

ABSTRACT
To investigate the role of the signal sequences of herpes simplex virus 1 (HSV-1) gK on virus replication and viral pathogenesis, we constructed recombinant viruses with or without mutations within the signal sequences of gK. These recombinant viruses expressed two additional copies of the mutated (MgK) or native (NgK) form of the gK gene in place of the latency-associated transcript with a myc epitope tag to facilitate detection at their 3′ ends. The replication of MgK virus was similar to that of NgK both in vitro and in vivo, as well as in the trigeminal ganglia (TG) of latently infected mice. The levels of gB and gK transcripts in the corneas, TG, and brains of infected mice on days 3 and 5 postinfection were markedly virus and time dependent, as well as tissue specific. Mutation in the signal sequence of gK in MgK virus blocked cell surface expression of gK-myc in rabbit skin cells, increased 50% lethal dose, and decreased corneal scarring in ocularly infected mice compared to the NgK or revertant (RgK) virus. MgK and NgK viruses, and not the RgK virus, showed a reduced extent of explant reactivation at the lower dose of ocular infection but not at the higher dose. However, the time of reactivation was not affected by overexpression of the different forms of gK. Taken together, these results strongly suggest that the 8mer peptide (ITAYGLVL) within the signal sequence of gK promotes cell surface expression of gK in infected cells and ocular pathogenesis in infected mice.
IMPORTANCE In this study, we show for the first time that mutations within the signal sequence of gK blocked cell surface expression of inserted recombinant gK in vitro. Furthermore, this blockage in cell surface expression was correlated with higher 50% lethal dose and less corneal scarring in vivo. Thus, these studies point to a key role for the 8mer within the signal sequence of gK in HSV-1-induced pathogenicity.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) infections are among the most frequent serious viral eye infections in the United States and are a major cause of virus-induced blindness (1–3). It is estimated that 70 to 90% of American adults have antibodies to HSV-1 and/or HSV-2 and ca. 25% of these individuals have clinical symptoms upon routine clinical inquiry. HSV-1 is responsible for >90% of ocular HSV infections (1, 2, 4–7). HSV-1-induced corneal scarring (CS), also broadly referred to as herpes stromal keratitis (HSK), can lead to blindness. Furthermore, HSV-1 is the leading cause of corneal blindness due to an infectious agent in developed countries (1, 2, 4–7). In addition to necrotizing HSK, ocular infection with HSV-1 can cause eye disease ranging in severity from blepharitis, conjunctivitis, and dendritic keratitis to disciform stromal edema (4, 7–11). In the United States, approximately 30,000 people per year suffer recurrent ocular HSV episodes, requiring doctor visits, medication, and in severe cases, corneal transplants.
Although it is well established that HSV-1-induced CS is due to immune responses to the virus, the exact identity of the immune responses that lead to CS is not yet clearly apparent. The finding that HSV-1 glycoproteins are the major inducers and targets of humoral and cell-mediated immune responses after infection (12–16) suggest that they may play a major role in the development of the immune responses that cause CS in infected individuals. We have investigated the role of the HSV-1 glycoproteins in protection from eye disease and have shown that immunization with glycoprotein B (gB), gC, gD, gE, or gI completely protected mice against lethal challenge with HSV-1 and eye disease, whereas no significant protection is seen on immunization with gG, gH, gL, or gJ (17–21). In marked contrast, immunization with gK leads to exacerbation of eye disease and facial dermatitis (14, 20). The gK-exacerbated CS occurs independently of the mouse strains or virus strains used in the analysis (22) and the pathology is due to the presence of CD8+ CD25+ T cells in the cornea of ocularly infected mice (23). We also have shown that the titers of anti-gK antibody are higher in the sera of patients with HSK than in the sera of individuals that are positive for HSV-1 but do not have HSK (24).
gK, one of the 12 known HSV-1 glycoproteins, is a multimembrane-spanning viral glycoprotein encoded by UL53 (16, 25–27). It is expressed on the virions and HSV-1 mutants that lack gK fail to acquire a cytoplasmic envelope efficiently and are unable to efficiently infect and establish latency in neurons (28–32). Deletion of gK has been shown to result in reductions in virus yield, plaque size, and translocation of the virus from the cytoplasm to the extracellular space (28). Moreover, viruses with deletion of gK are unable to undergo transport in either retrograde or anterograde directions in neuronal cell cultures (33). Therefore, to analyze the roles of gK in CS, we previously prepared a recombinant HSV-1 that expressed two additional copies of the gK gene. Compared to the wild-type (wt) HSV-1 strain McKrae, this recombinant virus exacerbated CS in two different strains of mice (34). Furthermore, we identified a peptide (8mer; ITAYGLVL) within the signal sequence of gK as being an immunodominant gK T-cell stimulatory region both in vitro and in vivo (35). Administration of this peptide as an eye drop resulted in significantly higher virus replication in the eyes of BALB/c mice, C57BL/6 mice, and New Zealand White (NZW) rabbits, as well as greater virus-induced CS in the ocularly infected mice (36). Moreover, in HSV-infected “humanized” HLA-A*0201 transgenic mice, this gK 8mer epitope induced strong gamma interferon-producing cytotoxic CD8+ T-cell responses (36).
In the present report, we investigated the effects of mutagenesis of the gK 8mer and overexpression of gK. We performed site-specific mutagenesis within the gK 8mer region and inserted the complete open reading frame (ORF) of the mutated form of the gK gene into both copies of the latency-associated transcript (LAT) regions and under the LAT promoter as we described previously (34, 37–40). Since suitable antibodies for detection of gK are not available, we inserted the myc epitope tag in-frame at the 3′ end of the mutated gK ORF for identification of the inserted gK. This virus was designated mutated gK (MgK). As a control and to explore the effects of overexpression, an additional recombinant virus was constructed that expresses two copies of the native form of the gK ORF with a myc tag in-frame at its 3′ end. This virus was designated native gK (NgK). This NgK virus is similar to the HSV-gK3 virus that we described previously (34) except for having the myc tag in-frame at its 3′ end. Revertant virus, designated RgK, also was used as a control. Both the NgK and the MgK viruses expressed the inserted gK in vitro as determined by Western blotting. However, although the NgK virus expressed the inserted gK on the cell surface of infected cells the MgK virus did not. The replication of the NgK, MgK, and RgK viruses in mouse tears were similar, as was latency in the trigeminal ganglia (TG), but the MgK virus had a higher 50% lethal dose (LD50) than NgK or RgK, with the NgK virus having the lowest LD50. Ocular infection with NgK resulted in more severe CS than infection with MgK or RgK virus. In summary, the presence of the 8mer within the signal sequence of gK blocked cell surface expression of the inserted form of gK that led to a higher LD50 and less CS in ocularly infected mice.
MATERIALS AND METHODS
Viruses, cells, and mice.
Triple plaque-purified wt McKrae, dLAT2903, HSV-gK3, MgK, NgK, and RgK viruses were used in the present study. Rabbit skin (RS) cells (used for the preparation of virus stocks, the culture of mouse tear films, and determination of growth kinetics) were grown in Eagle minimal essential medium supplemented with 5% fetal calf serum. Female 6-week-old inbred BALB/c and C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All animal procedures were performed in strict accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Animal research protocol was approved by the Institutional Animal Care and Use Committee of Cedars-Sinai Medical Center.
Construction of gK recombinant viruses.
The LAT plasmid (pLAT) was prepared using the BamHI B fragment of McKrae as we described previously (34, 41). The final construct lacked the 1.6-kb of LAT fragment and contained 880 bp upstream and 2,989 bp downstream of the deleted 1.6-kb of LAT fragment with a BamHI site to insert the gene of interest. The complete ORF of gK is 338 amino acids long (16). Two forms of full-length gK were synthesized by GenScript (Piscataway, NJ). One, designated native gK (NgK), contained the complete ORF of gK with a myc tag sequence in-frame at its 3′ end. The second, designated mutated gK (MgK), was synthesized such that it encoded a form of gK in which five amino acids within the 8mer peptide in the signal region were altered, i.e., ITAYGLVL to AGASGIVI, and had a myc tag sequence in-frame at its 3′ end (Fig. 1). For construction of the gK plasmids, the complete ORFs of NgK and MgK were cloned into the BamHI site of the pLAT expression vector as we described previously (34, 41). The NgK and MgK synthesized genes were inserted into the BamHI site of the pLAT, and the resulting plasmids were designated pLAT-NgK and pLAT-MgK, respectively. Each construct contained the 1,040-bp gK gene, 33 bp of c-myc (EQKLISEEDL), and the gK termination codon bounded by an 880-bp LAT fragment at the 5′ end of the gK construct and a 2,989-bp LAT fragment at the 3′ end.
FIG 1.
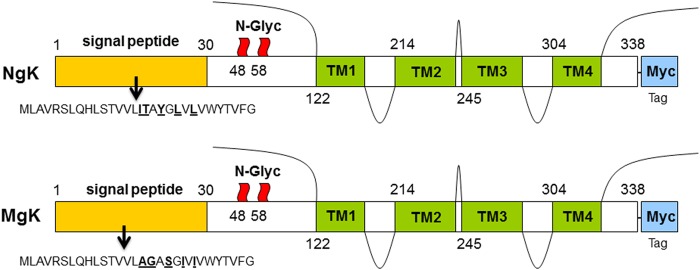
Myc-gK gene constructs used for the generation of NgK and MgK viruses. For the construction of recombinant NgK and MgK viruses, we synthesized the complete ORF of gK as follows. For NgK, the structure of the wt gK molecule of 338 amino acids is shown with an in-frame insertion of c-myc sequence on C terminus. Positions of the N-glycosylation sites are indicated at amino acid residues 48 and 58. The four possible gK transmembrane (TM) regions and the 30-amino-acid signal sequence of wt gK are shown. For MgK, the structure of mutated form of gK is similar to that of NgK, except that five of the amino acids within the signal sequences of wt gK were mutated (I16A, T17G, Y19S, L21I, and L23I). The NgK and MgK constructs were inserted into the BamHI site of pLAT as described in Materials and Methods.
NgK and MgK recombinant viruses were then generated by homologous recombination as we described previously (34, 41). Briefly, pLAT-NgK or pLAT-MgK was cotransfected with infectious dLAT2903 DNA using the calcium phosphate method. Viruses from the cotransfection were plated, and isolated plaques were picked and screened for gK gene insertion using restriction digestion and Southern blot analysis. Selected plaques that contained the inserted gK gene were plaque purified four times and reanalyzed by restriction digestion and Southern blot analysis to verify that the inserted gK gene was present in the LAT region. For each construct, a single plaque was then chosen for purification and designated NgK or MgK. These viruses contained the gK gene that is in the normal LAT location in the viral genome and are under the control of the LAT promoter. Thus, in addition to the endogenous HSV-1 gK gene, these viruses contained two additional copies of the gK gene under the LAT promoter (one in each viral long repeat).
Rescued viruses, in which the inserted gK gene was removed and the original deletion of the LAT gene was restored, were generated by cotransfection and homologous recombination using infectious NgK or MgK DNA and the pLAT plasmid. These rescued viruses behaved similarly to each other, as well as to the parental dLAT2903 virus. Thus, the rescued virus, designated RgK, generated from the MgK virus was used as the control virus for the subsequent studies.
Southern analyses.
RS cells were infected with 10 PFU of McKrae, HSV-gK3, NgK, or MgK/cell. Viral DNA was isolated and digested with EcoRV/EcoRI. Digested DNA was separated by a 0.9% agarose gel, and the gel was subsequently blotted onto a positively charged nylon membrane (Amersham Hybond–N+; GE Healthcare, Pittsburgh, PA). After transfer, the membrane was fixed by UV cross-linking (TL-2000 UV Cross-Linker). For, DNA hybridization and detection, the digoxigenin (DIG) nonradioactive nucleic acid labeling and detection system (DIG High Prime DNA labeling/detection kit for chemiluminescence; Roche Diagnostics, Indianapolis, IN) was used according to the manufacturer's protocol. The complete ORF of gK was isolated from pAC-gK1 construct that we described previously (26) by BamHI digestion. The gK complete ORF along with 7 nucleotides upstream of gK ATG and 15 noncoding nucleotides prior to the stop codon of gK is 1,040 bases long and is called gK-1040. The gK-1040 was used as probe to identify natural gK and inserted gK in NgK and MgK recombinant viruses. Briefly, the purified gK-1040 fragment was used as the template DNA for the hybridization probe and was random primer labeled with DIG overnight at 37°C. The following day, the membrane was prehybridized in the DIG Easy Hyb buffer (Roche Diagnostics) for 2 h at 65°C and subsequently hybridized with the GK probe overnight in the same solution at 65°C. The membrane was washed twice with a solution of 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) and 0.1% sodium dodecyl sulfate (SDS) for 5 min at room temperature, followed by two additional washes with a solution of 0.5× SSC and 0.1% SDS for 15 min at 65°C. The hybridized DIG-labeled gK probe was immunodetected with anti-DIG-alkaline phosphatase (AP) conjugate and visualized using the chemiluminescence substrate CSPD Ready-to-Use solution and X-ray film.
Virus replication in tissue culture.
RS cell monolayers at 70 to 80% confluence were infected with 0.1 or 0.01 PFU of NgK, MgK, or RgK/cell. Virus was harvested at various times by subjecting the cell monolayers to two freeze-thawing cycles. Virus titers were determined using standard plaque assays on RS cells as described previously (14).
Western analysis.
RS cell monolayers at 70 to 80% confluence were infected with 10 PFU of NgK, MgK, or RgK/cell or mock infected. At 16 h postinfection (p.i.), infected cells were lysed directly into gel sample buffer, and the protein concentrations in the cell lysates were determined by using a Pierce BCA protein assay kit according to the manufacturer's protocol (Thermo Scientific, Rockford, IL). Approximately 40 μg of protein was loaded per well, and samples were separated by SDS–10% PAGE, transferred to nitrocellulose, and reacted with anti-myc antibody (Cell Signaling Technology, Danvers, MA). The membrane was incubated with horseradish peroxidase-labeled secondary antibody (goat anti-rabbit IgG; 1:2,000) at 37°C for 1 h. The immunoreactive bands were detected using the enhanced chemiluminescence Western blotting system available from Thermo Scientific (Thermo Fisher Scientific, Waltham, MA).
Flow cytometric analysis.
RS cells were infected with 5 PFU of NgK, MgK, or RgK/cell or were mock infected. At 24 h p.i., the cells were harvested by centrifugation and stained with anti-myc-PE and anti-HSV-1 gC-fluorescein isothiocyanate (FITC) or anti-annexin V-phycoerythrin (PE) and -7-aminoactinomycin D (7-AAD) from BD Pharmingen (San Diego, CA) and Biolegend (San Diego, CA). Stained cells were washed twice with fluorescence-activated cell sorting (FACS) buffer (1× phosphate-buffered saline [PBS] with 0.1% sodium azide), resuspended in 4% paraformaldehyde, and analyzed using a multicolor five-laser LSR II instrument (Applied Biosystems, Foster City, CA).
Colocalization of gK expression.
RS cells were grown to confluence on Lab-Tek chamber slides and infected with 1 PFU of NgK, MgK, or RgK/cell or mock infected for 24 h as we described previously (42). The cells were fixed and permeabilized with 4% paraformaldehyde for 1 h at 4°C, followed by 20 min of incubation in serum-free protein block (Dako, Carpentaria, CA) at room temperature. The cells were then incubated with anti-myc-PE and anti-HSV-1 gC-FITC antibodies for 1 h at 4°C. Washed slides were air dried and mounted with DAPI (4′,6′-diamidino-2-phenylindole) Prolong Gold (Invitrogen). The fluorophores were imaged in separate channels with a Zeiss ApoTome-equipped Axio Imager Z1 (Carl Zeiss Microimaging) using a Nikon AIR laser scan confocal microscope.
Ocular infection.
Mice were infected ocularly with a 2-μl drop of tissue culture medium containing 2 × 106, 2 × 105, 2 × 104, 2 × 103, or 2 × 102 PFU of NgK, MgK, or RgK virus/eye. No corneal scarification was performed on the infected mice.
Monitoring CS.
The severity of CS was scored in a masked fashion by examination with a slit lamp biomicroscope. Disease was scored on a 0 to 4 scale (0 = no disease, 1 = 25%, 2 = 50%, 3 = 75%, and 4 = 100% involvement).
Titration of virus in tears of infected mice.
Tear films were collected from both eyes of 20 mice per group on days 1 to 5 after ocular infection using a Dacron-tipped swab. Each swab was placed in 0.5 ml of tissue culture medium and squeezed, and the amount of virus was determined using a standard plaque assay on RS cells (43).
Isolation of RNA from cornea, brain and TG of infected mice.
C57BL/6 mice were ocularly infected with 2 × 105 PFU of NgK, MgK, or RgK virus/eye. Corneas, TG, and brains from infected mice were collected on days 3 and 5 p.i. Isolated tissues were immersed in RNAlater RNA stabilization reagent and stored at −20°C until processing. The corneas, TG, or brain from each animal were processed for RNA extraction using TRIzol reagent as we described previously (44). Isolated total RNA was reverse-transcribed with random hexamer primers and murine leukemia virus reverse transcriptase provided in a high-capacity cDNA reverse transcription kit (Applied Biosystems) according to the manufacturer's recommendations. All isolated corneas were free of contamination from other parts of the mouse eye, vitreous fluid, and tears.
DNA extraction for HSV-1 gB and gK DNAs.
DNA was isolated from homogenized individual TG on day 28 p.i. using the commercially available DNeasy Blood & Tissue kit (Qiagen, Stanford, CA) according to the manufacturer's instructions.
TaqMan real-time PCR and PCR.
The gB and gK custom-made primers and probe set used in the present study were as follows: (i) gB-specific primers (forward, 5′-AACGCGACGCACATCAAG-3′; reverse, 5′-CTGGTACGCGATCAGAAAGC-3′; probe, 5′-FAM-CAGCCGCAGTACTACC-3′) and (ii) gK-specific primers (forward, 5′-GGCCACCTACCTCTTGAACTAC-3′; reverse, 5′-CAGGCGGGTAATTTTCGTGTAG-3′; probe, 5′-FAM-CAGGCCGCATCGTATC-3′). The amplicon length was 82 bp. As an internal control, a set of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) primers from Applied Biosystems (assay m999999.15_G1; amplicon length, 107 bp) was used.
The expression levels of gB and gK genes, as well as the expression of the endogenous control gene GAPDH, were evaluated using a commercially available TaqMan gene expression assay kit (Applied Biosystems) containing optimized primer and probe concentrations. In each experiment, an estimated relative copy number of gB and gK genes was calculated using standard curves generated from pAc-gB1 (45) and pGem-gK1040 (26), respectively. Briefly, each plasmid DNA template was serially diluted 10-fold such that 5 μl contained from 103 to 1011 copies of the desired gene and then subjected to TaqMan PCR with the same set of primers as the test samples. By comparing the normalized threshold cycle of each sample to the threshold cycle of the standards, the copy number for each reaction was determined. Quantitative PCR (qPCR) and quantitative reverse transcription-PCR (qRT-PCR) were performed using an ABI ViiA7 sequence detection system (Applied Biosystems) in 384-well plates. The threshold cycle (CT) values, which represent the PCR cycles at which there is a noticeable increase in the reporter fluorescence above baseline, were determined using ViiA7 RUO software.
In vitro explant reactivation assay.
Mice were sacrificed at 28 days p.i., and individual TG were removed and cultured in 1.5 ml of tissue culture medium as we described previously (46). Briefly, a 10-μl aliquot was removed from each culture daily for 20 days and used to infect RS cell monolayers. The RS cells were monitored daily for the appearance of cytopathic effect (CPE) for 5 days to determine the time of first appearance of reactivated virus from each TG. Since the media from the explanted TG cultures were plated daily, the times at which reactivated virus first appeared in the explanted TG cultures could be determined.
Statistical analysis.
Protective parameters were analyzed using the Student t test and the Fisher exact test with Instat software (GraphPad, San Diego, CA). The results were considered to be statistically significant if the P value was <0.05.
RESULTS
Structure of the HSV-1 NgK and MgK viruses.
To examine the effects of the 8mer in the signal region of gK on HSV-1 pathogenesis, we constructed two derivatives of HSV-1 strain McKrae that express two additional copies of the gK gene, with or without mutation, within the 8mer region of gK (Fig. 2). Since the wt strain McKrae was used as the original parental virus, a LAT-null mutant, dLAT2903, that was derived from the McKrae strain (41) was used to construct the NgK and MgK viruses. The genomic structure of the wt HSV-1 strain McKrae is shown in Fig. 2A and that of dLAT2903 in Fig. 2B. The location of the TATA box of the LAT promoter, which we described previously (47), is indicated. The NgK and MgK viruses were derived from the dLAT2903 strain by the insertion of the NgK and MgK genes and restoration of the LAT promoter so that the inserted NgK (Fig. 2C) or MgK (Fig. 2D) gene is under the control of the endogenous LAT promoter. The genomic structure of the NgK and MgK viruses was confirmed by restriction enzyme analysis, Southern blotting (Fig. 2E), and partial sequencing (not shown). The NgK and MgK viruses, in addition to their endogenous gK gene, have two additional copies of NgK and MgK genes, one of which is in each viral long repeat. The rescued viruses for NgK and MgK were found to be identical to each other and are similar to their parental dLAT2903 in all parameters examined (not shown). Thus, in all of the subsequent experiments we used the rescued virus derived from MgK, which we designated RgK.
FIG 2.

Construction and structure of the NgK and MgK mutant viruses. (A) The top schematic diagram shows the wt HSV-1 McKrae genome in the prototypic orientation. TRL and IRL represent the terminal and internal (or inverted) long repeats, respectively, and TRS and IRS represent the terminal and internal (or inverted) short repeats, respectively. UL and US represent the long and short unique regions, respectively. The solid rectangle represents the very stable 2-kb LAT. The start site for LAT transcription is indicated by an arrow at +1. “TATA” designates the relative location of the LAT promoter TATA box 28 nucleotides upstream of the start of transcription. (B) dLAT2903 has a deletion from LAT nucleotides −161 to +1667 in both copies of LAT and makes no LAT RNA. (C) NgK was constructed from dLAT2903 by homologous recombination between dLAT2903 DNA and a plasmid containing the complete LAT promoter and the entire structural NgK gene (including sequences for myc) signal. (D) MgK was constructed from dLAT2903 by homologous recombination between dLAT2903 DNA and a plasmid containing the complete LAT promoter and the entire structural MgK gene (including mutated sequences within the gK signal sequences and myc sequences). (E) NgK and MgK Southern blot analyses. RS cell monolayers were infected with 5 PFU of NgK, MgK, HSV-gK3, or McKrae/cell for 24 h. Viral DNAs were isolated, and 5 μg of DNA/virus was digested with EcoRV/EcoRI and hybridized to DIG-labeled HSV-1 gK (HSV-1 nucleotides 112170 to 113193). The lanes show results obtained for McKrae, HSV-gK3, NgK, and MgK. As expected, only one fragment is seen in McKrae lane. This band also is observed in HSV-gK3 and corresponds to HSV-1 nucleotides 110094 to 118640 surrounding the natural occurring gK (HSV-1 nucleotides 112170 to 113193). In contrast to the McKrae lane, in the HSV-gK3, NgK, and MgK lanes, in addition to the natural gK, there are two additional bands corresponding to two extra copies of gK gene inserted within IRL (HSV-1 nucleotides 110094 to 118640) and TRL (HSV-1 nucleotides 13 to 7727) regions of dLAT2903. Both regions within the IRL and TRL have a deletion of ∼1,600 bp of LAT.
Replication of NgK and MgK viruses in tissue culture.
To determine whether overexpression of gK or mutation within the signal sequence of gK affect viral replication, we quantified virus yield after infection of RS cells with 0.01 or 0.1 PFU of NgK, MgK, or RgK/cell. The cell monolayers were freeze-thawed at the indicated time points, and the yield of infectious virus was quantified by using a standard plaque assay. At the lowest multiplicity of infection (MOI), i.e., 0.01, the yield of viruses was time dependent. At 48 h p.i., the yield of NgK virus was lower than the yield of RgK. The yield of MgK was similar to that of the NgK virus, although not at earlier time points (Fig. 3A, P < 0.05, Student t test). At an MOI of 0.1, the yields of NgK and MgK viruses were similar at all tested time points and were lower than that of RgK virus at 0 to 48 h p.i. (Fig. 3B, P < 0.05, Student t test). At an MOI of 10, no significant differences in the replication of the viruses were detected (data not shown).
FIG 3.
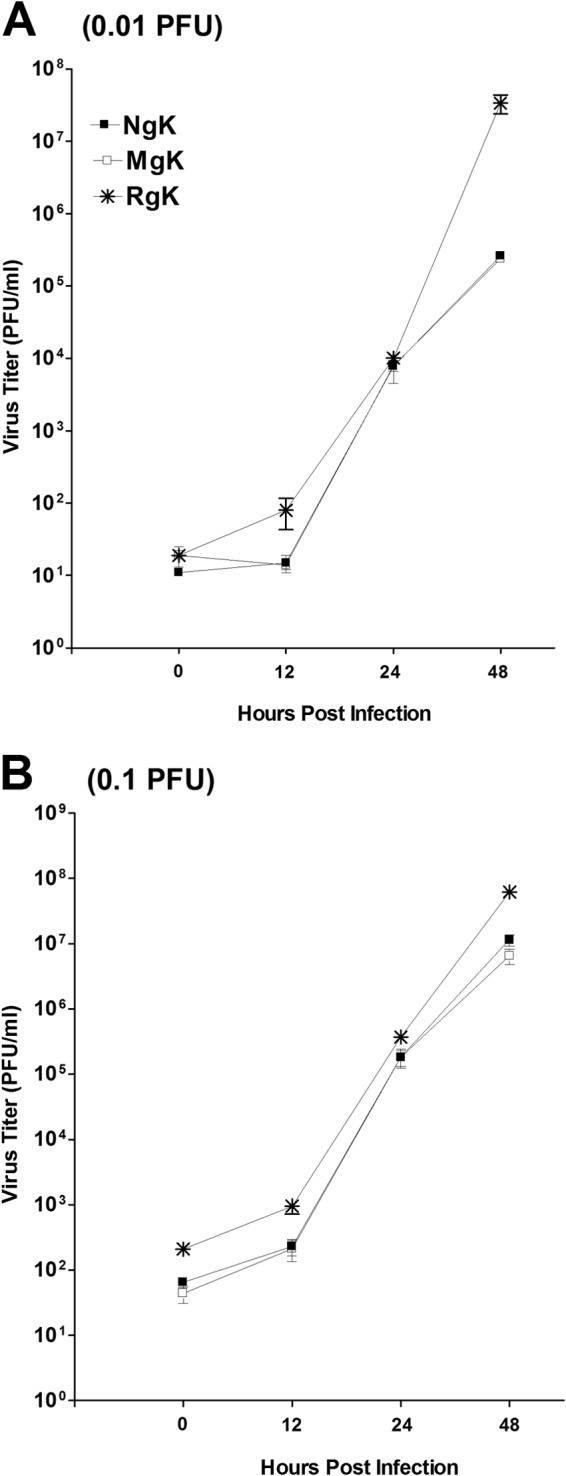
Replication of NgK and MgK viruses in vitro. Subconfluent RS cell monolayers were infected with 0.01 or 0.1 PFU of NgK, MgK, or RgK virus/cell as described in Materials and Methods. Total virus was harvested at the indicated times postinfection by two cycles of freeze-thawing. The amount of virus at each time point was determined by standard plaque assays on RS cells. Each point represents the mean ± the standard errors of the mean (SEM) from two separate experiments (n = 8). (A) 0.01 PFU; (B) 0.1 PFU.
Western blot analyses of NgK and MgK expression in tissue culture.
To verify that the recombinant viruses express the inserted genes, confluent monolayers of RS cells were infected for 24 h with MgK, NgK, or RgK at an MOI of 5 PFU/cell. Western blot analysis of total protein extracts using an anti-myc antibody revealed a band that was present in NgK-infected cells and MgK-infected cells (Fig. 4, lanes MgK and NgK), which was not detected in the RgK-infected cells (Fig. 4, lane RgK) or mock-infected RS cells (Fig. 4, lane Mock). In addition to this major band, a smaller and faintly reactive band was detected in the NgK-infected cells and the MgK-infected cells (Fig. 4, lanes MgK and NgK). Most likely, the major band represents the N-linked glycosylated form of gK, whereas the minor band represents the cleaved and partially glycosylated form of gK. Finally, the upper band that was detected in both infected and mock-infected groups may represent the internal myc protein (Fig. 4, upper band).
FIG 4.
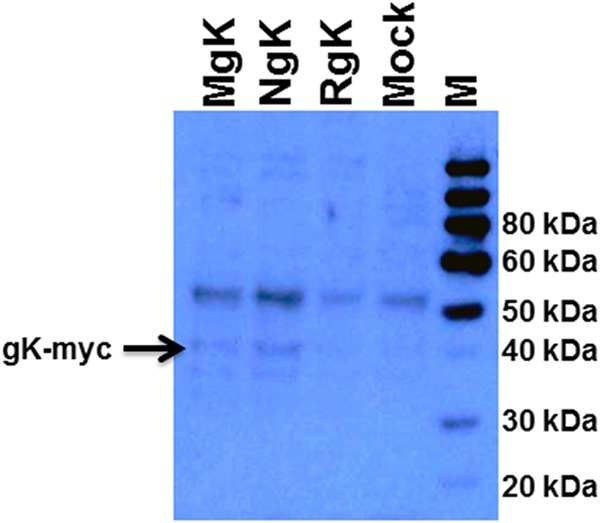
Western blot analyses of expressed NgK and MgK proteins. RS cells were infected with the NgK, MgK, or RgK at an MOI of 10 PFU/cell for 16 h. The cells were then lysed directly into gel sample buffer, similar amounts of protein were loaded into each well, and samples were separated by SDS–10% PAGE, transferred to nitrocellulose, and reacted with anti-myc peptide antibody as described in Materials and Methods. Bound antibody was detected with chemiluminescence detection, followed by autoradiography. The lanes show the results obtained for MgK, NgK, RgK, Mock, and M (molecular weight markers).
FACS analyses of MgK- and NgK-infected cells.
To further verify that the NgK and MgK viruses expressed the inserted genes, we used FACS analysis. RS cells were infected for 24 h with 5 PFU of NgK, MgK, or RgK/cell or were mock infected. The cells were then surface stained with anti-myc and anti-HSV-1 gC antibodies and subjected to FACS analysis in which stained cells were gated for expression of HSV-1 gC (Fig. 5A) or gK-myc (Fig. 5B). Infection of the cells was verified by the similar levels of expression of HSV-1 gC on cells infected with NgK, MgK, or RgK (Fig. 5A). In contrast to gC expression (Fig. 5A), the level of surface expression of gK-myc protein appeared to be >50% higher in NgK-infected cells compared to MgK- and RgK-infected cells (Fig. 5B). Thus, the presence of low levels of myc expression in the MgK- and RgK-infected cells most likely represent baseline expression of myc which is detected by the internalized anti-myc antibody.
FIG 5.
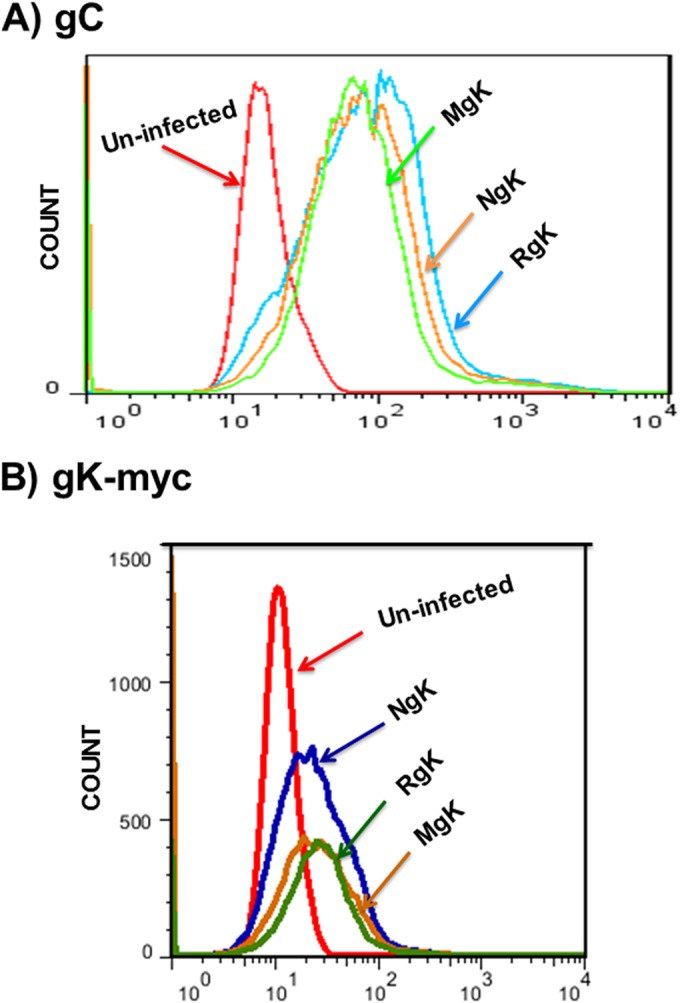
FACS analyses of NgK- or MgK-infected cells in vitro. RS cells were infected with the NgK, MgK, or RgK at an MOI of 5 PFU/cell for 24 h or were mock infected. At 24 h p.i., the cells were harvested, stained with anti-HSV-1 gC and anti-myc antibodies, fixed, and subjected to FACS analyses as described in Materials and Methods. Stained cells were gated for HSV-1 gC or gK-myc expression and are shown as an overlay. (A) gC; (B) gK-myc.
Cell surface expression of gC- and gK-myc by MgK and NgK in infected cells.
Although the results of the Western blotting (Fig. 4) indicated that gK is expressed by both NgK and MgK viruses, the results of the FACS analysis indicated that the two recombinant viruses differed in the cell surface expression of gK-myc (Fig. 5B). We therefore analyzed the surface expression of gK-myc expression using double-label immunofluorescence analysis. RS cells were grown on Lab-Tex chamber slides, infected with 5 PFU of NgK, MgK, or RgK/cell or mock infected for 24 h, and then incubated with fluorescently labeled antibodies against HSV-1 gC and the myc epitope. We found strong and similar levels of cell surface expression of gC for the cells infected with NgK, MgK, or RgK and, as expected, gC expression was not detected in the mock-infected cells (Fig. 6, gC-FITC). Also, as expected, gK-myc expression was not detectable in either the RgK-infected cells or mock-infected cells (Fig. 6, gK-myc). Cell surface expression of gK-myc was detected in the NgK-infected cells but not in the MgK-infected cells. Moreover, on merging the images of gC- and gK-myc staining, we found that the cell surface expression of gK-myc and gC colocalized in the NgK infected cells (Fig. 6, Merge). Collectively, these results suggested that mutation in the signal sequence of gK prevented cell surface expression of gK by MgK recombinant virus. This effect could be attributed to the mutation in the signal sequence as the NgK recombinant virus, which also overexpressed the myc sequence, was capable of cell surface expression.
FIG 6.
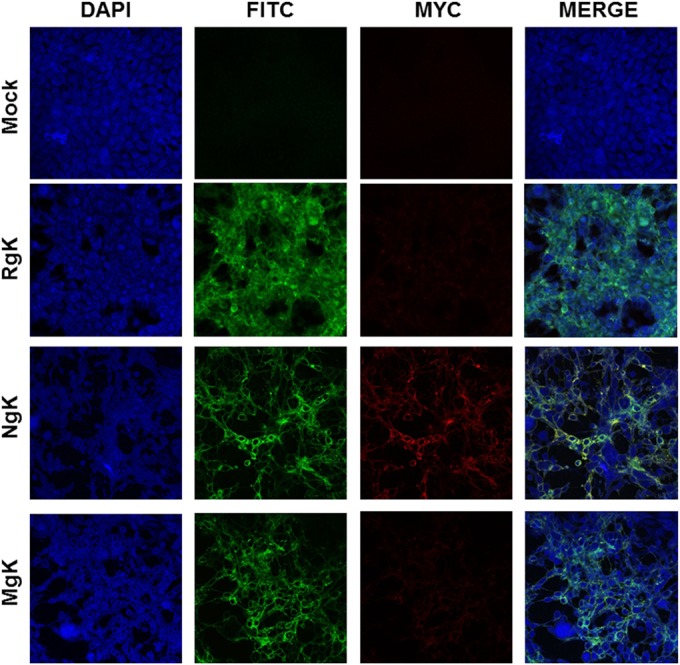
Immunostaining of NgK- or MgK-infected cells in vitro. RS cells were infected with the NgK, MgK, or RgK at an MOI of 5 PFU/cell for 24 h or were mock infected. At 24 h p.i., the cells were fixed, permeabilized, blocked, and stained with anti-HSV-1 gC (green), anti-myc (red), and DAPI nuclear stain (blue). The slides were fixed, and photomicrographs are shown at ×40 direct magnification. Colocalization is visualized as yellow.
Effect of gK overexpression on apoptosis and cell death of infected cells in culture.
To investigate the potential effects of gK overexpression and mutagenesis on cell death and apoptosis, we infected RS cells with 5 PFU of NgK, MgK, or RgK/cell or the cells were mock infected. At 24 h p.i., the infected cells were isolated, and FACS analyses were performed using a marker of cell death, propidium iodide, and an apoptosis marker, annexin V. The percentages of cells staining with propidium iodide and annexin V were similar in NgK-, MgK-, and RgK-infected cells and were not significantly different from the percentages observed in mock-infected cells (Fig. 7). Thus, we did not find evidence of an association between overexpression of gK and mutagenesis of gK with cell death or apoptosis.
FIG 7.

Effect of NgK or MgK on apoptosis and cell death in vitro. RS cells were infected with the NgK, MgK, or RgK at an MOI of 5 PFU/cell for 24 h or were mock infected. At 24 h p.i., the cells were harvested and reacted with annexin V and 7-AAD dye to analyze apoptosis and cell death, respectively. Stained cells were fixed, and FACS analysis was performed as described in Materials and Methods. The percentages of cells determined to be positive in each quadrant are shown for each virus or for uninfected cells.
Replication of NgK and MgK in mouse tear films.
Viral replication during ocular infection was assessed by determining the PFU in the tear films of infected mice. C57BL/6 mice were infected ocularly with 2 × 105 PFU of NgK, MgK, or RgK virus/eye and tear films collected on days 3 and 5 p.i. On day 3 p.i., the titers of virus were higher in the tear films from RgK- and MgK-infected mice than the NgK-infected mice, but these differences were not statistically significant (Fig. 8, P > 0.05). On day 5 p.i., the titers of virus in tear films of NgK- and MgK-infected mice were similar (Fig. 8, P > 0.05) and were significantly lower than the titers of virus in the tear films of RgK-infected mice (Fig. 8, P < 0.05). These results were consistent with the reduced virus replication with lower MOIs in NgK- and MgK-infected RS cells in tissue culture (see Fig. 3) and indicated that the presence of the two additional copies of gK significantly reduced the amount of HSV-1 found in the tear films of ocularly infected mice on day 5 p.i.
FIG 8.
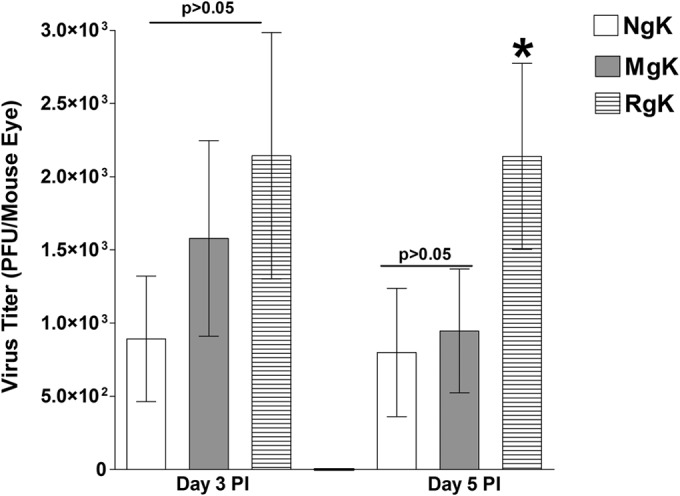
Virus replication in mouse tears. C57BL/6 mice were ocularly infected with 2 × 105 PFU of NgK, MgK, or RgK virus/eye. Tear films were collected on days 3 and 5, and virus titers were determined by standard plaque assays. Each point represents the mean titer for 20 eyes from two separate experiments.
Effect of gK overexpression on gB and gK expression during primary ocular infection.
To investigate the effect of gK overexpression and mutagenesis on viral transcription in vivo, C57BL/6 mice were infected with 2 × 105 PFU of NgK, MgK, or RgK virus/eye. The corneas, TG, and brain were collected on days 3 and 5 p.i., and the total RNA was isolated and subjected to TaqMan RT-PCR to estimate the levels of gK and gB mRNA using the level of GAPDH mRNA in each sample as an internal control. The results showed differences in expression among the various tissues. On day 3 p.i., the levels of gB (Fig. 9A) and gK (Fig. 9B) transcripts were significantly lower in the corneas from MgK-infected mice than in the corneas from NgK- or RgK-infected mice (P < 0.05). By day 5 p.i., the levels of gB and gK transcripts in the corneas of the MgK-infected mice had increased and were no longer significantly different from the levels in the corneas from NgK- or RgK-infected mice (Fig. 9A and B). On day 3 p.i., the levels of both gB (Fig. 9C, P < 0.05) and gK (Fig. 9D, P < 0.05) transcripts in the TG of mice infected with NgK or MgK were lower than the levels in the TG of mice infected with RgK. Again, by day 5 p.i., the levels of both gB and gK in the TG of mice infected with NgK, MgK, or RgK were not statistically significant (Fig. 9C and D, P > 0.05). On day 3 p.i., the levels of gB transcripts were similar in the brains of mice infected with either NgK or MgK but were significantly higher in the brains of RgK-infected mice (Fig. 9E, P < 0.05). By day 5 p.i., the levels of gB transcripts in the brains of mice infected with NgK, MgK, or RgK were similar (Fig. 9E, P > 0.05). In contrast, the levels of gK transcripts were similar in the brains of mice infected with NgK, MgK, or RgK virus on both days 3 and 5 p.i. (Fig. 9F, P > 0.05). These results indicated that the presence of two extra copies of gK influenced viral transcription in a time-dependent and tissue-specific manner and indicated that the levels of gB transcripts were significantly higher than the level of gK transcripts in all three tissues. Collectively, the results described above suggested that virus replication in the tears of infected mice, as well as the levels of gB and gK transcripts are affected by the presence two extra copies of the NgK or MgK genes.
FIG 9.

Expression of gB and gK in the corneas, TG, and brains of infected mice. C57BL/6 mice were ocularly infected with 2 × 105 PFU of NgK, MgK, or RgK virus/eye. gB and gK transcripts in corneas, TG, and brains were determined on days 3 and 5 p.i. by qRT-PCR. In each experiment, an estimated relative copy number of the HSV-1 gB and gK was calculated using standard curves generated from pAC-gB1 (45) and pAc-gK1 (26). Briefly, DNA template was serially diluted 10-fold such that 5 μl contained from 103 to 1011 copies of pAc-gB1 or pAc-gK1 and then subjected to TaqMan PCR using the same set of primers. By comparing the normalized threshold cycle of each sample to the threshold cycle of the standard, the copy number for each reaction was determined. GAPDH expression was used to normalize the relative expression of each transcript in corneas, TG, and brains of infected mice. Each bar represents the mean ± the SEM from six corneas or TG and three brains. (A) gB in cornea; (B) gK in cornea; (C) gB in TG; (D) gK in TG; (E) gB in brain; (F) gK in brain.
Viral latency in the TG of latently infected mice.
To determine whether the presence of NgK or MgK genes also influences the level of latency in the TG of latently infected mice, C57BL/6 mice were ocularly infected with NgK, MgK, or RgK virus and the TG isolated on day 28 p.i. Latency in individual mice was determined by qPCR using primers from the gB and gK regions of the HSV-1 genome. The levels of gB DNA were similar in mice infected with NgK, MgK, or RgK (Fig. 10A, P > 0.05). Similarly, no significant differences in the levels of gK in the TG of mice infected with NgK, MgK, or RgK were detected (Fig. 10B, P > 0.05). Thus, the level of latency in TG of infected mice was not affected by the presence of either the NgK or MgK gene.
FIG 10.
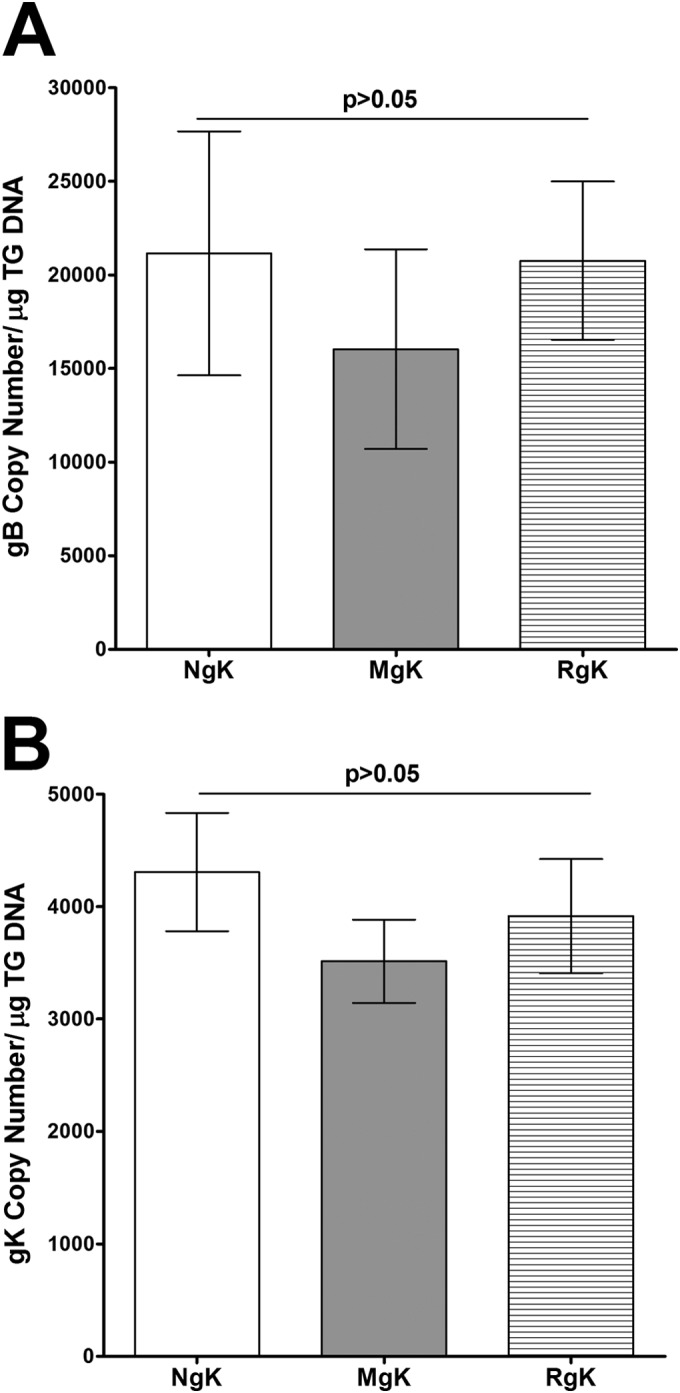
Detection of gK and gB DNA in TG of latently infected mice. C57BL/6 mice were ocularly infected with 2 × 105 PFU of NgK, MgK, or RgK virus/eye. TG from individual mice were isolated 28 days p.i., and TaqMan qPCR was performed as described in Materials and Methods. GAPDH was used as an endogenous control to normalize the relative expression of gK and gB DNA in infected TG. The relative copy numbers of the HSV-1 gB and gK genes were calculated using standard curves as described in Fig. 9. Each point represents the mean ± the SEM from 12 TG (six mice). (A) gB DNA; (B) gK DNA.
Explant reactivation in TG of latently infected mice.
To determine whether virus reactivation is affected differentially by NgK or MgK overexpression, C57BL/6 mice were ocularly infected with 2 × 105 PFU of NgK, MgK, or RgK virus/eye, and virus reactivation was analyzed by explanting individual TG. The time to reactivation was similar among the NgK, MgK, and RgK viruses (5.3 ± 0.2 days versus 5.8 ± 0.4 days versus 5.4 ± 0.4 days; Fig. 11, 2 × 105 PFU/eye, P > 0.05). Thus, the time to reactivation was not influenced by the presence of the NgK or MgK gene in mice infected with the recombinant viruses. In contrast, however, the presence of either the NgK gene or the MgK gene affected the numbers of TG showing reactivation. Although all of the TG from RgK-infected mice were reactivated during the monitoring period, 50% of TG from NgK-infected mice (6 of 12 TG) and 58% of TG from MgK-infected mice (5 of 12 TG) did not exhibit reactivation of the virus. With respect to the number of TG showing reactivation, the differences between NgK and MgK viruses compared to RgK virus were statistically significant (P < 0.002).
FIG 11.
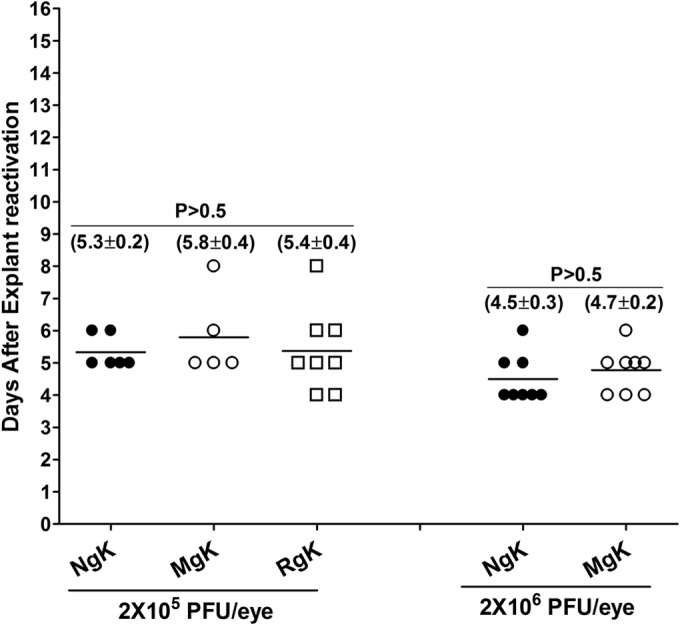
Effect of NgK and MgK on induced reactivation in latently infected mice. C57BL/6 mice were infected ocularly with 2 × 105 PFU of NgK, MgK, or RgK virus/eye, while some mice were infected with 2 × 106 PFU of NgK or MgK virus/eye. On day 28 p.i., TG from infected mice was isolated, and each individual TG was incubated in 1.5 ml of tissue culture medium at 37°C. A 100-μl aliquot was removed from each culture daily for 15 days and used to infect RS cell monolayers. The RS cells were monitored daily for the appearance of CPE for 5 days to determine the time of first appearance of reactivated virus from each TG. The results are plotted as the number of TG that reactivated daily. Numbers indicate the average time that the TG from each group first showed CPE ± the SEM. Each point represents the mean ± the SEM from 6 (NgK, 2 × 105 PFU/eye), 5 (MgK, 2 × 105 PFU/eye), 8 (RgK, 2 × 105 PFU/eye), 8 (NgK, 2 × 106 PFU/eye), and 8 (MgK, 2 × 106 PFU/eye). At 2 × 105 PFU/eye, 5 of 12 TG in NgK-infected mice, 7 of 12 TG in MgK-infected mice, and 2 of 10 TG in RgK-infected mice did not reactivate, whereas all 8 TG infected with 2 × 106 PFU of NgK or MgK/eye were reactivated.
To determine whether a higher infectious dose of virus would increase the numbers of TG showing reactivation of the viruses, we infected C57BL/6 mice with 2 × 106 PFU of NgK and MgK viruses/eye. Subsequent monitoring of explant reactivation indicated that the time to reactivation was similar in the TG of mice infected with NgK or MgK viruses (4.5 ± 0.3 days versus 4.7 ± 0.2 days; Fig. 11, 2 × 106 PFU/eye, P > 0.05). However, the time to reactivation was faster in the TG of mice infected with 2 × 106 PFU/eye compared to those infected with 2 × 105 PFU of NgK or MgK virus/eye (Fig. 11, compare the left side with the right side). Furthermore, all of the TG from mice infected with 2 × 106 PFU of NgK or MgK virus/eye were reactivated. Thus, at a lower infective dose the presence of the NgK or MgK gene does not affect time to reactivation of the virus but does affect the percentage of TG showing reactivation.
Virulence of NgK and MgK viruses in infected BALB/c and C57BL/6 mice.
To determine whether there is a difference in the virulence of the NgK and MgK viruses compared to the rescued RgK virus, we used BALB/c and C57BL/6 mice. BALB/c mice are sensitive to HSV-1 infection, whereas C57BL/6 mice are resistant. Groups of 5 BALB/c mice were infected ocularly in both eyes with 10-fold serial dilutions of each virus ranging from 2 × 102 to 2 × 105 PFU/eye, and the LD50 was determined for each group (Table 1). After ocular infection, the LD50 for NgK virus (15,642 PFU) was ∼2-fold lower than the LD50 for RgK virus (27,660 PFU) and significantly lower than the LD50 for MgK virus (2,115,650 PFU) (Table 1). Thus, the MgK virus appeared to be less virulent than either NgK or the marker rescued virus, as judged by the ability to kill BALB/c mice following ocular infection. This suggests that the higher mortality in the NgK-infected mice was due to the presence of NgK gene and that the lower mortality in the MgK-infected mice was due to the mutations in the signal sequence of the gK gene rather than to a defect in the recombinant virus.
TABLE 1.
Survival of BALB/c and C57BL/6 mice after ocular challenge with gK recombinant virusesa
| Virus | Mouse survival after virus infection at various doses (PFU/eye) |
LD50 (PFU) | ||||
|---|---|---|---|---|---|---|
| 2 × 106 | 2 × 105b | 2 × 104 | 2 × 103 | 2 × 102 | ||
| NgK | 4/5 | 12/12, 0/5 | 1/5 | 5/5 | 5/5 | 15,642 |
| MgK | 5/5 | 12/12, 4/5 | 4/5 | 5/5 | 5/5 | 2,115,650 |
| RgK | 5/5 | 10/10, 1/5 | 2/5 | 5/5 | 5/5 | 27,660 |
Mice were challenged ocularly with the specified amount of each virus, and survival was determined 28 days after challenge. All of the mice were BALB/c mice except as noted in column 3. Mouse survival is presented as the number of mice surviving/the number of mice tested.
The survival data in this column are presented for two mouse strains: C57BL/6, BALB/c.
LD50 studies performed using C57BL/6 mice indicated that at 2 × 105 PFU of NgK, MgK, or RgK virus/eye all of the infected mice survived ocular infection (Table 1). Similarly, at a dose of 2 × 106 PFU/eye, all mice infected with MgK and RgK survived (Table 1). At this dose, one mouse in the NgK group died as the result of infection, suggesting that even in C57BL/6 mice NgK is more pathogenic than the MgK or the RgK virus. However, since only one mouse died as the result of infection, it was not possible to calculate the LD50 of the recombinant viruses in C57BL/6 mice.
Overall, these results suggested that expression of NgK or MgK affected neurovirulence in infected mice compared to rescued virus. In addition, NgK gene insertion enhanced neurovirulence in infected mice, whereas the insertion of MgK reduced neurovirulence in infected mice.
Effect of NgK and MgK overexpression on CS in infected mice.
To determine the effect of NgK or MgK on eye disease, the severity of CS was evaluated following ocular infection of mice described in Table 1. CS was measured in the surviving mice on day 28 p.i. BALB/c mice infected with 2 × 104, 2 × 103, or 2 × 102 PFU of NgK, MgK, or RgK virus/eye developed similar levels of CS, and the differences were not statistically significant (Table 2, P > 0.05). CS also was examined in C57BL/6 mice infected ocularly with 2 × 105 PFU of NgK, MgK, or RgK virus/eye. C57BL/6 mice infected with 2 × 105 PFU of NgK, MgK, or RgK virus/eye displayed similar levels of CS (Table 2, C57BL/6, P > 0.1). To determine whether a higher number of PFU of virus/eye can increase CS, additional groups of C57BL/6 mice were infected with 2 × 106 PFU of each virus/eye. At this dose, CS was higher in the NgK- and MgK-infected mice than in RgK-infected mice (Table 2, 2 × 106, C57BL/6). The CS in NgK-infected mice was significantly higher than RgK-infected and MgK-infected mice (Table 2). Thus, overexpression of NgK also exacerbates virus-induced CS in infected C57BL/6 mice.
TABLE 2.
Corneal scarring of BALB/c and C57BL/6 mice after ocular infection with gK recombinant virusesa
| Virus | CS after virus infection at various doses (PFU/eye) |
||||
|---|---|---|---|---|---|
| C57BL/6 mice |
BALB/c mice |
||||
| 2 × 106b | 2 × 105 | 2 × 104 | 2 × 103 | 2 × 102 | |
| NgK | 2.4 ± 0.7 | 0.7 ± 0.3 | 0 | 0 | 0 |
| MgK | 1.1 ± 0.4 | 0.7 ± 0.3 | 0.4 ± 0.4 | 0 | 0 |
| RgK | 0.6 ± 0.3 | 0.5 ± 0.2 | 1 ± 1 | 1.1 ± 0.6 | 0.1 ± 0.1 |
Mice were infected ocularly with the specified amount of each virus, and CS was determined 28 days after challenge. CS values are indicated as the means ± the standard deviations.
These values were significantly different from RgK but not for MgK as determined using the Student t test.
DISCUSSION
Since gK is an essential HSV-1 gene and gK-null viruses are not efficiently propagated in vitro and in vivo (29, 32), to improve our understanding of the role of gK in HSV-1 pathogenesis, we previously constructed and characterized a recombinant HSV-1 that expresses two additional copies of the HSV-1 gK gene (34). This recombinant virus increased pathogenesis in infected mice. More recently, we reported that an 8mer within the signal sequence of gK also contributed to exacerbation of eye disease in different strains of mice and in rabbits (36). Using 33 peptides overlapping the entire 338 amino acids of gK, we established that no other region of gK contributed to this pathogenic effect (35, 36). The results of the current study support and extend our previous observations.
Deletion of gK has been shown to affect virus yield and plaque size and translocation of the virus from the cytoplasm to the extracellular space (28) and to play a role in virion entry, cytoplasmic virion envelopment, and virus-induced cell fusion (27–30, 48–52). These functions of HSV gK are similar to those reported for the gKs of pseudorabies and varicella-zoster virus (53–55). HSV-1 mutants that lack gK fail to acquire a cytoplasmic envelope efficiently. This leads to a drastic reduction in virion egress and spread; thus, HSV-1 mutants are unable to efficiently infect and establish latency in neurons (28–32). Our analysis of gK with mutated signal sequence indicated that the titers of the MgK virus in RS cells and the tears of ocularly infected C57BL/6 mice were similar to the titers of the NgK virus, indicating that the 8mer region of gK does not play a role in the replication of the virus. Despite having similar virus titers in the tear films of C57BL/6 mice, MgK-infected mice had less severe CS than mice infected with the NgK or RgK virus. Previously, we reported that immunization with gK, followed by infection with McKrae (or with the avirulent HSV-1 KOS virus), leads to CS in both BALB/c and C57BL/6 mice (22). The severity of CS in NgK-infected mice was similar to that of HSV-gK3 that lack the myc at its 3′ end (34). Overall, NgK virus behaved similarly to that of HSV-gK3, suggesting that in-frame expression of the myc tag by NgK had no deleterious effect on gK pathogenesis. Previously, we reported that exacerbation of CS in gK-immunized mice associated with the presence of CD8+ CD25+ T cells in the cornea of ocularly infected mice (23). In addition, our previous work showed that CD8+ T cells contribute to exacerbation of CS in HSV-gK3-infected mice (34) and that the pathogenic region of gK is located within the signal sequence of gK (35, 36). The gK 8mer-induced CD8+ T cell responses contributed to an increase in eye disease in BALB/c mice, C57BL/6 mice, and NZW rabbits (36). The findings of the present study are consistent with and extends our previous work by showing that the 8mer within the signal sequence of gK induces a CD8+ T cell response in cornea of ocularly infected mice, which contributes to the exacerbation of eye disease independent of the strain of mice.
The gK expressed on virions forms a complex with the membrane-associated UL20 viral protein (52, 56). HSV-1 mutants that lack gK fail to acquire a cytoplasmic envelope efficiently. This leads to a drastic reduction in virion egress and spread, and HSV-1 mutants are unable to efficiently infect and establish latency in neurons (28–32). In the present study, NgK was detected on the surface of infected cells when expressed by the LAT promoter. In marked contrast, we found that mutation within the signal sequence of gK prevented cell surface expression of gK by MgK. Thus, mutations in the signal sequence of gK driven by the LAT promoter and in place of LAT blocked transport of gK to the cell surface of infected cells. The lack of cell surface expression of gK in MgK virus-infected cells may be a contributing factor to its reduced pathogenesis in infected mice. The higher LD50 of the MgK virus in comparison to the NgK virus or rescued virus, as well as the reduced exacerbation of CS, may also be a result of the lack of gK cell surface expression. Alternatively, the differences in the total protein expression levels between NgK and MgK viruses and not mutation in the signal sequence of MgK may be responsible for the differences in the disease phenotype between NgK and MgK viruses. Taken together with our observation of the exacerbation of CS by overexpression of gK (i.e., NgK virus), these results raise the possibility that cell surface expression of gK and/or proper presentation of the 8mer peptide within the signal sequence of the native form of gK plays a key role in the pathogenesis of HSV-1. This would be consistent with a requirement for effective MHC-I presentation and the subsequent generation of CD8+ T cells that is required for exacerbation of CS in infected mice.
In addition, we found that the levels of latency in the TG of C57BL/6 mice infected with 2 × 105 PFU of each virus/eye was the same. However, at this dose of infection a significant number of TG in both NgK and MgK viruses were not reactivated compared to the rescued virus, although all TG in NgK and MgK viruses were reactivated when mice were infected with a 10-fold-higher dose of each virus. Thus, our results suggest that the presence of extra copies of gK and/or the presence of the myc tag affect the extent of reactivation but not the level of latency.
In summary, the results presented here support the hypothesis that overexpression of gK increases corneal scarring in mice. Moreover, the pathogenic role of gK in HSV-1-induced CS appears to be dependent on the cell surface expression of gK.
ACKNOWLEDGMENT
This study was supported by NIH grant 1 RO1 EY13615.
REFERENCES
- 1.Barron BA, Gee L, Hauck WW, Kurinij N, Dawson CR, Jones DB, Wilhelmus KR, Kaufman HE, Sugar J, Hyndiuk RA, et al. . 1994. Herpetic eye disease study: a controlled trial of oral acyclovir for herpes simplex stromal keratitis. Arch Ophthalmol 101:1871–1882. [DOI] [PubMed] [Google Scholar]
- 2.Wilhelmus KR, Dawson CR, Barron BA, Bacchetti P, Gee L, Jones DB, Kaufman HE, Sugar J, Hyndiuk RA, Laibson PR, Stulting RD, Asbell PA, Herpetic Eye Disease Study Group . 1996. Risk factors for herpes simplex virus epithelial keratitis recurring during treatment of stromal keratitis or iridocyclitis. Br J Ophthalmol 80:969–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farooq AV, Shukla D. 2012. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Survey Ophthalmol 57:448–462. doi: 10.1016/j.survophthal.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dawson CR. 1984. Ocular herpes simplex virus infections. Clin Dermatol 2:56–66. doi: 10.1016/0738-081X(84)90066-X. [DOI] [PubMed] [Google Scholar]
- 5.Liesegang TJ. 1999. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea 18:127–143. doi: 10.1097/00003226-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Liesegang TJ. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Hill TJ. 1987. Ocular pathogenicity of herpes simplex virus. Curr Eye Res 6:1–7. doi: 10.3109/02713688709020060. [DOI] [PubMed] [Google Scholar]
- 8.Binder PS. 1984. A review of the treatment of ocular herpes simplex infections in the neonate and immunocompromised host. Cornea 3:178–182. [PubMed] [Google Scholar]
- 9.Streilein JW, Dana MR, Ksander BR. 1997. Immunity causing blindness: five different paths to herpes stromal keratitis. Immunol Today 18:443–449. doi: 10.1016/S0167-5699(97)01114-6. [DOI] [PubMed] [Google Scholar]
- 10.Thomas J, Rouse BT. 1997. Immunopathogenesis of herpetic ocular disease. Immunol Res 16:375–386. doi: 10.1007/BF02786400. [DOI] [PubMed] [Google Scholar]
- 11.Branco BC, Gaudio PA, Margolis TP. 2004. Epidemiology and molecular analysis of herpes simplex keratitis requiring primary penetrating keratoplasty. Br J Ophthalmol 88:1285–1288. doi: 10.1136/bjo.2003.040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burke RL. 1991. Development of a herpes simplex virus subunit glycoprotein vaccine for prophylactic and therapeutic use. Rev Infect Dis 13:S906–S911. doi: 10.1093/clind/13.Supplement_11.S906. [DOI] [PubMed] [Google Scholar]
- 13.Burke RL. 1992. Contemporary approaches to vaccination against herpes simplex virus. Curr Top Microbiol Immunol 179:137–158. [DOI] [PubMed] [Google Scholar]
- 14.Ghiasi H, Kaiwar R, Nesburn AB, Slanina S, Wechsler SL. 1994. Expression of seven herpes simplex virus type 1 glycoproteins (gB, gC, gD, gE, gG, gH, and gI): comparative protection against lethal challenge in mice. J Virol 68:2118–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dix RD, Mills J. 1985. Acute and latent herpes simplex virus neurological disease in mice immunized with purified virus-specific glycoproteins gB or gD. J Med Virol 17:9–18. doi: 10.1002/jmv.1890170103. [DOI] [PubMed] [Google Scholar]
- 16.McGeoch DJ, Dalrymple MA, Davison AJ, Dolan A, Frame MC, McNab D, Perry LJ, Scott JE, Taylor P. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol 69:1531–1574. doi: 10.1099/0022-1317-69-7-1531. [DOI] [PubMed] [Google Scholar]
- 17.Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1992. Baculovirus-expressed glycoprotein G of herpes simplex virus type 1 partially protects vaccinated mice against lethal HSV-1 challenge. Virology 190:233–239. doi: 10.1016/0042-6822(92)91209-D. [DOI] [PubMed] [Google Scholar]
- 18.Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1992. Baculovirus-expressed glycoprotein H of herpes simplex virus type 1 (HSV-1) induces neutralizing antibody and delayed type hypersensitivity responses, but does not protect immunized mice against lethal HSV-1 challenge. J Gen Virol 73:719–722. doi: 10.1099/0022-1317-73-3-719. [DOI] [PubMed] [Google Scholar]
- 19.Ghiasi H, Kaiwar R, Slanina S, Nesburn AB, Wechsler SL. 1994. Expression and characterization of baculovirus expressed herpes simplex virus type 1 glycoprotein L. Arch Virol 138:199–212. [DOI] [PubMed] [Google Scholar]
- 20.Ghiasi H, Bahri S, Nesburn AB, Wechsler SL. 1995. Protection against herpes simplex virus-induced eye disease after vaccination with seven individually expressed herpes simplex virus 1 glycoproteins. Invest Ophthalmol Vis Sci 36:1352–1360. [PubMed] [Google Scholar]
- 21.Ghiasi H, Nesburn AB, Cai S, Wechsler SL. 1998. The US5 open reading frame of herpes simplex virus type 1 does encode a glycoprotein (gJ). Intervirology 41:91–97. doi: 10.1159/000024919. [DOI] [PubMed] [Google Scholar]
- 22.Ghiasi H, Cai S, Slanina S, Nesburn AB, Wechsler SL. 1997. Nonneutralizing antibody against the glycoprotein K of herpes simplex virus type-1 exacerbates herpes simplex virus type-1-induced corneal scarring in various virus-mouse strain combinations. Invest Ophthalmol Vis Sci 38:1213–1221. [PubMed] [Google Scholar]
- 23.Allen SJ, Mott KR, Ljubimov AV, Ghiasi H. 2010. Exacerbation of corneal scarring in HSV-1 gK-immunized mice correlates with elevation of CD8+ CD25+ T cells in corneas of ocularly infected mice. Virology 399:11–22. doi: 10.1016/j.virol.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mott KR, Osorio Y, Maguen E, Nesburn AB, Wittek AE, Cai S, Chattopadhyay S, Ghiasi H. 2007. Role of anti-glycoproteins D (anti-gD) and K (anti-gK) IgGs in pathology of herpes stromal keratitis in humans. Invest Ophthalmol Vis Sci 48:2185–2193. doi: 10.1167/iovs.06-1276. [DOI] [PubMed] [Google Scholar]
- 25.Hutchinson L, Goldsmith K, Snoddy D, Ghosh H, Graham FL, Johnson DC. 1992. Identification and characterization of a novel herpes simplex virus glycoprotein, gK, involved in cell fusion. J Virol 66:5603–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghiasi H, Slanina S, Nesburn AB, Wechsler SL. 1994. Characterization of baculovirus-expressed herpes simplex virus type 1 glycoprotein K. J Virol 68:2347–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foster TP, Rybachuk GV, Kousoulas KG. 2001. Glycoprotein K specified by herpes simplex virus type 1 is expressed on virions as a Golgi complex-dependent glycosylated species and functions in virion entry. J Virol 75:12431–12438. doi: 10.1128/JVI.75.24.12431-12438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foster TP, Kousoulas KG. 1999. Genetic analysis of the role of herpes simplex virus type 1 glycoprotein K in infectious virus production and egress. J Virol 73:8457–8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hutchinson L, Johnson DC. 1995. Herpes simplex virus glycoprotein K promotes egress of virus particles. J Virol 69:5401–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hutchinson L, Roop-Beauchamp C, Johnson DC. 1995. Herpes simplex virus glycoprotein K is known to influence fusion of infected cells, yet is not on the cell surface. J Virol 69:4556–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.David AT, Baghian A, Foster TP, Chouljenko VN, Kousoulas KG. 2008. The herpes simplex virus type 1 (HSV-1) glycoprotein K (gK) is essential for viral corneal spread and neuroinvasiveness. Curr Eye Res 33:455–467. doi: 10.1080/02713680802130362. [DOI] [PubMed] [Google Scholar]
- 32.Jayachandra S, Baghian A, Kousoulas KG. 1997. Herpes simplex virus type 1 glycoprotein K is not essential for infectious virus production in actively replicating cells but is required for efficient envelopment and translocation of infectious virions from the cytoplasm to the extracellular space. J Virol 71:5012–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.David AT, Saied A, Charles A, Subramanian R, Chouljenko VN, Kousoulas KG. 2012. A herpes simplex virus 1 (McKrae) mutant lacking the glycoprotein K gene is unable to infect via neuronal axons and egress from neuronal cell bodies. mBio 3:e00144-12. doi: 10.1128/mBio.00144-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mott KR, Perng GC, Osorio Y, Kousoulas KG, Ghiasi H. 2007. a recombinant herpes simplex virus type 1 expressing two additional copies of gK is more pathogenic than wild-type virus in two different strains of mice. J Virol 81:12962–12972. doi: 10.1128/JVI.01442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Osorio Y, Mott KR, Jabbar AM, Moreno A, Foster TP, Kousoulas KG, Ghiasi H. 2007. Epitope mapping of HSV-1 glycoprotein K (gK) reveals a T cell epitope located within the signal domain of gK. Virus Res 128:71–80. doi: 10.1016/j.virusres.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mott KR, Chentoufi AA, Carpenter D, Benmohamed L, Wechsler SL, Ghiasi H. 2009. The role of a glycoprotein K (gK) CD8+ T-cell epitope of herpes simplex virus on virus replication and pathogenicity. Invest Ophthalmol Vis Sci 50:2903–2912. doi: 10.1167/iovs.08-2957. [DOI] [PubMed] [Google Scholar]
- 37.Mott KR, Allen SJ, Zandian M, Akbari O, Hamrah P, Maazi H, Wechsler SL, Sharpe AH, Freeman GJ, Ghiasi H. 2014. Inclusion of CD80 in HSV targets the recombinant virus to PD-L1 on DCs and allows productive infection and robust immune responses. PLoS One 9:e87617. doi: 10.1371/journal.pone.0087617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghiasi H, Osorio Y, Perng GC, Nesburn AB, Wechsler SL. 2001. Recombinant herpes simplex virus type 1 expressing murine interleukin-4 is less virulent than wild-type virus in mice. J Virol 75:9029–9036. doi: 10.1128/JVI.75.19.9029-9036.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghiasi H, Osorio Y, Hedvat Y, Perng GC, Nesburn AB, Wechsler SL. 2002. Infection of BALB/c mice with a herpes simplex virus type 1 recombinant virus expressing IFN-γ driven by the LAT promoter. Virology 302:144–154. doi: 10.1006/viro.2002.1609. [DOI] [PubMed] [Google Scholar]
- 40.Ghiasi H, Osorio Y, Perng GC, Nesburn AB, Wechsler SL. 2002. Overexpression of interleukin-2 by a recombinant herpes simplex virus type 1 attenuates pathogenicity and enhances antiviral immunity. J Virol 76:9069–9078. doi: 10.1128/JVI.76.18.9069-9078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol 68:8045–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allen SJ, Mott KR, Matsuura Y, Moriishi K, Kousoulas KG, Ghiasi H. 2014. Binding of HSV-1 glycoprotein K (gK) to signal peptide peptidase (SPP) is required for virus infectivity. PLoS One 9:e85360. doi: 10.1371/journal.pone.0085360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghiasi H, Cai S, Slanina SM, Perng GC, Nesburn AB, Wechsler SL. 1999. The role of interleukin (IL)-2 and IL-4 in herpes simplex virus type 1 ocular replication and eye disease. J Infect Dis 179:1086–1093. doi: 10.1086/314736. [DOI] [PubMed] [Google Scholar]
- 44.Matundan H, Mott KR, Ghiasi H. 2014. Role of CD8+ T cells and myeloid DCs in protection from ocular HSV-1 challenge in immunized mice. J Virol 88:8016–8027. doi: 10.1128/JVI.00913-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1992. Expression of herpes simplex virus type 1 glycoprotein B in insect cells: initial analysis of its biochemical and immunological properties. Virus Res 22:25–39. [DOI] [PubMed] [Google Scholar]
- 46.Mott KR, Ghiasi H. 2008. Role of dendritic cells in enhancement of herpes simplex virus type 1 latency and reactivation in vaccinated mice. Clin Vaccine Immunol 15:1859–1867. doi: 10.1128/CVI.00318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zwaagstra JC, Ghiasi H, Nesburn AB, Wechsler SL. 1991. Identification of a major regulatory sequence in the latency associated transcript (LAT) promoter of herpes simplex virus type 1 (HSV-1). Virology 182:287–297. doi: 10.1016/0042-6822(91)90672-X. [DOI] [PubMed] [Google Scholar]
- 48.Debroy C, Pederson N, Person S. 1985. Nucleotide sequence of a herpes simplex virus type 1 gene that causes cell fusion. Virology 145:36–48. doi: 10.1016/0042-6822(85)90199-0. [DOI] [PubMed] [Google Scholar]
- 49.Bond VC, Person S. 1984. Fine structure physical map locations of alterations that affect cell fusion in herpes simplex virus type 1. Virology 132:368–376. doi: 10.1016/0042-6822(84)90042-4. [DOI] [PubMed] [Google Scholar]
- 50.Little SP, Schaffer PA. 1981. Expression of the syncytial (Syn) phenotype in HSV-1, strain KOS: genetic and phenotypic studies of mutants in two syn loci. Virology 112:686–702. doi: 10.1016/0042-6822(81)90314-7. [DOI] [PubMed] [Google Scholar]
- 51.Pogue-Geile KL, Spear PG. 1987. The single base pair substitution responsible for the Syn phenotype of herpes simplex virus type 1, strain MP. Virology 157:67–74. doi: 10.1016/0042-6822(87)90314-X. [DOI] [PubMed] [Google Scholar]
- 52.Foster TP, Melancon JM, Baines JD, Kousoulas KG. 2004. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J Virol 78:5347–5357. doi: 10.1128/JVI.78.10.5347-5357.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mo C, Suen J, Sommer M, Arvin A. 1999. Characterization of varicella-zoster virus glycoprotein K (open reading frame 5) and its role in virus growth. J Virol 73:4197–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klupp BG, Baumeister J, Dietz P, Granzow H, Mettenleiter TC. 1998. Pseudorabies virus glycoprotein gK is a virion structural component involved in virus release but is not required for entry. J Virol 72:1949–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dietz P, Klupp BG, Fuchs W, Kollner B, Weiland E, Mettenleiter TC. 2000. Pseudorabies virus glycoprotein K requires the UL20 gene product for processing. J Virol 74:5083–5090. doi: 10.1128/JVI.74.11.5083-5090.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Foster TP, Chouljenko VN, Kousoulas KG. 2008. Functional and physical interactions of the herpes simplex virus type 1 UL20 membrane protein with glycoprotein K. J Virol 82:6310–6323. doi: 10.1128/JVI.00147-08. [DOI] [PMC free article] [PubMed] [Google Scholar]