

Abstract
A major component of the protective antiviral host defense is contributed by the intracellular actions of the proteins encoded by interferon-stimulated genes (ISGs); among these are the interferon-induced proteins with tetratricopeptide repeats (IFITs), consisting of four members in human and three in mouse. IFIT proteins do not have any known enzyme activity. Instead, they inhibit virus replication by binding and regulating the functions of cellular and viral proteins and RNAs. Although all IFITs are comprised of multiple copies of the degenerate tetratricopeptide repeats, their distinct tertiary structures enable them to bind different partners and affect host-virus interactions differently. The recent use of Ifit knockout mouse models has revealed novel antiviral functions of these proteins and new insights into the specificities of ISG actions. This article focuses on human and murine IFIT1 and IFIT2 by reviewing their mechanisms of action, their critical roles in protecting mice from viral pathogenesis, and viral strategies to evade IFIT action.
INTRODUCTION
Control of virus replication and pathogenesis in mammals depends on rapid detection of the pathogen by the infected cell and its ability to mount an immediate antiviral response to impair virus replication and to limit virus spread to neighboring cells. The antiviral response is triggered by the activation of specific signaling cascades leading to transcriptional induction of many antiviral genes, which encode intracellular proteins to protect the infected cell as well as secreted cytokines that protect uninfected cells from the oncoming infection. The most important cytokines, in this context, are the type I interferons (IFNs). Their importance in protecting the host is revealed by the severe mortality among mice lacking IFNAR, the receptor that recognizes all type I IFNs, after infection with viruses that are only mildly pathogenic in wild-type mice (1). IFNs are synthesized and secreted by all host cells when their specific cell surface or cytoplasmic receptors detect viral molecular patterns (2). After secretion, the type I IFNs (in human and mouse, mainly beta IFN [IFN-β] and more than 10 subtypes of IFN-α) act by binding to IFNAR, thereby inducing the expression of more than 200 IFN-stimulated genes (ISGs) (3–5) whose products are nonlethal to host cells but can be detrimental to virus replication. Different ISGs inhibit different steps of viral life cycles, such as viral entry, transcription, translation, genome replication, assembly, and egress (6), thereby enabling the IFN system to orchestrate a multipronged attack on virus replication. This strategy, rather than the induction of one antiviral “magic bullet,” presumably accounts for the inability of an IFN-sensitive virus to quickly mutate into a resistant one. Another feature of ISG action is that one or a combination of ISGs targets a specific family of related viruses while having little effect on other viruses. IFNs are not essential for ISG induction; any stimulus capable of activating a member of the IFN regulatory factor (IRF) family of transcription factors has the potential to induce ISGs. Type I IFN-signaling uses IRF-9 for this purpose, whereas the membrane-bound Toll-like receptors (TLRs) or the cytoplasmic RIG-I-like receptors (RLRs) use IRF-3 or IRF-7 to induce ISGs (7–11). Consequently, ISGs are often induced in virus-infected cells in response to TLR or RLR activation, without an involvement of IFN. Since ISGs can be induced by a variety of cellular stress responses, their biological functions are likely to be broad and not necessarily restricted to antiviral effects. In this review, we focus on the IFIT1 and IFIT2 genes, members of the IFIT family of ISGs (which encode interferon-induced proteins with tetratricopeptide repeats) that has been discussed in several recent review articles as well (12–14).
THE IFIT GENE FAMILY
By convention, the human IFIT genes are spelled in all capital letters whereas the mouse Ifit genes are spelled with lowercase letters. Because of the lack of universal use of this convention, there is confusion in the literature about the identity of the gene product being discussed; this is compounded by the fact that the cognate human and mouse proteins are not necessarily equivalent in functions. Even at the amino acid sequence level, human IFIT1 is almost as different from murine Ifit1 as it is from human IFIT2. Hence, it is preferable to study their properties as those of distinct proteins, unless proven otherwise. Another source of confusion is the use of similar acronyms, such as IFITM, for other ISGs that are not at all related to IFIT genes. To avoid ambiguity between the mouse and the human IFIT gene families, we use herein “Ifit” for the murine and “IFIT” for the human members, respectively; however, IFIT is also used for referring to these gene families in general.
The first IFIT gene product to be discovered was human IFIT1. It was first identified as “P56,” a 56-kDa protein whose synthesis was strongly induced by IFN. The corresponding gene, ISG56 (also called IFIT1), was one of the first human ISGs to be cloned (15, 16), paving the way for the future cloning of other human and murine IFIT genes. Since IFIT mRNAs are very abundant in IFN-treated cells and are relatively short-lived, they have been used extensively as read-outs for studying transcriptional regulation of ISGs by IFN, TLRs, RLRs, and other signaling receptors; for example, reporter genes driven by the response elements in the promoter regions of the IFIT2 (ISG54, P54) or IFIT1 gene are popular reagents in the field. In contrast, functional studies of the IFIT proteins are relatively new; however, the recent expansion of the number of investigators interested in understanding IFIT functions has produced exciting observations and new insights.
IFIT genes have been identified for many mammals (human, monkey, mouse, rat, cow, dog, horse, dolphin, opossum), various birds and reptiles, amphibians (Xenopus frog), and bony fish. The number of IFIT genes and the composition of the family varies greatly from species to species. The human family comprises four canonical members, clustered together on chromosome 10: IFIT1 (ISG56), IFIT2 (ISG54), IFIT3 (ISG60 or IFIT4), and IFIT5 (ISG58) (17). Further, the uncharacterized IFIT1B as well as IFIT1P1, a pseudogene on chromosome 13, have been identified in human genome sequences. The murine Ifit family comprises three characterized members, clustered in a locus on chromosome 13: Ifit1 (Isg56), Ifit2 (Isg54), and Ifit3 (Isg49); Ifit1c (Gm14446) is an allelic variant of the Ifit1 gene (17, 18). The same locus also contains two additional members of unknown expression patterns and functions, Ifit1b (2010002M12Rik) and Ifit3b (I830012O16Rik). The IFIT genes possess a simple architecture with only 2 to 3 exons, and with the exception of IFIT1B, their stimulus-dependent expression is driven by the interferon-stimulated response elements (ISREs) present within the first 200 bp upstream of the transcriptional start site (17).
INDUCTION OF IFIT GENES UPON VIRUS INFECTION
IFIT genes are normally silent or expressed at very low constitutive levels. Induction of IFIT transcription is triggered by many stimuli, usually in context of viral and bacterial infections. The strongest IFIT inducers are type I IFNs (IFN-α/β) and type III IFNs (IFN-λs), whereas type II IFN (IFN-γ) is much weaker (4, 19). Type I and III IFNs activate the transcription factor ISGF3 (composed of STAT1, STAT2, and IRF-9), which translocates to the nucleus and binds to the ISRE in the IFIT promoters (20). Independently, IFIT genes are also induced in cells infected with different RNA viruses (such as West Nile virus [WNV], influenza viruses, respiratory syncytial virus, and vesicular stomatitis virus [VSV]), which are sensed by TLRs (TLR3 or TLR7/8) and RLRs (RIG-I, MDA5); similarly, DNA viruses (e.g., herpes simplex virus, adenovirus, and cytomegalovirus) activate DNA sensors (IFI16 or cyclic GMP-AMP synthase [cGAS]) (2, 21, 22). These virus-activated signaling pathways primarily engage IRF-3 or IRF-7, although in some cell types, IRF-1 or IRF-5 can be the relevant activated transcription factors (8–11). In the absence of infection, IFIT3 and IFIT5 induction caused by low-level production of IFN-α has been observed after prolonged treatment with all-trans-retinoic acid (ATRA) (23).
Although the presence of ISREs in the promoters of all IFIT genes predicts their concomitant induction, examples of differential expression patterns exist. For example, in IFN-α-injected mice, B cells and plasmacytoid dendritic cells (pDCs) express abundant Ifit2, but not much Ifit1, whereas T cells express high levels of both (24). Similarly, in mice infected with lymphocytic choriomeningitis virus (LCMV), neurons in different regions of the brain show differential expressions of specific Ifit genes. For example, the hippocampal CA1/CA2 pyramidal layers express primarily (but not exclusively) the Ifit1 gene, olfactory bulb neurons express the Ifit2 gene, and cerebellar granule cells neurons express the Ifit3 gene (25). Moreover, in the absence of infection, the basal levels of expression of Ifit1 and Ifit3 genes are higher in granule cell neurons than in cortical neurons (26). These observations suggest that different Ifit genes may have different cell-type-specific induction patterns.
IFIT PROTEIN STRUCTURES
The IFIT proteins, previously called P56, P54, P60 and P58, are cytoplasmic and do not possess enzyme activities. They are comprised of multiple copies of the eponymous tetratricopeptide repeat (TPR), a 34-amino-acid helix-turn-helix motif of degenerate sequence; only 9 residues in certain positions show limited conservation. Therefore, the primary structures of different TPRs within a given IFIT or in IFIT family members differ substantially from one another (17, 27); consequently, IFIT1 shares only about 44% identity with IFIT2 or IFIT3. The three-dimensional structures of IFIT2 and part of IFIT1 were recently determined. It is likely that IFIT1 exists as monomers, is shaped like a clamp, and possesses a positive-charged pocket which is responsible for RNA binding (28). IFIT2, on the other hand, is a homodimer, interconnected by intertwined, or “swapped,” domains; the overall shape is a twisted superhelix. The positive-charged cavity in IFIT2 is larger, forming a channel (29). Additional structural complexity derives from the formation of supercomplexes between IFITs: IFIT1, -2, and -3 can join in a big complex and may do so with other additional proteins (18, 30, 31). In contrast, Ifit1 was found to form a complex only with Ifit1c (18). The compositions and functions of all homo- and heteromeric IFIT complexes remain to be determined, but they likely enhance the spectrum of biological activities of the individual IFITs.
IFIT BINDING PARTNERS
IFITs, although devoid of enzymatic functions, are capable of specifically interacting with a range of cellular and viral RNAs and proteins; these interactions are instrumental in mediating their antiviral effects.
PROTEIN BINDING PARTNERS OF IFITs
IFIT1, IFIT2, Ifit1, and Ifit2 can inhibit mRNA translation initiation by binding to the multisubunit eukaryotic translation initiation factor 3 (eIF3) and interfering with the assembly of the preinitiation complex, consisting of the 40S ribosomal subunit, eIF3, eIF2/GTP/Met-tRNAi, and eIF4F (32–37). Interactions are subunit specific: IFIT1 and IFIT2 bind to eIF3e, whereas Ifit1 and Ifit2 bind to the eIF3c subunit. When IFIT1 is exogenously expressed, it inhibits overall cellular translation in HT1080 cells by 40% (32); this may slow down virus replication by slowing down the overall cellular metabolism. In addition, IFIT1 inhibits cap-independent in vitro translation driven by the hepatitis C virus (HCV) internal ribosome entry site (IRES), a complex RNA secondary structure which also requires eIF3 for ribosome recruitment (38, 39). Moreover, a specific polymorphism in the IFIT1 gene is strongly associated with sustained virologic response and better treatment efficacy in patients infected with HCV genotype 1 (40). An intriguing example of a virus-specific antiviral IFIT interaction evolved between IFIT1 and the human papillomavirus (HPV) E1 helicase. The sequestration of E1 by IFIT1 perturbs E1 function and inhibits viral DNA replication (41). Several studies suggest that the IFITs can modulate viral activation of the signaling pathways that lead to IFN induction (42–45), but the reported results are contradictory and confusing. Both IFIT1 and IFIT2 can apparently inhibit IFN induction by binding to STING, a signaling protein common to cytoplasmic sensing pathways used by DNA and RNA viruses to induce IFN (43). However, others reported that these IFITs have no effects on IFN induction (30).
IFIT/RNA INTERACTIONS
The RLRs (RIG-I and MDA5) recognize specific structural features of viral RNAs that are absent from cellular RNAs (2, 46). Similar molecular patterns are also recognized by the IFIT proteins: both IFIT1 and Ifit1 can specifically bind mRNA 5′ ends whose caps lack 2′-O-methylation of the first ribose, a hallmark of some viral but not cellular mRNAs (18, 47, 48). In consequence, IFIT1 competes with eIF4E for cap binding and prevents translation of the (viral) mRNA (18, 48). Importantly, the two different means of translation inhibition by IFIT1 (eIF3 and cap-RNA binding) are mediated by different regions of the protein: IFIT1 uses its N terminus and middle region to bind to RNA, whereas its C terminus binds eIF3 (28, 33). Another common feature of certain viral RNAs is a free 5′-triphosphate end (5′-ppp-RNA), whereas cellular RNAs are either capped or bear a 5′-monophosphate (such as tRNA) (49). IFIT1 and Ifit1 can bind and sequester 5′-ppp-RNA, thereby perturbing the replication of certain viruses (18, 28, 30). IFIT2 is able to bind AU-rich RNA in vitro (29).
INHIBITION OF VIRUS REPLICATION BY IFITs IN CELL CULTURES
Since IFIT1 and Ifit1 can bind and sequester 5′-ppp-RNAs (18, 30, 48), their effects on the replication of viruses which produce such RNAs were tested. In HeLa cells, the replication of VSV, whose genomic and leader RNAs carry 5′-ppp, was inhibited by the concomitant ectopic expression of all four IFITs. Human IFITs need to form a complex with each other to efficiently recognize 5′-ppp-RNA; therefore, the replication of VSV was restored markedly when the expression of IFIT1, IFIT2, or IFIT3 was knocked down individually in those cells. Similar findings were made with Rift valley fever virus (Bunyaviridae), which also does not get inhibited by the ectopic expression of a single IFIT. Influenza A virus (Orthomyxoviridae) showed restored replication when cells expressed a RNA binding-deficient mutant of IFIT1 instead of wild-type (wt) IFIT1, because only IFIT1 directly binds viral 5′-ppp-RNA, whereas IFIT2 and IFIT3 enhance the binding activity. When infected at a low multiplicity of infection, mouse embryo fibroblasts (MEFs) from Ifit1 knockout (Ifit1−/−) mouse yielded higher VSV titers than those for wt MEFs (30), because in contrast to human IFIT1, Ifit1 does not require other canonical Ifits to bind ppp-RNA (18).
Hepatitis C virus (HCV) is a hepatotropic flavivirus whose mRNA translation is driven by IRES-dependent recruitment of ribosomes, requiring eIF3 (38). In reporter plasmid-transfected cells, IFIT1 inhibits HCV IRES-dependent translation even more strongly than it does cap-dependent translation. This inhibition derives from IFIT1 binding to eIF3e, and consequently, IFIT1 is present, together with eIF3 and HCV IRES RNA, in ribosomal initiation complexes but not actively translating polysomes (39). Importantly, the expression construct used in these experiments was bicistronic; therefore, the IRES element was located in the middle of the RNA and was unaffected by the 5′-methylation/phosphorylation state of the RNA. Replication of infectious HCV (JFH1 strain) is inhibited by ectopic expression of IFIT1 in immortalized human hepatocytes and increased after knockdown of IFIT1 mRNA expression (50). Another flavivirus shows susceptibility to Ifit2 in cell culture (51): in Ifit2−/− cerebellar neurons and dendritic cells, West Nile virus (WNV) replicates to severalfold higher titers than those in wt cells. This advantage for WNV replication in Ifit2−/− cells is even stronger upon IFN pretreatment of wt versus Ifit2−/− MEFs and macrophages (51).
The replication of the paramyxovirus parainfluenzavirus 5 (PIV5; formerly simian virus 5 [SV5]), is suppressed by IFN pretreatment of infected cells. According to one report, this is due to selective inhibition of viral, but not cellular, mRNA translation (52). This inhibition is mediated mostly by IFIT1, as knockdown of IFIT1 restores viral mRNA translation and infectious virus production. Because PIV5 efficiently 2′-O-methylates its mRNA caps, the selective inhibition of PIV5 mRNA translation is cap-methylation independent and might involve binding of eIF3e by IFIT1 or result from sequestration of viral 5′-ppp-RNA (52).
INHIBITION OF VIRAL PATHOGENESIS BY MURINE Ifits
In the last few years, several Ifit knockout mouse lines have been established, thereby providing valuable model systems for investigating the role of Ifits in viral pathogenesis. The Ifit2−/− mice displayed robust changes in the susceptibility to specific viruses, whereas the Ifit1−/− mice revealed novel escape mechanisms used by viruses to evade the antiviral actions of Ifit1 (Fig. 1).
FIG 1.
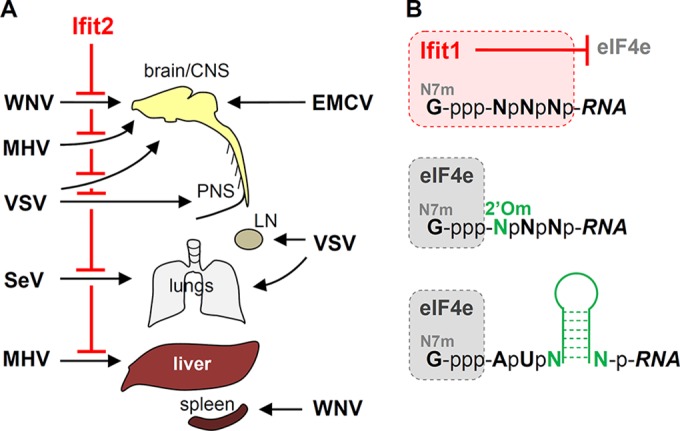
Functions of murine Ifit2 and Ifit1 in viral pathogenesis. (A) Ifit2 protects specific tissues from specific viruses (left) and has no effects on others (right). CNS, central nervous system; PNS, peripheral nervous system; LN, lymph node. (B) Viral strategies to evade Ifit1's antiviral activity. Ifit1 binds to the capped 5′ ends of viral mRNAs if the caps have no 2′-O-methylation and inhibits their translation by blocking the access of eIF4e to the mRNAs. Many viral mRNAs evade the inhibition by 2′-O-methylating their mRNA caps (2′Om), whereas others use RNA hairpin structures, near their 5′ ends, to prevent the access of Ifit1 to the mRNA caps.
Four Ifit2-susceptible viruses have been identified so far; three of them are neurotropic, whereas the fourth one is pneumotropic. VSV, a neurotropic virus, infects all cell types in culture and is very sensitive to antiviral IFN actions. Ifit2−/− mice are much more susceptible than wt mice to neuropathy caused by VSV infection of neurons of the central nervous system (CNS) as well as the peripheral nervous system (PNS) (53, 54). Intranasal VSV administration in wt mice leads to direct infection of the olfactory sensory neurons in the nasal cavity and results in quick entry of VSV into the olfactory bulb (OB) of the brain (55). In response to virus infection of the OB, type I IFN is induced; it diffuses into the caudal parts of the brain, inducing Ifit2 and other ISGs, before VSV reaches there (56). This endogenous IFN “pretreatment” inhibits VSV replication in the brain neurons and prevents neuropathogenesis. In Ifit2−/− mice, however, intranasal VSV infection is uniformly lethal due to high levels of virus replication in neurons in all regions of the brain, in spite of strong induction and action of IFN-β, indicating that, among all ISGs, only Ifit2 is responsible for inhibiting VSV replication in the neurons of the CNS (53). In support of this conclusion, mice lacking the IFNAR gene specifically in neurons are no more susceptible to VSV than Ifit2−/− mice. Preinduction of Ifit2 in brain neurons, preceding the oncoming virus infection, is required for its protective effect; accordingly, wt and Ifit2−/− mice are equally susceptible to intracranial injection of VSV.
Using another model of VSV pathogenesis, it was demonstrated that Ifit2 also protects the neurons of the PNS (54). In this model, VSV is administered subcutaneously in one hind footpad of a mouse, mimicking another natural route of infection (57). The virus replicates in the proximal lymph node and enters the PNS via the nodal nerve endings. In wt mice, type I IFN, produced in the lymph node, prevents further spread of the virus. In contrast, in Ifit2−/− mice, despite IFN induction, VSV replicates in the ipsilateral sciatic nerve of the PNS and then spreads to the CNS neurons of the spinal cord and the brain, causing paralysis and finally reaching the contralateral sciatic nerve as well. These studies firmly established the role of Ifit2 in protecting all neurons from VSV infection; however, the underlying mechanism remains elusive. It appears that Ifit2 impairs a specific feature of neuronal transmission of VSV in vivo, because in vitro, VSV replicates equally well in the presence or the absence of Ifit2 in primary fetal neuronal cultures, in a neuroblastoma cell line, or in mouse embryo fibroblasts (53).
In addition to establishing the role of Ifit2 in preventing neuropathy caused by VSV, the above studies revealed several aspects of specificity of antiviral actions of ISGs. ISG action is virus specific, because pathogenesis by another neurotropic virus, encephalomyocarditis virus, was not enhanced in Ifit2−/− mice (53). Even related ISGs, such as Ifit1 and Ifit2, have different antiviral properties: whereas 400 PFU of intranasal VSV is lethal to the Ifit2−/− mice, the Ifit1−/− mice are as resistant to high doses of the virus as wt mice (53). However, an independently derived Ifit1−/− mouse line is reported to be susceptible to 100,000 PFU of VSV (30). The underlying cause of the observed difference between the two Ifit1−/− lines is currently obscure. Finally, these studies revealed strong cell type specificity of ISG actions. The absence of only Ifit2, a single ISG among hundreds, makes neurons vulnerable to VSV infection. However, its absence does not affect other cell types in other organs, such as the liver and the lung, which are highly susceptible to intranasal or subcutaneous infection with VSV in IFNAR−/− mice, indicating that other ISGs protect other cell types from the same virus (53).
The lethality after footpad injection with another neurotropic virus, WNV, is increased in Ifit2−/− mice. Higher viral burden occurs in select brain regions (brain stem and cerebellum) but not in serum, kidney, or spleen, indicating a tissue-specific antiviral effect of Ifit2. In this system, the adaptive immune responses mediated by T and B cells are unaffected by Ifit2, and serum levels of inflammatory cytokine are similar between wt and Ifit2−/− mice. IFN-β levels are actually higher in the Ifit2−/− mice, indicating that, just as with VSV-infected mice, Ifit2 does not inhibit WNV indirectly by promoting IFN-β induction but probably has a direct effect, especially in neuronal tissues, on the life cycle of WNV within infected cells (51).
Mouse hepatitis virus (MHV) strain A59 is an RNA virus of the Coronaviridae with dual tropism for the CNS and the liver. MHV neuropathogenesis is studied using an intracranial injection model, in which wt mice lose weight but recover through the action of type I IFN. In contrast, 60% of Ifit2−/− mice succumb after showing increased clinical scores, with limb paralysis and greater weight losses. In the Ifit2−/− mice with higher clinical scores, MHV replicates to 100-fold-higher titers in the brain (in neurons and microglia); elevated virus burdens are also found in the liver. Cytokine production by microglia/macrophages in the brain was hardly affected in the absence of Ifit2, except that type I IFN production and ISG induction were significantly reduced in the brain microglia and macrophages of Ifit2−/− mice. These observations suggest that Ifit2 might promote type I IFN induction by MHV (58).
The last example of Ifit2 action against viral pathogenesis comes from intranasal infection of mice with a respirovirus, Sendai virus (SeV). Ifit2−/− mice, but not wt mice, succumb to infection with SeV strain 52 (59). In this model, the virus replicates only in lungs and there is a higher burden of SeV in the lungs of susceptible mice; the level of IFN-β in the lungs is higher as well. Surprisingly, IFNAR−/− mice are less susceptible to SeV than Ifit2−/− mice, although pulmonary virus replication is equally high. Using appropriate genetic crosses, it was established that higher pulmonary levels of both SeV and IFN are required for pathogenesis in Ifit2−/− mice (59).
VIRAL EVASION OF ANTIVIRAL IFIT ACTION
Through the use of viral mutants, investigators have obtained important insights into the nature of Ifit1's function in viral pathogenesis. The observation that the mutant viruses were virulent in Ifit1−/− but not wt mice made it possible to connect the activity of Ifit1 with specific mutations in the viral genome. This approach has revealed that Ifit1 specifically recognizes and binds mRNAs that are deficient in 2′-O-methylation of their caps and blocks their translation.
WNV mRNAs have fully methylated caps at their 5′ ends. The multifunctional viral NS5 protein acts as RNA polymerase and as cap guanine-N-7- and 2′-O-methyl-transferases; however, the different enzymatic activities of NS5 can be separately manipulated by introducing appropriate mutations in the viral genome. For example, when the catalytic active center of the 2′-O-methyltransferase in NS5 is mutated, viral mRNAs are capped and guanine-N-7-methylated, but not 2′-O-methylated. Such a mutant of WNV, E218A, is nonpathogenic in wt mice, although wt WNV is uniformly lethal; in contrast, the two viruses are equally lethal in Ifit1−/− mice (60, 61). The increased lethality of the mutant virus is accompanied by its ability to replicate better in the mouse brain and neuronal cultures in vitro. Similarly, the replication of a 2′-O-methyltransferase mutant of another flavivirus, Japanese encephalitis virus, is restored in Ifit1−/− MEFs and macrophages (47).
Coronaviruses, such as MHV, dedicate a separate nonstructural protein, nsp16, to the sole purpose of 2′-O-methylating viral mRNAs, and the D130A mutation in nsp16 destroys its enzyme activity. This mutant is unable to replicate in the spleen of wt mice but replicates well in the spleen and macrophages of Ifit1−/− mice (18, 46). Likewise, a respiratory coronavirus, severe acute respiratory syndrome coronavirus (SARS-CoV), is largely resistant to Ifit1 in the lungs of wt mice, whereas the replication of a mutant SARS-CoV, lacking nsp16 (Δnsp16), is suppressed 1,000-fold. In contrast, in Ifit1−/− mice, the mutant virus can replicate well and cause airway pathogenesis (62). Vaccinia virus (VACV), a cytoplasmic DNA poxvirus, encodes its own 2′-O-methyltransferase, and pathogenesis in wt mice with VACV depends on the virus's ability to 2′-O-methylate its mRNA caps (60). The above results, coupled with the previously discussed biochemical evidence for Ifit1's ability to selectively bind 2′-O-methyl-deficient capped RNAs, clearly indicate that this protein's biological function is to prevent translation of such viral mRNAs. Hence, active 2′-O-methylation of mRNAs can be viewed as an evasion mechanism used by many viruses to escape the inhibitory effect of Ifit1 on viral protein synthesis (Fig. 1).
How do viruses that do not synthesize 2′-O-methyl-capped mRNAs escape Ifit1? One such example is the neurotropic picornavirus encephalomyocarditis virus (EMCV), which is impervious to Ifit1 (53). The most likely reason is that the EMCV mRNA (which serves as the viral genome as well) is not capped at all and is masked at its 5′ end with a small viral protein. The most elegant Ifit1 evasion mechanism is used by alphaviruses (63). The alphaviral plus-sense RNA genome and subgenomic mRNAs are all capped, but since these viruses do not encode 2′-O-methyltransferases, they would be expected to be highly susceptible to inhibition by Ifit1, although in reality they are not (64). The mosquito-borne Venezuelan equine encephalitis virus (VEEV) is a neuropathic alphavirus. After subcutaneous footpad infection, the wild-type TRD strain is 100% lethal in wt mice even at a very low dose (63). In contrast, the derived, attenuated, more IFN-sensitive TC-83 strain is nonpathogenic in wt mice, even at high doses. However, in Ifit1−/− mice, TC-83 lethality is restored to 100%, with restored replication in spinal cord and brain in vivo as well as in MEFs in vitro. The only difference between the wt and the mutant virus are two point mutations, one of which is at the position 3 of the genome, adjacent to the non-2′-O-methylated 5′ cap (G3→A substitution); reversion of this mutation (A3→G) of TC-83 results in a virus as lethal as the wt VEEV. The mechanistic basis for the Ifit1 resistance of the wt VEEV lies in the hairpin secondary structure adjacent to the 5′ end of the RNA, which prevents Ifit1 from accessing the cap region. When nucleotide 3 is changed, as in the G3→A mutant, the distinct hairpin structure is lost and Ifit1 binds directly to the mutant RNA's capped 5′ end, thereby inhibiting translation of the mutant mRNA (63). The phenomenon holds true for other alphaviruses, such as Sindbis virus (attenuated strain Toto1101) (63). This escape mechanism, which is lost by mutation of a single nucleotide, demonstrates a strong antiviral selection pressure by Ifit1.
FUTURE PERSPECTIVES
Identification of the full repertoire of IFIT-sensitive viruses and the different molecular features of proteins and nucleic acids that are recognized by IFITs has only begun. As additional mice in the Ifit−/− family missing one or more Ifit genes become available, they should be challenged with a variety of RNA and DNA viruses. The property of IFITs to form complexes with each other as homo- or heterodimers, or as supercomplexes with other proteins, is expected to allow for a broad spectrum of binding partners, thereby minimizing the chances of viruses to escape the inhibitory actions of all IFIT family members. Additional complexity arises from the finding that the four IFITs are partially modified by a covalent linkage with ISG15 (“ISGylation”) (65), a small IFN-induced ubiquitin-like protein, which may change IFIT conformation and modify IFIT function or stability. It is tempting to speculate that cellular mRNAs may also be targets of IFIT binding. Usually, cellular mRNAs get degraded in the cytoplasm once they lose their 5′ cap. However, a few hundred decapped mRNAs are not degraded; instead, a cytoplasmic machinery recaps them, allowing for their translation (66). Importantly, these recapped messages likely lack 2′-O-methylation, making them possible targets of IFIT1. Since many of them also have AU-rich elements (AREs) in their 3′ end, they could be recognized by IFIT2 (29).
ACKNOWLEDGMENTS
The authors' research is supported by National Institutes of Health grants CA068782 and AI073303.
REFERENCES
- 1.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 2.Broz P, Monack DM. 2013. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol 13:551–565. doi: 10.1038/nri3479. [DOI] [PubMed] [Google Scholar]
- 3.Fensterl V, Sen GC. 2009. Interferons and viral infections. BioFactors 35:14–20. doi: 10.1002/biof.6. [DOI] [PubMed] [Google Scholar]
- 4.Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A 95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schoggins JW. 2014. Interferon-stimulated genes: roles in viral pathogenesis. Curr Opin Virol 6C:40–46. doi: 10.1016/j.coviro.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bandyopadhyay SK, Leonard GT Jr, Bandyopadhyay T, Stark GR, Sen GC. 1995. Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J Biol Chem 270:19624–19629. doi: 10.1074/jbc.270.33.19624. [DOI] [PubMed] [Google Scholar]
- 8.Barnes BJ, Richards J, Mancl M, Hanash S, Beretta L, Pitha PM. 2004. Global and distinct targets of IRF-5 and IRF-7 during innate response to viral infection. J Biol Chem 279:45194–45207. doi: 10.1074/jbc.M400726200. [DOI] [PubMed] [Google Scholar]
- 9.Elco CP, Guenther JM, Williams BR, Sen GC. 2005. Analysis of genes induced by Sendai virus infection of mutant cell lines reveals essential roles of interferon regulatory factor 3, NF-kappaB, and interferon but not Toll-like receptor 3. J Virol 79:3920–3929. doi: 10.1128/JVI.79.7.3920-3929.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grandvaux N, Servant MJ, ten Oever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J. 2002. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol 76:5532–5539. doi: 10.1128/JVI.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikushima H, Negishi H, Taniguchi T. 2013. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol 78:105–116. doi: 10.1101/sqb.2013.78.020321. [DOI] [PubMed] [Google Scholar]
- 12.Diamond MS. 2014. IFIT1: a dual sensor and effector molecule that detects non-2′-O methylated viral RNA and inhibits its translation. Cytokine Growth Factor Rev 25:543–550. doi: 10.1016/j.cytogfr.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diamond MS, Farzan M. 2013. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol 13:46–57. doi: 10.1038/nri3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vladimer GI, Gorna MW, Superti-Furga G. 2014. IFITs: emerging roles as key anti-viral proteins. Front Immunol 5:94. doi: 10.3389/fimmu.2014.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chebath J, Merlin G, Metz R, Benech P, Revel M. 1983. Interferon-induced 56,000 Mr protein and its mRNA in human cells: molecular cloning and partial sequence of the cDNA. Nucleic Acids Res 11:1213–1226. doi: 10.1093/nar/11.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kusari J, Sen GC. 1987. Transcriptional analyses of interferon-inducible mRNAs. Mol Cell Biol 7:528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fensterl V, Sen GC. 2011. The ISG56/IFIT1 gene family. J Interferon Cytokine Res 31:71–78. doi: 10.1089/jir.2010.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Habjan M, Hubel P, Lacerda L, Benda C, Holze C, Eberl CH, Mann A, Kindler E, Gil-Cruz C, Ziebuhr J, Thiel V, Pichlmair A. 2013. Sequestration by IFIT1 impairs translation of 2′O-unmethylated capped RNA. PLoS Pathog 9:e1003663. doi: 10.1371/journal.ppat.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kohli A, Zhang X, Yang J, Russell RS, Donnelly RP, Sheikh F, Sherman A, Young H, Imamichi T, Lempicki RA, Masur H, Kottilil S. 2012. Distinct and overlapping genomic profiles and antiviral effects of interferon-lambda and -alpha on HCV-infected and noninfected hepatoma cells. J Viral Hepat 19:843–853. doi: 10.1111/j.1365-2893.2012.01610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rauch I, Muller M, Decker T. 2013. The regulation of inflammation by interferons and their STATs. JAKSTAT 2:e23820. doi: 10.4161/jkst.23820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jakobsen MR, Bak RO, Andersen A, Berg RK, Jensen SB, Tengchuan J, Laustsen A, Hansen K, Ostergaard L, Fitzgerald KA, Xiao TS, Mikkelsen JG, Mogensen TH, Paludan SR. 2013. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc Natl Acad Sci U S A 110:E4571–E4580. doi: 10.1073/pnas.1311669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lam E, Stein S, Falck-Pedersen E. 2014. Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J Virol 88:974–981. doi: 10.1128/JVI.02702-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao S, Li D, Zhu HQ, Song MG, Pan XR, Jia PM, Peng LL, Dou AX, Chen GQ, Chen SJ, Chen Z, Tong JH. 2006. RIG-G as a key mediator of the antiproliferative activity of interferon-related pathways through enhancing p21 and p27 proteins. Proc Natl Acad Sci U S A 103:16448–16453. doi: 10.1073/pnas.0607830103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terenzi F, White C, Pal S, Williams BR, Sen GC. 2007. Tissue-specific and inducer-specific differential induction of ISG56 and ISG54 in mice. J Virol 81:8656–8665. doi: 10.1128/JVI.00322-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wacher C, Muller M, Hofer MJ, Getts DR, Zabaras R, Ousman SS, Terenzi F, Sen GC, King NJ, Campbell IL. 2007. Coordinated regulation and widespread cellular expression of interferon-stimulated genes (ISG) ISG-49, ISG-54, and ISG-56 in the central nervous system after infection with distinct viruses. J Virol 81:860–871. doi: 10.1128/JVI.01167-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho H, Proll SC, Szretter KJ, Katze MG, Gale M Jr, Diamond MS. 2013. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat Med 19:458–464. doi: 10.1038/nm.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D'Andrea LD, Regan L. 2003. TPR proteins: the versatile helix. Trends Biochem Sci 28:655–662. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 28.Abbas YM, Pichlmair A, Gorna MW, Superti-Furga G, Nagar B. 2013. Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature 494:60–64. doi: 10.1038/nature11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Z, Liang H, Zhou Q, Li Y, Chen H, Ye W, Chen D, Fleming J, Shu H, Liu Y. 2012. Crystal structure of ISG54 reveals a novel RNA binding structure and potential functional mechanisms. Cell Res 22:1328–1338. doi: 10.1038/cr.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pichlmair A, Lassnig C, Eberle CA, Gorna MW, Baumann CL, Burkard TR, Burckstummer T, Stefanovic A, Krieger S, Bennett KL, Rulicke T, Weber F, Colinge J, Muller M, Superti-Furga G. 2011. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat Immunol 12:624–630. doi: 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- 31.Stawowczyk M, Van Scoy S, Kumar KP, Reich NC. 2011. The interferon stimulated gene 54 promotes apoptosis. J Biol Chem 286:7257–7266. doi: 10.1074/jbc.M110.207068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo J, Hui DJ, Merrick WC, Sen GC. 2000. A new pathway of translational regulation mediated by eukaryotic initiation factor 3. EMBO J 19:6891–6899. doi: 10.1093/emboj/19.24.6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo J, Sen GC. 2000. Characterization of the interaction between the interferon-induced protein P56 and the Int6 protein encoded by a locus of insertion of the mouse mammary tumor virus. J Virol 74:1892–1899. doi: 10.1128/JVI.74.4.1892-1899.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hui DJ, Bhasker CR, Merrick WC, Sen GC. 2003. Viral stress-inducible protein p56 inhibits translation by blocking the interaction of eIF3 with the ternary complex eIF2.GTP.Met-tRNAi. J Biol Chem 278:39477–39482. doi: 10.1074/jbc.M305038200. [DOI] [PubMed] [Google Scholar]
- 35.Hui DJ, Terenzi F, Merrick WC, Sen GC. 2005. Mouse p56 blocks a distinct function of eukaryotic initiation factor 3 in translation initiation. J Biol Chem 280:3433–3440. doi: 10.1074/jbc.M406700200. [DOI] [PubMed] [Google Scholar]
- 36.Terenzi F, Hui DJ, Merrick WC, Sen GC. 2006. Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. J Biol Chem 281:34064–34071. doi: 10.1074/jbc.M605771200. [DOI] [PubMed] [Google Scholar]
- 37.Terenzi F, Pal S, Sen GC. 2005. Induction and mode of action of the viral stress-inducible murine proteins, P56 and P54. Virology 340:116–124. doi: 10.1016/j.virol.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 38.Fraser CS, Doudna JA. 2007. Structural and mechanistic insights into hepatitis C viral translation initiation. Nat Rev Microbiol 5:29–38. doi: 10.1038/nrmicro1558. [DOI] [PubMed] [Google Scholar]
- 39.Wang C, Pflugheber J, Sumpter R Jr, Sodora DL, Hui D, Sen GC, Gale M Jr. 2003. Alpha interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J Virol 77:3898–3912. doi: 10.1128/JVI.77.7.3898-3912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lopez-Rodriguez R, Trapero-Marugan M, Borque MJ, Roman M, Hernandez-Bartolome A, Rodriguez-Munoz Y, Martin-Vilchez S, Abad-Santos F, Munoz de Rueda P, Vidal-Castineira JR, Rodrigo L, Salmeron J, Moreno-Otero R, Sanz-Cameno P. 2011. Genetic variants of interferon-stimulated genes and IL-28B as host prognostic factors of response to combination treatment for chronic hepatitis C. Clin Pharmacol Ther 90:712–721. doi: 10.1038/clpt.2011.189. [DOI] [PubMed] [Google Scholar]
- 41.Terenzi F, Saikia P, Sen GC. 2008. Interferon-inducible protein, P56, inhibits HPV DNA replication by binding to the viral protein E1. EMBO J 27:3311–3321. doi: 10.1038/emboj.2008.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li C, Zhang W, Li Y, Guo L, Shu H, Liu Y. 2012. ISG60 negatively regulates cell antiviral responses by disrupting the VISA-associated complexes. Wuhan Univ J Nat Sci 17:1–6. doi: 10.1007/s11859-012-0795-6. [DOI] [Google Scholar]
- 43.Li Y, Li C, Xue P, Zhong B, Mao AP, Ran Y, Chen H, Wang YY, Yang F, Shu HB. 2009. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc Natl Acad Sci U S A 106:7945–7950. doi: 10.1073/pnas.0900818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu XY, Chen W, Wei B, Shan YF, Wang C. 2011. IFN-induced TPR protein IFIT3 potentiates antiviral signaling by bridging MAVS and TBK1. J Immunol 187:2559–2568. doi: 10.4049/jimmunol.1100963. [DOI] [PubMed] [Google Scholar]
- 45.Zhang B, Liu X, Chen W, Chen L. 2013. IFIT5 potentiates anti-viral response through enhancing innate immune signaling pathways. Acta Biochim Biophys Sin 45:867–874. doi: 10.1093/abbs/gmt088. [DOI] [PubMed] [Google Scholar]
- 46.Zust R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, Siddell SG, Ludewig B, Thiel V. 2011. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol 12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kimura T, Katoh H, Kayama H, Saiga H, Okuyama M, Okamoto T, Umemoto E, Matsuura Y, Yamamoto M, Takeda K. 2013. Ifit1 inhibits Japanese encephalitis virus replication through binding to 5′ capped 2′-O unmethylated RNA. J Virol 87:9997–10003. doi: 10.1128/JVI.00883-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar P, Sweeney TR, Skabkin MA, Skabkina OV, Hellen CU, Pestova TV. 2014. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp- mRNAs. Nucleic Acids Res 42:3228–3245. doi: 10.1093/nar/gkt1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerlier D, Lyles DS. 2011. Interplay between innate immunity and negative-strand RNA viruses: towards a rational model. Microbiol Mol Biol Rev 75:468–490. doi: 10.1128/MMBR.00007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raychoudhuri A, Shrivastava S, Steele R, Kim H, Ray R, Ray RB. 2011. ISG56 and IFITM1 proteins inhibit hepatitis C virus replication. J Virol 85:12881–12889. doi: 10.1128/JVI.05633-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cho H, Shrestha B, Sen GC, Diamond MS. 2013. A role for Ifit2 in restricting West Nile virus infection in the brain. J Virol 87:8363–8371. doi: 10.1128/JVI.01097-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andrejeva J, Norsted H, Habjan M, Thiel V, Goodbourn S, Randall RE. 2013. ISG56/IFIT1 is primarily responsible for interferon-induced changes to patterns of parainfluenza virus type 5 transcription and protein synthesis. J Gen Virol 94:59–68. doi: 10.1099/vir.0.046797-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, Diamond MS, Virgin HW, Sen GC. 2012. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog 8:e1002712. doi: 10.1371/journal.ppat.1002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fensterl V, Wetzel JL, Sen GC. 2014. Interferon-induced protein ifit2 protects mice from infection of the peripheral nervous system by vesicular stomatitis virus. J Virol 88:10303–10311. doi: 10.1128/JVI.01341-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Plakhov IV, Arlund EE, Aoki C, Reiss CS. 1995. The earliest events in vesicular stomatitis virus infection of the murine olfactory neuroepithelium and entry of the central nervous system. Virology 209:257–262. doi: 10.1006/viro.1995.1252. [DOI] [PubMed] [Google Scholar]
- 56.van den Pol AN, Ding S, Robek MD. 2014. Long-distance interferon signaling within the brain blocks virus spread. J Virol 88:3695–3704. doi: 10.1128/JVI.03509-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, Whelan SP, Guidotti LG, von Andrian UH. 2010. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature 465:1079–1083. doi: 10.1038/nature09118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Butchi NB, Hinton DR, Stohlman SA, Kapil P, Fensterl V, Sen GC, Bergmann CC. 2014. Ifit2 deficiency results in uncontrolled neurotropic coronavirus replication and enhanced encephalitis via impaired alpha/beta interferon induction in macrophages. J Virol 88:1051–1064. doi: 10.1128/JVI.02272-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wetzel JL, Fensterl V, Sen GC. 2014. Sendai virus pathogenesis in mice is prevented by Ifit2 and exacerbated by interferon. J Virol 88:13593–13601. doi: 10.1128/JVI.02201-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M Jr, Shi PY, Diamond MS. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Szretter KJ, Daniels BP, Cho H, Gainey MD, Yokoyama WM, Gale M Jr, Virgin HW, Klein RS, Sen GC, Diamond MS. 2012. 2′-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog 8:e1002698. doi: 10.1371/journal.ppat.1002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Menachery VD, Yount BL Jr, Josset L, Gralinski LE, Scobey T, Agnihothram S, Katze MG, Baric RS. 2014. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-O-methyltransferase activity. J Virol 88:4251–4264. doi: 10.1128/JVI.03571-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hyde JL, Gardner CL, Kimura T, White JP, Liu G, Trobaugh DW, Huang C, Tonelli M, Paessler S, Takeda K, Klimstra WB, Amarasinghe GK, Diamond MS. 2014. A viral RNA structural element alters host recognition of nonself RNA. Science 343:783–787. doi: 10.1126/science.1248465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y, Burke CW, Ryman KD, Klimstra WB. 2007. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J Virol 81:11246–11255. doi: 10.1128/JVI.01282-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao C, Denison C, Huibregtse JM, Gygi S, Krug RM. 2005. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc Natl Acad Sci U S A 102:10200–10205. doi: 10.1073/pnas.0504754102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mukherjee C, Patil DP, Kennedy BA, Bakthavachalu B, Bundschuh R, Schoenberg DR. 2012. Identification of cytoplasmic capping targets reveals a role for cap homeostasis in translation and mRNA stability. Cell Rep 2:674–684. doi: 10.1016/j.celrep.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]