

ABSTRACT
Numerous studies have focused on the regulatory functions of ICP27, an immediate-early (IE) protein of herpes simplex virus 1 (HSV-1). However, its homolog in HSV-2, termed ICP27t2, has been little studied. Here, we used two different approaches to functionally compare ICP27t2 and ICP27. In transfection-based assays, ICP27t2 closely resembled ICP27 in its capacity to enhance HSV-1 late gene expression, suppress the splicing of a viral intron, and complement the growth of an HSV-1 ICP27 null mutant. To study ICP27t2 in the context of viral infection, we engineered K2F1, an HSV-1 mutant that encodes ICP27t2 in place of ICP27. In Vero cells, K2F1 replicated with wild-type (WT) kinetics and yields, expressed delayed-early and late proteins normally, and was fully capable of activating several cellular signal transduction pathways that are ICP27 dependent. Thus, we conclude that ICP27t2 and ICP27 are functionally very similar and that ICP27t2 can mediate all ICP27 activities that are required for HSV-1 replication in cell culture. Surprisingly, however, we found that K2F1 forms plaques that are morphologically different from those of WT HSV-1. Investigation of this trait demonstrated that it results from the decreased release of progeny virions into the culture medium. This appears to be due to a reduction in the detachment of K2F1 progeny from the extracellular surface of the infected cell. We identified two HSV-1 ICP27 amino-terminal deletion mutants with a similar release defect. Together, these results demonstrate that ICP27 plays a heretofore-unappreciated role in modulating the efficiency of progeny virion release.
IMPORTANCE ICP27 is an essential, multifunctional regulatory protein that has a number of critical roles in the HSV-1 life cycle. Although ICP27 homologs are encoded by all known members of the Herpesviridae, previous work with several of these homologs has shown that they cannot substitute for ICP27 in the context of HSV-1-infected cells. Here, we identify ICP27t2 as the first homolog that can efficiently replace ICP27 in HSV-1 infection. Unexpectedly, our results also reveal that the sequence of the ICP27 gene can affect the release of HSV-1 progeny virions from the infected cell. Thus, our comparative study has revealed a novel function for ICP27 in the regulation of virus release.
INTRODUCTION
Herpes simplex virus 1 (HSV-1), a human alphaherpesvirus, is widespread in the human population, with greater than 57% of U.S. adults seropositive (1). The interaction of HSV-1 with the human host has been well studied (reviewed in reference 2). The virus is transmitted to new hosts via body secretions such as saliva, with the primary site of infection often being the oral mucosa. Following replication in epithelial cells, HSV-1 infects sensory neurons, where it establishes a lifelong latent infection in which the viral genome persists in the trigeminal ganglion as a semiquiescent episome. Periodically, the virus reactivates and returns to the oral cavity, where it can cause lesions (cold sores) and be transmitted to new hosts. Although such reactivated infections are not usually medically serious, HSV-1 can also spread to the brain and cause encephalitis, or to the eye, where it can result in blindness.
The replication cycle of HSV-1 in cultured cells has been studied in detail. This work has shown that the viral immediate-early (IE) proteins, the first viral polypeptides to be expressed during infection, are critical regulators of productive infection (2). In general, these polypeptides serve two functions. First, they activate expression of the viral delayed-early (DE) and late (L) genes, and second, they antagonize components of host intrinsic and adaptive immunity. One IE protein that has been studied in considerable detail is ICP27 (reviewed in reference 3). ICP27 is a multifunctional protein essential for replication, and homologs are conserved throughout the Herpesviridae. Although ICP27 is predominantly nuclear, it continuously shuttles between the nuclear and cytoplasmic compartments (4, 5). ICP27 binds to RNA, with a preference for G/C-rich sequences (6), and directly interacts with viral mRNA in infected cells (4). One of ICP27's most critical functions during infection is to activate the expression of several DE and L genes (7, 8). This function is complex, appearing to involve several distinct activities. Perhaps ICP27's best-characterized role in this regard is its ability to mediate the export of viral mRNAs from the nucleus to the cytoplasm (4). In this role, it is thought to bind intronless viral mRNAs in the nucleus and escort them through the nuclear pore to the cytoplasm via interactions with host mRNA export factors TAP/NXF1 and Aly/REF (9, 10). In addition to mediating mRNA export, ICP27 stimulates viral gene expression through other less-well-characterized mechanisms that enhance viral mRNA stability (4), polyadenyation (11), and translation (12, 13). ICP27 also inhibits host mRNA splicing (14), likely as a host shutoff mechanism, and suppresses removal of a cryptic intron in the viral L gene encoding glycoprotein C (15). Additionally, it carries out functions not directly related to gene expression, including the induction of several cellular signal transduction cascades, including the p38, JNK, and NF-κB pathways (16–18). ICP27 also regulates the localization of two other IE proteins, ICP0 and ICP4 (19–21). Early in infection, these proteins localize to the nucleus, but as infection proceeds, they move to the cytoplasm in an ICP27-dependent manner. We have shown that this relocalization is required for the packaging of these two IE proteins into progeny virion particles (22).
HSV-2 is a distinct human alphaherpesvirus that is also prevalent in the U.S. population (1). Genetically, it is highly related to HSV-1, showing 83% protein sequence identity and having a nearly identical arrangement of open reading frames (23). Moreover, HSV-1 and HSV-2 can readily recombine in tissue culture cells to form viable recombinants (24, 25). Notwithstanding their similarities, however, there are distinct biological differences between the two viruses, such as their preferred infection site in the human host (oral/facial for HSV-1, genital for HSV-2) (2). Moreover, despite their capacity to recombine in vitro, naturally occurring HSV-1/HSV-2 recombinants have not been described, suggesting that the two viruses rarely encounter one another in the host or that recombinants are at a replicative disadvantage in vivo.
To date, little work has been done on the HSV-2 homolog of ICP27, here referred to as ICP27t2. Both proteins are composed of 512 amino acid residues and are 79% identical in sequence (Fig. 1). However, the percent identity differs between the N- and C-terminal halves. Whereas the C-terminal halves are 93% identical, consistent with the fact that this is the portion of the protein conserved throughout the Herpesviridae (26, 27), the N-terminal halves exhibit only 65% identity. Several functions of ICP27 are dependent on sequences in the N-terminal half, including viral mRNA export (28), activation of cell signaling (17, 18), and the ability to alter localization of ICP0 and ICP4 (19). Thus, it is conceivable that the functions of ICP27t2 and ICP27 have diverged.
FIG 1.

Comparison of HSV-1 and HSV-2 ICP27. An alignment of the sequences of ICP27 (top) and ICP27t2 (bottom) from strains KOS and HG52, respectively, is shown. Yellow shading denotes amino acid differences. The sequences to which the H1113 and H1119 MAb epitopes on ICP27 have been mapped (40) are indicated.
In this study, we directly compared the activities of ICP27t2 and ICP27, using both transfection assays as well as an HSV-1 recombinant that encodes ICP27t2 in place of ICP27. Our results showed that ICP27t2 and ICP27 are functionally quite similar and that ICP27t2 can efficiently substitute for ICP27 in the context of an HSV-1 infection. Surprisingly, however, we found that the HSV-1 mutant expressing ICP27t2 forms plaques that have an altered morphology from those of the wild-type (WT) virus. Analysis of this trait has revealed a previously unrecognized role for ICP27 in the release of progeny virions from the infected cell.
MATERIALS AND METHODS
Cells, viruses, and infections.
Viral infections were carried out in Vero cells obtained from the American Type Culture Collection (ATCC). The cells were propagated in Dulbecco modified Eagle medium containing 5% heat-inactivated fetal calf serum, 50 units/ml penicillin, and 50 μg/ml streptomycin. Strain KOS1.1 (29) was the WT HSV-1 strain used in most experiments. Additional HSV-1 strains F and 17syn+ were obtained from Jim Lokensgard and Alistair McGregor, respectively. HSV-2 strains HG52 and MS were obtained from Jin-Young Han, and strain G was obtained from the ATCC. The engineering and isolation of HSV-1 mutant K2F1 (and an independent isolate, K2F1) were recently described (30). HSV-1 and HSV-2 infections were carried out in 199V medium, defined as medium 199 containing 50 units/ml penicillin, 50 μg/ml streptomycin, and 1% heat-inactivated newborn calf serum (NCS). For all virus yield assays (except the one shown below in Fig. 5A), infections were treated at 2 h postinfection (p.i.) with pH 3.0 acid-glycine buffer to inactivate extracellular virions (31). To harvest infections for analysis of total viral yields, a volume of sterilized milk equal to the volume of infected cell medium was added to each flask, and the entire culture was frozen at −80°C. The sterilized milk was prepared by reconstituting commercial instant nonfat dried milk (Carnation or a similar brand) in water to the manufacturer's specifications and autoclaving it twice for 20 min each cycle. Virus was released by three rounds of freeze-thawing prior to titration by plaque assay on Vero cells.
FIG 5.

K2F1 is deficient in release of virus to the extracellular medium. (A) Time course of WT HSV-1 extracellular release. Vero cells were infected with KOS1.1 at an MOI of 5 PFU/cell. At various times after infection, the supernatant and cells were harvested separately and titers were determined for the virus yields in each fraction. (B) K2F1 is deficient in extracellular release. Vero cells were infected in quadruplicate with KOS1.1, K2F1, or K2F1b at an MOI of 5 PFU/cell. At 12 and 24 h p.i., supernatant and cells were harvested separately and the percentage of the total yield that was released into the medium was determined. (C) Most cell-associated HSV-1 progeny are sensitive to pH 3.0 inactivation. Cultures of Vero cells were infected in duplicate at an MOI of 5 PFU/cell, and cells were incubated for 24 h. Cells were harvested by scraping into PBS and low-speed centrifugation. The scraped cells were either incubated for 2 min in PBS as a control or in pH 3.0 buffer to inactivate extracellular virions. After repelleting and suspension in fresh medium, samples were frozen and titers were determined. Prior to titration, one set of low-pH-treated samples was treated by pulsed microsonication to further release intracellular virus. The values represent the percentage of cell progeny that were inactivated by the treatment relative to the PBS control. (D) Most cell-associated HSV-1 progeny are sensitive to trypsin inactivation. Cultures of Vero cells were infected in triplicate and harvested as described for panel C. Cells were resuspended in either PBS, as a control, or in 0.05% trypsin–EDTA, and the resuspended cells were incubated at 37°C for up to 15 min. After repelleting and suspension in fresh medium, samples were frozen and titers were determined. The values represent the percentage of cell-associated progeny that were inactivated by the trypsin treatment relative to the 15-min PBS control. (E) K2F1 virions efficiently reach the cell surface. Vero cells were infected in quadruplicate with KOS1.1 or K2F1 at an MOI of 1 PFU/cell, and the infections were incubated for 20 h. After taking aliquots of the medium to determine release (middle), the scraped cells were treated for 2 min with PBS or pH 3.0 buffer as for panel C to determine sensitivity to low-pH treatment (right). The total viral yields are also shown (left). (F) K2F1 has a higher percentage of progeny virions on the cell surface than does WT HSV-1. Cells were infected in quadruplicate with KOS1.1 or K2F1 at an MOI of 5 PFU/cell and harvested at 24 h p.i. Aliquots of the medium were taken to determine release. The cells were then collecting by scraping and low-speed centrifugation and then extracted twice with TNE buffer. The combined salt washes and the washed cell fractions were frozen, and titers of all fractions were determined. Statistical analyses for results shown in panels B, E, and F were performed using the Student t test; error bars denote standard errors of the means. *, P < 0.05; **, P < 0.01; ns, not significant (P > 0.05).
Viral plaque assays were carried out as follows. Viral stocks were serially diluted in phosphate-buffered saline (PBS) containing 0.5 mM MgCl2, 0.9 mM CaCl2, 0.1% dextrose, and 1% heat-inactivated NCS. Aliquots were plated on 6- or 12-well trays of Vero cells for 1 h at 37°C. The inoculum was then replaced with 199V medium containing 1% (vol/vol) heat-inactivated pooled human serum (MP Biomedical) and reincubated at 37°C. HSV-1 plaque assays were incubated for 3 days prior to fixation with a 5-min methanol treatment. The monolayers were stained for ≥1 h with modified Giemsa stain (Sigma-Aldrich) diluted 10-fold in water. After removal of the stain, the trays were rinsed with water and dried, and plaques were counted. HSV-2 plaque assays were carried out identically, except the cells were fixed and stained at 2 days p.i. To compare the relative size of HSV-1 plaques, digital images of the stained plaques were obtained. The images were opened in ImageJ and outlined using the freehand tool. The number of pixels obtained was used as a quantitation of the plaque area; for the data shown in Fig. 4C, 30 plaques of each virus strain were analyzed.
FIG 4.

K2F1 forms plaques with an altered morphology. (A) Viral plaques were allowed to form on Vero cells under a liquid medium overlay containing 1% PHS. Plaques were fixed and stained with Giemsa stain at 3 days p.i. (B). K2F1 plaque phenotype depends on the concentration of PHS in the overlay. Equal numbers of PFU of the viral strains shown were subjected to plaque assays on Vero cells. The overlay medium contained differing concentrations of PHS (0 to 2.5%). Plaques were fixed and stained 3 days postinfection. (C) Cell-to-cell spread. Viral stocks were allowed to form plaques in the presence of 10% PHS to completely prevent extracellular spread. Plaques were fixed and stained 3 days postinfection. Digital images were obtained and analyzed by using ImageJ to determine the mean plaque area. Thirty plaques were measured for each virus. (D) K2F1 resembles WT HSV-1 in its sensitivity to neutralization by PHS. Triplicate aliquots containing 200 PFU of KOS1.1 or K2F1 were incubated for 30 min in medium lacking PHS (control) or in medium containing various concentrations of PHS. Following incubation, the virus samples were subjected to a standard plaque assay. Error bars represent standard errors of the means.
To determine the percentage of total viral progeny released into the medium, the following procedure was used. Infected cell medium was poured from the culture flask into a centrifuge tube which was spun for 5 min at 900 × g to pellet unattached cells. An aliquot of the supernatant (released fraction) was then mixed with an equal volume of sterile milk and frozen. The remaining supernatant and any pelleted cells were returned to the flask (uncorrected cell-associated fraction), an equal volume of sterile milk was added, and the flask was frozen. After determination of viral titers by plaque assay, the cell-associated fraction was determined by correcting for the amount of supernatant fraction that it contained. The percent released virus was determined by dividing the total viral yield by the released viral yield.
To determine the extent to which HSV-1 cell-associated progeny are inactivated by low-pH treatment, the following procedure was used. At the time of harvest, supernatants were collected as described above to determine viral release. Infected cells were then scraped in cold PBS, pelleted, and resuspended in 2 ml of either pH 3.0 acid-glycine buffer (31) or PBS. After incubation on ice for 2 min, the cells were repelleted and resuspended in 2 ml of 199V medium. Two milliliters of sterile milk was added, and the samples were frozen at −80°C. To quantitate the sensitivity of cell-associated virus to low pH, the titer of the acid-glycine buffer-treated samples was divided by the titer of the PBS-treated samples. These values were expressed as a percentage and subtracted from 100%. The sensitivity of cell-associated virus to trypsin was also examined. To do this, scraped, PBS-washed, and repelleted cells were incubated in 1 ml of either PBS, as a control, or 0.05% trypsin-EDTA (Life Technologies) for up to 15 min at 37°C. The cells were then pelleted, washed in normal medium, and resuspended in a 50% medium–50% sterile milk mixture prior to freezing. Quantitation of trypsin sensitivity (relative to the 15-min PBS-treated control) was performed as described above for the low-pH treatment.
To quantitate HSV-1 surface-attached progeny virions, we used the salt wash procedure of Newcomb and Brown (32) to remove viral progeny from the cell surface. Briefly, infected cells were scraped in PBS, pelleted, and resuspended in 2 ml TNE (0.01 M Tris-HCl, 0.5 M NaCl, 1 mM EDTA; pH 7.5). Following incubation on ice for 15 min, cells were repelleted, and the supernatant fraction was collected. The extraction was repeated, and the two salt wash fractions were combined, mixed with an equal volume of sterile milk and frozen at −80°C. For determination of the salt-resistant fraction, the final cell pellets were resuspended in a 50% medium–50% sterile milk and frozen.
Plasmids and transfection experiments.
The plasmid pT2-27, which contains the HSV-2 gene from strain HG52, was recently described (30). Additional plasmids used were the gC expression plasmid pgCΔpro (33), ICP27 expression plasmids pBH27 (34) and pC27 (5), ICP4 expression plasmid pK1-2 (35), and ICP0 expression plasmid pSHZ (36). Transfections were carried out using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. Transfection experiments to determine the effects of ICP27 on ICP0 and ICP4 localization were performed as previously described (19). Plasmid-based d27-1 complementation experiments were also carried out as described previously (37).
Protein analyses.
Immunoblotting and analysis of viral protein synthesis by metabolic pulse-labeling with [35S]methionine were performed as previously described (38). Several antibodies were used for immunoblotting. To detect ICP27, mouse monoclonal antibodies (MAbs) H1113 (Virusys) and H1119 (Rumbaugh-Goodwin Institute) were used. Mouse MAbs were also used to detect glycoproteins B (gB; HA-056; Virusys), C (gC; H1104; Rumbaugh-Goodwin Institute), and D (gD; H1103; Rumbaugh-Goodwin Institute). Rabbit polyclonal antisera, a kind gift of J. Diaz, was used to detect US11 (39). Cellular proteins were detected as follows: histone H3 was detected with rabbit MAb 1326-1 (Epitomics); EEA1 was detected by a mouse MAb (BD Biosciences); JNK was detected by rabbit serum 44-690G (BioSource); phospho-JNK was detected by rabbit serum 44-682G (BioSource); p38 was detected by rabbit serum 9212S (Cell Signaling); phospho-p38 was detected by rabbit serum 9211S (Cell Signaling); p38 was detected with rabbit serum 9212S (Cell Signaling); Iκb-α was detected by rabbit serum SC203 C-15 (Santa Cruz Biotechnology).
RESULTS
Analysis of the cloned ICP27t2 gene.
To compare the functions of ICP27t2 and ICP27, we first wished to study the activities of the cloned genes in uninfected cells. For expression of ICP27t2, we used the recently described plasmid pT2-27, which contains an intact ICP27t2 gene from HSV-2 strain HG52 (30). The insert includes the gene's promoter and polyadenylation site, and thus we expected that pT2-27 would express ICP27t2 upon transfection into mammalian cells. To verify this, Vero cells were transfected with pT2-27 or pBH27, an ICP27 expression plasmid (34). After 2 days, total cell proteins were collected and analyzed by immunoblotting using H1119, an ICP27-specific MAb that recognizes residues 1 to 11 (40), which are completely conserved in ICP27t2 (Fig. 1). The results (Fig. 2A) confirmed that ICP27t2 is efficiently expressed from pT2-27. We also noted that ICP27t2 is not recognized by MAb H1113, which binds to residues 109 to 137 (40) (data not shown). This is consistent with the fact that ICP27t2 differs substantially in this region (Fig. 1).
FIG 2.

Comparison of ICP27t2 and ICP27 in transfected cells. (A) Expression of ICP27t2 from pT2-27. Vero cells were transfected with pT2-27, harboring ICP27t2, or pBH27, harboring ICP27. At 2 days posttransfection, total protein extracts were prepared and analyzed by immunoblotting using MAb H1119. (B) Stimulation of gC expression from a transfected plasmid. Vero cells were transfected with vector DNA only or with pgCΔpro minus or plus pBH27 or pT2-27. Total proteins were harvested after 2 days, and gC levels were analyzed by immunoblotting. (C) Regulation of gC mRNA. Vero cells were transfected as shown, and total cell RNA was prepared 2 days posttransfection and subjected to Northern analysis with a gC gene-specific probe. The band labeled “u” corresponds to unspliced gC mRNA, while the band labeled “s” corresponds to the spliced form. (D) Analysis of effects on ICP0 and ICP4 localization. Vero cells growing on coverslips were transfected with an ICP0-carrying or ICP4-carrying plasmid, minus or plus plasmids harboring ICP27 (pC27) or ICP27t2 (pT2-27). The cells were fixed 1 day later and analyzed by immunofluorescence using MAb specific for ICP0 or ICP4. (E). Complementation of HSV-1 ICP27 null mutant d27-1. Vero cells were transfected in triplicate with pUC19 DNA as a control, or with pBH27 or pT2-27 DNA. One day posttransfection, the cells were infected with ICP27 mutant d27-1 and incubated for a further day. Viral titers were determined by plaque assay on V27 cells. Error bars denote standard errors of the means.
One of ICP27's most critical functions during productive infection is to enhance the expression of several DE and L genes (7, 41, 42). One L gene that is highly dependent on ICP27 is that encoding gC (also known as UL44). We previously showed that ICP27's stimulatory effects on this gene can be demonstrated in a plasmid cotransfection assay using plasmid pgCΔpro, a construct in which the gC gene is transcribed under the control of the human cytomegalovirus (HCMV) IE promoter (33). To test whether ICP27t2 can activate this gene, we carried out a cotransfection assay and found that it resembles ICP27 in its capacity to enhance gC protein expression (Fig. 2B). We next looked at mRNA expression. In this regard, we have previously shown that ICP27 has two distinct effects on gC mRNA expressed from this plasmid (15). First, it enhances the accumulation of the mRNA. Second, it promotes the retention of a 225-nucleotide intron that when spliced out leads to a transcript encoding an alternate, secreted form of gC. To examine both RNA effects, we cotransfected pgCΔpro with either the ICP27t2 or ICP27 plasmids and analyzed the gC transcripts by Northern blotting. A plasmid carrying the BHV-4 ICP27 homolog was included as a control, as we have previously shown that this homolog enhances gC mRNA accumulation but is unable to promote intron retention (15). The results (Fig. 2C) indicated that ICP27t2 was similar to ICP27 and BHV-4 ICP27 in its ability to enhance accumulation of gC mRNA. Furthermore, it resembled ICP27 in being able to promote the retention of the gC intron, leading to the appearance of predominantly unspliced gC transcript (“u” in Fig. 2C). In contrast, BHV-4 ICP27 was unable to promote intron retention, leading to predominant expression of the spliced transcript (“s” in Fig. 2C). Thus, we conclude that ICP27t2 is similar to ICP27 in having the ability to both enhance the accumulation of gC mRNA as well as promote retention of its intron.
In addition to its effects on viral gene expression, ICP27 can alter the nucleocytoplasmic distribution of the HSV-1 IE proteins ICP0 and ICP4. As this ICP27 function can be assayed in transfected cells (20, 21), we compared ICP27t2 and ICP27 for their effects on the localization of these two proteins. To do this, Vero cells were transfected with ICP0-expressing or ICP4-expressing plasmids alone or together with the ICP27 or ICP27t2 expression constructs. The cells were fixed 1 day later and processed for immunofluorescence. The nucleocytoplasmic distribution of ICP0 or ICP4 was then assessed in a blinded fashion and divided into five categories (N, N > C, N = C, C > N, and C) (Fig. 2D). As expected, ICP0 was predominantly nuclear when expressed on its own, and coexpression of ICP27 shifted its localization to the cytoplasm. ICP27t2 had a similar effect on ICP0. Similarly, ICP4 was nuclear when expressed on its own, and ICP27 shifted its localization to the cytoplasm. However, ICP27t2 was notably less efficient than ICP27 in promoting ICP4 cytoplasmic localization. A repeat experiment confirmed this result, and in this case, parallel immunoblotting analysis showed that the difference in ICP4 localization was not due to expression differences between ICP27 and ICP27t2, as these were expressed comparably in the transfected cells (data not shown). These results indicated that ICP27t2 is deficient in its ability to promote the cytoplasmic localization of HSV-1 ICP4 in transfected cells.
Given the several similarities between ICP27t2 and ICP27, we next investigated whether ICP27t2 is able to substitute for ICP27 in the context of an HSV-1 infection. To test this, we asked whether the cloned ICP27t2 gene can complement the growth of HSV-1 ICP27 deletion mutant d27-1. Thus, Vero cells were transfected with vector DNA only or with the plasmids carrying ICP27 or ICP27t2. The next day, the cells were infected with d27-1 and incubated for an additional day. The amount of viral progeny was then determined by plaque assay on V27 cells, which are derivatives of Vero cells that express ICP27 upon infection and complement the growth of HSV-1 ICP27 mutants. As expected, d27-1 was completely unable to replicate in Vero cells, and this defect was efficiently complemented by expression of ICP27 (Fig. 2E). Notably, ICP27t2 performed as well as ICP27 in enhancing replication. These results showed that ICP27t2 can substitute functionally for ICP27 in the context of infected Vero cells.
Characterization of K2F1, an HSV-1 mutant encoding ICP27t2.
To study ICP27t2 further, we wanted to examine its activities in infected cells when expressed from its natural context in the viral genome. In a recent publication, we described the isolation of K2F1, an HSV-1 mutant that encodes ICP27t2 in place of ICP27 (30). Aside from showing that the virus is viable in Vero cells and that it is sensitive to the drug leptomycin B (similar to WT HSV-1), the phenotype of K2F1 was not further characterized. Here, we report a detailed phenotypic analysis of K2F1. It should be mentioned that in many of the experiments described below we included a second genetically independent isolate of K2F1, designated K2F1b (30). This was done to ensure that the phenotype of the mutant was the result of the engineered ICP27 gene change, rather than the consequence of an additional unknown mutation.
We first asked whether K2F1 replicates similarly to WT HSV-1. Vero cells were infected at a low multiplicity of infection (MOI; 0.01 PFU/cell), and viral yields at various time points were determined (Fig. 3A). The results indicated that both K2F1 isolates replicated indistinguishably from WT HSV-1 in terms of kinetics and yield. We also found that K2F1 and K2F1b grew similarly to WT HSV-1 in another cell line, human HEp-2 cells (L. Sanders III and S. Rice, unpublished data). Thus, as suggested by the plasmid complementation experiment results, ICP27t2 can carry out all functions of ICP27 that are required for viral replication in cell culture.
FIG 3.

Phenotypic analysis of K2F1. (A) Growth analysis. Vero cells were infected in triplicate at an MOI of 0.05 PFU/cell as shown, and infections were harvested at various time points. Yields were determined based on titers of the harvested infections by plaque assay on Vero cells. Error bars represent standard errors of the means. (B) Viral protein synthesis. Vero cells were infected at an MOI of 10 PFU/cell as shown, and at various time points the cells were labeled for 30 min with [35S]methionine. The proteins were then analyzed by SDS-PAGE and autoradiography. The asterisk indicates the position of a polypeptide expressed at 4 h p.i. in the K2F1 infections; this protein likely corresponds to ICP27t2. (C) Accumulation of viral proteins. Vero cells were infected as shown at an MOI of 10 PFU/cell. Total proteins were collected at 9 h p.i. and analyzed for viral protein expression by immunoblotting. Histone H3 was analyzed as a loading control. (D) Activation of cellular signaling pathways after HSV-1 infection. Vero cells were infected at an MOI of 10 PFU/cell as shown, and total viral proteins were harvested at 7 h p.i. Activation of cellular signaling pathways was assessed by immunoblotting.
Although K2F1 replicates efficiently, it is possible that it expresses viral genes with different kinetics or at different levels than WT HSV-1. To examine this possibility, we carried out a metabolic labeling experiment wherein infected Vero cells were pulse-labeled for 30 min at 4, 8, and 12 h p.i. The proteins were then analyzed by SDS-PAGE and autoradiography (Fig. 3B). At all time points analyzed, both K2F1 isolates gave a pattern of protein synthesis that was nearly identical to that of the WT infection. The only difference noted was at the 4-h time point, where K2F1 and K2F1b expressed an ∼65-kDa protein (indicated by the asterisk in Fig. 3B) that was absent in the corresponding position of the WT infection. This band likely corresponds to ICP27t2 itself, which runs on SDS-PAGE at a position slightly higher than ICP27 (e.g., see Fig. 2A). To study viral gene expression further, we also looked at accumulation of viral proteins by immunoblotting, focusing on ICP27 itself as well as several L proteins at an intermediate time point of infection (9 h p.i.) (Fig. 3C). Both K2F1 isolates were indistinguishable from WT HSV-1 in their accumulation of these proteins. Together, the pulse-labeling and immunoblotting experiments indicated that K2F1 is very similar to WT HSV-1 in the expression of viral genes.
HSV-1 infection results in the activation of several cellular signal transduction cascades, including the JNK, p38, and NF-κB pathways, and ICP27 is required for the induction (17, 18). To see if ICP27t2 can similarly induce these pathways, we compared the activation of these systems in K2F1- and WT-infected cells. For comparison, we also assessed cells infected with HSV-2 strain HG52 (the source of the ICP27t2 gene in K2F1). Total protein samples were made from infected Vero cells at 7 h p.i. and analyzed by immunoblotting. Analysis of total and phosphorylated JNK indicated that K2F1 activated this pathway similarly to KOS1.1 (Fig. 3D). Interestingly, HSV-2 failed to show robust activation of JNK at this time point, suggesting a possible type-specific difference in signaling. Examination of total and phosphorylated p38 indicated that K2F1 activated this pathway as efficiently as WT HSV-1. HSV-2 also efficiently activated the p38 system at this time point. Lastly, K2F1 and WT HSV-1 and HSV-2 all induced NF-κB signaling comparably, based on the loss of IκB-α, which is indicative of the activation of this signal transduction cascade. Based on these data, we conclude that ICP27t2 is similar to ICP27 in its ability to induce the JNK, p38, and NF-κB signaling systems in the context of an HSV-1 infection.
Given that ICP27t2 was not as efficient as ICP27 in promoting cytoplasmic ICP4 in transfected cells (Fig. 2D), we predicted that ICP4 would be more restricted to the nucleus in K2F1-infected cells. However, immunofluorescence analyses at several time points after infection failed to show any difference between ICP4 localization in K2F1- and WT HSV-1-infected cells (data not shown). Thus, in the context of HSV-1 infection, the substitution of ICP27t2 for ICP27 does not appear to alter ICP4 localization. Additionally, we found that ICP27t2 localization in K2F1 infection closely resembled that of ICP27 localization in WT HSV-1 infections (data not shown).
K2F1 plaques have an altered morphology.
Based on the phenotypic resemblance of K2F1 to WT HSV-1, it is clear that ICP27 and ICP27t2 are functionally very similar. However, in the course of our analyses, we noted that both K2F1 isolates showed a clear phenotypic difference from WT HSV-1: they formed plaques with a different morphology. It should be noted that our standard plaque assays are carried out using a liquid overlay containing 1% normal pooled human serum (PHS). This reagent is included because the anti-HSV antibodies routinely present in PHS neutralize extracellular virus and inhibit spread of infection through the medium, allowing the formation of discrete plaques via cell-to-cell spread (43). However, in the case of WT HSV-1, we routinely observed that the plaques had a ragged and often a comet-like appearance (Fig. 4A), suggesting some extracellular spread. In contrast, K2F1 and K2F1b plaques exhibited a much more well-delineated, round morphology, as shown for K2F1 in Fig. 4A.
When plaque assays were performed with a semisolid agarose overlay, no differences in plaque morphology were seen (data not shown). This suggested that the K2F1 plaquing phenotype depends on the liquid overlay. To investigate this further, we carried out a plaque assay in which the concentration of PHS in the liquid overlay was varied from 0 to 2.5% (Fig. 4B). When no serum was present, both K2F1 isolates and WT HSV-1 gave rise to similar comet-shaped plaques, although careful examination of the plaques in this and repeat experiments indicated that the K2F1 comet-like plaques were usually smaller than those of the WT. At intermediate HS concentrations (0.5 to 1.5%), the morphology difference between mutant and WT was readily evident, with the mutant plaques having a much rounder appearance. However, at high PHS concentrations (2 to 2.5%), both mutant and WT viruses formed similar round plaques. These results indicated that the plaquing phenotype of K2F1 is dependent on the concentration of PHS in the overlay medium, with K2F1 showing a reduced tendency to form comet-like plaques at low PHS concentrations. Comet-like plaques have been studied extensively in the vaccinia virus system (44, 45) and are believed to result from the extracellular spread of infection via convection currents that become established in the culture dish. Our results thus suggest that, under the conditions of our plaque assay, K2F1 spreads less efficiently through the medium than does WT HSV-1.
In addition to being released into the extracellular milieu, HSV-1 progeny virions can infect neighboring cells via cell-cell junctions that are shielded from the external environment (46). Such spread is typical in highly polarized epithelial cells, but it is also observed in nonpolarized epithelial cells, such as Vero cells. To determine whether K2F1 has a defect in cell-to-cell spread in Vero cells, we allowed K2F1, K2F1b, and WT HSV-1 to form plaques in liquid medium containing a very high concentration of HS (10%), in order to strongly inhibit extracellular spread. After 3 days, the monolayers were fixed and stained. Quantitative analysis of plaque areas (Fig. 4C) indicated that K2F1 and K2F1b plaques were only slightly smaller than WT plaques (89.5% and 91.1% of the WT size, respectively). Thus, alteration of the ICP27 gene in K2F1 has only a marginal effect on cell-to-cell spread.
As discussed above, the comet-like nature of WT HSV-1 plaques in our standard plaque assay suggests that the virus can spread extracellularly in the presence of 1% PHS, implying that it is incompletely neutralized by this concentration of PHS. If so, one explanation for K2F1's round plaque phenotype is that its progeny virions are more susceptible to neutralization by PHS than are WT virions. To test this, we carried out a virus neutralization assay in which aliquots containing 200 PFU of either WT HSV-1 or K2F1 were left untreated or were treated for 30 min at 37°C with 2-fold serial dilutions of PHS. The samples were then plated on Vero cells under standard plaquing conditions. The results (Fig. 4D) indicated that KOS1.1 and K2F1 are very similar in their sensitivities to PHS neutralization, with both viruses showing 50% neutralization at ∼2.5% PHS.
K2F1 is defective in extracellular release.
Another explanation for the plaquing phenotype of K2F1 is that, as it replicates, it releases less virus into the medium than does WT HSV-1. To investigate this possibility, we first analyzed the time course of extracellular virus release for WT HSV-1. Vero cells were infected with KOS1.1 at an MOI of 5 PFU/cell, and at various time points, the medium and cells were separately harvested and titers were determined. The results (Fig. 5A) indicated that at an intermediate time point in infection (11 h p.i.), only 1.7% of the progeny viruses were present in the medium. However, this level rose to 24% at 24 h p.i. and was maintained at this approximate level throughout the remaining time points. We next compared the extracellular release of K2F1 and K2F1b to WT HSV-1 (Fig. 5B). At 12 h p.i., K2F1 and K2F1b release was reduced more than 2-fold compared to KOS1.1. At 24 h p.i., the release of K2F1 and K2F1b relative to WT HSV-1 had increased but was still reduced to 72% and 63% of the WT level, respectively. These data indicate that K2F1 is deficient in the release of infectious virus into the medium.
K2F1 virions are deficient in detachment from the cell surface.
To be released, HSV-1 virions must first be assembled in their fully infectious form (see references 46 and 47 for reviews of HSV-1 assembly and release). This likely occurs when tegumented DNA-bearing capsids bud into cytoplasmic vesicles, possibly derived from the trans-Golgi network, and acquire their final envelope. The subsequent release of virions into the extracellular milieu may involve two distinct steps. First, the virion-containing vesicles must undergo exocytosis, i.e., they must travel to and fuse with the plasma membrane, introducing the progeny virions into the extracellular environment. However, at that point, viral progeny may still be tethered to the outside surface of the cell, and indeed microscopic evidence suggests that this commonly occurs in HSV-1 infection (48, 49). Therefore, for complete release, a second step is required in which the virions detach from the cell surface.
Although our results indicate that K2F1 is deficient in extracellular release, they do not indicate whether mutant progeny are defective in reaching the cell surface, or in detaching from the surface once there (or both). To study this, we first characterized the WT HSV-1 infection, in order to establish to what degree progeny virions are intracellular versus extracellular at the completion of infection. To address this, we took advantage of the finding that HSV-1 virions are readily inactivated by brief exposure to pH 3.0-containing buffers, while cells are resistant to this treatment (50). Thus, WT HSV-1-infected Vero cells were collected at 24 h p.i. by scraping into PBS and low-speed centrifugation. Cells were resuspended in pH 3.0 acid-glycine buffer (50), or in PBS as a control. After a 2-min incubation, all samples were repelleted, resuspended in medium to restore the pH to neutrality, and frozen at −80°C. Plaque assays were then carried out to quantitate the level of infectious virus (Fig. 5C). This analysis showed that ∼99.9% of the WT HSV-1 cell-associated progeny were inactivated by brief exposure to pH 3.0, suggesting that the overwhelming majority of progeny virions are extracellular. We considered the possibility that intracellular progeny were not efficiently detected in the pH 3.0-treated samples because they were inside intracellular vesicles that were not efficiently disrupted by our standard procedure of freeze-thawing. To address this possibility, we additionally treated one set of pH 3.0-exposed samples by pulsed microsonication in an attempt to further disrupt vesicles. However, this treatment only minimally (∼3-fold) increased the titer of the pH 3.0-treated samples, still leaving >99.5% of the cell-associated progeny sensitive to low pH (Fig. 5C).
To confirm that the majority of cell-associated virions are extracellular, we next tested the sensitivity of viral progeny to trypsin. WT HSV-1-infected Vero cells were harvested at 24 h p.i., and the scraped, washed, and pelleted cells were resuspended in either PBS, as a control, or in trypsin-EDTA at the same concentration we routinely use to passage our Vero cell stocks. After 5, 10, or 15 min, cells were collected by centrifugation, washed in normal medium to remove trypsin, and frozen at −80°C. Titration of these fractions by plaque assay revealed that the 5-min trypsin treatment inactivated 82.4% of the cell-associated virus, while the 10- and 15-min treatments inactivated 99.4% (Fig. 5D). Moreover, trypsin treatment of up to 15 min did not appear to lyse the infected cells, as judged by trypan blue exclusion analysis (data not shown). Together, the results of this and the previous experiment strongly suggest that, at the completion of WT HSV-1 infection of Vero cells, >99% of the cell-associated progeny are attached to the outside surface of the infected cell.
Given the above results, the release defect of K2F1 could be explained if its progeny virions are not as efficiently transported to the cell surface as WT HSV-1 virions. To investigate this, we used the low-pH sensitivity assay described above to directly compare the efficiency with which K2F1 and WT HSV-1 virions reach the cell surface. We also quantified the total viral yields, as well as the percentage of total virus released to the medium. The results are shown in Fig. 5E. As expected, K2F1 and WT HSV-1 infections were similar in total yields, and ∼2-fold-fewer progeny were released from the K2F1-infected cells (5.4% versus 11.8%). Notably, similar to the WT HSV-1 infection, >99% of the remaining K2F1 cell-associated progeny were extracellular, based on their sensitivity to low pH. This indicated that K2F1 virions are able to efficiently reach the cell surface.
We next considered whether K2F1 progeny are defective in detaching from the cell surface. If so, one would predict that K2F1-infected cells would have a greater fraction of progeny virions on the surface than do WT HSV-1-infected cells. To test this, we took advantage of the finding of Newcomb and Brown, that surface-attached HSV-1 progeny can be efficiently eluted from cells, in an infectious state, by washing the cells with a buffer containing 0.5 M NaCl (32). Thus, Vero cells were infected with WT HSV-1 or K2F1 at an MOI of 5 PFU/cell. At 24 h p.i., aliquots of the medium were taken for determination of viral release. The cells were then scraped, pelleted, and washed twice with TNE buffer (0.01 M Tris-HCl, 0.5 M NaCl, 1 mM EDTA; pH 7.5) as described previously (32). Titers of the released fractions, the combined salt washes, and the residual washed cell fractions were then determined. The compiled results (Fig. 5F) showed that, as expected, K2F1 released less progeny into the medium than did WT HSV-1 (7.6% versus 12.6%). Furthermore, based on the titers of the combined salt wash fractions, K2F1-infected cells had a higher fraction of surface-attached viral progeny than did WT HSV-1-infected cells (89.0% versus 84.6%). In fact, this difference of 4.4% corresponded closely with the 5.0% difference in release. In contrast, there was not a significant difference in the fraction of viruses remaining associated with the cells after the salt wash (3.4% versus 2.8%). These results strongly suggest that K2F1 progeny virions are more efficiently retained on the cell surface than are WT virions, i.e., they do not detach as readily. This decrease can explain the release defect of K2F1.
HSV-1 ICP27 mutants d2-3 and dAc exhibit release defects.
Our results with K2F1 demonstrated that changes to the sequence of ICP27 can reduce the extent to which HSV-1 progeny virions are released from cells. In previous work, we characterized several replication-competent HSV-1 ICP27 mutants that make WT-sized plaques (M50T, dAc, d2-3, d5-6, and d6-7) (19, 31) (Fig. 6A). We considered the possibility that some of these might have previously unrecognized release defects similar to that of K2F1. Since this trait correlates with plaque morphology, we first analyzed the mutants in our standard plaque assay. Three of the mutants (d5-6, d6-7, and M50T) formed comet-like plaques similar to the WT (data not shown). However, dAc and d2-3, which are deleted of residues 21 to 63 and 64 to 108, respectively, formed round plaques similar to those of K2F1. In the case of d2-3, the round plaque phenotype was more pronounced than that of K2F1. This was evident when the plaque assays were extended from the usual 3 days to 4 days (Fig. 6B). Under these conditions, KOS1.1 plaques were clearly comet-like, while K2F1 plaques also showed some cometing. In contrast, d2-3 plaques remained round with few signs of comets.
FIG 6.

HSV-1 ICP27 deletion mutants dAc and d2-3 are deficient in viral release. (A) Schematic diagram of mutations in viable HSV-1 ICP27 mutants. Dashed lines represent deletions, and the vertical line in the M50T bar represents a point mutation at residue 50. The capacities of the viruses to form comet-like plaques are indicated. (B) Plaque morphology phenotype of d2-3. Virus stocks were allowed to form plaques on Vero cells in liquid medium containing 1% HS. The monolayers were fixed and stained 4 days p.i. (C) dAc and d2-3 are defective in release. Vero cells were infected in triplicate with 5 PFU/cell of KOS1.1 and dAc. At 24 h p.i., supernatant and cells were harvested and titers were determined separately. The percent released and total yields were determined. (D) d2-3 is defective in release. Analysis was performed as for panel C. Statistical analyses in panels C and D were performed using the Student t test; error bars denote standard errors of the means. **, P < 0.01; ***, P < 0.001.
To see if the round plaque phenotypes of dAc and d2-3 correlated with reduced release levels, we quantitated the extracellular release of these mutants. Whereas dAc showed a 2.6-fold release defect compared to WT HSV-1 (Fig. 6C), d2-3 was more reduced, exhibiting a 4.0-fold defect (Fig. 6D). Thus, we conclude that certain in-frame deletions in the N-terminal portion of ICP27 result in a viral release defect similar to that of K2F1. It should be noted that, unlike K2F1, which replicates as efficiently as WT HSV-1, both dAc and d2-3 showed modest but significant reductions in total viral yields, with dAc being reduced 3.1-fold compared to WT HSV-1 and d2-3 being reduced 11-fold. Tian et al. also recently noted a growth defect for d2-3 in Vero cells (10).
HSV-1 and HSV-2 laboratory strains vary in the efficiency of viral release.
Our analysis of K2F1 demonstrated that the ICP27t2 gene of HSV-2 strain HG52 reduces the efficiency of viral release when it is expressed in the context of the HSV-1 genome. To see if strain HG52 normally has a low release efficiency, we quantitated its release relative to HSV-1 strain KOS1.1 (Fig. 7). After a 24-h infection, KOS1.1 released 7.8% of its progeny into the medium, whereas HG52 released only 0.48%, a 16-fold difference. To see whether HSV-1 and HSV-2 strains generally differ in release, we repeated the analysis, this time including two additional strains of each virus type. While KOS1.1 and HG52 gave results similar to those seen in the first experiment (13.1% and 0.76% release, respectively), the other strains of HSV-1 and HSV-2 showed intermediate levels of release (ranging from 2.6% for HSV-2 strain G to 5.3% for HSV-1 17+). These results do not indicate a general type-specific difference in the release efficiency of laboratory strains of HSV-1 and HSV-2. On the other hand, the results show that the release efficiencies of HSV-1 and -2 laboratory strains can vary quite significantly and that HSV-2 strain HG52 is inefficient in release compared to several other strains.
FIG 7.
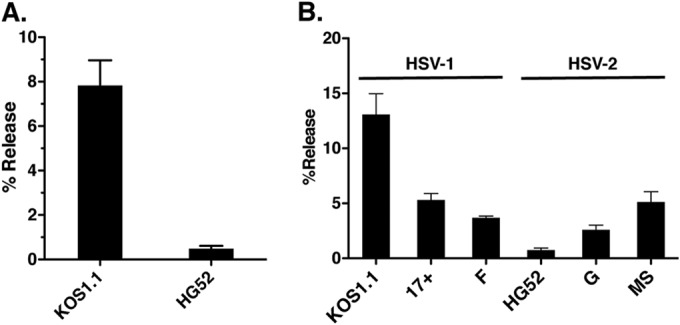
HSV-1 and HSV-2 strains vary significantly in the level of extracellular virion release. (A) Comparison of HSV-1 strain KOS1.1 and HSV-2 strain HG52. Vero cells were infected at an MOI of 5 PFU/cell, and infections were incubated for 24 h. Supernatant and cell fractions were harvested and titers were determined separately, and then the percent release was determined. Error bars denote standard errors of the means. (B) Comparison of multiple HSV-1 and HSV-2 strains. The experiment was performed as for panel A except infection was carried out at an MOI of 1 PFU/cell.
DISCUSSION
ICP27t2 and ICP27 are functionally very similar.
The ICP27 gene belongs to the set of 43 to 44 “core” herpesvirus genes which are conserved in all members of the Herpesviridae (23). For the most part, the protein products of these genes mediate functions that are fundamental to herpesviral replication and/or host interactions. Despite this conservation, the functions of the ICP27 gene family appear to have diverged over time, as the ICP27 homologs from HCMV (51), varicella-zoster virus (52, 53), and pseudorabies virus (54) are completely unable to complement the replication defect of an HSV-1 ICP27 null mutant when expressed in trans. The Epstein-Barr virus homolog can provide partial complementation, but the level is reduced 100-fold compared to the level provided by ICP27 itself (53). In contrast to these past findings, here we showed that ICP27t2 can fully complement the growth of the HSV-1 null mutant d27-1. Furthermore, when the ICP27t2 gene is inserted into the HSV-1 genome in place of the type 1 gene, the resulting mutant, known as K2F1, grows with WT kinetics and yields. Moreover, K2F1 exhibits a normal pattern of viral gene expression and efficiently induces the p38, JNK, and NF-κb signaling pathways, which are known to depend upon ICP27 for their activation. Together, these results demonstrate that ICP27t2 and ICP27 are functionally very similar and that ICP27t2 is able to carry out all of the ICP27 functions required for HSV-1 replication in cell culture. From an evolutionary perspective, the high degree of functional similarity between ICP27 and ICP27t2 is not surprising, as HSV-1 and HSV-2 are close relatives that diverged only recently in evolutionary time (∼8 million years ago) (23). Moreover, the two viruses undergo very similar interactions with their human host and cause clinically similar diseases (2).
Although ICP27t2 has not been the subject of many previous studies, some investigators have looked at its effects on mRNA splicing. Nojima et al. showed that ICP27t2 alters the splicing of the cellular transcript encoding the promyelocytic leukemia (PML) protein, specifically inhibiting removal of its intron 7a (55). More recently, Tang et al. demonstrated that ICP27t2 suppresses splicing of the HSV-2 ICP34.5 mRNA, allowing the virus to express a variant form of ICP34.5 (56). Here, we directly compared ICP27 and ICP27t2 in their effects on splicing of the HSV-1 gC transcript. Both proteins functioned similarly to suppress removal of the small intron in the gC gene (Fig. 2C). Likewise, Korom et al. recently showed that ICP27t2 and ICP27 have similar suppressive effects on HSV-2 ICP34.5 mRNA splicing (57). Together, these data demonstrate that ICP27t2 is functionally very similar to ICP27 in its inhibitory effects on mRNA splicing.
We did observe one functional difference between ICP27t2 and ICP27 in our gene transfection studies. Whereas WT ICP27 efficiently promoted the cytoplasmic localization of ICP4, ICP27t2 was relatively inefficient in this activity (Fig. 2D). Based on this finding, we predicted that K2F1-infected cells would show a decrease in cytoplasmic ICP4 at late times of infection. However, this was not found to be the case, as ICP4 was localized normally in the K2F1 infections. This may be because other HSV-1 factors also regulate ICP4 localization during infection and can compensate for the ICP27 alteration in K2F1. Consistent with this, a recent study showed that HSV-1 tegument protein VP22 promotes the cytoplasmic localization of ICP4 (58).
ICP27 modulates the efficiency of HSV-1 virion release.
Although the K2F1 mutant is indistinguishable from WT HSV-1 in most respects, we were surprised to find that it forms morphologically distinct plaques on Vero cells when the assays are carried out in a liquid medium overlay containing 1% PHS. Whereas WT plaques are comet shaped with ragged edges, K2F1 plaques are round and have well-delineated edges. The comet-like appearance of WT plaques is very likely due to secondary infections that arise from the spread of free virus through the medium, with the directionality driven by convective flows in the medium (45, 59). Similar comet-shaped plaques have been observed in several other viral systems when liquid overlays are used. These include vaccinia virus (44, 45), vesicular stomatitis virus (59), and influenza virus (60). Secondary spread of WT HSV-1 in the presence of 1% PHS is not surprising, as our neutralization analysis (Fig. 4D) showed that this concentration of PHS is insufficient to neutralize the majority of HSV-1 virions after a 30-min incubation.
K2F1 tends to form round and not comet-like plaques because it releases approximately 2-fold less progeny into the medium during each infectious cycle than does WT HSV-1 (Fig. 5B, E, and F). This makes secondary infections less likely. Results of our studies with HSV-1 ICP27 mutants dAc and d2-3 were consistent with this conclusion. We identified these two mutants as ones that form round plaques, similar to K2F1. Both were subsequently found to be deficient in release, arguing that the plaquing and release phenotypes are mechanistically linked. Moreover, in the case of d2-3, both plaquing (Fig. 6B) and release (Fig. 6D) phenotypes were more extreme than those of K2F1, again suggesting a linkage between these traits. We also note that, in the vaccinia virus system, similar 2- to 4-fold reductions in viral release efficiency can have similar negative effects on the formation of comet-like plaques (61). The deletions in the dAc and d2-3 mutants encompass ICP27 residues 20 to 63 and 64 to 108, respectively. Interestingly, ICP27t2 differs considerably from ICP27 in this same region (Fig. 1). This suggests that the N-terminal portion of ICP27, which is dispensable for replication in Vero cells (31), is involved in ICP27's release function. This idea could be tested through further mutagenesis experiments, including the construction of HSV-1 mutants bearing hybrid ICP27t2/ICP27 genes.
Regarding dAc and d2-3, we previously reported that these mutants are replication competent in Vero cells (31). However, in this study, we found that both viruses had modest replication defects, with dAc showing a 3-fold growth deficit compared to WT HSV-1 and d2-3 having an 11-fold defect. Tian et al. also recently reported that d2-3 has a modest growth defect in Vero cells (10). Although it is possible that the replication defects of dAc and d2-3 contribute to their plaque morphology phenotype, it is important to note that our virus release experiments measure percent release, and thus our analysis takes into account reductions in yields. It is interesting that, despite their reduced yields, both dAc and d2-3 produce near-WT-sized plaques when the assays are carried out in a liquid medium overlay containing high levels of PHS (5%) (data not shown). This indicates that these mutants exhibit normal or near-normal cell-to-cell spread. Consistent with this observation, Roller et al. noted that several other HSV-1 mutants with up to 50-fold reductions in virus yield can produce normal-sized plaques on epithelial cell monolayers (62).
From the perspective of viral genetics, our analysis of K2F1 clearly identifies ICP27 as a genetic determinant of HSV virion release efficiency. It should be pointed out that this conclusion is made in the context of a specific genetic analysis involving parental strains HSV-1 KOS1.1 and HSV-2 HG52. Perhaps fortuitously for our study, these strains differ quite dramatically (by more than 15-fold) in virion release. It is noteworthy that a wider analysis that included additional HSV-1 and -2 strains failed to reveal a general difference between HSV-1 and -2 release efficiency (Fig. 7). Thus, the release phenotype of HSV strains is not simply dictated by the ICP27 gene. Indeed, it is formally possible that in some HSV strains, ICP27 does not contribute to determining viral release efficiency. However, a more likely scenario is that ICP27 is just one of multiple HSV genes that influence extracellular virion release. Identifying such genes will represent an important advance in our understanding of this understudied aspect of HSV infection.
ICP27 modulates the interaction of progeny virions with the surface of infected cells.
Although the mechanism by which ICP27 affects viral release is not known, our experiments with K2F1 provide some clues. First, we found that K2F1 progeny virions efficiently reach the extracellular surface of the infected cell (Fig. 5E), indicating that ICP27's effects are after exocytosis of virion-containing vesicles. Second, we were able to use a salt wash procedure to artificially release progeny virions from the cell surface. In doing so, we found that K2F1-infected cells had a higher percentage of total progeny on the cell surface than did WT HSV-1-infected cells (Fig. 5F). This suggests that K2F1 virions are more tightly associated with the cell surface than are WT HSV-1 virions, which appear to detach more readily. Thus, ICP27 appears to modulate the efficiency with which HSV-1 progeny virions attach to the surface of the infected cell.
At present, we can only speculate on how ICP27 modulates the interaction between progeny virions and the cell surface. However, as there is no evidence that ICP27 is localized to the cell surface, it is likely that ICP27's role is indirect. We can envision two general models. First, as ICP27 has a well-described role in viral gene regulation, it is possible that its effects are due to altered expression of one or more viral gene products that are involved in virion release. We do not favor this model because our phenotypic characterization of K2F1 showed that its gene expression profile is nearly indistinguishable from WT HSV-1. The second model is that ICP27 carries out an activity unrelated to its gene expression function that affects attachment of virions to cells. One potential clue in this regard is that ICP27 N-terminal sequences appear to be important in release, as discussed above. Given that N-terminal sequences can dramatically affect the cellular distribution of ICP0 and ICP4 (19, 22), it is possible that ICP27 also regulates the localization of one or more viral proteins directly involved in release. Interestingly, recent work has shown that the extracellular release of HSV-1 from Vero cells occurs at specific membrane patches enriched for viral glycoproteins (49). With this in mind, it would be interesting to compare the extent and glycoprotein content of these patches in cells infected with WT HSV-1 versus ICP27 release mutants.
To further understand how ICP27 affects virion detachment, it will be important to define the molecular interactions that tether progeny HSV-1 virions to the surfaces of infected cells. To our knowledge, little is known about this subject. Attachment may be mediated by viral envelope proteins gC and gB, which bind to heparan sulfate- or chondroitin sulfate-containing proteoglycans widely present on mammalian cell surfaces (63). Indeed, such binding is important at the beginning of infection for attachment of incoming virions. Supporting a role for these interactions in release, it has been shown that polyanionic mimics of heparan sulfate can elute HSV-2 progeny virions from the surface of infected cells (64). It is also possible that progeny virions use the viral envelope protein gD to interact with cellular receptors, such as HVEM or nectin-1 (63). Another possibility is that the cellular restriction factor tetherin (65) plays a role in adhering progeny virions to the cell surface. Consistent with this, recent work showed that tetherin is capable of inhibiting HSV-1 release (66, 67). On the other hand, Vero cells are reported to not express tetherin (66), suggesting that this molecule did not play a role in our studies.
Implications for HSV-1 pathogenesis.
Our work shows that changes in the sequence of ICP27 can alter, up to 4-fold, the efficiency with which HSV-1 progeny virions are released into the extracellular milieu. The release of HSV-1 is likely to play an important role in viral pathogenesis. However, it is doubtful whether highly efficient release of free virus is favored during natural HSV-1 infections in vivo. In this setting, HSV-1 is known to be adept at cell-cell spread via cellular junctions, with the virus using this method to move stealthily between epithelial cells and into and out of neurons (46). The tethering of progeny virions to the surface of the producer cell may promote this by ensuring that virions are situated in close proximity to their adjacent target cells. At the same time, the tethering of progeny would be expected to limit dissemination of infection and thus help the virus avoid a strong host immune response. On the other hand, in the context of a reactivated HSV-1 infection, release of free virions may be essential for transmission of the virus to new hosts. Thus, it is possible that in vivo, HSV-1 must carefully modulate the release of extracellular virus. Our work suggests that ICP27 could be one component of such a regulatory system.
It is clear from our work that the release efficiency of a particular HSV strain is a genetically determined trait that can differ over a wide range. It is quite possible that this trait significantly affects the results of some types of virological analysis, e.g., the analysis of HSV pathogenesis in experimental animals. Thus, the inherent release efficiency of a given viral strain is a property that should be considered by HSV researchers. Complicating the picture, however, is the likelihood that HSV release rates can evolve in vitro, according to culture and harvest conditions, as viral strains are passaged in the laboratory. For this reason, it will be both interesting and important to study release in low-passage-number clinical isolates of HSV-1 and HSV-2.
ACKNOWLEDGMENTS
We thank Leon Sanders III for analysis of K2F1 replication in HEp-2 cells. We are also indebted to Leslie Schiff for many insightful discussions and to Jin-Young Han, Jim Lokensgard, and Alistair McGregor for providing HSV-1 and HSV-2 strains.
This research was supported by a grant from the NIH (R01-AI42737).
REFERENCES
- 1.Xu F, Sternberg MR, Kottiri BJ, McQuillan GM, Lee FK, Nahmias AJ, Berman SM, Markowitz LE. 2006. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 296:964–973. doi: 10.1001/jama.296.8.964. [DOI] [PubMed] [Google Scholar]
- 2.Roizman B, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses, p 2501–2601 InKnipe DM, Howley PM (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Sandri-Goldin RM. 2011. The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol 6:1261–1277. doi: 10.2217/fmb.11.119. [DOI] [PubMed] [Google Scholar]
- 4.Sandri-Goldin RM. 1998. ICP27 mediates HSV RNA export by shuttling through a leucine-rich nuclear export signal and binding viral intronless RNAs through an RGG motif. Genes Dev 12:868–879. doi: 10.1101/gad.12.6.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mears WE, Rice SA. 1998. The herpes simplex virus immediate-early protein ICP27 shuttles between nucleus and cytoplasm. Virology 242:128–137. doi: 10.1006/viro.1997.9006. [DOI] [PubMed] [Google Scholar]
- 6.Corbin-Lickfett KA, Chen IH, Cocco MJ, Sandri-Goldin RM. 2009. The HSV-1 ICP27 RGG box specifically binds flexible, GC-rich sequences but not G-quartet structures. Nucleic Acids Res 37:7290–7301. doi: 10.1093/nar/gkp793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rice SA, Knipe DM. 1990. Genetic evidence for two distinct transactivation functions of the herpes simplex virus alpha protein ICP27. J Virol 64:1704–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uprichard SL, Knipe DM. 1996. Herpes simplex ICP27 mutant viruses exhibit reduced expression of specific DNA replication genes. J Virol 70:1969–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson LA, Li L, Sandri-Goldin RM. 2009. The cellular RNA export receptor TAP/NXF1 is required for ICP27-mediated export of herpes simplex virus 1 RNA, but the TREX complex adaptor protein Aly/REF appears to be dispensable. J Virol 83:6335–6346. doi: 10.1128/JVI.00375-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian X, Devi-Rao G, Golovanov AP, Sandri-Goldin RM. 2013. The interaction of the cellular export adaptor protein Aly/REF with ICP27 contributes to the efficiency of herpes simplex virus 1 mRNA export. J Virol 87:7210–7217. doi: 10.1128/JVI.00738-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McLauchlan J, Phelan A, Loney C, Sandri-Goldin RM, Clements JB. 1992. Herpes simplex virus IE63 acts at the posttranscriptional level to stimulate viral mRNA 3′ processing. J Virol 66:6939–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellison KS, Maranchuk RA, Mottet KL, Smiley JR. 2005. Control of VP16 translation by the herpes simplex virus type 1 immediate-early protein ICP27. J Virol 79:4120–4131. doi: 10.1128/JVI.79.7.4120-4131.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fontaine-Rodriguez EC, Knipe DM. 2008. Herpes simplex virus ICP27 increases translation of a subset of viral late mRNAs. J Virol 82:3538–3405. doi: 10.1128/JVI.02395-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardy WR, Sandri-Goldin RM. 1994. Herpes simplex virus inhibits host cell splicing, and regulatory protein ICP27 is required for this effect. J Virol 68:7790–7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sedlackova L, Perkins KD, Lengyel J, Strain AK, van Santen VL, Rice SA. 2008. Herpes simplex virus type 1 ICP27 regulates expression of a variant, secreted form of glycoprotein C by an intron retention mechanism. J Virol 82:7443–7455. doi: 10.1128/JVI.00388-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gillis PA, Okagaki LH, Rice SA. 2009. Herpes simplex virus type 1 ICP27 induces p38 mitogen-activated protein kinase signaling and apoptosis in HeLa cells. J Virol 83:1767–1777. doi: 10.1128/JVI.01944-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hargett D, McLean T, Bachenheimer SL. 2005. Herpes simplex virus ICP27 activation of stress kinases JNK and p38. J Virol 79:8348–8360. doi: 10.1128/JVI.79.13.8348-8360.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hargett D, Rice S, Bachenheimer SL. 2006. Herpes simplex virus type 1 ICP27-dependent activation of NF-κB. J Virol 80:10565–10578. doi: 10.1128/JVI.01119-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lengyel J, Strain AK, Perkins KD, Rice SA. 2006. ICP27-dependent resistance of herpes simplex virus type 1 to leptomycin B is associated with enhanced nuclear localization of ICP4 and ICP0. Virology 352:368–379. doi: 10.1016/j.virol.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 20.Zhu Z, Cai W, Schaffer PA. 1994. Cooperativity among herpes simplex virus type 1 immediate-early regulatory proteins: ICP4 and ICP27 affect the intracellular localization of ICP0. J Virol 68:3027–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Z, Schaffer PA. 1995. Intracellular localization of the herpes simplex virus type 1 major transcriptional regulatory protein, ICP4, is affected by ICP27. J Virol 69:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sedlackova L, Rice SA. 2008. Herpes simplex virus type 1 immediate-early protein ICP27 is required for efficient incorporation of ICP0 and ICP4 into virions. J Virol 82:268–277. doi: 10.1128/JVI.01588-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davison AJ. 2011. Evolution of sexually transmitted and sexually transmissible human herpesviruses. Ann N Y Acad Sci 1230:E37–E49. doi: 10.1111/j.1749-6632.2011.06358.x. [DOI] [PubMed] [Google Scholar]
- 24.Morse LS, Buchman TG, Roizman B, Schaffer PA. 1977. Anatomy of herpes simplex virus DNA. IX. Apparent exclusion of some parental DNA arrangements in the generation of intertypic (HSV-1 X HSV-2) recombinants. J Virol 24:231–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Preston VG, Davison AJ, Marsden HS, Timbury MC, Subak-Sharpe JH, Wilkie NM. 1978. Recombinants between herpes simplex virus types 1 and 2: analyses of genome structures and expression of immediate early polypeptides. J Virol 28:499–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soliman TM, Silverstein SJ. 2000. Herpesvirus mRNAs are sorted for export via Crm1-dependent and -independent pathways. J Virol 74:2814–2825. doi: 10.1128/JVI.74.6.2814-2825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lischka P, Thomas M, Toth Z, Mueller R, Stamminger T. 2007. Multimerization of human cytomegalovirus regulatory protein UL69 via a domain that is conserved within its herpesvirus homologues. J Gen Virol 88:405–410. doi: 10.1099/vir.0.82480-0. [DOI] [PubMed] [Google Scholar]
- 28.Chen IH, Sciabica KS, Sandri-Goldin RM. 2002. ICP27 interacts with the RNA export factor Aly/REF to direct herpes simplex virus type 1 intronless mRNAs to the TAP export pathway. J Virol 76:12877–12889. doi: 10.1128/JVI.76.24.12877-12889.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughes RG Jr, Munyon WH. 1975. Temperature-sensitive mutants of herpes simplex virus type 1 defective in lysis but not in transformation. J Virol 16:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park D, Lengyel J, Rice SA. 2013. Role of immediate early protein ICP27 in the differential sensitivity of herpes simplex viruses 1 and 2 to leptomycin B. J Virol 87:8940–8951. doi: 10.1128/JVI.00633-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lengyel J, Guy C, Leong V, Borge S, Rice SA. 2002. Mapping of functional regions in the amino-terminal portion of the herpes simplex virus ICP27 regulatory protein: importance of the leucine-rich nuclear export signal and RGG box RNA-binding domain. J Virol 76:11866–11879. doi: 10.1128/JVI.76.23.11866-11879.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newcomb WW, Brown JC. 2010. Structure and capsid association of the herpesvirus large tegument protein UL36. J Virol 84:9408–9414. doi: 10.1128/JVI.00361-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perkins KD, Gregonis J, Borge S, Rice SA. 2003. Transactivation of a viral target gene by herpes simplex virus ICP27 is posttranscriptional and does not require the endogenous promoter or polyadenylation site. J Virol 77:9872–9884. doi: 10.1128/JVI.77.18.9872-9884.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rice SA, Knipe DM. 1988. Gene-specific transactivation by herpes simplex virus type 1 alpha protein ICP27. J Virol 62:3814–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeLuca NA, Schaffer PA. 1987. Activities of herpes simplex virus type 1 (HSV-1) ICP4 genes specifying nonsense peptides. Nucleic Acids Res 15:4491–4511. doi: 10.1093/nar/15.11.4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nabel GJ, Rice SA, Knipe DM, Baltimore D. 1988. Alternative mechanisms for activation of human immunodeficiency virus enhancer in T cells. Science 239:1299–1302. doi: 10.1126/science.2830675. [DOI] [PubMed] [Google Scholar]
- 37.Rice SA, Lam V, Knipe DM. 1993. The acidic amino-terminal region of herpes simplex virus type 1 alpha protein ICP27 is required for an essential lytic function. J Virol 67:1778–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rice SA, Lam V. 1994. Amino acid substitution mutations in the herpes simplex virus ICP27 protein define an essential gene regulation function. J Virol 68:823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diaz JJ, Simonin D, Masse T, Deviller P, Kindbeiter K, Denoroy L, Madjar JJ. 1993. The herpes simplex virus type 1 US11 gene product is a phosphorylated protein found to be non-specifically associated with both ribosomal subunits. J Gen Virol 74:397–406. doi: 10.1099/0022-1317-74-3-397. [DOI] [PubMed] [Google Scholar]
- 40.Mears WE, Lam V, Rice SA. 1995. Identification of nuclear and nucleolar localization signals in the herpes simplex virus regulatory protein ICP27. J Virol 69:935–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sacks WR, Greene CC, Aschman DP, Schaffer PA. 1985. Herpes simplex virus type 1 ICP27 is an essential regulatory protein. J Virol 55:796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McMahan L, Schaffer PA. 1990. The repressing and enhancing functions of the herpes simplex virus regulatory protein ICP27 map to C-terminal regions and are required to modulate viral gene expression very early in infection. J Virol 64:3471–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blaho JA, Morton ER, Yedowitz JC. 2005. Herpes simplex virus: propagation, quantification, and storage. Curr Protoc Microbiol Chapter 14:Unit 14E.11. [DOI] [PubMed] [Google Scholar]
- 44.Blasco R, Sisler JR, Moss B. 1993. Dissociation of progeny vaccinia virus from the cell membrane is regulated by a viral envelope glycoprotein: effect of a point mutation in the lectin homology domain of the A34R gene. J Virol 67:3319–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Law M, Hollinshead R, Smith GL. 2002. Antibody-sensitive and antibody-resistant cell-to-cell spread by vaccinia virus: role of the A33R protein in antibody-resistant spread. J Gen Virol 83:209–222. [DOI] [PubMed] [Google Scholar]
- 46.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 47.Sattentau Q. 2008. Avoiding the void: cell-to-cell spread of human viruses. Nat Rev Microbiol 6:815–826. doi: 10.1038/nrmicro1972. [DOI] [PubMed] [Google Scholar]
- 48.Johnson DC, Webb M, Wisner TW, Brunetti C. 2001. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J Virol 75:821–833. doi: 10.1128/JVI.75.2.821-833.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mingo RM, Han J, Newcomb WW, Brown JC. 2012. Replication of herpes simplex virus: egress of progeny virus at specialized cell membrane sites. J Virol 86:7084–7097. doi: 10.1128/JVI.00463-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai WZ, Person S, DebRoy C, Gu BH. 1988. Functional regions and structural features of the gB glycoprotein of herpes simplex virus type 1. An analysis of linker insertion mutants. J Mol Biol 201:575–588. [DOI] [PubMed] [Google Scholar]
- 51.Winkler M, Rice SA, Stamminger T. 1994. UL69 of human cytomegalovirus, an open reading frame with homology to ICP27 of herpes simplex virus, encodes a transactivator of gene expression. J Virol 68:3943–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moriuchi H, Moriuchi M, Smith HA, Cohen JI. 1994. Varicella-zoster virus open reading frame 4 protein is functionally distinct from and does not complement its herpes simplex virus type 1 homolog, ICP27. J Virol 68:1987–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boyer JL, Swaminathan S, Silverstein SJ. 2002. The Epstein-Barr virus SM protein is functionally similar to ICP27 from herpes simplex virus in viral infections. J Virol 76:9420–9433. doi: 10.1128/JVI.76.18.9420-9433.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schwartz JA, Brittle EE, Reynolds AE, Enquist LW, Silverstein SJ. 2006. UL54-null pseudorabies virus is attenuated in mice but productively infects cells in culture. J Virol 80:769–784. doi: 10.1128/JVI.80.2.769-784.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nojima T, Oshiro-Ideue T, Nakanoya H, Kawamura H, Morimoto T, Kawaguchi Y, Kataoka N, Hagiwara M. 2009. Herpesvirus protein ICP27 switches PML isoform by altering mRNA splicing. Nucleic Acids Res 37:6515–6527. doi: 10.1093/nar.gkp633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang S, Guo N, Patel A, Krause PR. 2013. Herpes simplex virus 2 expresses a novel form of ICP34.5, a major viral neurovirulence factor, through regulated alternative splicing. J Virol 87:5820–5830. doi: 10.1128/JVI.03500-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Korom M, Davis KL, Morrison LA. 2014. Up to four distinct polypeptides are produced from the γ34.5 open reading frame of herpes simplex virus 2. J Virol 88:11284–11296. doi: 10.1128/JVI.01284-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tanaka M, Kato A, Satoh Y, Ide T, Sagou K, Kimura K, Hasegawa H, Kawaguchi Y. 2012. Herpes simplex virus 1 VP22 regulates translocation of multiple viral and cellular proteins and promotes neurovirulence. J Virol 86:5264–5277. doi: 10.1128/JVI.06913-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu Y, Yin J. 2007. A quantitative comet assay: imaging and analysis of virus plaques formed with a liquid overlay. J Virol Methods 139:100–102. doi: 10.1016/j.jviromet.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 60.Lindsay SM, Timm A, Yin J. 2012. A quantitative comet infection assay for influenza virus. J Virol Methods 179:351–358. doi: 10.1016/j.jviromet.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horsington J, Lynn H, Turnbull L, Cheng D, Braet F, Diefenbach RJ, Whitchurch CB, Karupiah G, Newsome TP. 2013. A36-dependent actin filament nucleation promotes release of vaccinia virus. PLoS Pathog 9:e1003239. doi: 10.1371/journal.ppat.1003239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roller RJ, Haugo AC, Yang K, Baines JD. 2014. The herpes simplex virus 1 UL51 gene product has cell type-specific functions in cell-to-cell spread. J Virol 88:4058–4068. doi: 10.1128/JVI.03707-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krummenacher C, Carfi A, Eisenberg RJ, Cohen GH. 2013. Entry of herpesviruses into cells: the enigma variations. Adv Exp Med Biol 790:178–195. doi: 10.1007/978-1-4614-7651-1_10. [DOI] [PubMed] [Google Scholar]
- 64.O'Keeffe R, Johnston MD, Slater NK. 1998. The primary production of an infectious recombinant herpes simplex virus vaccine. Biotechnol Bioeng 57:262–271. doi:. [DOI] [PubMed] [Google Scholar]
- 65.Sauter D. 2014. Counteraction of the multifunctional restriction factor tetherin. Front Microbiol 5:163. doi: 10.3389/fmicb.2014.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Blondeau C, Pelchen-Matthews A, Mlcochova P, Marsh M, Milne RS, Towers GJ. 2013. Tetherin restricts herpes simplex virus 1 and is antagonized by glycoprotein M. J Virol 87:13124–13133. doi: 10.1128/JVI.02250-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zenner HL, Mauricio R, Banting G, Crump CM. 2013. Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J Virol 87:13115–13123. doi: 10.1128/JVI.02167-13. [DOI] [PMC free article] [PubMed] [Google Scholar]