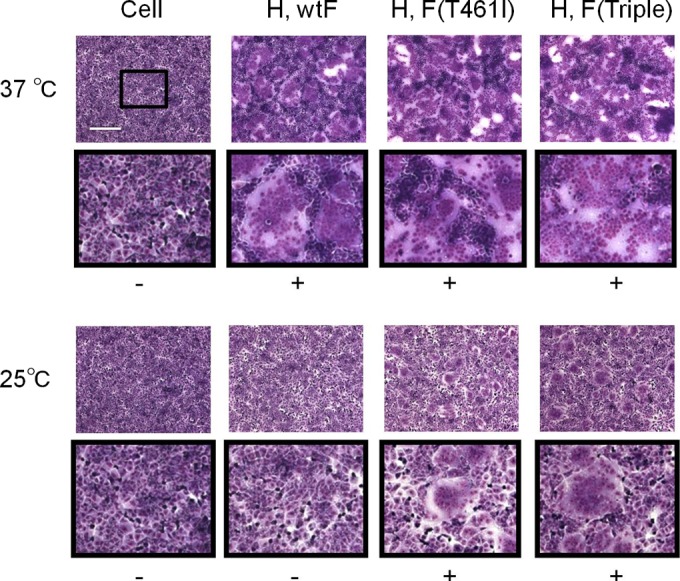
Syncytium formation assay at different temperatures. Vero/hSLAM cells were transfected with expression plasmids encoding the wild-type (wt) or mutant F protein with T461I or triple substitutions (S103I/N462S/N465S), together with that encoding the wt H protein. The cells were incubated at 37°C or 25°C. At 24 h (37°C) or 48 h (25°C) posttransfection, the cells were stained with a Giemsa solution and observed under a light microscope. Scale bar, 200 μm. To show the syncytia better, the central rectangle area enclosed in the upper panel is shown below in the lower panel after 3-fold enlargement. –, No syncytium formation; +, syncytium formation.
We also performed the fusion assay in Vero cells expressing neither SLAM nor nectin 4. The F proteins with the T461I or S103I/N462S/N465S substitution, but not the wild-type F protein, induced cell-cell fusion at 37°C, in conjunction with the wild-type H protein (29). The wild-type F protein did not induce syncytium formation even at 50°C, nor did the mutant F proteins exhibit higher fusion activity at 50°C than at 37°C (data not shown).
Growth and cytopathogenicity of hyperfusogenic viruses in non-neuronal cells.
Although these hyperfusogenic viruses appear to have a growth advantage over the wild-type MV, amino acid sequences of the F protein are highly conserved among clinical isolates, and viruses with fusion-enhancing mutations have been only isolated from SSPE patients. To understand this paradox, we examined the growth curves of these mutant viruses and wild-type MV in Vero/hSLAM cells infected at an MOI of 0.01. IC323-F(T461I)-EGFP and IC323-F(S103I/N462S/N465S)-EGFP produced severalfold higher titers at 24 h p.i. and comparable titers at 48 h p.i. compared to IC323-EGFP (Fig. 3A). However, titers of these hyperfusogenic viruses rapidly declined after 48 h p.i., whereas IC323-EGFP continued to grow and maintained high titers up to 96 h p.i. Since virus-induced membrane fusion can cause cell death (36), we next evaluated the cytopathogenicity of these hyperfusogenic viruses by quantifying LDH release in culture supernatants from virus-infected Vero/hSLAM cells. Both hyperfusogenic viruses exhibited stronger cytotoxicity at 72 h p.i. than IC323-EGFP (Fig. 3B). Moreover, IC323-F(S103I/N462S/N465S)-EGFP induced strong cytotoxicity even at 48 h p.i. when the wild-type MV hardly caused cell death. These results suggest that hyperfusogenic viruses induce stronger cytopathology and exhibit eventual growth disadvantage in cultured cells. IC323-F(S103I/N462S/N465S)-EGFP appears to display higher cytotoxicity than IC323-F(T461I)-EGFP. In fact, we found that a fusion inhibitory peptide could inhibit the cell-cell fusion induced by the latter virus, but not by the former (data not shown). Thus, the two mutant F proteins appear to have somewhat different properties. We also examined viral growth in nectin 4-expressing NCI-H358 cells. The hyperfusogenic viruses grew comparably to IC3232-EGFP until 24 h p.i., but titers of these viruses were lower thereafter compared to IC3232-EGFP (Fig. 3C).
FIG 3.

Growth and cytotoxicity of the hyperfusogenic viruses in cultured cell lines. (A) Growth kinetics of recombinant viruses in Vero/hSLAM cells. Vero/hSLAM cells were infected with IC323-EGFP, IC323-F(T461I)-EGFP, or IC323-F(S103I/N462S/N465S)-EGFP) at an MOI of 0.01. At various time points postinfection, viruses were harvested from cells and supernatants, and their PFU counts were determined. The averages and standard deviations (SD) of the titers of three experiments are shown. (B) Cytotoxicity assay for Vero/hSLAM cells infected with the recombinant viruses. Cells were infected with IC323-EGFP, IC323-F(T461I)-EGFP, or IC323-F(S103I/N462S/N465S)-EGFP, and the cytotoxicity was measured at various time points postinfection by measuring the LDH release into the culture medium. The data are expressed as a percentage of maximum LDH release. The averages and SD of three experiments are shown. (C) Growth kinetics of recombinant viruses in NCI-H358 cells examined as in panel A.
Spread of hyperfusogenic viruses in SLAM knock-in mice.
We previously demonstrated that IC323-F(T461I)-EGFP and IC323-F(S103I/N462S/N465S)-EGFP spread widely in the brains of suckling hamsters and killed them ∼1 week after intracerebral inoculation, while hamsters inoculated with the wild-type MV did not show any symptom and survived (29). To examine growth and spread of MV under a more physiological condition where receptor-expressing cells are present, we used SLAM knock-in mice crossed with mice lacking the type I IFN receptor, which reproduce lymphotropism and immunosuppression in human MV infection (30, 37). Three groups of four IFNAR−/− SLAM knock-in mice were, respectively, inoculated intracerebrally with 104 PFU/mice of IC323-EGFP, IC323-F(T461I)-EGFP, or IC323-F(S103I/N462S/N465S)-EGFP. At 6 days p.i., all mice were euthanized for microscopic observation, although they had not exhibited any symptoms. Although no EGFP was detected in the brains of mice inoculated with IC323-EGFP, hyperfusogenic viruses were found to spread in the brains of all infected mice [IC323-F(S103I/N462S/N465S)-EGFP did more efficiently than IC323-F(T461I)-EGFP] (Fig. 4A). To quantitate growth of MV in peripheral tissues, the Renilla luciferase-expressing recombinant MVs with the wild-type or mutant F protein were generated by using reverse genetics [IC323-Luci and IC323-F(T461I)-Luci, respectively]. An aliquot containing 1.5 × 105 PFU of IC323-Luci or IC323-F(T461I)-Luci was injected into the peritoneal cavity of each IFNAR−/− SLAM knock-in mouse (8 mice for each virus). At 7 days p.i., the spleens were collected, homogenized, and subjected to the quantification for their Renilla luciferase activities. In our previous studies with IFNAR−/− SLAM knock-in mouse (30, 38), MV was found to grow well in the spleens where many SLAM-positive cells are present. The luciferase activities in the spleens of mice infected with IC323-F(T461I)-Luci were nearly at the detection limit and significantly lower than those with IC323-Luci (Fig. 4B).
FIG 4.
Spread of hyperfusogenic viruses in the brains and spleens of SLAM knock-in mice. (A) Spread of EGFP-expressing viruses in the brains of infected IFNAR−/− SLAM knock-in mice. Four mice were infected with 104 PFU of IC323-EGFP, IC323-F(T461I)-EGFP, or IC323-F(S103I/N462S/N465S)-EGFP (Triple), intracerebrally. At 6 days p.i., the brains of euthanized mice were observed under a fluorescent stereomicroscope. Light and EGFP images of the brains were photographed, and top and sagittal views are shown. (B) Eight IFNAR−/− SLAM knock-in mice were inoculated intraperitoneally with 1.5 × 105 PFU of IC323-Luci or IC323-F(T461I)-Luci. At 7 days p.i., the spleens were removed and analyzed for their Renilla luciferase activities. Each circular symbol indicates the sample from a single mouse; bars indicate the mean values, and asterisks indicate statistically significant difference (P < 0.05). RLU, relative light unit.
DISCUSSION
We have demonstrated that substitutions T461I and S103I/N462S/N465S in the extracellular domain of the F protein found in multiple SSPE strains conferred enhanced fusion activity on the viruses (29) and that these substitutions promoted MV spread in human neuroblastoma cell lines and brains of suckling hamsters (29), as well as in human primary neuron cultures (the present study). However, the growth of these hyperfusogenic viruses rapidly declined at later time points in SLAM-positive Vero/hSLAM, and A549/hSLAM cells and in nectin 4-positive NCI-H358 cells, unlike that of the wild-type virus (the present study and data not shown). Furthermore, virus load was significantly lower in the spleens of IFNAR−/− SLAM knock-in mice inoculated intraperitoneally with a hyperfusogenic virus than that with the parental virus, although the former virus, but not the latter, spread in the brains after intracerebral inoculation. All of these findings suggest that hyperfusogenic viruses spread efficiently in neuronal cells, but their growth is reduced in SLAM- or nectin 4-expressing cell lines, as well as in SLAM-positive peripheral tissues in vivo.
The enhanced cytopathology, as revealed by higher levels of LDH release, likely explains the observed lower titers of hyperfusogenic viruses in cultured cells at later time points, as well as in the spleens of IFNAR−/− SLAM knock-in mice inoculated intraperitoneally. It was reported that another hyperfusogenic MV mutant also induced strong cytopathology in infected cells, resulting in inefficient growth (39). Furthermore, hyperfusogenic mutants of other paramyxoviruses were found to exhibit enhanced cytopathogenicity (40, 41) and show decreased growth (41). These hyperfusogenic mutants of paramyxoviruses and other fusogenic viral proteins were suggested to be useful for cancer therapy due to their strong cytopathogenicity (36, 39–41).
Demyelination and infection of neurons are characteristic features in the brains of patients with SSPE (42, 43). Although small numbers of astrocytes, endothelial cells, and oligodendrocytes have been found to be infected, neurons are thought to be the main target for MV in the CNS (42–45). However, neuronal cells do not express MV receptors SLAM and nectin 4. Indeed, MV entry into neuronal cells is inefficient, since the wild-type MV infected human neuronal cell lines and primary cultured neurons only rarely even at high MOIs (29; the present study). On the other hand, hyperfusogenic viruses spread efficiently in neuronal cells, although they exhibited reduced levels of growth in SLAM- or nectin 4-positive cells largely because of their strong cytopathogenicity. These results indicate that enhanced fusion activity allows MV to spread efficiently in neuronal cells independently of SLAM and nectin 4. Importantly, syncytia were not visible in primary neurons infected with hyperfusogenic viruses, a finding consistent with clinical observations that syncytia are not present in the brains of SSPE patients (43). Furthermore, syncytium formation was not apparent in the brains of suckling hamsters after intracerebral inoculation with these hyperfusogenic viruses, even though the viruses spread to the whole cerebrum (29). The cell-cell contacts between neurons may be limited to small areas such as synapses and mostly hindered by other supporting cells and myelinated nerve fibers in the brain. This spatial arrangement may be a reason why syncytia are not induced in MV-infected brains or primary cultured neurons even if the virus spread extensively. Primary neurons used in the present study tended to grow in clusters, and these clusters were loosely connected to each other by extended axon-like structures. It is likely that MV efficiently spread to other cells mainly within the same clusters through the cell-cell fusion (without apparent syncytium formation) for a few days but failed to spread thereafter due to a paucity of nearby contacting cells. Furthermore, the absence of apparent syncytia may explain the survival of virus-infected primary neurons for a month.
Avila et al. have recently reported that substitutions in a central pocket domain of the globular head of the F protein of canine distemper virus, a morbillivirus closely related to MV, decreased the stability of its prefusion state and enhanced its fusion activity (46). These researchers found, however, that the substitution of a corresponding residue in the MV F protein adversely affected its fusion activity. We confirmed that the plasmid-mediated expression of the MV H protein and the wild-type F protein did not induce syncytia at 25°C, unlike that at 37°C. In contrast, the F protein with the T461I or S103I/N462S/N465S substitution, when expressed with the H protein, caused syncytium formation even at 25°C. We found that the MV F proteins containing the single amino acid substitution (M94V, S262R, L354M, and N462K) that exhibit enhanced fusion activity (29) also induced syncytia at 25°C (data not shown). The results indicate that these fusion-enhancing substitutions in the extracellular domain of the F protein decrease the conformational stability of the prefusion F protein trimer. The decreased stability likely allows the F protein to be more easily triggered by the H protein upon its receptor-binding. Thus, whereas the wild-type F protein is only triggered by authentic receptors (SLAM and nectin 4) at 37°C, the F proteins with the T461I or S103I/N462S/N465S substitution may also be triggered by unknown molecules other than SLAM and nectin 4. Furthermore, these F mutants can be triggered by the authentic receptors at lower temperatures.
The mutations in the M gene are thought to play a central role in SSPE pathogenesis (43, 47–52). These mutations lead to the lack of virus particle formation, and may allow the virus to escape from host immune responses, resulting in establishment of persistent infection in the CNS (47, 53). In addition, these characteristic mutations in the M protein had been thought to be important for MV spread in the CNS (53–55). However, we previously demonstrated that the mutations in the M protein or in the cytoplasmic domain of the F protein per se were not sufficient for MV spread in neuronal cells lacking SLAM and nectin 4 (29). On the other hand, substitutions in the extracellular domain of the F protein, T461I and S103I/N462S/N465S, promoted MV spread in neuronal cells. There are 15 complete F gene sequences from SSPE strains available in the NCBI database (33, 47, 49, 56–60). All of these sequences contain the alteration in the cytoplasmic domain of the F protein (48–50). Notably, five of them also contain the substitution T461I (47, 49, 58), while two of them contain the substitution S103I/N462S/N465S (56, 60). Furthermore, other two strains contain the substitution M94V (49, 57), which is known to enhance fusion activity (61). However, the substitution M94V by itself was not sufficient for killing suckling hamsters probably because of its moderately enhanced fusion activity (29). Thus, many of the SSPE strains indeed have fusion-enhancing mutations in the extracellular domain of the F protein, in addition to the alteration in its cytoplasmic domain. From our findings, we propose that MV comes to exhibit neuropathogenicity and cause SSPE by acquiring fusion-enhancing mutations in the extracellular domain of the F protein (Fig. 5).
FIG 5.

Model for the role of mutations in the extracellular domain of the F protein in MV growth and spread in the CNS and peripheral organs.
ACKNOWLEDGMENTS
We thank S. Ikegame and M. Ito for providing technical information.
This study was supported by grants from the Ministry of Health, Labor, and Welfare (the Research Committee of Prion Disease and Slow Virus Infection) of Japan, and by JSPS Kakenhi grants 21249032, 24115005, and 24790444.
REFERENCES
- 1.Griffin DE. 2007. Measles virus, p 1551–1585 In Knipe DM, Howley PM (ed), Fields virology, 5th ed Lippincott/The Williams & Wilkins Co, Philadelphia, PA. [Google Scholar]
- 2.Ferreira CS, Frenzke M, Leonard VH, Welstead GG, Richardson CD, Cattaneo R. 2010. Measles virus infection of alveolar macrophages and dendritic cells precedes spread to lymphatic organs in transgenic mice expressing human signaling lymphocytic activation molecule (SLAM, CD150). J Virol 84:3033–3042. doi: 10.1128/JVI.01559-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lemon K, de Vries RD, Mesman AW, McQuaid S, van Amerongen G, Yuksel S, Ludlow M, Rennick LJ, Kuiken T, Rima BK, Geijtenbeek TB, Osterhaus AD, Duprex WP, de Swart RL. 2011. Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog 7:e1001263. doi: 10.1371/journal.ppat.1001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frenzke M, Sawatsky B, Wong XX, Delpeut S, Mateo M, Cattaneo R, von Messling V. 2013. Nectin-4-dependent measles virus spread to the cynomolgus monkey tracheal epithelium: role of infected immune cells infiltrating the lamina propria. J Virol 87:2526–2534. doi: 10.1128/JVI.03037-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ludlow M, Lemon K, de Vries RD, McQuaid S, Millar EL, van Amerongen G, Yuksel S, Verburgh RJ, Osterhaus AD, de Swart RL, Duprex WP. 2013. Measles virus infection of epithelial cells in the macaque upper respiratory tract is mediated by subepithelial immune cells. J Virol 87:4033–4042. doi: 10.1128/JVI.03258-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muhlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB, Cichutek K, von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480:530–533. doi: 10.1038/nature10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. 2011. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 7:e1002240. doi: 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leonard VH, Sinn PL, Hodge G, Miest T, Devaux P, Oezguen N, Braun W, McCray PB Jr, McChesney MB, Cattaneo R. 2008. Measles virus blind to its epithelial cell receptor remains virulent in rhesus monkeys but cannot cross the airway epithelium and is not shed. J Clin Invest 118:2448–2458. doi: 10.1172/JCI35454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 10.Ono N, Tatsuo H, Hidaka Y, Aoki T, Minagawa H, Yanagi Y. 2001. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J Virol 75:4399–4401. doi: 10.1128/JVI.75.9.4399-4401.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erlenhoefer C, Wurzer WJ, Loffler S, Schneider-Schaulies S, ter Meulen V, Schneider-Schaulies J. 2001. CD150 (SLAM) is a receptor for measles virus but is not involved in viral contact-mediated proliferation inhibition. J Virol 75:4499–4505. doi: 10.1128/JVI.75.10.4499-4505.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu EC, Iorio C, Sarangi F, Khine AA, Richardson CD. 2001. CDw150(SLAM) is a receptor for a lymphotropic strain of measles virus and may account for the immunosuppressive properties of this virus. Virology 279:9–21. [DOI] [PubMed] [Google Scholar]
- 13.Manchester M, Liszewski MK, Atkinson JP, Oldstone MB. 1994. Multiple isoforms of CD46 (membrane cofactor protein) serve as receptors for measles virus. Proc Natl Acad Sci U S A 91:2161–2165. doi: 10.1073/pnas.91.6.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naniche D, Varior-Krishnan G, Cervoni F, Wild TF, Rossi B, Rabourdin-Combe C, Gerlier D. 1993. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J Virol 67:6025–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dorig RE, Marcil A, Chopra A, Richardson CD. 1993. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 75:295–305. doi: 10.1016/0092-8674(93)80071-L. [DOI] [PubMed] [Google Scholar]
- 16.Masse N, Barrett T, Muller CP, Wild TF, Buckland R. 2002. Identification of a second major site for CD46 binding in the hemagglutinin protein from a laboratory strain of measles virus (MV): potential consequences for wild-type MV infection. J Virol 76:13034–13038. doi: 10.1128/JVI.76.24.13034-13038.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tahara M, Takeda M, Seki F, Hashiguchi T, Yanagi Y. 2007. Multiple amino acid substitutions in hemagglutinin are necessary for wild-type measles virus to acquire the ability to use receptor CD46 efficiently. J Virol 81:2564–2572. doi: 10.1128/JVI.02449-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rima BK, Duprex WP. 2006. Morbilliviruses and human disease. J Pathol 208:199–214. doi: 10.1002/path.1873. [DOI] [PubMed] [Google Scholar]
- 19.Bellini WJ, Rota JS, Lowe LE, Katz RS, Dyken PR, Zaki SR, Shieh WJ, Rota PA. 2005. Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than was previously recognized. J Infect Dis 192:1686–1693. doi: 10.1086/497169. [DOI] [PubMed] [Google Scholar]
- 20.Gutierrez J, Issacson RS, Koppel BS. 2010. Subacute sclerosing panencephalitis: an update. Dev Med Child Neurol 52:901–907. doi: 10.1111/j.1469-8749.2010.03717.x. [DOI] [PubMed] [Google Scholar]
- 21.Brancati F, Fortugno P, Bottillo I, Lopez M, Josselin E, Boudghene-Stambouli O, Agolini E, Bernardini L, Bellacchio E, Iannicelli M, Rossi A, Dib-Lachachi A, Stuppia L, Palka G, Mundlos S, Stricker S, Kornak U, Zambruno G, Dallapiccola B. 2010. Mutations in PVRL4, encoding cell adhesion molecule nectin-4, cause ectodermal dysplasia-syndactyly syndrome. Am J Hum Genet 87:265–273. doi: 10.1016/j.ajhg.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McQuaid S, Cosby SL. 2002. An immunohistochemical study of the distribution of the measles virus receptors, CD46 and SLAM, in normal human tissues and subacute sclerosing panencephalitis. Lab Invest 82:403–409. doi: 10.1038/labinvest.3780434. [DOI] [PubMed] [Google Scholar]
- 23.Reymond N, Fabre S, Lecocq E, Adelaide J, Dubreuil P, Lopez M. 2001. Nectin4/PRR4, a new afadin-associated member of the nectin family that trans-interacts with nectin1/PRR1 through V domain interaction. J Biol Chem 276:43205–43215. doi: 10.1074/jbc.M103810200. [DOI] [PubMed] [Google Scholar]
- 24.Prussia AJ, Plemper RK, Snyder JP. 2008. Measles virus entry inhibitors: a structural proposal for mechanism of action and the development of resistance. Biochemistry 47:13573–13583. doi: 10.1021/bi801513p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plemper RK, Brindley MA, Iorio RM. 2011. Structural and mechanistic studies of measles virus illuminate paramyxovirus entry. PLoS Pathog 7:e1002058. doi: 10.1371/journal.ppat.1002058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rota JS, Hummel KB, Rota PA, Bellini WJ. 1992. Genetic variability of the glycoprotein genes of current wild-type measles isolates. Virology 188:135–142. doi: 10.1016/0042-6822(92)90742-8. [DOI] [PubMed] [Google Scholar]
- 28.Rota JS, Wang ZD, Rota PA, Bellini WJ. 1994. Comparison of sequences of the H, F, and N coding genes of measles virus vaccine strains. Virus Res 31:317–330. doi: 10.1016/0168-1702(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe S, Shirogane Y, Suzuki SO, Ikegame S, Koga R, Yanagi Y. 2013. Mutant fusion proteins with enhanced fusion activity promote measles virus spread in human neuronal cells and brains of suckling hamsters. J Virol 87:2648–2659. doi: 10.1128/JVI.02632-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohno S, Ono N, Seki F, Takeda M, Kura S, Tsuzuki T, Yanagi Y. 2007. Measles virus infection of SLAM (CD150) knock-in mice reproduces tropism and immunosuppression in human infection. J Virol 81:1650–1659. doi: 10.1128/JVI.02134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takeda M, Ohno S, Seki F, Nakatsu Y, Tahara M, Yanagi Y. 2005. Long untranslated regions of the measles virus M and F genes control virus replication and cytopathogenicity. J Virol 79:14346–14354. doi: 10.1128/JVI.79.22.14346-14354.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- 33.Seki F, Yamada K, Nakatsu Y, Okamura K, Yanagi Y, Nakayama T, Komase K, Takeda M. 2011. The si strain of measles virus derived from a patient with subacute sclerosing panencephalitis possesses typical genome alterations and unique amino Acid changes that modulate receptor specificity and reduce membrane fusion activity. J Virol 85:11871–11882. doi: 10.1128/JVI.05067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hashimoto K, Ono N, Tatsuo H, Minagawa H, Takeda M, Takeuchi K, Yanagi Y. 2002. SLAM (CD150)-independent measles virus entry as revealed by recombinant virus expressing green fluorescent protein. J Virol 76:6743–6749. doi: 10.1128/JVI.76.13.6743-6749.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harrison SC. 2008. Viral membrane fusion. Nat Struct Mol Biol 15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Higuchi H, Bronk SF, Bateman A, Harrington K, Vile RG, Gores GJ. 2000. Viral fusogenic membrane glycoprotein expression causes syncytia formation with bioenergetic cell death: implications for gene therapy. Cancer Res 60:6396–6402. [PubMed] [Google Scholar]
- 37.Koga R, Ohno S, Ikegame S, Yanagi Y. 2010. Measles virus-induced immunosuppression in SLAM knock-in mice. J Virol 84:5360–5367. doi: 10.1128/JVI.02525-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takeda M, Ohno S, Tahara M, Takeuchi H, Shirogane Y, Ohmura H, Nakamura T, Yanagi Y. 2008. Measles viruses possessing the polymerase protein genes of the Edmonton vaccine strain exhibit attenuated gene expression and growth in cultured cells and SLAM knock-in mice. J Virol 82:11979–11984. doi: 10.1128/JVI.00867-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heidmeier S, Hanauer JR, Friedrich K, Prufer S, Schneider IC, Buchholz CJ, Cichutek K, Muhlebach MD. 2014. A single amino acid substitution in the measles virus F(2) protein reciprocally modulates membrane fusion activity in pathogenic and oncolytic strains. Virus Res 180:43–48. doi: 10.1016/j.virusres.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 40.Altomonte J, Marozin S, Schmid RM, Ebert O. 2010. Engineered Newcastle disease virus as an improved oncolytic agent against hepatocellular carcinoma. Mol Ther 18:275–284. doi: 10.1038/mt.2009.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gainey MD, Manuse MJ, Parks GD. 2008. A hyperfusogenic F protein enhances the oncolytic potency of a paramyxovirus simian virus 5 P/V mutant without compromising sensitivity to type I interferon. J Virol 82:9369–9380. doi: 10.1128/JVI.01054-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rima BK, Duprex WP. 2005. Molecular mechanisms of measles virus persistence. Virus Res 111:132–147. doi: 10.1016/j.virusres.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 43.Schneider-Schaulies J, Meulen V, Schneider-Schaulies S. 2003. Measles infection of the central nervous system. J Neurovirol 9:247–252. doi: 10.1080/13550280390193993, doi:. [DOI] [PubMed] [Google Scholar]
- 44.Allen IV, McQuaid S, McMahon J, Kirk J, McConnell R. 1996. The significance of measles virus antigen and genome distribution in the CNS in SSPE for mechanisms of viral spread and demyelination. J Neuropathol Exp Neurol 55:471–480. doi: 10.1097/00005072-199604000-00010. [DOI] [PubMed] [Google Scholar]
- 45.Kirk J, Zhou AL, McQuaid S, Cosby SL, Allen IV. 1991. Cerebral endothelial cell infection by measles virus in subacute sclerosing panencephalitis: ultrastructural and in situ hybridization evidence. Neuropathol Appl Neurobiol 17:289–297. doi: 10.1111/j.1365-2990.1991.tb00726.x. [DOI] [PubMed] [Google Scholar]
- 46.Avila M, Alves L, Khosravi M, Ader-Ebert N, Origgi F, Schneider-Schaulies J, Zurbriggen A, Plemper RK, Plattet P. 2014. Molecular determinants defining the triggering range of prefusion F complexes of canine distemper virus. J Virol 88:2951–2966. doi: 10.1128/JVI.03123-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cattaneo R, Schmid A, Eschle D, Baczko K, ter Meulen V, Billeter MA. 1988. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell 55:255–265. doi: 10.1016/0092-8674(88)90048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Billeter MA, Cattaneo R, Spielhofer P, Kaelin K, Huber M, Schmid A, Baczko K, ter Meulen V. 1994. Generation and properties of measles virus mutations typically associated with subacute sclerosing panencephalitis. Ann N Y Acad Sci 724:367–377. doi: 10.1111/j.1749-6632.1994.tb38934.x. [DOI] [PubMed] [Google Scholar]
- 49.Ning X, Ayata M, Kimura M, Komase K, Furukawa K, Seto T, Ito N, Shingai M, Matsunaga I, Yamano T, Ogura H. 2002. Alterations and diversity in the cytoplasmic tail of the fusion protein of subacute sclerosing panencephalitis virus strains isolated in Osaka, Japan. Virus Res 86:123–131. doi: 10.1016/S0168-1702(02)00042-4. [DOI] [PubMed] [Google Scholar]
- 50.Schmid A, Spielhofer P, Cattaneo R, Baczko K, ter Meulen V, Billeter MA. 1992. Subacute sclerosing panencephalitis is typically characterized by alterations in the fusion protein cytoplasmic domain of the persisting measles virus. Virology 188:910–915. doi: 10.1016/0042-6822(92)90552-Z. [DOI] [PubMed] [Google Scholar]
- 51.Ayata M, Komase K, Shingai M, Matsunaga I, Katayama Y, Ogura H. 2002. Mutations affecting transcriptional termination in the p gene end of subacute sclerosing panencephalitis viruses. J Virol 76:13062–13068. doi: 10.1128/JVI.76.24.13062-13068.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cattaneo R, Schmid A, Rebmann G, Baczko K, Ter Meulen V, Bellini WJ, Rozenblatt S, Billeter MA. 1986. Accumulated measles virus mutations in a case of subacute sclerosing panencephalitis: interrupted matrix protein reading frame and transcription alteration. Virology 154:97–107. doi: 10.1016/0042-6822(86)90433-2. [DOI] [PubMed] [Google Scholar]
- 53.Cathomen T, Mrkic B, Spehner D, Drillien R, Naef R, Pavlovic J, Aguzzi A, Billeter MA, Cattaneo R. 1998. A matrix-less measles virus is infectious and elicits extensive cell fusion: consequences for propagation in the brain. EMBO J 17:3899–3908. doi: 10.1093/emboj/17.14.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cathomen T, Naim HY, Cattaneo R. 1998. Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. J Virol 72:1224–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tahara M, Takeda M, Yanagi Y. 2007. Altered interaction of the matrix protein with the cytoplasmic tail of hemagglutinin modulates measles virus growth by affecting virus assembly and cell-cell fusion. J Virol 81:6827–6836. doi: 10.1128/JVI.00248-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Makino S, Sasaki K, Nakagawa M, Saito M, Shinohara Y. 1977. Isolation and biological characterization of a measles virus-like agent from the brain of an autopsied case of subacute sclerosing panencephalitis (SSPE). Microbiol Immunol 21:193–205. doi: 10.1111/j.1348-0421.1977.tb00281.x. [DOI] [PubMed] [Google Scholar]
- 57.Moulin E, Beal V, Jeantet D, Horvat B, Wild TF, Waku-Kouomou D. 2011. Molecular characterization of measles virus strains causing subacute sclerosing panencephalitis in France in 1977 and 2007. J Med Virol 83:1614–1623. doi: 10.1002/jmv.22152. [DOI] [PubMed] [Google Scholar]
- 58.Baricevic M, Forcic D, Santak M, Mazuran R. 2007. A comparison of complete untranslated regions of measles virus genomes derived from wild-type viruses and SSPE brain tissues. Virus Genes 35:17–27. doi: 10.1007/s11262-006-0035-2. [DOI] [PubMed] [Google Scholar]
- 59.Hotta H, Nihei K, Abe Y, Kato S, Jiang DP, Nagano-Fujii M, Sada K. 2006. Full-length sequence analysis of subacute sclerosing panencephalitis (SSPE) virus, a mutant of measles virus, isolated from brain tissues of a patient shortly after onset of SSPE. Microbiol Immunol 50:525–534. doi: 10.1111/j.1348-0421.2006.tb03822.x. [DOI] [PubMed] [Google Scholar]
- 60.Homma M, Tashiro M, Konno H, Ohara Y, Hino M, Takase S. 1982. Isolation and characterization of subacute sclerosing panencephalitis virus (Yamagata-1 strain) from a brain autopsy. Microbiol Immunol 26:1195–1202. doi: 10.1111/j.1348-0421.1982.tb00270.x. [DOI] [PubMed] [Google Scholar]
- 61.Plemper RK, Compans RW. 2003. Mutations in the putative HR-C region of the measles virus F2 glycoprotein modulate syncytium formation. J Virol 77:4181–4190. doi: 10.1128/JVI.77.7.4181-4190.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]