

ABSTRACT
Influenza A virus (IAV) infections are influenced by type 1 interferon-mediated antiviral defenses and by viral countermeasures to these defenses. When IAV NS1 protein is disabled, RNase L restricts virus replication; however, the RNAs targeted for cleavage by RNase L under these conditions have not been defined. In this study, we used deep-sequencing methods to identify RNase L cleavage sites within host and viral RNAs from IAV PR8ΔNS1-infected A549 cells. Short hairpin RNA knockdown of RNase L allowed us to distinguish between RNase L-dependent and RNase L-independent cleavage sites. RNase L-dependent cleavage sites were evident at discrete locations in IAV RNA segments (both positive and negative strands). Cleavage in PB2, PB1, and PA genomic RNAs suggests that viral RNPs are susceptible to cleavage by RNase L. Prominent amounts of cleavage mapped to specific regions within IAV RNAs, including some areas of increased synonymous-site conservation. Among cellular RNAs, RNase L-dependent cleavage was most frequent at precise locations in rRNAs. Our data show that RNase L targets specific sites in both host and viral RNAs to restrict influenza virus replication when NS1 protein is disabled.
IMPORTANCE RNase L is a critical component of interferon-regulated and double-stranded-RNA-activated antiviral host responses. We sought to determine how RNase L exerts its antiviral activity during influenza virus infection. We enhanced the antiviral activity of RNase L by disabling a viral protein, NS1, that inhibits the activation of RNase L. Then, using deep-sequencing methods, we identified the host and viral RNAs targeted by RNase L. We found that RNase L cleaved viral RNAs and rRNAs at very precise locations. The direct cleavage of IAV RNAs by RNase L highlights an intimate battle between viral RNAs and an antiviral endonuclease.
INTRODUCTION
Influenza A virus (IAV) impacts human populations worldwide. Seasonal transmission results in substantial morbidity and mortality rates despite the availability of vaccines and antiviral drugs. In the 2013-2014 season, IAV was widespread in all 50 U.S. states, accounting for deaths of children and adults (1). Because of these notable disease burdens, IAV continues to be studied intensively (2). Recent discoveries of new IAV mRNAs and proteins highlight progress in our understanding of viral replication and pathogenesis (3, 4). Nonetheless, gaps in our understanding of IAV remain. For instance, we still do not appreciate how new pandemic strains arise and develop sustained transmission in human populations. Reassortment of segmented IAV RNA genomes is clearly involved; however, the precise conditions and constraints associated with this process remain somewhat mysterious (5).
IAV has eight negative-sense genomic RNA segments, each encoding one or more viral proteins (PB2, PB1, PA, HA, NA, NP, M, and NS RNA segments) that contribute to virus replication and pathogenesis (6). The IAV genomic RNAs, which are present as ribonucleoprotein (RNP) complexes containing nucleoprotein (NP) and viral polymerase (PB2, PB1, and PA proteins) (7, 8), function as templates for viral transcription and RNA replication in the nuclei of infected cells. During RNA replication, both genomic and complementary antigenomic IAV RNAs are maintained in RNPs (7, 8), precluding the formation of large double-stranded RNA (dsRNA) intermediates. Nonetheless, viral dsRNAs play critical roles during infections and viruses often encode mechanisms to inhibit dsRNA-activated antiviral pathways.
Influenza virus NS1 protein is a critical determinant of pathogenesis, in large part because of its ability to bind dsRNA (reviewed in reference 9). When the dsRNA binding domain of NS1 protein is disabled by mutation or deletion, virus replication is restricted by interferon-regulated antiviral pathways (10). RNase L, an endoribonuclease involved in the interferon-regulated and dsRNA-activated antiviral immune response, is especially important in the restriction of IAV when NS1 protein is disabled (11). An experimental antiviral drug targeting NS1 exploits these pathways, inhibiting virus replication in an RNase L-dependent manner (12). RNase L also contributes to IAV-induced immunopathology in the lung (13). Despite the impact of RNase L during IAV infections, the exact mechanisms by which RNase L restricts IAV replication remain to be determined. In particular, we sought to determine whether RNase L cleaves IAV RNAs within infected cells or whether RNase L-mediated cleavage of rRNAs is sufficient to inhibit viral gene expression and replication. Furthermore, it is unknown whether IAV genomes, which are maintained within helical nucleoprotein complexes (RNPs) (7), are susceptible to cleavage by RNase L.
To address these issues, we used cDNA synthesis and deep-sequencing methods to detect RNase L-mediated cleavage sites within host and viral RNAs (14). At a rudimentary level, RNase L targets single-stranded UpN dinucleotides (15), consistent with the specific accommodation of uridine in the substrate binding site of RNase L (16, 17). Nonetheless, we found that RNase L exhibits much more specificity in the cleavage of host and viral RNAs; wherein RNase L cleaves after single-stranded UA and UU dinucleotides much more frequently than after UG and UC dinucleotides (UA/UU > UG > UC) (14). We observed that activation of RNase L in poliovirus-infected HeLa cells leads to cleavage at two specific sites within the 18S rRNA (UU743 and UU541) (14). In this study, we analyzed RNA from IAV PR8ΔNS1-infected A549 cells. Endoribonuclease cleavage sites were detected in each segment of IAV RNA, in both genomic and antigenomic strands. Unexpectedly, large amounts of cleavage mapped to some regions of increased synonymous-site conservation within IAV RNAs; RNA sequences associated with overlapping alternate reading frames (ARFs) (3, 4, 18), alternative splicing (3), and virion RNA packaging (19, 20). We discuss the significance of these findings as they relate to the evolution of influenza viruses and the antiviral activities of RNase L.
MATERIALS AND METHODS
Cell culture.
Canine kidney epithelium (MDCK) and human lung epithelium (A549) cell lines were purchased from ATCC. MDCK-NS1-GFP (where GFP is green fluorescent protein) cells were described previously (21). A549 cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA) and antibiotics. MDCK cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS. MDCK-NS1-GFP cells were grown in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, and 0.5 mg/ml hygromycin (Invitrogen).
Viruses.
We used mouse-adapted H1N1 strain A/PR/8/34 of IAV and the IAVΔNS1 mutant with the NS1-encoding gene deleted (10). Wild-type IAV was prepared as a stock grown in 11-day-old embryonated chicken eggs as previously described (10). IAVΔNS1 was grown in MDCK-NS1 cells in DMEM supplemented with 0.3% bovine albumin (Sigma-Aldrich), 1% penicillin-streptomycin, and 1 μg/ml of l-1-tosylamide-2-phenylmethyl chloromethyl ketone (TPCK)-treated trypsin (Sigma-Aldrich) (21).
Stable depletion of RNase L in A549 cells.
Lentiviruses expressing either control nonmammalian short hairpin RNA (shRNA) or shRNA targeting the 3′ untranslated region of RNase L (Sigma-Aldrich products SHC002 and TRCN0000226437, respectively) along with lentivirus packaging plasmids were transfected into HEK293T cells with Lipofectamine 2000 (Life Technologies). Virus-containing medium was collected 48 h after transfection. A549 cells were infected with the lentiviruses in the presence of 8 μg/ml Polybrene (Life Technologies). A549 cells stably expressing control or RNase L shRNAs were selected with puromycin (2.5 μg/ml). Twenty clones expressing RNase L shRNA were screened as follows. RNase L activity in intact cells was determined by transfection of poly(rI)·poly(rC) [poly(I·C); Sigma-Aldrich] at 2 μg/ml for 5 h, followed by isolation of total RNA and analysis of rRNA for RNase L-specific cleavage products with RNA chips (and an Agilent 2100 Bioanalyzer) (22). RNase L expression was detected by immunoblotting with a monoclonal antibody (23). Three clones in which RNase L expression was depleted with shRNA were combined into a single culture for IAV infections.
IAV infections.
For RNA analysis, A549 cells expressing control or RNase L shRNA (2 × 106/10-cm dish) were washed twice in phosphate-buffered saline (PBS) and placed in serum-free RPMI 1640 medium. Cells were infected with IAV or IAVΔNS1 at a multiplicity of infection (MOI) of 10 and incubated for 1 h with gentle agitation every 10 min. The medium was removed, cells were washed twice with PBS, medium containing 10% FBS was added, and cells were incubated for 24 h. The cells were washed with PBS (five times), trypsinized, and resuspended in 1 ml PBS per dish. Cell suspensions from 10 identical biological replicates (control or RNase L shRNA) were mixed together. Total RNA was isolated with TRIzol (Life Technologies). For viral plaque assays, A549 cells (2.5 × 106/10-cm dish) expressing control or RNase L shRNA were infected with IAV or IAVΔNS1 at an MOI of 1 for 24 h.
Plaque assays.
IAV and IAVΔNS1 titers were determined on MDCK or MDCK-NS1-GFP cells, respectively, in six-well plates (106 cells/well). Cells were washed twice with PBS and incubated with virus (diluted in DMEM) for 1 h at 37°C with frequent shaking (24). After incubation, the virus inoculum was removed and cells were overlaid with DMEM containing 0.6% Oxoid agar and 1 μg/ml TPCK-treated trypsin (both from Sigma-Aldrich). After 72 h, the plaques were visualized by staining with crystal violet.
rRNA cleavage assays.
rRNA cleavage was monitored on RNA chips with an Agilent 2100 Bioanalyzer as described previously (22).
Immunoblotting.
Cell lysates (30 μg protein) were fractionated by SDS–10% polyacrylamide gel electrophoresis (PAGE), and proteins were transferred to polyvinylidene difluoride membranes (0.45 μm; Bio-Rad) and probed with RNase L (23) or β-actin antibodies (Sigma-Aldrich).
Cyclic phosphate cDNA synthesis and Illumina sequencing.
Cyclic phosphate cDNA libraries were synthesized and Illumina sequencing was performed as previously described (14). For sequencing on the Illumina MiSeq platform, 2.5 nM each cDNA library (uninfected plus control shRNA/uninfected plus RNase L shRNA/IAVΔNS1-infected plus control shRNA/IAVΔNS1-infected plus RNase L shRNA) were pooled into one run.
Bioinformatics.
Illumina sequence data were trimmed of residual cDNA linker sequence and then aligned in a sequential manner with the following sequences with Bowtie2 (25) with the –un option: IAV [A/Puerto Rico/8/34/Mount Sinai(H1N1)] segment 1 (GenBank accession no. AF389115.1), segment 2 (AF389116.1), segment 3 (AF389117.1), segment 4 (AF389118.1), segment 5 (AF389119.1), segment 6 (AF389120.1), segment 7 (AF389121.1), and segment 8 (AF389122.1) with nucleotides 57 to 528 deleted, 45S pre-rRNA (NR_046235.1), 5S rRNA (NR_023371.1), U6 snRNA (NR_046491.1) and mRNA and other 16S and 12S mitochondrial rRNAs, U1/U2/U4/U5/U6atac snRNAs, U3 snoRNA, 7SL and 7SK RNAs, RNase MRP RNA, hg18 tRNAs (Genomic tRNA database), CD/HACA box and Cajal body snoRNAs (snoRNABase), and the human genome build 36.1 (hg18). Aligned data were converted to bedgraph (26). The number of RNA cleavage sites in each cDNA library was determined by counting 8-base unique molecular identifiers (UMIs) (UMItools; GitHub, Inc.) (27). The frequency of endonuclease cleavage sites was plotted against the nucleotide position of each genome with Microsoft Excel or R (28). The amount of cleavage at each site in host and viral RNAs was normalized as follows: % total cDNA reads = (number of UMI cDNA reads at each position or RNA of interest/total aligned reads in each cDNA library) × 100.
Synonymous-site conservation.
Synonymous-site conservation was analyzed with synplot2 (http://www.firthlab.path.cam.ac.uk/vindex.html) (29). Plots are based on alignments of large numbers of IAV RNA sequences (from 960 to 1,600, depending on the segment) selected via Blastclust to sample the diversity of all available sequences.
Microarray data accession number.
Deep-sequencing data were deposited in the NCBI Gene Expression Omnibus under GEO Series accession number GSE60581 (30).
RESULTS
RNase L restricts IAV replication and leads to rRNA cleavage when NS1 protein is deleted.
A cell culture system was used to analyze RNase L-mediated rRNA cleavage products during IAV infections (Fig. 1). shRNAs were used to reduce the expression of endogenous RNase L compared to that in control shRNA-treated cells (Fig. 1A). RNase L, when activated in virus-infected cells, produces characteristic rRNA fragments (31, 32). RNase L-mediated rRNA fragments were evident in IAVΔNS1-infected A549 cells and in cells transfected with poly(I·C), a synthetic dsRNA (Fig. 1B, control shRNA-treated cells, mobility of rRNA fragments indicated by arrowheads). rRNA fragments were absent from both uninfected cells and wild-type IAV-infected cells, where NS1 was expressed normally (Fig. 1B). Furthermore, knockdown of RNase L expression dramatically diminished the cleavage of rRNA in IAVΔNS1-infected cells and in poly(I·C)-transfected cells, compared to that in control shRNA-treated cells (Fig. 1B). The magnitudes of IAV replication were consistent with these observations. Wild-type IAV replicated equally well in both control and RNase L shRNA-treated cells, whereas IAVΔNS1 replication was restricted in control shRNA-treated cells (Fig. 1C). Knockdown of RNase L in A549 cells increased IAVΔNS1 replication 17-fold (Fig. 1C). These results, which show that RNase L is activated during infection when IAV NS1 protein is deleted, are consistent with those reported by others (11, 12).
FIG 1.

NS1 prevents RNase L activation during IAV infections of A549 cells. (A) RNase L levels determined by immunoblotting in control and RNase L shRNA-treated A549 cells. Results of biological triplicates are shown. (B) RNase L activity by rRNA cleavage was determined on RNA chips (Agilent) with RNA isolated from control or RNase L shRNA-treated cells infected with IAV or IAVΔNS1 at an MOI of 10 for 24 h or transfected with poly(I·C) (2 μg/ml) for 5 h. Arrowheads (to the right) indicate positions of RNase L-mediated degradation products of rRNA. The RNAs used for 2′,3′-cyclic phosphate cDNA synthesis and Illumina sequencing are indicated by check marks. (C) IAV yields following infection (MOI of 1, 24 hpi) (24). The data shown are mean results ± standard deviations from three biological replicates. Two-tailed t tests were done. ns, not significant; *, P < 0.05.
Endoribonuclease cleavage sites in host and viral RNAs.
Cyclic phosphate cDNA synthesis and Illumina sequencing, in conjunction with bioinformatic analyses, revealed the frequency and location of cleavage sites within host and viral RNAs (14). These methods exploit a specific chemical signature (cyclic phosphates) found at the 3′ end of RNA fragments produced by RNase L and other metal ion-independent endonucleases. Cyclic phosphate cDNA libraries were made with RNA from uninfected and IAVΔNS1-infected A549 cells (Fig. 1B, check marks). We compared data from four cDNA libraries, (i) uninfected control shRNA-treated cells, (ii) uninfected RNase L shRNA-treated cells, (iii) IAVΔNS1-infected control shRNA-treated cells, and (iv) IAVΔNS1-infected RNase L shRNA-treated cells (Fig. 2).
FIG 2.
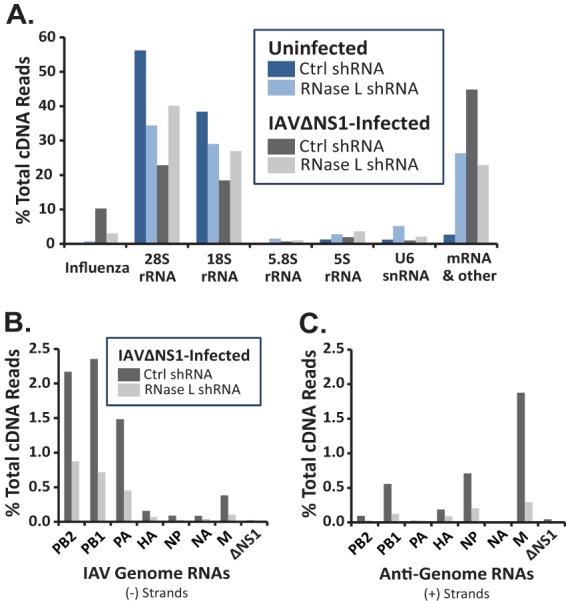
Frequencies of endonucleolytic cleavage in host and viral RNAs. Cyclic phosphate cDNA synthesis and Illumina sequencing were used to detect endoribonucleolytic cleavage of host and viral RNAs (14). cDNA libraries from uninfected and IAVΔNS1-infected A549 cells/control (Ctrl) and RNase L shRNA-treated cells. (A) Amounts of cleavage in host and viral RNAs. Frequencies of cleavage in individual IAV RNA segments: genome (B) and antigenome/mRNA (C).
Endoribonuclease cleavage sites were found in host and viral RNAs (Fig. 2A). UMIs were used during cDNA synthesis (27), allowing us to count the cleavage events in host and viral RNAs (14). We normalized the data (% total cDNA reads) in order to quantitatively compare the amounts of cleavage in particular RNAs across samples. 28S, 18S, 5.8S, and 5S rRNAs and U6 snRNA accounted for a large portion of the cleavage sites in all four samples (Fig. 2A). More than 10% of all cleavage events were in IAV RNAs in control shRNA-treated cells (Fig. 2A, Influenza, dark gray bar). Cleavage within IAV RNAs was reduced by RNase L shRNA (Fig. 2A, Influenza, light gray bar). It is important to note that IAV PA and PA-X endonucleases are metal-dependent enzymes that do not produce cyclic phosphates at RNA cleavage sites (33, 34). Consequently, the cyclic phosphate cDNA libraries that we used to identify RNase L cleavage sites do not contain confounding PA endonuclease cleavage sites.
Endoribonuclease cleavage sites were detected in each segment of IAV RNA, in both negative and positive strands (Fig. 2B and C; Table 1). PB2, PB1, and PA genomic RNAs were cleaved more frequently than other IAV genomic RNAs (Fig. 2B; Table 1). Cyclic phosphate cDNA synthesis and Illumina sequencing methods detect cleavage sites as a function of RNA abundance/RNA fragment abundance. Therefore, cleavage sites tend to be detected more frequently in large, abundant RNAs. For instance, rRNAs have more cleavage sites than any other RNA in cells (Fig. 2A). We do not know the exact abundance of IAV RNAs at 24 hpi in our experiments; however, PB2, PB1, and PA RNAs are likely to be abundant at these times and these are relatively large RNAs compared to other IAV RNA segments. Consequently, it is not surprising to see relatively large amounts of cleavage in these large IAV RNA segments (Table 1; Fig. 2B).
TABLE 1.
Endonucleolytic cleavage of IAV RNAs
| IAV RNA and strand | No. of cDNA reads |
% of total cDNA reads |
|||
|---|---|---|---|---|---|
| Control shRNA | RNase L shRNA | Control shRNA | RNase L shRNA | Fold change | |
| PB2 | |||||
| − | 689 | 383 | 2.17 | 0.87 | 2.48 |
| + | 30 | 12 | 0.09 | 0.03 | 3.45 |
| PB1 | |||||
| − | 748 | 313 | 2.36 | 0.71 | 3.29 |
| + | 177 | 54 | 0.56 | 0.12 | 4.52 |
| PA | |||||
| − | 471 | 199 | 1.48 | 0.45 | 3.26 |
| + | 7 | 11 | 0.02 | 0.03 | 0.88 |
| HA | |||||
| − | 50 | 30 | 0.16 | 0.07 | 2.30 |
| + | 59 | 39 | 0.19 | 0.09 | 2.09 |
| NP | |||||
| − | 28 | 12 | 0.09 | 0.03 | 3.22 |
| + | 225 | 89 | 0.71 | 0.20 | 3.49 |
| NA | |||||
| − | 27 | 19 | 0.09 | 0.04 | 1.96 |
| + | 3 | 3 | 0.01 | 0.01 | 1.38 |
| M | |||||
| − | 121 | 45 | 0.38 | 0.10 | 3.71 |
| + | 595 | 128 | 1.87 | 0.29 | 6.41 |
| ΔNS1 | |||||
| − | 6 | 8 | 0.02 | 0.02 | 1.03 |
| + | 14 | 3 | 0.04 | 0.01 | 6.43 |
| Sum | |||||
| − | 2,140 | 1,009 | 6.74 | 2.30 | 2.92 |
| + | 1,096 | 336 | 3.50 | 0.77 | 4.51 |
| Total | 3,250 | 1,348 | 10.23 | 3.08 | 3.32 |
Among IAV positive-strand RNAs, M was cleaved most frequently (Fig. 2C; Table 1). As noted above, RNase L knockdown reduced the amounts of cleavage in each IAV RNA segment (Fig. 2B and C; Table 1). UA, UU, and UG dinucleotides, characteristic RNase L targets, were frequent sites of cleavage within IAV RNAs (Fig. 3 and 4). RNase L knockdown reduced cleavage predominantly at UA and UU dinucleotides, especially within IAV positive-strand RNAs (Fig. 3 and 4). RNase L knockdown reduced cleavage at UA and UU dinucleotides in IAV positive strands by 9- and 11-fold, respectively (Fig. 4). Significant amounts of cleavage were evident after pyrimidines (UU, UC, AU, AC, GU, GC, CU, and CC dinucleotides), most notably within IAV genomic RNAs (Fig. 3 and 4). Cleavage after pyrimidines is characteristic of RNase A family enzymes (35), including angiogenin and RNase 4, enzymes found in the nuclei of cells (36, 37).
FIG 3.

Dinucleotides at endoribonuclease cleavage sites in IAV RNAs (IAV RNAs 1 to 8). IAVΔNS1-infected A549 cells were treated with control shRNA (dark gray bars) or RNase L shRNA (light gray bars). Shown are percentages of the total cDNA reads at each of 16 dinucleotides in IAV genomic RNA segments (left) and antigenomic/mRNA segments (right).
FIG 4.
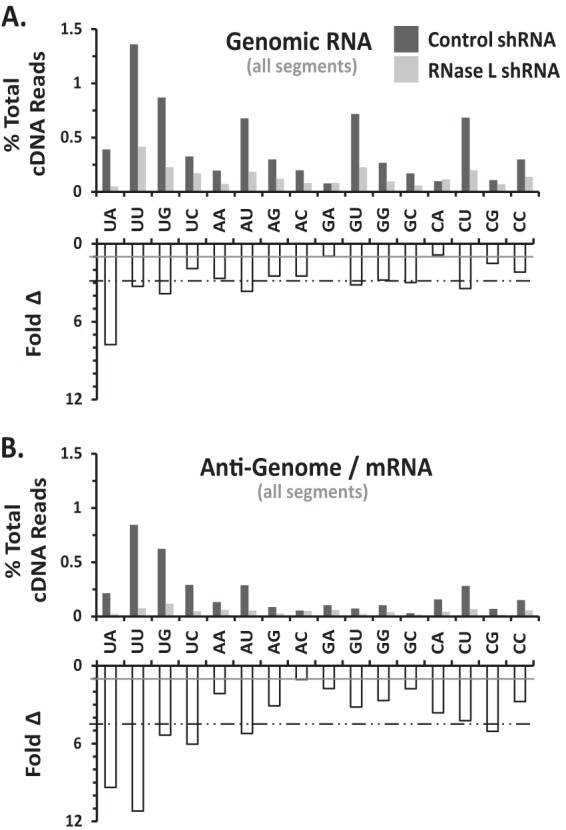
Dinucleotides at endoribonuclease cleavage sites in IAV RNAs (genome wide). IAVΔNS1-infected A549 cells were treated with control shRNA (dark gray bars) or RNase L shRNA (light gray bars). (A) IAV negative-strand RNAs. (B) IAV positive-strand RNAs. Panels: top, percentage of the total cDNA reads at each of 16 dinucleotides in IAV RNAs; bottom, fold reduction in cleavage due to RNase L knockdown (ratio of the percentages of the total cDNA reads in control and RNase L shRNA-treated samples). Gray line, ratio of 1 (no change). Dashed line, average reduction for all dinucleotides.
Cleavage sites mapped to discrete regions within IAV RNAs, often within and adjacent to areas of increased synonymous-site conservation.
Discrete regions of PB2, PB1, PA, NP, and M RNA segments accounted for the majority of the cleavage events within IAV RNAs (Fig. 5 and 6). RNase L knockdown greatly reduced the magnitude of cleavage in each region of IAV RNA (Fig. 6). Notably, large amounts of cleavage were observed within and adjacent to some areas of increased synonymous-site conservation (Fig. 5). The observed to expected (obs/exp) ratio of synonymous-site conservation allows direct comparison across different IAV RNA segments (Fig. 5). The IAV NS RNA segment was excluded from these analyses because of the deletion in this RNA and the low number of cleavage events detected in the residual ΔNS1 RNA (Fig. 2B and C; Table 1). Prominent cleavage sites within and near regions of increased synonymous-site conservation in PB1 and M RNA segments were mapped to base-by-base sequences (Fig. 7 and 8).
FIG 5.

Frequency and location of endonucleolytic cleavage sites in IAV RNAs. Cleavage sites (UMIs) in IAV negative-strand (blue bars) and positive-strand (red bars) RNAs are plotted alongside synonymous-site conservation (SSC). SSC (gray lines), observed number of substitutions in a 15-codon sliding window/expected number of substitutions along the window. A dashed line indicates an SSC obs/exp ratio of 1. Cleavage sites from IAVΔNS1-infected control shRNA-treated A549 cells are shown.
FIG 6.

Frequency and location of cleavage sites in IAV RNAs. Shown are the percentages of the total cDNA reads detected at each nucleotide in IAV RNAs from IAVΔNS1-infected control shRNA-treated cells (red bars) and RNase L shRNA-treated cells (blue bars). (A) IAV negative strands. (B) IAV positive strands.
FIG 7.

Prominent cleavage sites in M positive-strand RNA nucleotides 100 to 259. IAVΔNS1-infected A549 cells were treated with control shRNA (dark gray bars) or RNase L shRNA (light gray bars). Shown are the percentages of the total cDNA reads at the indicated positions in M positive-strand RNA. UA and UU dinucleotides are underlined. The locations of the M1 open reading frame (ORF), the M42 ARF, the M4 intron, the M4 splice donor (m4SD) sequence, and the RNA structure described by Moss et al. (38) are shown.
FIG 8.

Prominent cleavage sites in PB1 RNA segment. IAVΔNS1-infected A549 cells were treated with control (dark gray bars) or RNase L (light gray bars) shRNA. Shown are the percentages of the total cDNA reads at the indicated positions in IAV RNA. (A) The 160-base-long sequence at the 3′ end of the PB1 negative strand. The promoter, viral RNA packaging sequences, and coding regions are annotated according to Wise et al. (53). UA and UU dinucleotides are underlined. (B) The 80-base-long sequence of the PB1 positive strand.
PB2 negative-strand RNA was cleaved in a discrete region spanning nucleotides 250 to 650, sequences without increased synonymous-site conservation (Fig. 5). PB1 RNA was cleaved frequently in both the negative and positive strands, with substantial amounts of cleavage at the 3′ end of the negative strand and the 5′ end of the positive strand (Fig. 5). The endonuclease cleavage sites in PB1 coincide with areas of increased synonymous-site conservation, including the areas associated with the PB1-F2 ARF (Fig. 5). PA RNA was cleaved at discrete locations in the negative strand, within and adjacent to areas of increased synonymous-site conservation (Fig. 5). HA and NA RNAs, which have relatively small amounts of synonymous-site conservation, had limited amounts of endonucleolytic cleavage compared to other IAV RNA segments (Fig. 5; Table 1). NP and M RNAs were cleaved predominantly in positive strands, within and adjacent to areas of increased synonymous-site conservation (Fig. 5 and 7). Cleavage sites within M positive strands were predominantly at UpN dinucleotides (UA, UU, UG, UC), and these cleavage sites were almost completely eliminated by RNase L knockdown (Fig. 3 and 7). The IAV RNA sequence most frequently cleaved by RNase L was present in M positive-strand RNA; a 15-base sequence (200AUUUUAGGAUUUGUG214) with more than 200 cDNA reads predominantly at UA, UU, and UG dinucleotides (Fig. 7). These RNase L cleavage sites lie within an area of increased synonymous-site conservation (Fig. 5, M RNA), 50 bases downstream from the M4 splice donor sequence (Fig. 7), within an RNA structure of unknown function (38).
It is important to note that while endoribonucleolytic cleavage was prominent in some areas of increased synonymous-site conservation (e.g., PB1-F2 ARF, M segment splice junction, PA-X ARF in negative strand), other areas of increased synonymous-site conservation had little to no endonucleolytic cleavage (e.g., 3′ ends of PB2, PB1, and M segments). Thus, there was no absolute correlation between increased synonymous-site conservation and increased endonucleolytic cleavage (Fig. 5).
RNase L-dependent cleavage sites within rRNAs.
RNase L-dependent and RNase L-independent cleavage sites within rRNAs were clearly apparent in the cDNA libraries (Fig. 9; Tables 2 and 3). RNase L-mediated cleavage within 18S rRNA was frequently detected at UU743 and UU541 (Fig. 9A; Table 2), consistent with experimental data from HeLa cells (14). Less frequently detected RNase L-dependent cleavage sites in 18S rRNA were detected at UC136, UC199, CU540, CU742, and UU1297 (Fig. 9A; Table 2). 28S rRNA was cleaved in an RNase L-dependent manner at UG2486, UU2741, and UG2762 (Fig. 9B; Table 3). 28S rRNA cleavage at CU4031 and UG4032 was provoked specifically in IAVΔNS1-infected cells; however, cleavage at these sites was not diminished significantly by RNase L knockdown, preventing attribution to RNase L activity (Table 3). RNase L-dependent cleavage sites were on the surface of ribosomes (Fig. 9C; see Movie S1 in the supplemental material), as might be expected.
FIG 9.
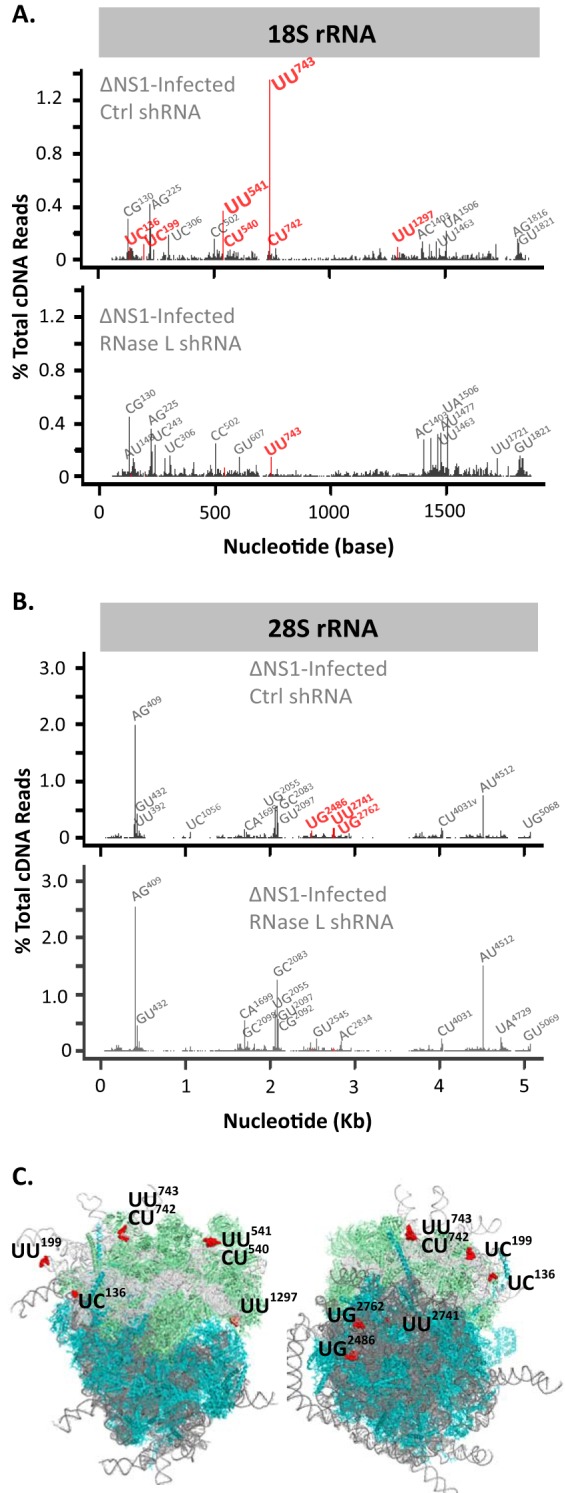
RNase L-dependent cleavage sites in rRNA/80S ribosomes. The human ribosome structures shown are from Anger et al. (54). The frequency and location of cleavage sites in the 18S (A) and 28S (B) rRNAs are shown. (C) Locations of RNase L cleavage sites in 80S ribosomes.
TABLE 2.
RNase L-dependent and RNase L-independent cleavage sites in 18S rRNAa
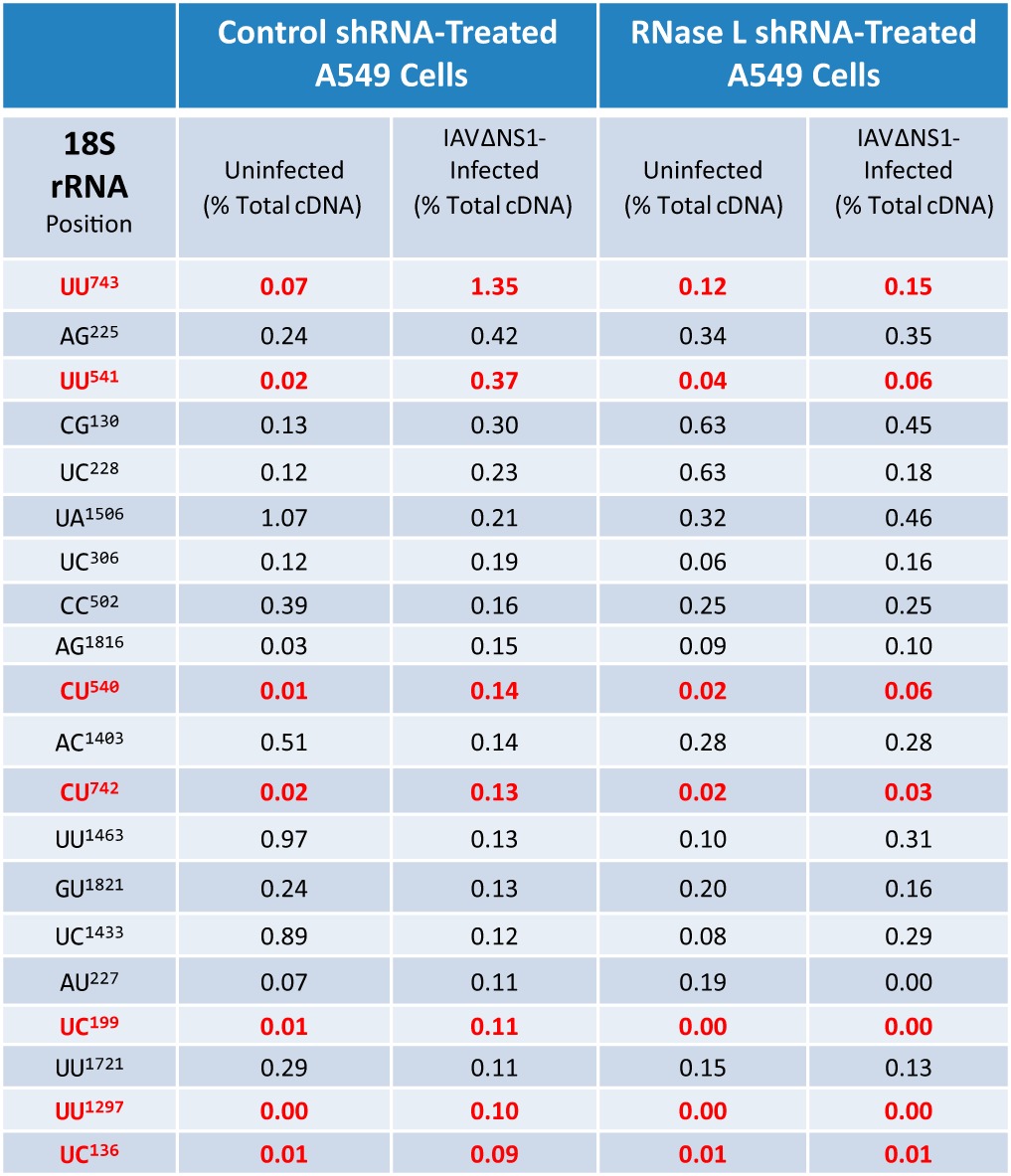
The percentages of the total cDNA reads at the 20 most frequent cleavage sites in the 18S rRNA (benchmarked to IAVΔNS1-infected control shRNA-treated A549 cells) are shown. Red values are for RNase L-dependent cleavage sites. In IAVΔNS1-infected control shRNA-treated A549 cells, there was ≥5-fold greater cleavage than in uninfected control shRNA-treated cells and uninfected RNase L shRNA-treated cells and ≥2.5-fold increased greater cleavage than in IAVΔNS1-infected RNase L shRNA-treated cells.
TABLE 3.
RNase L-dependent and RNase L-independent cleavage sites in 28S rRNAa
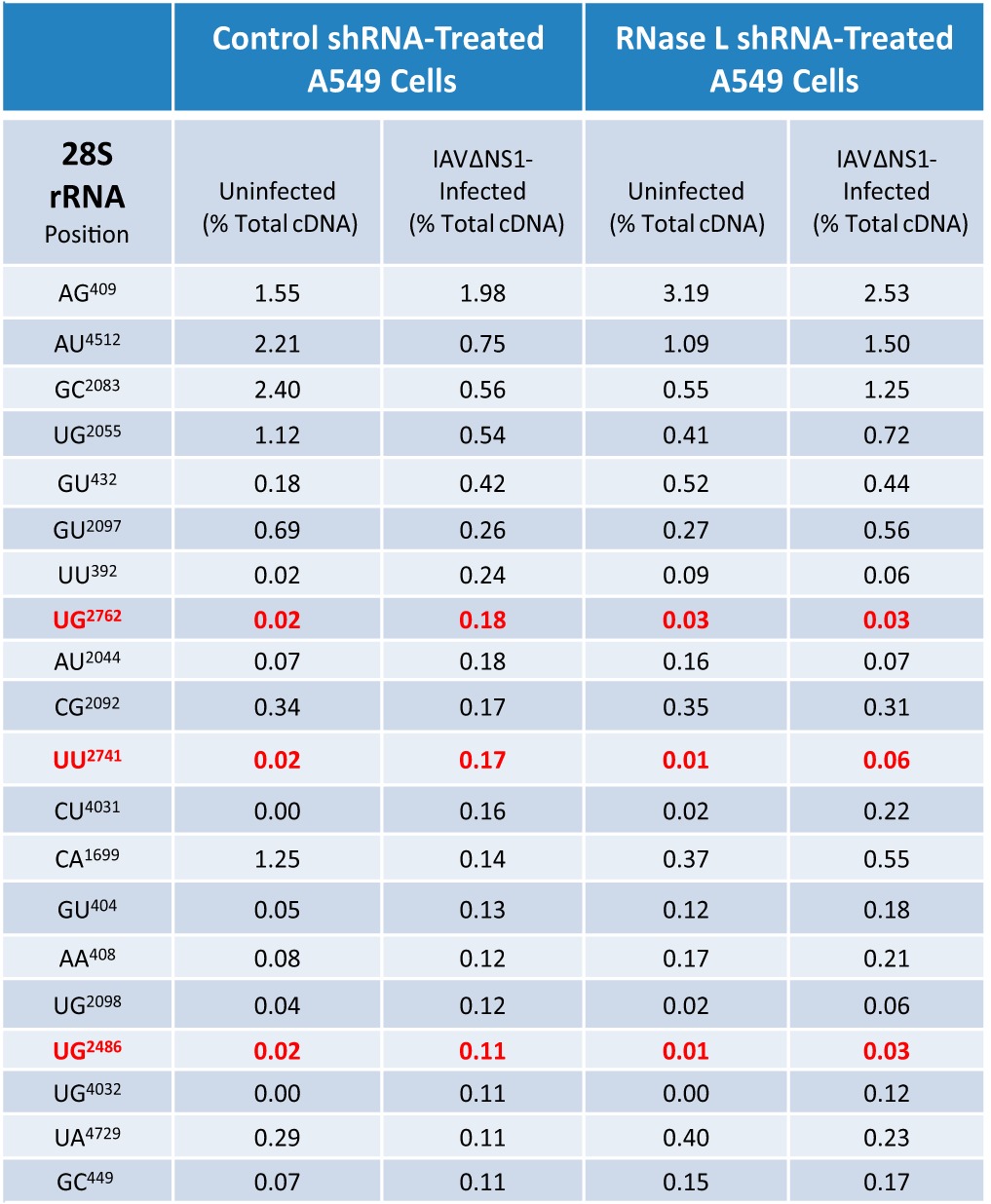
The percentages of the total cDNA reads at the 20 most frequent cleavage sites in the 18S rRNA (benchmarked to IAVΔNS1-infected control shRNA-treated A549 cells) are shown. Red values are for RNase L-dependent cleavage sites. In IAVΔNS1-infected control shRNA-treated A549 cells, there was ≥5-fold greater cleavage than in uninfected control shRNA-treated cells and uninfected RNase L shRNA-treated cells and ≥2.5-fold increased greater cleavage than in IAVΔNS1-infected RNase L shRNA-treated cells.
DISCUSSION
RNase L is a key effecter of type I interferon-regulated antiviral responses (39). In recent years, viral countermeasures to RNase L have been described, reinforcing the important nature of this host defense mechanism for picornaviruses (40, 41), coronaviruses (42), rotaviruses (43), and influenza viruses (11). Nonetheless, our understanding of RNase L antiviral mechanisms is incomplete because of the absence of information regarding precise RNA targets of RNase L during infections. To address this gap in our knowledge, we used deep-sequencing methods (14) to identify the targets of RNase L in IAVΔNS1-infected A549 cells.
Potential bias in the cDNA synthesis and sequencing analyses are important considerations. Potential sources of bias in the cDNA libraries include the Arabidopsis thaliana tRNA ligase and the variable sequences and structures of host and viral RNA fragments. In theory, the tRNA ligase could favor RNA fragments with particular features (one or another 3′-terminal nucleotide, dinucleotide, or structure). Another potential source of bias in Illumina sequencing involves GC-rich RNAs, which are sometimes underrepresented in Illumina cDNA sequencing (44). On the basis of our validation of these methods (14), any potential bias in the tRNA ligase reactions or Illumina sequencing results is negligible. Cyclic phosphate cDNA synthesis and Illumina sequencing methods detect cyclic phosphates on RNA fragments terminating in any base or dinucleotide (Fig. 3 and 4). These methods reliably distinguish between RNase A and RNase L cleavage sites in viral RNAs (14). RNase A cleaves viral RNA at single-stranded pyrimidines, whereas RNase L cleaves RNA predominantly at single-stranded UA and UU dinucleotides, with some cleavage at single-stranded UG dinucleotides and extremely small amounts of cleavage at UC, AU, CA, and CU dinucleotides (14). We cannot exclude potential bias due to an unfavorable or favorable RNA structure(s) at the 3′ ends of RNA fragments; however, we have no evidence to suggest that RNA structures impact tRNA ligase reactions, cDNA synthesis, or Illumina sequencing data sets. We have detected cleavage sites in single-stranded bulges and loops throughout large structured host and viral RNAs (reference 14 and this report). Thus, while it is possible that A. thaliana tRNA ligase has biased ligation efficiency for or against some structured RNAs, the ligase works on all sorts of structured RNAs with cyclic phosphates to the extent that we detect them. The most frequent RNase L-dependent cleavage site within cells is found within a GC-rich expansion sequence on the surface of 40S ribosomal subunits (UU743 in 18S rRNA; Fig. 9). While GC-rich RNAs have been reported by others to be underrepresented within Illumina sequencing data sets (44), we have had no trouble reproducibly detecting an RNase L-dependent cleavage site within a GC-rich sequence of 18S rRNA (Fig. 9).
The IAV cDNA libraries used for this study were prepared once, without replicates. However, several quality control indicators were evident in each cDNA library, compared to other well-validated cDNA libraries. U6 snRNA has abundant amounts of cyclic phosphate at its 3′ end because of the enzymatic activity of hUSB1 in cells or in vivo (14, 45). Cyclic phosphates specifically at the 3′ end of this cellular RNA function as a positive control in each cDNA library. Consistent with this metric, prominent amounts of cyclic phosphate were evident at this site in U6 snRNA in the four cDNA libraries described here (data not shown). In addition, we also detected prominent amounts of cyclic phosphate near the 3′ end of the 5S rRNA in our cDNA libraries, consistent with the metrics observed from cyclic phosphate cDNA libraries from HeLa cells (14). Another indicator of library quality includes the RNase L-induced cleavage sites in rRNAs that accumulate in cDNA libraries from cells where RNase L is activated. These RNase L-dependent cleavage sites are located on the surface of 80S ribosomes, consistent with cleavage occurring in vivo (14). Degradation of rRNAs during the preparation of libraries would likely result in cyclic phosphates throughout rRNAs. Thus, on the basis of these metrics, we are confident that the cDNA libraries described herein are of high quality.
The data in Fig. 2A account for the relative numbers of UMI reads in host and viral RNAs (% total cDNA reads), with the cumulative amount being 100% for each cDNA library. Consistent with this data normalization, increased UMI reads in one category correspond to relative decreases in one or another category. For instance, increased amounts of signal in 28S and 18S rRNAs tend to correspond to less signal in the mRNA and other categories (Fig. 2A). This is most evident when comparing the data for uninfected control shRNA-treated A549 cells (Fig. 2A, dark blue bars) with those for uninfected RNase L shRNA-treated A549 cells (Fig. 2A, light blue bars). These data alone do not define targets of RNase L. Rather, it is critical that % total cDNA reads be compared at individual sites in each RNA. RNase L-dependent cleavage sites are evident when magnitudes of cleavage (% total UMI reads) are consistently greater in control shRNA-treated A549 cells than those in RNase L shRNA-treated cells for specific regions in IAV RNAs (Fig. 6 to 8) and rRNAs (Fig. 9; Tables 2 and 3). We saw no evidence of RNase L activity in normal, uninfected cells (Fig. 1 and 9; Tables 2 and 3). However, small amounts of RNase L activity could be obscured by other endoribonucleases in normal, uninfected cells, as measured by cyclic phosphate cDNA libraries. Thus, we cannot draw any conclusions at this time regarding the potential roles of RNase L in basal RNA turnover.
It is well established that IAV NS1 prevents the activation of RNase L (11), among other functions (9). When NS1 is disabled, rRNAs are cleaved in an RNase L-dependent manner (Fig. 1). Cyclic phosphate cDNA synthesis and Illumina sequencing, in conjunction with bioinformatic analyses, revealed the frequency, location, and dinucleotide specificity of endoribonuclease cleavage sites in host and viral RNAs from these cells (Fig. 2 to 9; Tables 1 to 3). RNase L shRNA treatment reduced the cleavage of host and viral RNAs predominantly at UA and UU dinucleotides (Fig. 3 and 4), consistent with the specificity of RNase L (14, 15). It is noteworthy to compare the obvious RNase L-dependent cleavage sites within rRNAs with those in IAV RNAs. 18S rRNA UU743 (1.35% of the total cDNA reads) and UU541 (0.37% of the total cDNA reads) are the two most frequent RNase L-dependent cleavage sites among all of the RNAs from IAVΔNS1-infected A549 cells (Table 2). The IAV RNA sequence most frequently cleaved by RNase L was present in M positive-strand RNA, a uridine-rich 15-base sequence (200AUUUUAGGAUUUGUG214) with 0.7% of the total cDNA reads at predominantly UA, UU, and UG dinucleotides (Fig. 7). Thus, in comparison with 18S rRNA, these combined sites within IAV M RNA are among the most frequently cleaved targets of RNase L in IAVΔNS1-infected A549 cells. Intriguingly, these RNase L-dependent cleavage sites are coincident with an area of increased synonymous-site conservation within IAV M RNA (Fig. 5). The methods used cannot distinguish between IAV mRNA and cRNA as it relates to endoribonuclease cleavage sites. However, we suspect that cleavage detected in IAV positive-strand segments likely originate from mRNA, since IAV mRNAs are more abundant than IAV cRNAs. In addition, IAV mRNAs are present in the cytoplasm, where we observed RNase L-dependent rRNA cleavage. Consequently, the abundant amounts of cleavage detected in the M RNA segment may be due to cleavage of M mRNA rather than M cRNA.
Of note, RNase L appeared to cleave other IAV RNAs within and adjacent to some areas of increased synonymous-site conservation, including PB1 positive and negative strands, NP positive strands, and PA negative strands (Fig. 5). In a reciprocal manner, HA and NA RNAs, which have, in comparison, smaller amounts of synonymous-site conservation, exhibited little cleavage by RNase L or other endonucleases (Fig. 5). On the basis of the patterns of synonymous-site conservation and endonucleolytic cleavage that we have observed, it might be important to consider the potential roles of endonucleases in viral RNA packaging and reassortment. Because HA and NA RNA segments are more refractory to endonucleases, they might reassort more successfully than other RNA segments. Viral RNA packaging signals (19, 20) tend to be uridine-rich sequences within areas of increased synonymous-site conservation, potential targets of RNase L. By preventing the activation of the OAS/RNase L pathway, NS1 protects these conserved regions of IAV RNA from cleavage by RNase L.
In a previous investigation, we found that poliovirus RNA was cleaved by RNase L and other metal ion-independent endoribonucleases; however, only the positive strand of poliovirus RNA was cleaved in infected cells (14). Endonucleolytic cleavage sites were conspicuously absent from poliovirus negative strands (14), perhaps because of the nature of membranous viral replication complexes (46). The membranous replication complexes of positive-strand viruses sequester replicating viral RNA and provide a compartment wherein a virus-encoded competitive inhibitor of RNase L can overwhelm small amounts of RNase L, preventing the cleavage of viral negative-strand templates and nascent positive strands (40, 47, 48). In contrast to RNAs of positive-strand viruses like poliovirus, IAV RNAs are localized intermittently in both the nucleus and the cytoplasm and are not sequestered within membranous replication complexes. In the present study, we found that IAV RNAs were cleaved by RNase L and other metal ion-independent endoribonucleases in both negative and positive strands (Fig. 2 to 6; Table 1). In fact, PB2, PB1, and PA negative strands were among the most frequently cleaved IAV RNAs (Fig. 2B and 3; Table 1), suggesting that nucleocapsid does not protect IAV RNAs from cleavage within RNPs, consistent with structural studies of RNPs (7).
Frequent cleavage at pyrimidines within IAV RNAs (Fig. 3 and 4) is consistent with the specificity of angiogenin, RNase 4, or related RNase A family enzymes (35–37). Importantly, angiogenin is found in the nuclei of cells (37), where it could potentially cleave IAV genomic RNAs. IAV RNAs, especially IAV genomic RNAs, exhibited cleavage at pyrimidines (Fig. 3 and 4). Cleavage at these sites was not reduced significantly by RNase L shRNA. Thus, on the basis of the nature of the cleavage sites (pyrimidines) and their resistance to RNase L knockdown, it is reasonable to implicate RNase A family enzymes in the cleavage of IAV RNAs.
rRNAs are obvious targets of RNase L (Fig. 1B). In this study, we identified RNase L-dependent cleavage sites in 18S and 28S rRNAs (Fig. 9; Tables 2 and 3). We observed ≥5-fold-more cleavage at particular sites in rRNAs in IAVΔNS1-infected A549 cells than in uninfected cells (Tables 2 and 3). RNase L knockdown reduced the amounts of cleavage specifically at these RNase L-dependent sites (Tables 2 and 3). 18S rRNA UU743 and UU541 are the most frequent targets of RNase L among the host and viral RNAs from A549 cells (Fig. 9; Table 2). These are the same RNase L-dependent 18S rRNA cleavage sites identified in poliovirus-infected HeLa cells (14). The antiviral activity of RNase L likely involves cleavage at these sites in 18S rRNA. Cleavage at these sites in ribosomes could inhibit mRNA translation, thereby contributing to the inhibition of viral gene expression.
In addition to rRNAs, endoribonuclease cleavage sites were detected in host mRNAs and various noncoding RNAs (Fig. 2A, mRNA and other). Cleavage sites in individual host mRNAs were most frequently associated with small numbers of cDNA reads, making it difficult to attribute the cleavage sites to specific endoribonucleases. Furthermore, we did not detect cyclic phosphates in a large intergenic noncoding RNA associated with IAV replication (49), suggesting that this cellular RNA was not cleaved frequently by RNase L or other metal ion-independent endoribonucleases in uninfected or IAVΔNS1-infected A549 cells.
Now that we have defined a constellation of RNase L-dependent cleavage sites in host and viral RNAs in IAVΔNS1-infected A549 cells, we expect that cyclic phosphate cDNA libraries can be used to monitor RNase L activity in vitro and in vivo. There are several important questions to address. (i) Does NS1 expression in the context of wild-type virus completely block RNase L activity? (ii) Does RNase L cleave host and viral RNA in vivo? Evidence from mice suggests that RNase L contributes to IAV-induced immunopathology (13), indicating that NS1 may not be able to completely block the activation of RNase L in vivo. Cyclic phosphate cDNA libraries should be able to reveal endoribonuclease cleavage sites in host and viral RNAs from tissues, including lung tissue from IAV-infected mice (13). Future experiments can be used to examine the kinetics of host and viral RNA cleavage in IAV-infected cells, to assess the impact of rRNA cleavage on host and viral mRNA translation (ribosome function), and to determine whether RIG-I ligands are generated by RNase L activity during the course of IAV infections, as predicted by other studies (50).
Summary.
RNase L targeted a precise subset of UA and UU dinucleotides in both host and viral RNAs when influenza virus NS1 protein was disabled. Uridine-rich influenza virus RNA sequences in some areas of increased synonymous-site conservation were particularly susceptible to cleavage by RNase L. On the basis of these data, it is reasonable to conclude that RNase L- and NS1-mediated countermeasures influence the evolution of RNA sequences in influenza viruses (51), with UA- and UU-rich sequences arising in viral genomes to support viral RNA packaging (19, 20), ARFs (4), splicing (3), and other functions when NS1 protein effectively blocks RNase L activity. With this in mind, the antiviral activity and sequence specificity of RNase L can be considered in the design of novel codon-deoptimized IAV vaccines (52). We suspect that codon deoptimization can be used to increase the susceptibility of viral RNAs to endoribonucleases.
Supplementary Material
ACKNOWLEDGMENTS
We thank Andrew E. Firth (University of Cambridge) for providing IAV synonymous-site conservation data and Brian Kempf for critical evaluation of the manuscript.
D.A.C. and S.B. performed the experiments. A.C. and A.G.-S. contributed reagents. R.H.S. and D.J.B. designed the study. D.A.C., S.B., J.R.H., R.H.S., and D.J.B. analyzed the data. D.A.C., S.B., R.H.S., and D.J.B. wrote the paper.
This work was supported by Mucosal and Vaccine Research Colorado (MAVRC), Golfers Against Cancer, the University of Colorado Cancer Center, March of Dimes Basil O'Conner Starter Award (J.R.H.), Damon Runyon-Rachleff Innovation Award (J.R.H.), American Cancer Society Research Scholar Award (J.R.H.), United States Public Health Service National Institutes of Health grants T32-AI052066 (D.A.C.), U19AI106754 (A.G.-S.), CA044059 (R.H.S.), and AI042189 (D.J.B.). This work was also partly supported by CRIP (Center for Research on Influenza Pathogenesis), and NIAID funded the Center of Excellence for Influenza Research and Surveillance (CEIRS) under contract HHSN272201400008C (A.G.-S.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02953-14.
REFERENCES
- 1.Arriola CS, Brammer L, Epperson S, Blanton L, Kniss K, Mustaquim D, Steffens C, Dhara R, Leon M, Perez A, Chaves SS, Katz J, Wallis T, Villanueva J, Xu X, Abd Elal AI, Gubareva L, Cox N, Finelli L, Bresee J, Jhung M. 2014. Update: influenza activity—United States, September 29, 2013–February 8, 2014. MMWR Morb Mortal Wkly Rep 63:148–154 http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6307a3.htm. [PMC free article] [PubMed] [Google Scholar]
- 2.Medina RA, García-Sastre A. 2011. Influenza A viruses: new research developments. Nat Rev Microbiol 9:590–603. doi: 10.1038/nrmicro2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wise HM, Hutchinson EC, Jagger BW, Stuart AD, Kang ZH, Robb N, Schwartzman LM, Kash JC, Fodor E, Firth AE, Gog JR, Taubenberger JK, Digard P. 2012. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain. PLoS Pathog 8:e1002998. doi: 10.1371/journal.ppat.1002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, Dalton RM, Lo A, Efstathiou S, Atkins JF, Firth AE, Taubenberger JK, Digard P. 2012. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 337:199–204. doi: 10.1126/science.1222213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma W, Liu Q, Qiao C, Del Real G, García-Sastre A, Webby RJ, Richt JA. 2014. North American triple reassortant and Eurasian H1N1 swine influenza viruses do not readily reassort to generate a 2009 pandemic H1N1-like virus. mBio 5:e00919–13. doi: 10.1128/mBio.00919-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horimoto T, Kawaoka Y. 2005. Influenza: lessons from past pandemics, warnings from current incidents. Nat Rev Microbiol 3:591–600. doi: 10.1038/nrmicro1208. [DOI] [PubMed] [Google Scholar]
- 7.Arranz R, Coloma R, Chichon FJ, Conesa JJ, Carrascosa JL, Valpuesta JM, Ortin J, Martin-Benito J. 2012. The structure of native influenza virion ribonucleoproteins. Science 338:1634–1637. doi: 10.1126/science.1228172. [DOI] [PubMed] [Google Scholar]
- 8.Portela A, Digard P. 2002. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J Gen Virol 83:723–734. doi: 10.1099/vir.0.18133-0. [DOI] [PubMed] [Google Scholar]
- 9.Engel DA. 2013. The influenza virus NS1 protein as a therapeutic target. Antiviral Res 99:409–416. doi: 10.1016/j.antiviral.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.García-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, Palese P, Muster T. 1998. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252:324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- 11.Min JY, Krug RM. 2006. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: inhibiting the 2′-5′ oligo(A) synthetase/RNase L pathway. Proc Natl Acad Sci U S A 103:7100–7105. doi: 10.1073/pnas.0602184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walkiewicz MP, Basu D, Jablonski JJ, Geysen HM, Engel DA. 2011. Novel inhibitor of influenza non-structural protein 1 blocks multi-cycle replication in an RNase L-dependent manner. J Gen Virol 92:60–70. doi: 10.1099/vir.0.025015-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Y, Kang MJ, Jha BK, Silverman RH, Lee CG, Elias JA. 2013. Role of ribonuclease L in viral pathogen-associated molecular pattern/influenza virus and cigarette smoke-induced inflammation and remodeling. J Immunol 191:2637–2646. doi: 10.4049/jimmunol.1300082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper DA, Jha BK, Silverman RH, Hesselberth JR, Barton DJ. 2014. Ribonuclease L and metal ion-independent endoribonuclease cleavage sites in host and viral RNAs. Nucleic Acids Res 42:5202–5216. doi: 10.1093/nar/gku118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wreschner DH, McCauley JW, Skehel JJ, Kerr IM. 1981. Interferon action—sequence specificity of the ppp(A2′p)nA-dependent ribonuclease. Nature 289:414–417. doi: 10.1038/289414a0. [DOI] [PubMed] [Google Scholar]
- 16.Han Y, Donovan J, Rath S, Whitney G, Chitrakar A, Korennykh A. 2014. Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 343:1244–1248. doi: 10.1126/science.1249845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang H, Zeqiraj E, Dong B, Jha BK, Duffy NM, Orlicky S, Thevakumaran N, Talukdar M, Pillon MC, Ceccarelli DF, Wan LC, Juang YC, Mao DY, Gaughan C, Brinton MA, Perelygin AA, Kourinov I, Guarne A, Silverman RH, Sicheri F. 2014. Dimeric structure of pseudokinase RNase L bound to 2-5A reveals a basis for interferon-induced antiviral activity. Mol Cell 53:221–234. doi: 10.1016/j.molcel.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O'Neill R, Schickli J, Palese P, Henklein P, Bennink JR, Yewdell JW. 2001. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med 7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- 19.Gog JR, Afonso Edos S, Dalton RM, Leclercq I, Tiley L, Elton D, von Kirchbach JC, Naffakh N, Escriou N, Digard P. 2007. Codon conservation in the influenza A virus genome defines RNA packaging signals. Nucleic Acids Res 35:1897–1907. doi: 10.1093/nar/gkm087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hutchinson EC, von Kirchbach JC, Gog JR, Digard P. 2010. Genome packaging in influenza A virus. J Gen Virol 91:313–328. doi: 10.1099/vir.0.017608-0. [DOI] [PubMed] [Google Scholar]
- 21.Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, García-Sastre A. 2009. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiang Y, Wang Z, Murakami J, Plummer S, Klein EA, Carpten JD, Trent JM, Isaacs WB, Casey G, Silverman RH. 2003. Effects of RNase L mutations associated with prostate cancer on apoptosis induced by 2′,5′-oligoadenylates. Cancer Res 63:6795–6801. [PubMed] [Google Scholar]
- 23.Dong B, Silverman RH. 1995. 2-5A-dependent RNase molecules dimerize during activation by 2-5A. J Biol Chem 270:4133–4137. doi: 10.1074/jbc.270.8.4133. [DOI] [PubMed] [Google Scholar]
- 24.Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, García-Sastre A. 2010. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A 107:11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kivioja T, Vaharautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J. 2011. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 9:72–74. doi: 10.1038/nmeth.1778. [DOI] [PubMed] [Google Scholar]
- 28.R Development Core Team. 2012. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 29.Firth AE. 2014. Mapping overlapping functional elements embedded within the protein-coding regions of RNA viruses. Nucleic Acids Res 42:12425–12439. doi: 10.1093/nar/gku981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A. 2013. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res 41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silverman RH, Skehel JJ, James TC, Wreschner DH, Kerr IM. 1983. rRNA cleavage as an index of ppp(A2′p)nA activity in interferon-treated encephalomyocarditis virus-infected cells. J Virol 46:1051–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wreschner DH, James TC, Silverman RH, Kerr IM. 1981. Ribosomal RNA cleavage, nuclease activation and 2-5A(ppp(A2′p)nA) in interferon-treated cells. Nucleic Acids Res 9:1571–1581. doi: 10.1093/nar/9.7.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crépin T, Dias A, Palencia A, Swale C, Cusack S, Ruigrok RWH. 2010. Mutational and metal binding analysis of the endonuclease domain of the influenza virus polymerase PA subunit. J Virol 84:9096–9104. doi: 10.1128/JVI.00995-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Lv Z, Ge R, Li X, Deng T, Fodor E, Rao Z, Liu Y. 2009. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 458:909–913. doi: 10.1038/nature07720. [DOI] [PubMed] [Google Scholar]
- 35.Hofsteenge J, Moldow C, Vicentini AM, Zelenko O, Jarai-Kote Z, Neumann U. 1998. A single amino acid substitution changes ribonuclease 4 from a uridine-specific to a cytidine-specific enzyme. Biochemistry 37:9250–9257. doi: 10.1021/bi9803832. [DOI] [PubMed] [Google Scholar]
- 36.Li S, Sheng J, Hu JK, Yu W, Kishikawa H, Hu MG, Shima K, Wu D, Xu Z, Xin W, Sims KB, Landers JE, Brown RH Jr, Hu GF. 2013. Ribonuclease 4 protects neuron degeneration by promoting angiogenesis, neurogenesis, and neuronal survival under stress. Angiogenesis 16:387–404. doi: 10.1007/s10456-012-9322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsuji T, Sun Y, Kishimoto K, Olson KA, Liu S, Hirukawa S, Hu GF. 2005. Angiogenin is translocated to the nucleus of HeLa cells and is involved in ribosomal RNA transcription and cell proliferation. Cancer Res 65:1352–1360. doi: 10.1158/0008-5472.CAN-04-2058. [DOI] [PubMed] [Google Scholar]
- 38.Moss WN, Priore SF, Turner DH. 2011. Identification of potential conserved RNA secondary structure throughout influenza A coding regions. RNA 17:991–1011. doi: 10.1261/rna.2619511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Silverman RH. 2007. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol 81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han JQ, Townsend HL, Jha BK, Paranjape JM, Silverman RH, Barton DJ. 2007. A phylogenetically conserved RNA structure in the poliovirus open reading frame inhibits the antiviral endoribonuclease RNase L. J Virol 81:5561–5572. doi: 10.1128/JVI.01857-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sorgeloos F, Jha BK, Silverman RH, Michiels T. 2013. Evasion of antiviral innate immunity by Theiler's virus L* protein through direct inhibition of RNase L. PLoS Pathog 9:e1003474. doi: 10.1371/journal.ppat.1003474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao L, Jha BK, Wu A, Elliott R, Ziebuhr J, Gorbalenya AE, Silverman RH, Weiss SR. 2012. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 11:607–616. doi: 10.1016/j.chom.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang R, Jha BK, Ogden KM, Dong B, Zhao L, Elliott R, Patton JT, Silverman RH, Weiss SR. 2013. Homologous 2′,5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc Natl Acad Sci U S A 110:13114–13119. doi: 10.1073/pnas.1306917110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benjamini Y, Speed TP. 2012. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res 40:e72. doi: 10.1093/nar/gks001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schutz K, Hesselberth JR, Fields S. 2010. Capture and sequence analysis of RNAs with terminal 2′,3′-cyclic phosphates. RNA 16:621–631. doi: 10.1261/rna.1934910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahlquist P. 2006. Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses. Nat Rev Microbiol 4:371–382. doi: 10.1038/nrmicro1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Townsend HL, Jha BK, Han JQ, Maluf NK, Silverman RH, Barton DJ. 2008. A viral RNA competitively inhibits the antiviral endoribonuclease domain of RNase L. RNA 14:1026–1036. doi: 10.1261/rna.958908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Townsend HL, Jha BK, Silverman RH, Barton DJ. 2008. A putative loop E motif and an H-H kissing loop interaction are conserved and functional features in a group C enterovirus RNA that inhibits ribonuclease L. RNA Biol 5:263–272. doi: 10.4161/rna.7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winterling C, Koch M, Koeppel M, García-Alcalde F, Karlas A, Meyer TF. 2014. Evidence for a crucial role of a host non-coding RNA in influenza A virus replication. RNA Biol 11:66–75. doi: 10.4161/rna.27504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malathi K, Dong B, Gale M Jr, Silverman RH. 2007. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448:816–819. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Belalov IS, Lukashev AN. 2013. Causes and implications of codon usage bias in RNA viruses. PLoS One 8:e56642. doi: 10.1371/journal.pone.0056642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nogales A, Baker SF, Ortiz-Riano E, Dewhurst S, Topham DJ, Martinez-Sobrido L. 2014. Influenza A virus attenuation by codon deoptimization of the NS gene for vaccine development. J Virol 88:10525–10540. doi: 10.1128/JVI.01565-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wise HM, Barbezange C, Jagger BW, Dalton RM, Gog JR, Curran MD, Taubenberger JK, Anderson EC, Digard P. 2011. Overlapping signals for translational regulation and packaging of influenza A virus segment 2. Nucleic Acids Res 39:7775–7790. doi: 10.1093/nar/gkr487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anger AM, Armache JP, Berninghausen O, Habeck M, Subklewe M, Wilson DN, Beckmann R. 2013. Structures of the human and Drosophila 80S ribosome. Nature 497:80–85. doi: 10.1038/nature12104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.