

ABSTRACT
Human papillomavirus (HPV) is a significant oncogenic virus, but the innate immune response to HPV is poorly understood. Human α-defensin 5 (HD5) is an innate immune effector peptide secreted by epithelial cells in the genitourinary tract. HD5 is broadly antimicrobial, exhibiting potent antiviral activity against HPV at physiologic concentrations; however, the specific mechanism of HD5-mediated inhibition against HPV is unknown. During infection, the HPV capsid undergoes several critical cell-mediated viral protein processing steps, including unfolding and cleavage of the minor capsid protein L2 by host cyclophilin B and furin. Using HPV16 pseudovirus, we show that HD5 interacts directly with the virus and inhibits the furin-mediated cleavage of L2 at the cell surface during infection at a step downstream of the cyclophilin B-mediated unfolding of L2. Importantly, HD5 does not affect the enzymatic activity of furin directly. Thus, our data support a model in which HD5 prevents furin from accessing L2 by occluding the furin cleavage site via direct binding to the viral capsid.
IMPORTANCE Our study elucidates a new antiviral action for α-defensins against nonenveloped viruses in which HD5 directly interferes with a critical host-mediated viral processing step, furin cleavage of L2, at the cell surface. Blocking this key event has deleterious effects on the intracellular steps of virus infection. Thus, in addition to informing the antiviral mechanisms of α-defensins, our studies highlight the critical role of furin cleavage in HPV entry. Innate immune control, mediated in part by α-defensins expressed in the genital mucosa, may influence susceptibility to HPV infections that lead to cervical cancer. Moreover, understanding the mechanism of these natural antivirals may inform the design of therapeutics to limit HPV infection.
INTRODUCTION
Defensins are effector peptides of the human innate immune system. They are divided into two classes, α- and β-defensins, based on the pattern of disulfide bonds that stabilize their tertiary structure (1, 2). HD5 is one of six human α-defensins and is constitutively expressed and secreted in the female and male genitourinary tracts (3–5). Concentrations of HD5 in vaginal lavage fluid of healthy women have been reported to be 16.5 ± 10.5 μM (3). Although originally discovered due to their antibacterial activity, defensin antiviral activity against both enveloped and nonenveloped viruses has also been described. Neutralization of enveloped viruses, such as human immunodeficiency virus 1 (HIV-1), herpes simplex virus (HSV), and respiratory syncytial virus (RSV), is largely dependent on direct interactions of defensins with both viral attachment proteins and cellular receptors, as well as envelope damage, fusion inhibition, and modulation of host responses (6). Inhibition of these viruses may be due to multiple defensin actions rather than a single overriding inhibitory mechanism. While less is known about the mechanisms of defensin antiviral activity against nonenveloped viruses, human adenoviruses (HAdVs), papillomaviruses, and polyomaviruses (PyVs) are all neutralized by specific α-defensins at physiologic concentrations (7–11). Of these viruses, only PyV infection is inhibited by β-defensins (8). The α-defensin-mediated neutralization mechanisms of HAdV, JC PyV, and BK PyV have been described in some detail. In each case, α-defensins bind to the viral capsids outside the cell to block infection. For both HAdV and JC PyV, α-defensin binding alters intracellular trafficking of the incoming virion (9, 10). In the case of HAdV, escape of the virus from the endosome is blocked due to a failure to uncoat (10, 12). For JC PyV, trafficking to the endoplasmic reticulum (ER) is reduced (9). In contrast, extracellular aggregation of BK PyV through α-defensin interactions alone is sufficient to attenuate infection (8). Although some aspects of human papillomavirus (HPV) neutralization by α-defensins have been described, the step in the viral entry pathway that is blocked has not been identified (7).
The cellular entry pathway of HPV is complex. The capsid is comprised of two structural proteins, the major capsid protein L1 and the minor protein L2. The majority of L2 is protected within the L1 capsid, although there is a portion of L2 at the N terminus that is thought to be surface exposed (7, 13). During infection, the virus initially binds to heparin sulfate proteoglycans (HSPGs) on the extracellular matrix (ECM) through L1 (14, 15). The virus then passes to HSPGs on basal keratinocytes, and L1 undergoes a conformational change that exposes more of L2 (16). It is unclear if the L1 change happens while the virus is still attached to the basal membrane or to the cell surface (17). On the cell surface, host cyclophilin B (CyPB) binds to the exposed portion of L2 and unfolds a region of the N terminus that contains a highly conserved furin cleavage site (18). Extracellular host furin then cleaves L2, and the virus is passed to an unknown secondary receptor that mediates internalization (19, 20). After internalization, the virus traffics through the endosomal system, requiring interactions with sorting nexin 17 in the endosome as well as the retromer complex and γ-secretase in the trans-Golgi network (21–27). During this process, acidification and further conformational changes induced by CyPB result in L1 and L2 dissociation and viral uncoating (28, 29). L2 then interacts with and permeabilizes the host cell membrane, actions that are dependent upon prior furin cleavage at the cell surface (30, 31). The L2-genome complex then travels to the nucleus via dynein-mediated transport along microtubules and localizes to nuclear PML/ND10 domains, where viral mRNA is transcribed (32–35). Overall, the HPV16 entry pathway is best characterized, and there are multiple indications that some of these steps differ among HPV serotypes (36, 37). As such, we focused our studies on HPV16.
Previous work indicated that HD5 is most effective when present with HPV16 at the cell surface (7). Thus, we hypothesized that binding to the virus in the extracellular milieu would alter or inhibit cell surface steps in the viral life cycle, resulting in deleterious effects on entry. Consistent with this model, treatment of cells with HD5 before infection or after the virus has been internalized has minimal effect on infection. We first demonstrated that purified HD5 can aggregate HPV16 PsV, confirming a direct interaction that was suggested by previous studies. By assaying the steps in HPV16 entry that occur on the cell surface, we found that furin cleavage and exposure of the RG-1 epitope in L2 is blocked by HD5 binding. Disruption of this conserved, critical step in HPV entry is consistent with the previously described failure of the genome to escape the endosomal pathway due to HD5 inhibition and provides a rationale for the broad activity of HD5 against mucosal and cutaneous HPV types.
MATERIALS AND METHODS
Cell culture and pseudovirus (PsV) production.
293TT cells were a kind gift from Denise Galloway (University of Washington, Seattle, WA). HaCaT cells were a kind gift from Paul Nghiem (University of Washington, Seattle, WA). HeLa cells were purchased from American Type Culture Collection (ATCC). Cell culture reagents were purchased from Corning CellGro. All cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (Sigma-Aldrich), 100 units/ml penicillin, 100 μg/ml streptomycin, 4 mM l-glutamine, and 0.1 mM nonessential amino acids (complete medium). 293TT culture medium was supplemented with 0.4 mg/ml hygromycin B (Sigma-Aldrich).
HPV16 PsV was made as previously described via viral propagation and the improved maturation method (38, 39) (http://home.ccr.cancer.gov/lco/ImprovedMaturation.htm). PsV seed stock was made by cotransfecting 293TT cells with plasmids encoding codon-optimized HPV16 L1 and L2 (p16L1L2; kind gift of Martin Müller, GCRC) and an enhanced green fluorescent protein (eGFP) reporter (pfwB; kind gift of John Schiller, National Cancer Institute, Bethesda, MD). Cleared lysate from this transfection contained mature PsV and was then used to infect additional 293TT cells. Mature PsV from the lysate of these cells was purified by ultracentrifugation on a discontinuous Optiprep (Sigma-Aldrich) density gradient (steps of 27% to 33% to 39% Optiprep). To make 16L2-GP-N HPV16 PsV (18), L2 G99A and P100A mutations were introduced by short overlap extension PCR into the L2 open reading frame (ORF) of the p16L1L2 vector. The mutated plasmid was then used to create PsVs by viral propagation as described above. Similarly, Myc-16L2-HA HPV16 PsV was made by fusing a Myc epitope tag (EQKLISEEDL) to the amino terminus and a hemagglutinin (HA) tag (YPVYDVPDYA) to the carboxyl terminus of L2 via PCR in the p16L1L2 vector. In all cases, the L1 content of purified PsVs was quantified by SDS-PAGE and SYPRO ruby (Life Technologies) stain compared to a bovine serum albumin (BSA) standard. L1 protein content was converted to particle number using a conversion factor of 3.0 × 107 particles/ng L1 (http://home.ccr.cancer.gov/lco/pseudovirusproduction.htm).
Dynamic light scattering.
Folded HD5 was made from a synthesized 80% pure linearized peptide (CPC Scientific, Sunnyvale, CA) and purified by reverse-phase high-pressure liquid chromatography, and an HD5 derivative containing l-α-aminobutyric acid in place of cysteine (HD5 Abu) was chemically synthesized, as previously described (11, 40). HD5 or HD5 Abu was serially diluted in 10 mM Tris and 150 mM NaCl, pH 7.5, and mixed with 1.3 × 1010 p/ml of HPV16 PsV in a final volume of 50 μl in disposable cuvettes (Malvern Instruments). Control samples of HPV16 PsV alone were diluted in the same buffer. Samples were incubated on ice for 45 min and equilibrated at 37°C for 3 min before analysis. Each sample was measured using a Zetasizer Nano ZS (Malvern Instruments), and the volume mean diameter was obtained from the manufacturer's software. The fold change was calculated by dividing the average diameter of the defensin-treated samples by the average diameter of the untreated controls for each of four independent replicates. Experiments were analyzed by two-way analysis of variance (ANOVA) with Bonferroni posttests to compare each concentration of the HD5 isoform to the control sample using Prism (version 5.0d; GraphPad Software, Inc., La Jolla, CA). For all tests, P values of <0.05 were considered significant.
Immunofluorescence microscopy.
To label PsV with Alexa Fluor 555 (AF555), the standard PsV maturation and lysis buffers were altered to omit ammonium sulfate (41, 42). After PsV maturation, cleared lysate (750 μl) was diluted with an equal volume of Dulbecco's phosphate-buffered saline (DPBS), 1 M NaH3CO2 was added to a final concentration of 100 mM, and 35 μg of Alexa Fluor 555 dye dissolved in dimethyl sulfoxide (DMSO) was added. After 1 h of incubation at room temperature (RT), the labeling reaction was neutralized by the addition of 13 mM NaPO4. Labeled PsV was purified from the lysate by ultracentrifugation through an Optiprep gradient, and HPV16 particle number was determined as described above. A total of 2.5 × 109 particles of AF555-HPV16 PsV were incubated with or without 5 μM HD5 or 5.4 μg/ml RG-1 (43) (kind gift of Richard Roden, Johns Hopkins University, Baltimore, MD) alone or in combination for 45 min on ice in complete medium. As a control, 5 μM HD5 and 5.4 μg/ml RG-1 were also incubated together in complete medium without AF555-HPV16 PsV. The samples were then added to HaCaT cells plated on coverslips (seeded at 3 × 105 cells/coverslip 24 h prior to infection) and allowed to infect for 12 h. Assuming that the cells doubled overnight, this is equivalent to a multiplicity of infection (MOI) of 4 × 103 PsV particles/cell. Cells were fixed using 4% paraformaldehyde (PFA) and then quenched and permeabilized in 20 mM glycine-0.5% Triton X-100 in PBS. All samples were stained using goat anti-mouse Alex Fluor 488 (Life Technologies) in blocking buffer (1% BSA-0.05% Tween 80) for 45 min and TO-PRO (Life Technologies) in PBS-1% Tween 20. Samples were mounted using ProLong Gold (Life Technologies). Three fields of view were captured for each sample on a Zeiss 510 Meta laser scanning confocal microscope.
Image analysis was performed using ImageJ of maximum-intensity z-profiles of cells, excluding images in the z-series above or below the plane of the nucleus (44). Cell boundaries were defined using bright-field images. Thresholds for green (antibody) and red (PsV) channels above the background were determined using uninfected cells. Manders coefficients were obtained using the JaCoP plugin of ImageJ for 40 to 50 individual cells per condition (45). M1 coefficients for individual cells were converted to percentages for Fig. 2D and 4B. M2 coefficients for individual cells were converted to percentages for Fig. 2E and 4C. Experiments were analyzed by unpaired t test using Prism. For all tests, P values of <0.05 were considered significant.
FIG 2.

(A) HD5 neutralizes AF555-HPV16 PsV in complete medium. HeLa cells were infected with AF555-HPV16 PsV incubated with increasing concentrations of HD5 in complete medium. Data are from three independent experiments normalized to control infection in the absence of inhibitor ± SD. IC50 = 1.1 μM, 95% CI = 0.93 to 1.32 μM. (B) RG-1 antibody neutralizes AF555-HPV16 PsV. HeLa cells were infected with AF555-HPV16 PsV incubated with increasing concentrations of RG-1 antibody in complete medium. Data are from three independent experiments normalized to control infection in the absence of inhibitor ± SD. IC50 = 1,759-fold dilution, 95% CI = 1,428- to 2,167-fold dilution. (C) The presence of HD5 prevents binding of the RG-1 antibody to HPV16 during cell entry. Images of HaCaT cells 12 h p.i. with [(+) HPV16] or without [(−) HPV16] AF555-HPV16 PsV in the presence of no inhibitor (virus alone), 5 μM HD5 (HD5), 5.4 μg/ml RG-1 antibody equivalent to a 200-fold dilution (RG-1), or 5 μM HD5 and 5.4 μg/ml RG-1 together (RG-1 + HD5). Individual panels depict maximum intensity z-projections of signal above the threshold for images in the z-stack that are coplanar with the nucleus for HPV16 (red) and RG-1 (2° Ab, green). In the merged images, the nucleus is blue. Scale bar is 10 μm. Manders coefficient values M1 (D) and M2 (E) are plotted as percentages of RG-1 colocalized with HPV16 and percentages of HPV16 colocalized with RG-1, respectively, for 50 to 60 cells for each condition. Whiskers are 5 to 95%, the horizontal line is the median, and outliers are depicted as individual points. ***, P < 0.0001. (F) HPV16 PsV treated with a combination of 5.4 μg/ml RG-1 and 5 μM HD5 is neutralized. Data are the means ± SD from 3 independent experiments normalized to infection in the absence of inhibitor.
FIG 4.

The cyclophilin B-independent HPV16 mutant (16L2-GP-N) remains sensitive to HD5 neutralization. (A) Images of HaCaT cells 12 h p.i. with AF555-16L2-GP-N PsV in the presence of no inhibitor (virus alone), 5 μM HD5 (HD5), 5.4 μg/ml RG-1 antibody (RG-1), or 5 μM HD5 and 5.4 μg/ml RG-1 together (RG-1 + HD5). Individual panels depict maximum intensity z-projections of signal above the threshold for images in the z-stack that are coplanar with the nucleus for 16L2-GP-N (red) and RG-1 (2° Ab; green). In the merged images, the nucleus is blue. Scale bar is 10 μm. Manders coefficient values M1 (B) and M2 (C) are plotted as percentages of RG-1 colocalized with 16L2-GP-N and percentages of 16L2-GP-N colocalized with RG-1, respectively, for 45 to 60 cells for each condition. Whiskers are 5 to 95%, the horizontal line is the median, and outliers are depicted as individual points. ***, P < 0.0001. (D) HD5 neutralizes 16L2-GP-N. HeLa cells were infected with WT HPV16 PsV (black circles, IC50 = 0.88 μM, 95% CI = 0.78 to 0.99 μM) or 16L2-GP-N PsV (open circles, IC50 = 0.89 μM, 95% CI = 0.72 to 0.92 μM) incubated with increasing concentrations of HD5. Data are the means ± SD from three independent experiments compared to control infection in the absence of inhibitor.
Neutralization assays.
Serial dilutions of HPV16 PsVs in serum-free DMEM (SFM) were used to infect HeLa cells in 96-well plates for 4 h. The cells were washed and cultured in complete medium for ∼44 h. Total eGFP expression was quantified 48 h postinfection (p.i.) with a Typhoon 9400 variable mode imager (GE Healthcare) and ImageJ software. A standard curve of infection was constructed in Prism software. A virus concentration resulting in ∼80% total signal was used in inhibition studies.
To determine antiviral concentrations of HD5 or RG-1, increasing concentrations of either inhibitor were incubated with HPV16 PsV on ice for 45 min in SFM (Fig. 4D and 5A) or complete medium (Fig. 2A and B and 5A). The mixture was added to HeLa cells in a 96-well plate and incubated at 37°C for 4 h. Cells were washed and cultured with complete medium for ∼44 h. Total eGFP fluorescence was quantified as described above and normalized to a control sample infected without inhibitors using ImageJ. Fifty-percent inhibitory concentrations (IC50) were determined using nonlinear regression in Prism.
FIG 5.

(A) Myc-L2-HA HPV16 PsV is sensitive to HD5. HeLa cells were infected with WT HPV16 PsV in SFM (black circles, IC50 = 1.47 μM, 95% CI = 1.35 to 1.6 μM), Myc-L2-HA PsV in SFM (open circles, IC50 = 1.71 μM, 95% CI = 1.6 to 1.8 μM), or 10 times as much Myc-L2-HA PsV in complete medium (gray circles, IC50 = 0.79 μM, 95% CI = 0.69 to 0.9 μM) incubated with increasing concentrations of HD5. Data are means ± SD from three independent experiments normalized to infection in the absence of inhibitor. (B) HD5 inhibits furin cleavage of L2. HeLa cells were infected with Myc-16L2-HA HPV16 PsV in the presence of 40 μM furin inhibitor (FI), 1 to 10 μM HD5, or no inhibitor (−). L2 cleavage was assessed by immunoblotting of cell lysates 16 h p.i. using an anti-HA antibody. Cleaved L2 (arrow) is visible as a faster-migrating band below uncleaved L2. Shown are three independent experiments. (C) Analysis of a greater amount of lysate confirms the inhibition of furin cleavage. Different amounts of HD5-treated HPV16 PsV lysate, indicated by fold change relative to the amounts loaded in panel B, were assessed by immunoblotting with anti-HA antibody. (D) HD5 does not directly affect the enzymatic activity of furin. A total of 1.8 ng rL2:1–160 was digested with 1 U of furin in the presence or absence of the indicated inhibitors for 1 h at 30°C. Samples were immunoblotted using an anti-His antibody. Cleaved rL2:1–160 is the faster-migrating band. Shown are two independent experiments. (E) Titration of furin required for rL2:1–160 cleavage. rL2:1–160 was digested with a 3-fold dilution series of furin, starting at 1 U of total furin. Samples were resolved on a reducing gel and immunoblotted using anti-His antibody. Cleaved rL2:1–160 is the faster-migrating band.
rL2:13–36 and rL2:1–160 production.
To generate rL2:13–36, an ORF encoding amino acids 13 to 36 of HPV16 L2 with a C-terminal HA epitope tag, ProTEV enzyme cleavage site, and 6× His epitope tag was cloned into pRSET-A (Life Technologies). The rL2:1–160 construct encoded an N-terminal Myc tag and residues 1 to 160 of HPV16 L2 with a C-terminal 6× His tag. A total of 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) was used to induce protein expression in BL21 CodonPlus(DE3)-RIPL or BL21-Gold(DE3) Escherichia coli (Stratagene), and the protein was purified using TALON resin (Clontech) according to the manufacturer's instructions. Purified protein was exchanged into 50 mM NaPO4-130 mM NaCl via dialysis and stored at −80°C.
RG-1 immunoprecipitation.
A total of 10.8 μg/ml RG-1 was incubated alone, with 14 ng of rL2:13–36, or with 14 ng of rL2:13–36 and 5 μM HD5 in binding buffer (50 mM NaPO4, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [PMSF] [pH 8; 50 μl total]) at 4°C for 1.5 h. These concentrations were calculated to approximate the conditions under which HD5 neutralizes viral infection in our immunofluorescence competition assay. A total of 20 μl 50% TALON resin in binding buffer was added to each sample and incubated at 4°C for an additional 1 h. Flowthrough was saved, and beads were washed 3 times with wash buffer (50 mM NaPO4, 150 mM NaCl, 1 mM PMSF, 0.1% Triton X-100, 10 mM imidazole). Samples were eluted using wash buffer without 0.1% Triton X-100 and containing 250 mM imidazole. Samples were resolved on a 15% SDS-PAGE gel. Antibody heavy chain was visualized using goat anti-mouse horseradish peroxidase (HRP; 1:5,000; Thermo-Fisher) and enhanced chemiluminescence (Bio-Rad).
rL2:13–36 competition assay.
We first calculated the concentration of HPV16 L2 molecules in the control wells of our infection assay, assuming a maximal 72 copies of L2 per PsV. Based on this number, we incubated a 500-fold molar excess of rL2:13–36 with or without 5.4 μg/ml RG-1, 5 μg/ml mouse IgG1 (Sigma-Aldrich), or 5 μM HD5 in serum-free medium for 1 h on ice. Total protein in all samples was equalized using BSA. HPV16 PsV was then added to the samples and incubated on ice for an additional hour. These mixtures were added to HeLa cells that had been washed with SFM and allowed to infect at 37°C for 4 h. Inoculum was removed, cells were washed once, and complete medium was added. Infection was quantified 48 h p.i. as described above. Treated samples were normalized to control infections in the absence of inhibitors. Experiments were analyzed by one-way ANOVA with Bonferroni posttests to compare selected pairs of samples (see Fig. 3B) or with Dunnett's posttests to compare each sample to the control (see Fig. 3C) using Prism software. For all tests, P values of <0.05 were considered significant.
FIG 3.

HD5 does not block RG-1-L2 epitope binding. (A) RG-1 was immunoprecipitated by rL2:13–36 in the presence or absence of 5 μM HD5. Shown are bound antibody from three independent experiments (Exp 1 to 3) and a representative unbound fraction (FT) from one experiment visualized by immunoblotting. (B) Excess rL2:13–36 rescues HPV16 from RG-1 neutralization. Infection of HeLa cells by HPV16 PsV incubated with RG-1 alone or in competition with a 500-fold molar excess of rL2:13–36 was quantified relative to infection in the absence of inhibitor. BSA was used to normalize protein levels in all samples, and mouse IgG1 was used as an isotype control for RG-1. Data are means ± SD from three independent experiments. ***, P < 0.0001. (C) rL2:13–36 does not rescue HPV16 infection from HD5 neutralization. Infection of HeLa cells by HPV16 PsV incubated with 5 μM HD5 alone or in competition with a 500-fold molar excess of rL2:13–36 was quantified relative to infection in the absence of inhibitor. BSA was used to normalize protein levels in all samples. Data are means ± SD from three independent experiments. ***, P < 0.0001.
Myc-16L2-HA HPV16 PsV cleavage assay.
A total of 4.8 × 1010 particles of Myc-16L2-HA HPV16 PsV were incubated with or without 1, 5, or 10 μM HD5 or 40 μM furin inhibitor (dec.-RVKR-cmk; Calbiochem) in complete medium for 1 h on ice. Samples were added to HeLa cells, and infection was allowed to proceed at 37°C for 16 h. Medium was removed, and cells were washed with serum-free medium. Samples were lysed with 2× SDS loading buffer (Bio-Rad) with β-mercaptoethanol and heated at 95°C. Samples were resolved on 7.5% SDS-PAGE gels and immunoblotted using mouse anti-HA (1:1,000; Thermo-Fisher) and goat anti-mouse HRP (1:5,000; Thermo-Fisher).
Furin cleavage assay.
A total of 1.8 ng of rL2:1–160 was incubated with or without 1, 5, or 10 μM HD5 or 40 μM furin inhibitor in 20 μl furin cleavage buffer (100 mM HEPES-1 mM CaCl2, pH 7.4) on ice for 45 min. One unit of furin (NEB) was added, and the samples were incubated at 30°C for 1 h. Reduced, denatured samples were resolved on 15% SDS-PAGE gels and analyzed by immunoblotting with mouse anti-His antibody (1:1,000; Thermo-Fisher) and goat anti-mouse HRP (1:5,000; Thermo-Fisher).
RESULTS
HD5 interacts directly with HPV16 PsV.
For other nonenveloped viruses, direct binding of defensins to the viral capsid is required for antiviral activity (8, 10, 40, 46). As the antiviral activity of HD5 is highest when HD5 and HPV16 PsV are first coincubated outside the cell (7), it is likely that inhibition of HPV16 infection is also due to direct binding of the defensin to the viral capsid. To assess this interaction, we quantified viral aggregation as a change in mean particle diameter using dynamic light scattering (DLS), which was a correlate for defensin binding in our previous studies of HAdV (40). HD5 treatment increased the average diameter of HPV16 PsV in a dose-dependent manner (Fig. 1). No change in diameter was observed in samples treated with 0.6 μM HD5, consistent with minimal antiviral activity at this concentration (see Fig. 4D). To assess the importance of defensin tertiary structure in HPV binding, we analyzed a linear HD5 mutant (HD5 Abu), which has the same charge as HD5 but no regular structure (11). Like for HAdV, HD5 Abu has no antiviral activity against HPV16 PsV (data not shown) and did not aggregate HPV16 PsV (Fig. 1). These data support a model in which HD5 interacts directly with the HPV16 capsid to neutralize infection.
FIG 1.
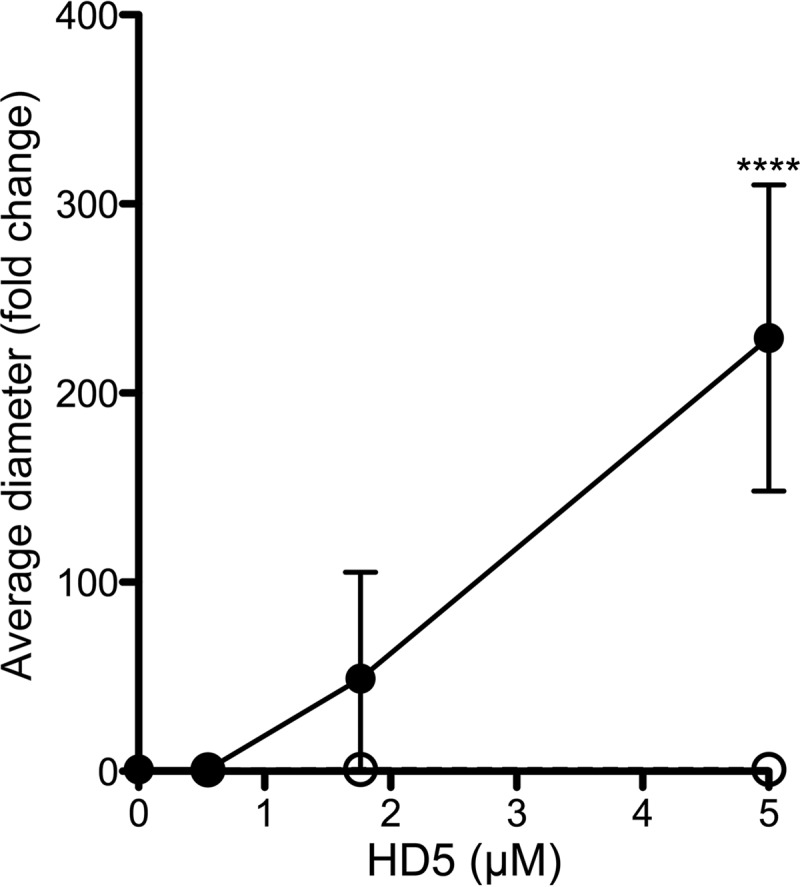
HD5 binding aggregates HPV16 PsV. The mean diameter of HPV15 PsV was measured upon incubation with increasing concentrations of wild-type HD5 (black circles) or HD5 Abu (open circles). Data are the average fold increases in diameter for each condition compared to untreated controls from 4 independent experiments ± standard deviations (SD). ****, P < 0.0001.
HD5 inhibits exposure of an L2-neutralizing antibody epitope.
To identify the step in HPV entry that is blocked by HD5, we assessed the capacity of the anti-L2-neutralizing antibody RG-1 to bind its epitope. RG-1 is known to bind to L2 only after furin cleavage (43, 47). Accordingly, binding of the RG-1 antibody can be used to assess the completion of necessary processing at the cell surface. HaCaT cells were infected with Alexa Fluor-labeled HPV16 PsV that had been incubated with HD5, RG-1, or HD5 and RG-1 together prior to infection. For these experiments, we used inhibitory concentrations based on the dose-response effects of these agents on HPV16 infection alone and in combination (Fig. 2A, B, and F). Cells were fixed at 12 h p.i., and an Alexa Fluor 488-conjugated secondary antibody was added to visualize RG-1 that was bound before fixation. In the absence of both HD5 and RG-1, HPV16 was well dispersed in the cytoplasm. HD5 treatment alone had no effect on the intracellular distribution of the virus. However, RG-1 treatment aggregated the virus, which remained in proximity to the plasma membrane. Consistent with this effect, most of the RG-1 signal colocalized with virus (median, ∼89%; Fig. 2D), and a significant portion of the total viral signal colocalized with RG-1 (median, 28%; Fig. 2E). Treatment with both HD5 and RG-1 together resulted in an intracellular distribution of virus comparable to that of HD5 alone. Some RG-1 signal was observed in these samples; however, RG-1 colocalization with the virus (median, ∼22%; Fig. 2D) and viral colocalization with RG-1 (median, ∼5%; Fig. 2E) were both dramatically decreased upon the addition of HD5. We observed some internalization of RG-1 even in the absence of virus (Fig. 2C, top), which likely explains the presence of RG-1 that is not colocalized with virus in these samples. Thus, HD5 prevents RG-1 from binding to the virus.
HD5 does not directly interfere with RG-1 binding to L2.
One possible reason that RG-1 cannot bind to the virus in the presence of HD5 is that HD5 binds to either RG-1 itself or to the RG-1 epitope on L2 and directly interferes with the antibody-epitope interaction. To address this, we made a C-terminally 6× His- and HA-tagged recombinant L2 peptide (rL2:13–36) containing the RG-1 epitope (residues 17 to 36) (43). RG-1 binds to rL2:13–36 and could be precipitated through the 6× His tag using TALON beads, while the antibody alone did not bind to the beads (Fig. 3A). The addition of HD5 did not interfere with the ability of the peptide to precipitate the RG-1 antibody. Importantly, these experiments were performed using HD5, RG-1, and L2 concentrations that were calculated to closely approximate those used in the immunofluorescence studies and under which we have observed neutralization of infection (Fig. 2A to C). These experiments indicate that HD5 does not directly interfere with the RG-1–L2 interaction.
To confirm this interpretation, we designed an assay to rescue viral infection by competing HD5 or RG-1 with a molar excess of rL2:13–36. The amount of rL2:13–36 was calculated to be 500-fold greater than the maximum amount of L2 that could be present in PsVs in the sample. As proof of concept, we first assessed the ability of the L2 peptide to rescue HPV16 infection in the presence of RG-1. In the absence of rL2:13–36, RG-1 neutralized the virus and decreased infection to approximately 20% of the control infection (Fig. 3B). However, incubation of the RG-1 antibody with the rL2:13–36 peptide before addition of the virus successfully competed the antibody away from the virus and rescued viral infection to ∼70%. We then determined whether excess rL2:13–36 could similarly compete with the virus for HD5 binding. As expected, HPV16 PsV infection was unaffected by the presence of excess rL2:13–36, and HD5 alone potently inhibited infection (Fig. 3C). Incubation of HD5 with excess rL2:13–36 prior to the addition of the mixture to HPV16 PsV did not attenuate HD5 antiviral activity, indicating that HD5 does not bind to residues 13 to 36 of L2. Taken together, these results indicate that HD5 does not bind to either the antibody or the epitope on L2. Therefore, the defensin may be indirectly inhibiting RG-1 binding to the virus by interfering with exposure of the L2 epitope at the cell surface.
Bypassing the CyPB-mediated unfolding of L2 does not relieve the HD5 block.
Prior to RG-1 epitope exposure, the virus undergoes an L1 conformational change (16). Inhibition of this step by neutralizing antibodies blocks viral internalization (48). Defensin-treated HPV16 is still able to enter the cell, suggesting that the defensin-dependent block is after this L1 conformational change (7). The next step is an L2 conformational change mediated by host CyPB that results in exposure of L2 for subsequent furin cleavage (18). Accordingly, inhibition of CyPB with cyclosporine (CsA) blocks furin cleavage. CsA inhibition is bypassed by mutation of the CyPB binding site in L2 (G99A and P100A), likely by altering the flexibility of L2 and increasing exposure of the furin cleavage site, as demonstrated by detection of the RG-1 epitope during infection (18). To assess the possibility that HD5 inhibits the CyPB-mediated unfolding of L2, we engineered these mutations into HPV16 PsVs to make them CyPB-independent (16L2-GP-N PsVs). We first found that the mutant 16L2-GP-N is as sensitive to HD5 as wild-type (WT) HPV16, with an almost identical IC50 (Fig. 4D), indicating that the mutations in L2 were unable to bypass the HD5 block. Next, we repeated the RG-1/HD5 competition assay to determine the state of L2. We found that the intracellular distribution of Alexa Fluor 555-labeled 16L2-GP-N in the presence of HD5, RG-1, or both HD5 and RG-1 was equivalent to that of the WT under each condition (compare Fig. 2C and 4A). Furthermore, the ability of HD5 to reduce RG-1 colocalization with virus (Fig. 4B) and virus colocalization with RG-1 (Fig. 4C) was unchanged in 16L2-GP-N compared to that of the WT. Thus, mutation of the virus to bypass the requirement for a CyPB-mediated conformational change in L2 at the cell surface was not sufficient to alleviate the HD5 block in infection, suggesting that this block was at a subsequent step.
HD5 inhibits cleavage of L2.
Cleavage of L2 by cellular furin occurs after the CyPB-induced conformational change (19). In order to directly assess the cleavage state of L2 during infection, we made an L2 construct containing a Myc tag on the N terminus and an HA tag on the C terminus. The HA tag facilitates detection of L2 in cellular lysates, while the Myc tag extends the N terminus of L2 and increases our ability to resolve the cleaved and uncleaved forms of L2 by SDS-PAGE. We generated PsVs incorporating this L2 construct in place of WT L2 (Myc-16L2-HA). The sensitivity of Myc-16L2-HA PsV to HD5 inhibition was equivalent to that of the WT (Fig. 5A). We then assessed the effect of HD5 on the cleavage state of L2 during infection of HeLa cells. As this biochemical assay required ∼10 times more input virus (4.8 × 1010 particles/sample) than we used in previous experiments, we verified the IC50 of HD5 in complete medium against this higher virus concentration. Although the dose-responsive effect of HD5 was slightly altered by the higher viral concentration, 10 μM HD5 was still inhibitory (Fig. 5A). PsV was incubated with or without increasing concentrations (1 to 10 μM) of HD5, and the mixture was added to HeLa cells. Furin inhibitor (40 μM) was used as a positive control for inhibition of L2 cleavage. Cells were lysed 16 h p.i., and clarified lysates were resolved by SDS-PAGE and immunoblotted for L2. In untreated samples, a minor band with faster mobility than uncleaved L2 was observed, consistent with removal of the N terminus of L2, including the Myc tag (Fig. 5B). In samples treated with furin inhibitor, this cleavage product was absent. HD5 also completely inhibited cleavage of L2, and this effect was dose responsive in three independent experiments. To confirm the disappearance of the L2 cleavage product, we ran twice the amount of lysate from the 10 μM HD5 sample of experiment 1 and did not detect the cleaved L2 band (Fig. 5C). Thus, HD5 blocks L2 cleavage by furin during cell entry.
We surmised that HD5 was either blocking the furin enzyme directly or interfering with the ability of furin to access the L2 cleavage site on the virus. To investigate the first possibility, we purified a truncated L2 protein comprising the first 160 residues of L2 (rL2:1–160). Using a minimal amount of furin and an amount of rL2:1–160 (1.8 ng) that on a molar basis was below the total L2 content of the PsVs in the infection assay, we found that antiviral concentrations of HD5 had no effect on furin cleavage of this unencapsidated L2 (Fig. 5D). Importantly, the assay remained sensitive to inhibition by furin inhibitor. And L2 remained largely uncleaved in samples treated with 3-fold less furin, suggesting that even small changes in furin activity would be detectable in this assay (Fig. 5E). Thus, HD5 does not directly attenuate the protease activity of furin. Rather, HD5 obstructs L2 cleavage during infection, likely through steric hindrance of L2 cleavage sites on the virus.
DISCUSSION
We have uncovered a unique mechanism by which α-defensin binding to a viral capsid at the cell surface alters host-mediated processing of viral capsid proteins, which manifests as a block in cellular trafficking further downstream in the viral entry pathway. Furin cleavage of L2 is a key step in HPV entry, as inhibition of furin cleavage during infection results in retention of PsVs in the endosomal pathway (19). Our finding that HD5 binds to HPV16 PsV and directly and specifically blocks L2 furin cleavage is consistent with the initial description of α-defensin inhibition of HPV16, which resembled the uncleaved PsV phenotype (7). This is an appealing model that may explain the broad inhibitory activity of defensins against both mucosal and cutaneous PV serotypes from both humans and other mammals, as the requirement for furin cleavage is widely conserved, although the generalization of this mechanism to other HPV serotypes remains to be formally demonstrated. Our data exclude an effect of HD5 on the enzymatic activity of furin itself. Rather, they suggest that the ability of furin to access L2 in the context of the incoming capsid is compromised through steric hindrance, likely imposed by HD5 binding to L1. Although the molecular mechanisms differ, our data support a general theme of α-defensin-mediated alteration in intracellular trafficking as an inhibitory mechanism of diverse nonenveloped viruses.
The most probable scenario is that the antiviral activity of HD5 is due to a direct interaction between the virus and HD5. Our current data (Fig. 1) and previous studies strongly support this notion. First, HD5 aggregates HPV16 PsV, indicating a direct interaction between the defensin and virus. In addition, α-defensin mutants that were attenuated for anti-HAdV activity due to direct alterations in capsid binding were also deleterious for anti-HPV16 activity (40, 46). Finally, pretreatment of cells with HD5 was not sufficient to neutralize infection, arguing against a cellular rather than a viral target that mediates inhibition (7). We speculate that HD5 likely binds L1 specifically and blocks access of the furin enzyme to the cleavage site on L2 via steric hindrance, as our data show that HD5 does not interfere with the interaction between the RG-1 antibody and the epitope on L2 nor does a molar excess of rL2:13–36 rescue viral infection, making L2 itself an unlikely binding partner. This peptide comprises a significant amount of L2 that is thought to be exposed at the capsid surface even after the L1 conformational change (13). Moreover, rL2:1–160 was also unable to compete with HD5 for binding to the capsid in experiments analogous to those in Fig. 3 (data not shown). While our data indicate that L2 is not the sole binding partner, the N terminus of L2 may still comprise part of the interaction site with HD5, as the exact binding site on the capsid is still unknown. It remains to be determined exactly how the defensin interacts with the capsid to block furin cleavage at the molecular level. One possibility is that the defensins bind to an element of the HPV capsid that facilitates multimeric defensin-defensin interactions, effectively blanketing the virus and blocking access of furin to the N terminus of L2 that is exposed following the CyPB interaction. This would be similar to the AdV-HD5 interaction, which is highly multivalent (11, 46). Indeed, mutations that affect defensin-defensin interactions are markedly detrimental for defensin activity against both HAdV and HPV16 (40, 46). Alternatively, the specific location of the defensin interaction with the capsid may be deleterious to furin access. These models might be distinguished by future biophysical measurements of defensin binding or by high-resolution structures of defensins in complex with HPV.
Beyond a molecular explanation for inhibition of furin cleavage, a more detailed delineation of the defensin-virus interface may provide an explanation for the ability of defensins to bind to capsids of unrelated viruses. On the virus side of the interaction, it is unclear if the defensins recognize a conserved sequence or structural element common to multiple viruses. Our previous studies have identified critical determinants at the junction of two capsid proteins of AdV that dictate HD5 binding (11). Analogous experiments using HPV16 or PyVs have not yet been performed. On the defensin side, much more is known about the specific molecular features of both HD5 and HNP1 that dictate their antiviral activity against AdV and HPV16. Alanine scan mutagenesis studies of HD5 and HNP1 have shown that neutralization of AdV and HPV16 is reliant on sequence, charge, and specific hydrophobic residues of each defensin (40, 46). Importantly, most HD5 mutants that alter binding to AdV capsomers are also attenuated for HPV16 neutralization, indicating that HPV16 inhibition is likely dependent on a similar binding principle. Thus, future studies of HPV and other viral systems may reveal common principles that dictate α-defensin activity against nonenveloped viruses, defining a mechanism distinct from the more completely characterized activity of defensins against bacteria and enveloped viruses (6, 49).
We noted a discrepancy between the concentration of HD5 that completely blocks furin cleavage and the concentration required to maximally block infection. This may be explained by differences in the sensitivities of the two assays or by differences in the total amount of virus in each assay. Alternatively, partial prevention of L2 cleavage may be sufficient to completely block infection, since the proportion of L2 in each virion that must be cleaved by furin for effective endosome lysis is unknown. A third possible explanation is that HD5 inhibits the virus via a secondary mechanism, possibly by altering intracellular trafficking. Consistent with this hypothesis, α-defensins do not block HPV16 uncoating, perhaps because the virus has entered a noninfectious pathway where the uncoated virion is unable to deliver its genome to the nucleus (7). HD5 could mediate this effect via targeting the virus to defensin-specific receptors, analogous to binding of serum growth factors to HPV16 that allow the virus to enter cells through growth factor receptors (50). Alternatively, defensins could block viral protein interactions with cellular proteins involved in endosomal function such as the retromer complex and γ-secretase, which were recently shown to be critical for moving the viral genome through the endosomal system to the nucleus (23, 24, 26, 27). Thus, future studies to address the impact of HD5 on intracellular trafficking of HPV are warranted.
Our data reveal a striking contrast between the inhibitory mechanisms of a secreted innate immune effector, α-defensins, and secreted effectors of the adaptive immune response, neutralizing antibodies. The major difference is that α-defensins block a critical step in the viral entry pathway but still allow virus internalization, whereas most neutralizing antibodies specific to L1 or L2 inhibit viral entry altogether (47, 48, 51). Indeed, the very fact that defensin-treated HPV still enters cells indicated to us that the defensin acts after the L1 conformational change induced by HSPG binding and through a mechanism distinct from those previously described for neutralizing antibodies. This difference likely has ramifications for subsequent immune control of infection, as internalized virus may be exposed to intracellular sensors of the innate immune response (e.g., Toll-like receptors, the inflammasome, and interferon stimulatory DNA pathways) inaccessible to antibody-neutralized virus, leading to the induction of an antiviral state in neighboring cells (52–54).
α-Defensin-mediated control of HPV infection in the female and male genitourinary tract may play a direct and indirect role in preventing HPV-associated cancers, by both inhibiting virus infection and modulating the antiviral immune response. HPV is associated with roughly 5% of all cancers worldwide and is thought to be the predominant cause of cervical cancer, with 60% of cervical carcinomas associated with HPV16 alone (55). Increasingly, HPV has also been linked to anal, oral, and throat cancers (56). New data suggest that cervical carcinoma originates at cells of the squamocolumnar junction (57). Recent evidence for reduced HD5 expression at these sites could render them more permissive to HPV infection (58). Moreover, α-defensins are known to act as cytokines and chemokines, and immune responses to HPV at these sites could be attenuated (6, 59, 60). Thus, better understanding of the molecular mechanisms underlying the inhibition of HPV infection and the potential role for α-defensins in HPV infection in vivo may lead to improvements in antiviral therapies and vaccine design against these clinically relevant viruses.
ACKNOWLEDGMENTS
This work was supported by Public Health Service, National Research Service, award T32 GM007270 from the National Institute of General Medical Sciences (to M.E.W.) and R01 AI104920 from the National Institute of Allergy and Infectious Diseases (to J.G.S.).
We thank Denise Galloway, Christopher Buck, Richard Roden, Patricia Day, and John Schiller for reagents and advice. We also thank John Scott for use of his confocal microscope.
REFERENCES
- 1.Hazlett L, Wu M. 2011. Defensins in innate immunity. Cell Tissue Res 343:175–188. doi: 10.1007/s00441-010-1022-4. [DOI] [PubMed] [Google Scholar]
- 2.Lehrer RI, Lu W. 2012. Alpha-defensins in human innate immunity. Immunol Rev 245:84–112. doi: 10.1111/j.1600-065X.2011.01082.x. [DOI] [PubMed] [Google Scholar]
- 3.Fan SR, Liu XP, Liao QP. 2008. Human defensins and cytokines in vaginal lavage fluid of women with bacterial vaginosis. Int J Gynaecol Obstet 103:50–54. doi: 10.1016/j.ijgo.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 4.Com E, Bourgeon F, Evrard B, Ganz T, Colleu D, Jegou B, Pineau C. 2003. Expression of antimicrobial defensins in the male reproductive tract of rats, mice, and humans. Biol Reprod 68:95–104. doi: 10.1095/biolreprod.102.005389. [DOI] [PubMed] [Google Scholar]
- 5.Spencer JD, Hains DS, Porter E, Bevins CL, DiRosario J, Becknell B, Wang H, Schwaderer AL. 2012. Human alpha defensin 5 expression in the human kidney and urinary tract. PLoS One 7:e31712. doi: 10.1371/journal.pone.0031712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson SS, Wiens ME, Smith JG. 2013. Antiviral mechanisms of human defensins. J Mol Biol 425:4965–4980. doi: 10.1016/j.jmb.2013.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buck CB, Day PM, Thompson CD, Lubkowski J, Lu W, Lowy DR, Schiller JT. 2006. Human alpha-defensins block papillomavirus infection. Proc Natl Acad Sci U S A 103:1516–1521. doi: 10.1073/pnas.0508033103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dugan AS, Maginnis MS, Jordan JA, Gasparovic ML, Manley K, Page R, Williams G, Porter E, O'Hara BA, Atwood WJ. 2008. Human alpha-defensins inhibit BK virus infection by aggregating virions and blocking binding to host cells. J Biol Chem 283:31125–31132. doi: 10.1074/jbc.M805902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zins SR, Nelson CD, Maginnis MS, Banerjee R, O'Hara BA, Atwood WJ. 2013. The human alpha defensin HD5 neutralizes JC polyomavirus infection by reducing ER traffic and stabilizing the viral capsid. J Virol 88:948–960. doi: 10.1128/JVI.02766-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith JG, Nemerow GR. 2008. Mechanism of adenovirus neutralization by human alpha-defensins. Cell Host Microbe 3:11–19. doi: 10.1016/j.chom.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Smith JG, Silvestry M, Lindert S, Lu W, Nemerow GR, Stewart PL. 2010. Insight into the mechanisms of adenovirus capsid disassembly from studies of defensin neutralization. PLoS Pathog 6:e1000959. doi: 10.1371/journal.ppat.1000959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen EK, Nemerow GR, Smith JG. 2010. Direct evidence from single-cell analysis that human alpha-defensins block adenovirus uncoating to neutralize infection. J Virol 84:4041–4049. doi: 10.1128/JVI.02471-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang JW, Roden RB. 2013. L2, the minor capsid protein of papillomavirus. Virology 445:175–186. doi: 10.1016/j.virol.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joyce JG, Tung JS, Przysiecki CT, Cook JC, Lehman ED, Sands JA, Jansen KU, Keller PM. 1999. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J Biol Chem 274:5810–5822. doi: 10.1074/jbc.274.9.5810. [DOI] [PubMed] [Google Scholar]
- 15.Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J Virol 75:1565–1570. doi: 10.1128/JVI.75.3.1565-1570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selinka H-C, Giroglou T, Nowak T, Christensen N, Sapp M. 2003. Further evidence that papillomavirus capsids exist in two distinct conformations. J Virol 77:12961–12968. doi: 10.1128/JVI.77.24.12961-12967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kines RC, Thompson CD, Lowy DR, Schiller JT, Day PM. 2009. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc Natl Acad Sci U S A 106:20458–20463. doi: 10.1073/pnas.0908502106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bienkowska-Haba M, Patel HD, Sapp M. 2009. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog 5:e1000524. doi: 10.1371/journal.ppat.1000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richards RM, Lowy DR, Schiller JT, Day PM. 2006. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci U S A 103:1522–1527. doi: 10.1073/pnas.0508815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Day PM, Lowy DR, Schiller JT. 2008. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J Virol 82:12565–12568. doi: 10.1128/JVI.01631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergant Marusic M, Ozbun MA, Campos SK, Myers MP, Banks L. 2012. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 13:455–467. doi: 10.1111/j.1600-0854.2011.01320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergant M, Banks L. 2013. SNX17 facilitates infection with diverse papillomavirus types. J Virol 87:1270–1273. doi: 10.1128/JVI.01991-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipovsky A, Popa A, Pimienta G, Wyler M, Bhan A, Kuruvilla L, Guie MA, Poffenberger AC, Nelson CD, Atwood WJ, DiMaio D. 2013. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc Natl Acad Sci U S A 110:7452–7457. doi: 10.1073/pnas.1302164110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Day PM, Thompson CD, Schowalter RM, Lowy DR, Schiller JT. 2013. Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J Virol 87:3862–3870. doi: 10.1128/JVI.03222-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DiGiuseppe S, Bienkowska-Haba M, Hilbig L, Sapp M. 2014. The nuclear retention signal of HPV16 L2 protein is essential for incoming viral genome to transverse the trans-Golgi network. Virology 458–459:93–105. doi: 10.1016/j.virol.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W, Kazakov T, Popa A, DiMaio D. 2014. Vesicular trafficking of incoming human papillomavirus 16 to the Golgi apparatus and endoplasmic reticulum requires gamma-secretase activity. mBio 5(5):e01777-14. doi: 10.1128/mBio.01777-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karanam B, Peng S, Li T, Buck C, Day PM, Roden RB. 2010. Papillomavirus infection requires gamma secretase. J Virol 84:10661–10670. doi: 10.1128/JVI.01081-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bienkowska-Haba M, Williams C, Kim SM, Garcea RL, Sapp M. 2012. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J Virol 86:9875–9887. doi: 10.1128/JVI.00980-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muller KH, Spoden GA, Scheffer KD, Brunnhofer R, De Brabander JK, Maier ME, Florin L, Muller CP. 2014. Inhibition by cellular vacuolar ATPase impairs human papillomavirus uncoating and infection. Antimicrob Agents Chemother 58:2905–2911. doi: 10.1128/AAC.02284-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamper N, Day PM, Nowak T, Selinka HC, Florin L, Bolscher J, Hilbig L, Schiller JT, Sapp M. 2006. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J Virol 80:759–768. doi: 10.1128/JVI.80.2.759-768.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bronnimann MP, Chapman JA, Park CK, Campos SK. 2013. A transmembrane domain and GxxxG motifs within L2 are essential for papillomavirus infection. J Virol 87:464–473. doi: 10.1128/JVI.01539-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Florin L, Becker KA, Lambert C, Nowak T, Sapp C, Strand D, Streeck RE, Sapp M. 2006. Identification of a dynein interacting domain in the papillomavirus minor capsid protein l2. J Virol 80:6691–6696. doi: 10.1128/JVI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Day PM, Baker CC, Lowy DR, Schiller JT. 2004. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc Natl Acad Sci U S A 101:14252–14257. doi: 10.1073/pnas.0404229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF. 2009. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog 5:e1000318. doi: 10.1371/journal.ppat.1000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneider MA, Spoden GA, Florin L, Lambert C. 2011. Identification of the dynein light chains required for human papillomavirus infection. Cell Microbiol 13:32–46. doi: 10.1111/j.1462-5822.2010.01515.x. [DOI] [PubMed] [Google Scholar]
- 36.Raff AB, Woodham AW, Raff LM, Skeate JG, Yan L, Da Silva DM, Schelhaas M, Kast WM. 2013. The evolving field of human papillomavirus receptor research: a review of binding and entry. J Virol 87:6062–6072. doi: 10.1128/JVI.00330-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Florin L, Sapp M, Spoden GA. 2012. Host-cell factors involved in papillomavirus entry. Med Microbiol Immunol 201:437–448. doi: 10.1007/s00430-012-0270-1. [DOI] [PubMed] [Google Scholar]
- 38.Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, Trus BL. 2008. Arrangement of L2 within the papillomavirus capsid. J Virol 82:5190–5197. doi: 10.1128/JVI.02726-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cardone G, Moyer AL, Cheng N, Thompson CD, Dvoretzky I, Lowy DR, Schiller JT, Steven AC, Buck CB, Trus BL. 2014. Maturation of the human papillomavirus 16 capsid. mBio 5(4):e01104-14. doi: 10.1128/mBio.01104-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gounder AP, Wiens ME, Wilson SS, Lu W, Smith JG. 2012. Critical determinants of human alpha-defensin 5 activity against non-enveloped viruses. J Biol Chem 287:24554–24562. doi: 10.1074/jbc.M112.354068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts JN, Buck CB, Thompson CD, Kines R, Bernardo M, Choyke PL, Lowy DR, Schiller JT. 2007. Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat Med 13:857–861. doi: 10.1038/nm1598. [DOI] [PubMed] [Google Scholar]
- 42.Schelhaas M, Shah B, Holzer M, Blattmann P, Kuhling L, Day PM, Schiller JT, Helenius A. 2012. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog 8:e1002657. doi: 10.1371/journal.ppat.1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gambhira R, Karanam B, Jagu S, Roberts JN, Buck CB, Bossis I, Alphs H, Culp T, Christensen ND, Roden RB. 2007. A protective and broadly cross-neutralizing epitope of human papillomavirus L2. J Virol 81:13927–13931. doi: 10.1128/JVI.00936-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolte S, Cordelieres FP. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 46.Tenge VR, Gounder AP, Wiens ME, Lu W, Smith JG. 2014. Delineation of interfaces on human alpha-defensins critical for human adenovirus and human papillomavirus inhibition. PLoS Pathog 10:e1004360. doi: 10.1371/journal.ppat.1004360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Day PM, Gambhira R, Roden RB, Lowy DR, Schiller JT. 2008. Mechanisms of human papillomavirus type 16 neutralization by l2 cross-neutralizing and l1 type-specific antibodies. J Virol 82:4638–4646. doi: 10.1128/JVI.00143-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Day PM, Thompson CD, Buck CB, Pang YY, Lowy DR, Schiller JT. 2007. Neutralization of human papillomavirus with monoclonal antibodies reveals different mechanisms of inhibition. J Virol 81:8784–8792. doi: 10.1128/JVI.00552-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wiens ME, Wilson SS, Lucero CM, Smith JG. 2014. Defensins and viral infection: dispelling common misconceptions. PLoS Pathog 10:e1004186. doi: 10.1371/journal.ppat.1004186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Surviladze Z, Dziduszko A, Ozbun MA. 2012. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog 8:e1002519. doi: 10.1371/journal.ppat.1002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roden RB, Weissinger EM, Henderson DW, Booy F, Kirnbauer R, Mushinski JF, Lowy DR, Schiller JT. 1994. Neutralization of bovine papillomavirus by antibodies to L1 and L2 capsid proteins. J Virol 68:7570–7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lamkanfi M, Dixit VM. 2014. Mechanisms and functions of inflammasomes. Cell 157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 53.Rathinam VA, Fitzgerald KA. 2011. Cytosolic surveillance and antiviral immunity. Curr Opin Virol 1:455–462. doi: 10.1016/j.coviro.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arpaia N, Barton GM. 2011. Toll-like receptors: key players in antiviral immunity. Curr Opin Virol 1:447–454. doi: 10.1016/j.coviro.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parkin DM, Bray F. 2006. Chapter 2: the burden of HPV-related cancers. Vaccine 24:S11–S25. doi: 10.1016/j.vaccine.2006.05.111. [DOI] [PubMed] [Google Scholar]
- 56.D'Souza G, Dempsey A. 2011. The role of HPV in head and neck cancer and review of the HPV vaccine. Prev Med 53(Suppl 1):S5–S11. doi: 10.1016/j.ypmed.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Herfs M, Yamamoto Y, Laury A, Wang X, Nucci MR, McLaughlin-Drubin ME, Munger K, Feldman S, McKeon FD, Xian W, Crum CP. 2012. A discrete population of squamocolumnar junction cells implicated in the pathogenesis of cervical cancer. Proc Natl Acad Sci U S A 109:10516–10521. doi: 10.1073/pnas.1202684109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hubert P, Herman L, Roncarati P, Maillard C, Renoux V, Demoulin S, Erpicum C, Foidart JM, Boniver J, Noel A, Delvenne P, Herfs M. 6October2014. Altered alpha-defensin 5 expression in cervical squamocolumnar junction: implication in the formation of a viral/tumor permissive microenvironment. J Pathol doi: 10.1002/path.4435. [DOI] [PubMed] [Google Scholar]
- 59.Grigat J, Soruri A, Forssmann U, Riggert J, Zwirner J. 2007. Chemoattraction of macrophages, T lymphocytes, and mast cells is evolutionarily conserved within the human alpha-defensin family. J Immunol 179:3958–3965. doi: 10.4049/jimmunol.179.6.3958. [DOI] [PubMed] [Google Scholar]
- 60.Bowdish DM, Davidson DJ, Hancock RE. 2006. Immunomodulatory properties of defensins and cathelicidins. Curr Top Microbiol Immunol 306:27–66. doi: 10.1007/3-540-29916-5_2. [DOI] [PMC free article] [PubMed] [Google Scholar]