

Abstract
VX-787 is a novel inhibitor of influenza virus replication that blocks the PB2 cap-snatching activity of the influenza viral polymerase complex. Viral genetics and X-ray crystallography studies provide support for the idea that VX-787 occupies the 7-methyl GTP (m7GTP) cap-binding site of PB2. VX-787 binds the cap-binding domain of the PB2 subunit with a KD (dissociation constant) of 24 nM as determined by isothermal titration calorimetry (ITC). The cell-based EC50 (the concentration of compound that ensures 50% cell viability of an uninfected control) for VX-787 is 1.6 nM in a cytopathic effect (CPE) assay, with a similar EC50 in a viral RNA replication assay. VX-787 is active against a diverse panel of influenza A virus strains, including H1N1pdm09 and H5N1 strains, as well as strains with reduced susceptibility to neuraminidase inhibitors (NAIs). VX-787 was highly efficacious in both prophylaxis and treatment models of mouse influenza and was superior to the neuraminidase inhibitor, oseltamivir, including in delayed-start-to-treat experiments, with 100% survival at up to 96 h postinfection and partial survival in groups where the initiation of therapy was delayed up to 120 h postinfection. At different doses, VX-787 showed a 1-log to >5-log reduction in viral load (relative to vehicle controls) in mouse lungs. Overall, these favorable findings validate the PB2 subunit of the viral polymerase as a drug target for influenza therapy and support the continued development of VX-787 as a novel antiviral agent for the treatment of influenza infection.
INTRODUCTION
Influenza is a potentially deadly infectious disease that has imposed a substantial burden in terms of morbidity and mortality on human populations (1). Recent statistics suggest that, each year in the United States, 5% to 20% of the population becomes infected with influenza virus, with more than 200,000 hospitalizations for respiratory and heart-related complications and an annual mortality rate ranging from ∼3,000 to 49,000 deaths (2, 3).
Vaccination has become the mainstay of efforts to minimize the impact of seasonal influenza (4, 5), and while generally effective in healthy adults, it is often less effective in elderly individuals, and there have been recent examples where predictions of which viral strains to include have been inadequate (6, 7). Further, the 2009 H1N1 pandemic demonstrated that the logistics required to rapidly isolate and identify the correct strain and to produce enough vaccine worldwide was quite challenging (8–10). The continued incidence of human infection by avian influenza virus strains, namely, either the highly pathogenic H5N1 or H7N7 subtypes or the recently emerging low pathogenic H7N9 and H10N9 strains, represents an additional challenge for vaccine development strategies (11–15)
Antiviral agents may be used for the prophylaxis of influenza virus infection (mainly in high-risk settings) or in a treatment modality for the reduction of illness duration. They may also provide an option for rapid deployment during a pandemic situation (16). The current standard of care is treatment using the neuraminidase inhibitors oseltamivir and zanamivir. It is recommended that treatment begin as early as possible after the onset of illness, ideally within 48 h. The benefit of these agents has recently received considerable scrutiny, with several observational studies reporting reduced mortality in treated individuals during the recent H1N1pdm09 pandemic (17–19) whereas other reviews have questioned this observation (20, 21). In addition to the limited treatment window, another concern for this class of antivirals is the possibility of neuraminidase inhibitor resistance, as occurred during the 2008 to 2009 season when oseltamivir-resistant H1N1 was prevalent (22, 23). This highlights the need for new classes of antiviral agents with novel mechanisms of action (24–27).
In order to provide alternative therapeutic options for patients with influenza infection, we initiated a phenotypic-assay-based drug discovery effort. Subsequently, the molecular target of a series of azaindole hits resulting from these screening efforts was determined to be the PB2 cap-binding domain of the influenza viral polymerase complex (28). The viral polymerase contains three subunits, PB1, PB2, and PA, that are responsible for replication and transcription of the viral RNA (vRNA) genome in the nuclei of infected cells. The heterotrimeric viral polymerase synthesizes viral mRNAs via a cap-snatching mechanism wherein the virus utilizes host pre-mRNA as a primer for transcription. The PB2 subunit contains a cap-binding domain for 7-methyl GTP (m7GTP) on the 5′ end of the host pre-mRNA. Once bound to PB2, the PA endonuclease subunit cleaves the host RNA strand, leaving a 10-to-13-nucleotide primer. The PB1 subunit contains the conserved polymerase domain and utilizes the primer for RNA elongation (29–31).
In this report, we describe the preclinical characterization of VX-787, a novel PB2 inhibitor for influenza A virus that resulted from extensive lead optimization (28) of hits that emerged from the aforementioned phenotypic screen.
MATERIALS AND METHODS
Viruses, cell culture, and viral infection.
All experiments involving infectious virus were performed under appropriate, i.e., animal biosafety level 2 (ABSL2) or ABSL3 conditions. Madin Darby canine kidney (MDCK) cells, HEK293T cells, and influenza virus strains A/Puerto Rico/8/34 (H1N1), A/WS/33 (H1N1), A/Hong Kong/8/68 (H3N2), and A/Victoria/3/75 (H3N2) were purchased from the America Type Culture Collection (ATCC). A/California/07/2009 (H1N1pdm09) was obtained through the Influenza Reagent Resource (Influenza Division, World Health Organization, Collaborating Center for Surveillance, Epidemiology and Control of Influenza, Centers for Disease Control and Prevention, Atlanta, GA). A/Georgia/17/2006 (H1N1), A/Georgia/20/2006 (H1N1), A/Nebraska/1/2006 (H3N2), A/Texas/12/2007 (H3N2), and A/Texas/48/2009 (H1N1) were acquired from the Virus Surveillance and Diagnosis Branch, Influenza Division, National Center for Immunization and Respiratory Diseases (NCIRD)/Coordinating Center for Infectious Diseases (CCID) (Centers of Disease Control and Prevention, Atlanta, GA). All studies were performed with low-passage-number MDCK cells maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with l-glutamine (Invitrogen), penicillin-streptomycin (Invitrogen), and HEPES (Invitrogen), referred to here as cDMEM, with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO, or Invitrogen). Influenza virus stocks were propagated in MDCK cells using standard methods (26) and stocks frozen at −80°C. MDCK cells were infected with influenza virus in cDMEM–1 μg/ml TPCK (N-tosyl-l-phenylalanine chloromethyl ketone)-treated trypsin (USB Corporation/Affymetrix, Santa Clara, CA) (viral growth media [VGM]).
Chemicals.
VX-787 was synthesized at Vertex Pharmaceuticals Incorporated (28), and 10 mM stock solutions were prepared in dimethyl sulfoxide (DMSO). Zanamivir hydrate (Sequoia Research Products, Pangbourne, United Kingdom) was dissolved in DMEM at a concentration of 10 mM. For in vitro experiments, the ethyl ester prodrug of oseltamivir (Focus Synthesis, San Diego, CA, USA) was hydrolyzed by base catalysis of the ethyl ester, releasing the active form, oseltamivir carboxylate, and dissolved at 10 mM in DMSO. For in vivo experiments with oseltamivir, the oral suspension was used. Favipiravir (T-705) was synthesized at Syncom, based on published methods (32). Amantadine was obtained from Sigma-Aldrich Chemicals.
MDCK cell protection and cytotoxicity assays.
Compound antiviral activity and cytotoxicity were evaluated in MDCK cells using a modification of standard methods (33, 34); cell viability was determined by measurement of ATP levels (CellTiter-Glo; Promega, Madison, WI). MDCK cells were plated into black, clear-bottom, 384-well plates at a density of 2 × 104 cells per well in 50 μl of cDMEM. Cells were incubated at 37°C and 5% CO2 with saturated humidity to allow cells to adhere and form a monolayer. After 5 h, 40 μl of cDMEM was removed, and 25 μl of VGM containing diluted drug (final DMSO concentration of 0.5%) and 15 μl of influenza virus at a concentration of 100 50% tissue culture infective doses (TCID50)/well were added. The cytotoxic effect of compounds was similarly determined, in the absence of virus. Internal controls consisted of wells containing cells only or untreated cells infected with virus. Plates were incubated for 72 h, followed by addition of 20 μl of CellTiter-Glo and incubation at room temperature for 10 min. Luminescence was measured using an EnVision Multilabel Reader (PerkinElmer, Waltham, MA). The EC50 (the concentration of compound that ensures 50% cell viability of an uninfected control) and CC50 (the concentration of compound that reduces cell viability by 50% compared to an untreated control) were calculated by fitting the compound dose versus the response data (4-parameter curve-fitting method employing a Levenburg Marquardt algorithm) (Condoseo software; Genedata, Basel, Switzerland). Viral titers were determined by preparation of 10-fold serial dilutions of the sample into VGM, followed by a 4-day cytopathic effect (CPE) assay, and are expressed as TCID50 values (35). For evaluation of lung titers, lungs were homogenized in a total volume of 1 ml DMEM using a 1-min burst of a Mini-16 bead beater. The homogenates were clarified by centrifugation at 900 × g for 10 min; supernatants were diluted as described above.
MDCK cell plaque assay.
MDCK cells were plated in a 6-well plate at a density of 5 × 105 cells per well and incubated at 37°C and 5% CO2 with saturated humidity overnight. On the day of infection, cells were washed once with DMEM and then infected with virus in 1 ml VGM for 60 min. Virus-containing media were aspirated, and the cell monolayer was overlaid with 3 ml of 0.8% low-melting agarose (SeaPlaque; Lonza Inc., Rockland, ME) at 37°C in VGM. Plates were left at room temperature for 15 min to allow the agarose to solidify and were then incubated at 37°C and 5% CO2 with saturated humidity for 48 to 72 h. Following incubation, the agarose plug was removed and the cell monolayer fixed with 2 ml of 10% formaldehyde for 1 h. The cell monolayer was then rinsed once with water and stained with 2.5% Coomassie blue (Sigma-Aldrich) for 5 min to visualize the plaque.
bDNA assay.
Compound activity was determined by measuring A/Puerto Rico/8/34 hemagglutinin negative-strand [HA(−)] RNA synthesis (36) in infected MDCK cells using custom branched DNA (bDNA) QuantiGene with influenza virus A HA(−) oligonucleotide probe sets (Affymetrix). In clear, tissue-culture-treated 96-well plates, MDCK cells were seeded at 4 × 104 cells per well in duplicate plates, in 100 μl of VGM. Cells were incubated 5 h to allow cells to adhere and form a monolayer. A 90-μl volume of media was removed, and 50 μl of VGM containing diluted drug (final DMSO concentration of 0.5%) and 40 μl of A/Puerto Rico/8/34 virus at a concentration of 8 × 103 TCID50/well at a multiplicity of infection (MOI) of 0.2 were added to duplicate plates. Internal controls consisted of wells containing cells only or virus-infected cells. Plates were incubated at 37°C and 5% CO2 with saturated humidity for 22 h. Cells were lysed with the addition of 140 μl of lysis buffer/well and incubated at 53°C for 1 h. Following incubation, 20 μl of lysate was transferred to bDNA capture plates and combined with 80 μl of lysis buffer containing HA(−) probes. Plates were incubated overnight at 53°C to allow hybridization of RNA to the probe. After aspiration of the lysate, plates were sequentially washed and incubated for 1 h at 45°C with 100 μl of a 1:1,000 dilution of amplifier solution followed by 100 μl of a 1:1,000 dilution of signal probe. After a final round of aspiration and washing, 100 μl of 2.0 substrate solution was added to each plate and luminescence was quantitated using a Wallac Victor 1420 multilabel counter (PerkinElmer, Waltham, MA). Curves were fitted to data points using nonlinear regression analysis, and EC50s were interpolated from the resulting curve using GraphPad Prism software, version 5.0 (GraphPad Software Inc., San Diego, CA), and SoftMax Pro 5.2 (Molecular Devices, Sunnyvale, CA).
Sequencing of the influenza A virus polymerase complex genes from viral stocks or infected MDCK cells.
Sequence analysis of influenza A virus utilized reverse transcriptase PCR (RT-PCR) amplification of approximately 3-kb RNA fragments for the PB2, PB1, and PA coding regions. Viral RNA was extracted from 100 μl of viral stock or from 2 × 106 infected MDCK cells in 300 μl of lysis buffer under denaturing conditions. Viral RNA was isolated using either the RNeasy Plus Mini method (Qiagen, Valencia, CA, USA) or the QIAamp Virus RNA Mini method (Qiagen). cDNA was synthesized from vRNA in a 50-μl reaction mixture containing 2.5 μM Universal 12 primer (AGCRAAAGCAGG), 400 U of Superscript III reverse transcriptase (Invitrogen), 40 U of RNase OUT (Invitrogen), PC2 reaction buffer (50 mM Tris-HCl [pH 9.1], 16 mM ammonium sulfate, 3.5 mM MgCl2, 150 μg/ml bovine serum albumin [BSA]) (AB Peptides, St. Louis, MO, USA), 500 μM deoxynucleoside triphosphates (dNTPs) (Clontech, Mountain View, CA, USA), and 5 mM dithiothreitol (DTT) (Invitrogen). The mixture was incubated for 5 min at 65°C followed by ramping extension temperatures (25°C for 10 min, 42°C for 10 min, 50°C for 20 min, 55°C for 10 min, and 70°C for 15 min). Amplification of cDNA was performed on 5 μl of the completed RT reaction mixture with PC2 reaction buffer, 200 μM (each) dNTPs, 1.5 M betaine (Sigma-Aldrich), 3.2 U Klentaq DNA polymerase (AB Peptides), 1.6 U Pfu DNA polymerase (Stratagene, La Jolla, CA, USA), and a 400 μM concentration of each primer for a final reaction volume of 50 μl (for the PA segment, 5′-CGTCTCNGGGAGCGAAAGCAGGTACTGATCCAAAAT [Forward] and 5′-CGTCTCNTATTAGTAGAAACAAGGTACTTTTTTGGA [Reverse]; for the PB1 segment, 5′-CGTCTCNGGGAGCGAAAGCAGGCAAACCATTTGAA [Forward] and 5′-CGTCTCNTATTAGTAGGAACAAGGCATTTTTTCATG [Reverse]; for the PB2 segment, 5′-CGTCTCNGGGAGCGAAAGCAGGTCAATTATATTCAA [Forward] and 5′-CGTCTCNTATTAGTAGAAACAAGGTCGTTTTTAAAC [Reverse]). The reaction mixture was incubated at 94°C for 2 min, followed by 40 cycles of 94°C for 15 s, 68°C to 0.4°C/cycle (“touchdown” PCR) for 20 s, and 68°C for 3.5 min, followed by incubation at 68°C for 7 min. The PCR product was purified using a QIAquick 96 PCR purification kit (Qiagen), and an aliquot was analyzed by 1% agarose gel electrophoresis for quality. Purified DNA was sequenced using Sanger sequencing.
Construction of plasmids for reverse genetics.
Plasmid constructs encoding viral genome segments were created as described previously (37). Influenza A/Puerto Rico/8/34 cDNA virus was amplified using oligonucleotides containing terminal BsmB1 restriction endonuclease sites, and the amplified product was cloned into pCR-XL-TOPO shuttle vector using a TOPO XL PCR cloning kit (Invitrogen). The cDNA-containing TOPO vectors were digested with BsmB1 and insertions ligated into pHH21 RNA polymerase I-driven expression vector (38). Plasmids were sequenced to confirm proper integration of the cDNA fragment and the absence of unwanted mutations.
Mutations encoding amino acid changes identified from resistance selection experiments were cloned into the corresponding pHH21-A/Puerto Rico/8/34 plasmid using a QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, Inc., Wilmington, DE, USA) following the manufacturer's protocol. Briefly, 10 ng of wild-type plasmid and 150 ng of each forward- and reverse-mutation-containing primer were combined with 5 μl 10× reaction buffer, 1 μl dNTP mix, 3 μl QuikSolution, and 2.5 U of PfuUltra high-fidelity (HF) DNA polymerase in PCR-grade water to reach 50 μl, and mutations were introduced by PCR. Thermal cycling was performed with an initial denaturation at 95°C for 1 min, 18 cycles of 95°C for 50 s, 60°C for 50 s, and 68°C for 5 min, and a final extension for 7 min at 68°C. Residual wild-type plasmid was digested with DpnI restriction enzyme, and mutation-containing plasmid was transformed into bacteria and plasmid was isolated using a Qiagen Plasmid Midi kit (Qiagen). Incorporation of point mutations into the plasmid was confirmed by Sanger sequencing.
Generation of recombinant viruses.
Recombinant viruses were generated by transient transfection of HEK293T cells with a plasmid mixture containing 1.25 μg of the pHH21 mutation-containing plasmid, each of the seven remaining pHH21 wild-type plasmids, and the four RNP complex expression plasmids (38) using TransIT-LT1 transfection reagent (MirusBio, Madison, WI, USA). Briefly, a 1:2 (wt/vol) mixture of the plasmid mixture and transfection reagent was incubated in 1.5 ml of Opti-MEM I media (Invitrogen) at room temperature for 15 min. The transfection mixture was then added to T-75 flasks containing HEK293T cells at 50% confluence maintained in 15 ml cDMEM supplemented with 10% FBS. Cells were incubated at 37°C in 5% CO2 at saturated humidity for 24 h, washed once with cDMEM, and incubated for an additional 48 h in 20 ml VGM. Following incubation, the supernatant was harvested and centrifuged at 650 × g for 10 min to remove cell debris and virus was further expanded in MDCK cells infected at a low MOI, after which viral stocks were harvested and titers were determined as previously described.
Neuraminidase assays.
Virus sensitivity to neuraminidase inhibitors was determined by the chemiluminescent neuraminidase inhibitor assay using an NA-XTD kit (Applied Biosystems, Foster City, CA, USA) per the manufacturer's recommendations. Viruses were diluted in NA-XTD assay buffer (26 mM morpholineethanesulfonic acid, 4 mM CaCl2 [pH 6.0]) such that the signal-to-noise ratio was greater than 40:1. Resistance was defined as determination of a 50% inhibitory concentration greater than 10-fold the mean for the type/subtype (39).
ITC and surface plasmon resonance (SPR).
Isothermal titration calorimetry (ITC) experiments were carried out using an Auto iTC200 calorimeter (GE Healthcare, Piscataway, NJ). The protein solution (50 μM) in the calorimetric cell was titrated at 25°C with ligand dissolved in the same buffer (10 mM HEPES [pH 7.4], 150 mM NaCl, 5% DMSO) at a concentration of 500 μM in the injection syringe. The heat released after each injection was determined by integrating the calorimetric signal. The heat due to the binding reaction between the inhibitor and the protein was determined as the difference between the heat of the reaction and the corresponding heat of the dilution.
SPR data from VX-787 binding with the PB2 cap-binding domain were determined by immobilizing the protein to ∼1,000 response units (RUs) via amine coupling (20 μg/ml in acetate buffer, pH 5.5) on a carboxymethylated dextran surface using a Biacore T200 instrument (GE Healthcare, Piscataway, NJ). VX-787 was titrated at 3.1 to 100 nM and 25°C in 10 mM HEPES (pH 7.4)–150 mM NaCl–0.05% surfactant P20–5% DMSO, and the data were corrected for the DMSO concentration. Data were fitted using either kinetic (1:1) or steady-state analysis to obtain the binding parameters.
Synergy/antagonism experiments.
VX-787 was tested in a 3-day MDCK cell CPE-based assay in which cells were infected with A/Puerto Rico/8/34 at an MOI of 0.01 in combination experiments with oseltamivir carboxylate, zanamivir, or favipiravir (40). For Bliss independence experiments, decreasing concentrations of each inhibitor were combined in a full factorial combination, and the results were analyzed using Macsynergy II software (41). For Loewe additivity experiments, a fixed ratio of combined inhibitors was serially diluted and the combination index (C.I.) calculated using the median-effect approach (42) (Calcusyn software, Biosoft, Cambridge, United Kingdom). Experiments were also performed using combinations of VX-787 with itself as a control, confirming additivity. Cell viability was determined using CellTiter-Glo.
Mouse infection experiments.
All mouse studies used male 5-to-7-week-old BALB/c mice (Charles River Laboratories, Wilmington, MA). All mouse studies utilizing A/Puerto Rico/8/34 were conducted at Vertex Pharmaceuticals Incorporated under an approved Institutional Animal Care and Use protocol. For all studies utilizing A/Puerto Rico/8/34, mice were housed in groups of four animals per cage. Eight mice were enrolled per study group. Mice were anesthetized with ketamine/xylazine, and virus was introduced intranasally at 25 μl/nostril for a final challenge dose of 5 × 103 TCID50/mouse. Mice were weighed and observed for signs of morbidity daily for 21 days postinfection. Any mouse that scored positive for four of the following observations (>35% body weight [BW] loss, ruffled fur, hunched posture, respiratory distress, reduced mobility, or hypothermia) was deemed moribund, euthanized, and scored as a death in accordance with guidelines established with the Vertex Institutional Animal Care and Use Committee.
Mouse studies with H1N1pdm09 or influenza A/Vietnam/1203/2004 (H5N1) virus were performed in ABSL3 facilities at the Institute for Antiviral Research, Utah State University, Logan, UT, under an approved Institutional Animal Care and Use protocol. Intranasal challenge doses were 3 × 104 and 2 × 102 TCID50/mouse for A/California/04/2009 and A/Vietnam/1203/2004, respectively. Mouse-adapted A/California/07/2009 was obtained from Elena Govorkova, St. Jude's Children's Research Hospital, Memphis, TN (43), and A/Vietnam/1203/2004 was obtained from Jackie Katz of the Centers for Disease Control, Atlanta, Georgia. Ten to 20 mice were enrolled per group. The mice were observed daily for morbidity and weighed every other day to assess weight changes. All of the described experiments utilized either a prophylactic dosing regimen where compound treatment was started 2 h prior to infection or therapeutic dosing regimens where treatment was initiated 24 to 96 h postinfection. Survival data were analyzed using the Kaplan Meier method, and BW was analyzed using two-way analysis of variance (ANOVA) and a Bonferroni posttest to compare groups. P values of less than 0.05 were considered significant.
Lung viral titer determinations.
Five or six mice were enrolled per study group. Treatment with VX-787, oseltamivir, or vehicle was initiated 2 h prior or 24 h postinfection and continued until study day 6. On study day 6, cohorts of mice were euthanized and lungs collected and homogenized. Lung homogenates were serially diluted and added to MDCK cells to establish the lung viral burden, expressed as TCID50/lung. Data were analyzed using a two-way ANOVA and a Bonferroni posttest to compare groups. P values of <0.05 were considered significant.
RESULTS
Binding of VX-787 to the PB2 domain.
Binding of VX-787 (Fig. 1) to PB2 was analyzed using ITC and SPR (Fig. 2). The KD for VX-787 is approximately 60-fold lower than that of m7GTP (Fig. 1), the natural ligand for PB2 (see Table S1 in the supplemental material). ITC (Fig. 2A; see also Table S1) and SPR (Fig. 2B; see also Table S1) data for VX-787 confirm tight binding of VX-787 to the PB2 domain.
FIG 1.
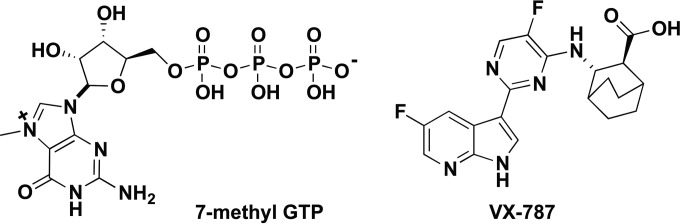
Chemical structure of m7GTP and VX-787.
FIG 2.

Binding data for VX-787 to PB2 cap-binding domain. (A) Isothermal titration calorimetric data: the upper panel shows raw ITC data, and the lower panel shows integrated ITC data fitted to a single-binding-site model. (B) Surface plasmon resonance (SPR) binding data and kinetic/steady-state analysis for VX-787 with the PB2 cap-binding domain.
X-ray crystal structures of the complex of m7GTP and VX-787 with PB2.
The X-ray crystal structures of m7GTP and VX-787 provide further evidence of VX-787 binding to the central cap-binding domain of PB2 (Fig. 3) (28).The structure of the PB2 cap-binding fragment was consistent with that previously described for complexes with m7GTP (29). These structures revealed that VX-787 occupies the m7GTP binding site and produces interactions with the PB2 fragment similar to those seen with the m7GTP guanine base (Fig. 3). Both m7GTP and VX-787 form hydrogen bonds to the side chains of Glu361 and Lys376. The azaindole ring of VX-787 is sandwiched between the aromatic side chains of His357 and Phe404, while the pyrimidine ring forms an attractive aromatic pi-stacking interaction with the phenyl ring of Phe323. The carboxylic acid of VX-787 shows water-mediated interactions with the epsilon nitrogen of His357 and with Gln406, as well as with the main chain carbonyl of Arg355.
FIG 3.

X-ray structure of PB2 cap-binding domain. (A) Contact residues with m7GTP (left) or VX-787 (right). (B) Ribbon diagram and surface depicting cap-binding domain of PB2 bound to VX-787 (PyMOL Molecular Graphics System, version 1.7; Schrödinger, LLC).
In vitro antiviral activity and mechanistic studies with VX-787.
Key in vitro antiviral activities of VX-787 are summarized (Table 1), including inhibition of production of influenza virus-specific RNAs. Further, VX-787 showed no cytotoxicity when incubated with cells in the absence of virus. VX-787 showed potent activity against all influenza A virus strains tested, with an EC50 range of 0.13 to 3.2 nM (Table 2). The tested strains includes H1N1 and H3N2 reference strains from 1933 to 2009, including the pandemic 2009 H1N1 A/California/07/2009 and A/Texas/48/2009 strains and the highly pathogenic avian H5N1 A/Viet Nam/1203/2004 strain. VX-787 was equally effective against all strains, including those that were resistant to amantadine and neuraminidase inhibitors. The activity appears to be specific for influenza A viruses, as negligible activity against influenza B virus was observed (see Table S5 in the supplemental material).
TABLE 1.
Summary of key in vitro activities of VX-787 with influenza virus type A reference strain A/Puerto Rico/8/34a
| Assay | Mean assay result (nM), no. of expts |
|---|---|
| CPE (ATP) (VX-787 EC50) | 1.6 ± 0.77, n = 16 |
| Viral RNA (bDNA) (VX-787 EC50) | 2.6 ± 0.88, n = 11 |
| Viral RNA (bDNA) (VX-787 EC99) | 11 ± 9.2, n = 10 |
| Cytotoxicity (ATP) (VX-787 CC50) | ≥10,000 ± ND, n = 6 |
Data shown represent means ± standard deviations of the results of n independent experiments. EC50, effective concentration at which the ATP or viral RNA level is half the maximum; bDNA, branched DNA; EC99, effective concentration at which the viral RNA level is 99% maximum; CC50, concentration at which the cytopathic effect, as measured by ATP levels, is half the maximum in the 3-day CPE-based assay; ND, not determined.
TABLE 2.
In vitro activity of VX-787 against influenza virus type A strains that are susceptible or resistant to oseltamivir carboxylate or amantadine
| Virus | Type/subtype | Phenotype |
Mean VX-787 EC50 (nM), no. of exptsc | |
|---|---|---|---|---|
| Oseltamivir carboxylatea | Amantadineb | |||
| A/WS/33 | A/H1N1 | S | R | 3.2 ± 4.3, n = 4 |
| A/Hong Kong/8/68 | A/H3N2 | S | S | 0.60 ± 0.11, n = 3 |
| A/Victoria/3/75 | A/H3N2 | S | S | 1.3 ± 0.63, n = 5 |
| A/Georgia/17/2006 | A/H1N1 | S | S | 0.13 ± 0.048, n = 3 |
| A/Georgia/20/2006 | A/H1N1 | R | S | 2.6 ± 3.8, n = 4 |
| A/Nebraska/1/2006 | A/H3N2 | S | R | 2.1 ± 1.3, n = 4 |
| A/Texas/12/2007 | A/H3N2 | R | R | 0.65 ± 0.22, n = 4 |
| A/California/07/2009 | A/H1N1pdm09 | S | R | 1.8 ± 1.6, n = 7 |
| A/Texas/48/2009 | A/H1N1pdm09 | R | R | 2.8 ± 3.2, n = 6 |
| A/Viet Nam/1203/2004 | A/H5N1 | S | R | <1.5, n = 1d |
R or S, neuraminidase inhibitor resistant or susceptible, respectively, as determined by enzymatic assay and publicly available sequences.
R or S, amantadine resistant (M2 31N mutation) or susceptible, respectively, as determined by analysis of publicly available sequences.
Data shown represent means ± standard deviations of the results of n independent experiments.
The experiment was performed at Southern Research Institute.
In vitro selection of variants resistant to VX-787.
Preliminary in vitro selection experiments were performed using VX-787 and the influenza A/Puerto Rico/8/34 virus strain, with the goal of this experiment having mainly been mechanism of action/target confirmation and not a detailed analysis of resistance. Variants were selected using a range of concentrations of inhibitor, and the effects of any observed amino acid substitutions in the PA, PB1, or PB2 genes were evaluated in the reverse-genetics system. Variants conferring reduced sensitivity to VX-787 were restricted to the PB2 gene, and none were observed in PA or PB1. Six PB2 variants, Q306H, S324I, S324N, S324R, F404Y, and N510T, were identified in two or more independent cultures and showed a greater than 10-fold change in VX-787 sensitivity (Table 3). The observed PB2 sequence variants are rarely observed in naturally occurring human isolates (Table 3). These isolates maintained wild-type sensitivity to the neuraminidase inhibitors oseltamivir and zanamivir.
TABLE 3.
VX-787-selected PB2 variant viruses maintain susceptibility to neuraminidase inhibitors in vitro
| Influenza virus variant | VX-787 EC50 (μM), no. of exptsa | Fold shift |
Frequency of variant in human data set | ||
|---|---|---|---|---|---|
| VX-787 EC50b | Oseltamivir carboxylate IC50c | Zanamivir IC50c | |||
| WT | 0.0030 ± 0.0018, n = 18 | 1.0 | 1.0 | 1.0 | |
| PB2-Q306H | 0.56 ± 0.71, n = 9 | 186 | <2 | <2 | 0/8,919 |
| PB2-S324I | 0.47 ± 0.071, n = 3 | 157 | <2 | <2 | 0/8,942 |
| PB2-S324N | 0.38 ± 0.68, n = 5 | 127 | <2 | <2 | 1/8,942 |
| PB2-S324R | 0.19 ± 0.23, n = 12 | 63 | <2 | <2 | 0/8,942 |
| PB2-F404Y | 0.77 ± 0.50, n = 8 | 257 | <2 | <2 | 3/8,912 |
| PB2-N510T | 0.40 ± 0.050, n = 3 | 133 | <2 | <2 | 1/8,926 |
Data shown represent means ± standard deviations of the results of n independent experiments.
EC50 determined using 3-day CPE assay.
IC50 determined using enzymatic neuraminidase assay.
Time-of-addition experiments with VX-787.
Time course experiments were performed to demonstrate the effects of VX-787 on viral RNA production in infected cells (Fig. 4). We analyzed the intracellular viral positive [(+)]-strand and negative [(−)]-strand RNA to monitor the impact of VX-787 on viral mRNA and genomic RNA synthesis. Under normal conditions, viral (+)-strand RNA levels were observed to increase rapidly starting around 5 h postinfection, reaching a peak at 6 to 8 h postinfection. The (−)-strand RNA appeared 6 to 7 h postinfection, and levels continued to increase to 11 h postinfection (the last time point sampled). When VX-787 was present at the initiation of infection, no measureable viral (+)-strand or (−)-strand RNA was observed. When VX-787 was added 6 h postinfection, the production of (+)-strand viral RNA was immediately arrested, and the rate of production of (−)-strand viral RNA was reduced. This result clearly indicates that VX-787 directly impacts mRNA and genomic RNA synthesis. Neither oseltamivir nor zanamivir had a significant effect on (+)-strand or (−)-strand viral RNA production, either when added at the initiation of infection or when added 6 h postinfection. Favipiravir demonstrated effects on influenza viral (+)-strand RNA and early (−)-strand RNA synthesis similar to those of VX-787 although to a lower extent under these conditions. When treatment was initiated at 6 h postinfection, favipiravir no longer had an effect on (−)-strand synthesis.
FIG 4.

Time-of-addition experiment. MDCK cells were infected with influenza A/Puerto Rico/8/34 virus at an MOI of 2. Arrows indicate times at which test compounds were added, at 45 min (left) or 6 h (right) postinfection. The concentrations and compounds tested were 0.04 μM VX-787, 50 μM favipiravir, 22 μM oseltamivir carboxylate, and 1 μM zanamivir, each approximately 25-fold the EC50 in a 3-day CPE assay. Additionally, the concentrations of oseltamivir carboxylate and zanamivir chosen were >1,000-fold the enzymatic IC50 for these compounds. Samples were harvested every 2 or 3 h posttreatment to monitor the levels of viral (+)-strand RNA and viral (−)-strand RNA using a bDNA assay.
Effect of VX-787 on influenza cytopathic effect.
MDCK cells, plated in a 96-well plate, were infected with A/Puerto Rico/8/34 virus at an MOI of 2 in the presence or absence of inhibitors. Viability of the cultures was monitored by measuring ATP content at different times after infection (Fig. 5A). Virus-infected control wells showed loss of viability beginning approximately 24 h after infection, and CPE progressed to >95% by 48 h after infection. The wells treated with VX-787 maintained viability over a period of up to 72 h, showing no significant difference from uninfected control wells. Cells treated with favipiravir provided protection similar to the VX-787 results. Neuraminidase inhibitor-treated wells showed no protection and lost viability at the same rate as control infected wells. Similarly, control wells containing MDCK cells infected at an MOI of 2 in a plaque assay format showed loss of viability at 48 h and at 72 h but remained viable when incubated in the presence of VX-787 or favipiravir. Neuraminidase inhibitor-treated wells showed a CPE and became negative for Coomassie blue staining in a manner similar to that seen with control infected wells (Fig. 5B).
FIG 5.
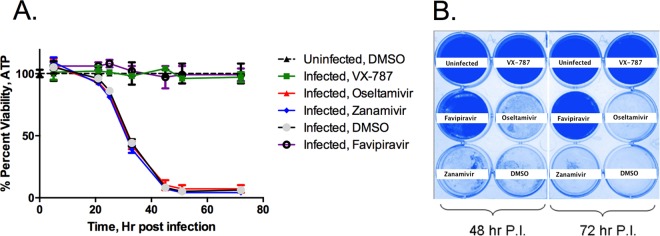
Evaluation of VX-787 cytopathic effect. MDCK cells were infected with influenza A/Puerto Rico/8/34 virus at an MOI of 2 in the presence of inhibitor. The infected cells were harvested at different times postinfection to determine cell viability by analysis of ATP content using the CPE assay (A) or stained with Coomassie blue in a plaque assay (B). Viability is graphed as percent means ± standard deviations of the results of three independent experiments.
Effect of MOI on antiviral activity of VX-787.
Infection experiments were performed with MDCK cells and A/Puerto Rico/8/34 virus using an MOI range of 0.001 to 10. VX-787 was effective at maintaining the viability of the cells for 3 days at all MOIs tested, with only a small shift in the dose-response curve (Fig. 6). This is in marked contrast to the loss in viral susceptibility to oseltamivir above an MOI of 0.1.
FIG 6.
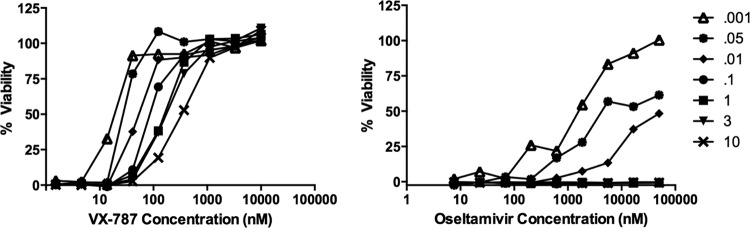
Antiviral activity of VX-787 (left panel) and oseltamivir carboxylate (right panel) against influenza A/Puerto Rico/8/34 virus in MDCK cells was determined across a range of MOIs. MDCK cells in 384-well plates were infected with the indicated range of virus and incubated with 3-fold dilutions of VX-787 ranging from 0.00015 to 1 μM or with oseltamivir carboxylate at from 0.0076 to 50 μM. After incubation at 37°C in 5% CO2 for 3 days, the antiviral activity was investigated by analysis of ATP levels of infected cells. Results are presented as the mean percentage values normalized to noninfected controls, obtained from quadruplicate wells in the same experiment.
Combination experiments with VX-787 and oseltamivir, zanamivir, or favipiravir.
Combination experiments were performed with VX-787 and the neuraminidase inhibitor oseltamivir carboxylate or zanamivir, or the polymerase inhibitor favipiravir, using both the Bliss independence method (MacSynergy) and the Loewe additivity method (median effect). The Bliss independence method resulted in synergy volumes of 312 and 268 for oseltamivir carboxylate and zanamivir, respectively (Fig. 7). A synergy volume of 317 was obtained for favipiravir (results not shown). The Loewe additivity method produced C.I. (combination index) values of 0.58, 0.64, and 0.89 at the 50% effect level for oseltamivir, zanamivir, and favipiravir, respectively. These data indicate that VX-787 is synergistic with these influenza inhibitors, which work through different mechanisms of action.
FIG 7.
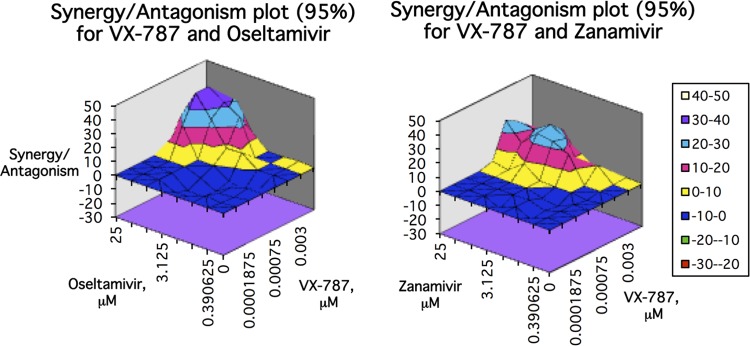
In vitro antiviral combination studies with VX-787 and oseltamivir carboxylate or zanamivir. Inhibitor pairs were tested in full factorial combination, and the expected additive antiviral effect of each combination was calculated according to the Bliss independence method. Data shown were assessed at the 95% confidence level. The simple additivity result is indicated by the zero plane, on the vertical axis. Synergy is indicated by values above the additivity plane and antagonism by values below the plane.
Efficacy experiments with VX-787 in the mouse influenza A virus infection model.
Multiple laboratory mouse strains can be experimentally infected with influenza virus. BALB/c mice represent the most commonly used mouse strain for the preclinical evaluation of small-molecule influenza virus therapeutics (reviewed in reference 33). Oseltamivir is often used as a control, with doses of 10 mg/kg of body weight in mice resulting in an area under the plasma concentration-time curve (AUC) equivalent to that of the human clinical dose of 75 mg twice a day (BID) (results not shown). VX-787 has favorable mouse pharmacokinetic properties following a single oral administration of 10 mg/kg: AUC from 0 h to infinity (AUC0–∞), 4.98 μg · h/ml; maximum concentration of drug in serum (Cmax), 2.37 μg/ml; time to maximum concentration of drug in serum (Tmax), 0.25 h; half-life (t1/2), 6.7 h (28).
A dose-response study in the mouse prophylactic model was performed using A/Puerto Rico/8/34. Dosing with vehicle, oseltamivir, or VX-787 was initiated 2 h prior to infection and continued twice daily for 10 days (Fig. 8A). The dosing duration of 10 days was determined from pilot studies using oseltamivir regimens of from 5 to 12 days; the optimum efficacy for oseltamivir was observed after treatment for 7 to 10 days (data not shown). All of the mice that received vehicle alone succumbed to the infection by study day 9 and had lost, on average, ∼32% of their BW. While VX-787 administered at 1, 3, or 10 mg/kg BID provided complete survival, there was a dose-dependent reduction in BW loss (Fig. 8A; see also Table S2 in the supplemental material). In separate experiments, the minimal effective dose was 0.3 mg/kg BID, which provided 25% survival and significantly greater BW loss (see Table S2).
FIG 8.

Prophylactic or delayed-start-to-treat effectiveness of VX-787 in the mouse influenza A virus infection model. Male BALB/c mice (8/group) were infected with a lethal challenge of influenza virus (A/Puerto Rico/8/34; 3 × 105 TCID50/mouse) followed by administration of vehicle, oseltamivir, or VX-787 at the indicated doses and start times. The 21-day survival rate and percent BW loss (means ± standard errors of the means [SEM]) are shown. The shaded area represents the treatment period. Oseltamivir was not included in studies beyond 48 h postinfection.
The extent to which VX-787 administration could be delayed and still provide effectiveness in this model was investigated by challenging mice with influenza A virus and dosing with vehicle, oseltamivir, or VX-787 starting at 24, 48, 72, 96, or 120 h postinfection, with continued BID dosing for 10 days (Fig. 8B to F). All vehicle control mice succumbed to disease by study day 8 or 9. VX-787 administered at 1, 3, or 10 mg/kg BID provided complete protection from death and reduced BW loss when dosing was initiated at up to 96 h postinfection compared with vehicle control results. Dosing of oseltamivir at 10 mg/kg BID provided complete protection only when dosing was initiated at 24 h postinfection or earlier.
Activity of VX-787 in the mouse infection model using H1N1pdm09 or avian/highly pathogenic influenza virus.
The efficacy of VX-787 was studied in mice infected with the 2009 pandemic A/California/04/2009 (H1N1pdm09) strain. This strain is distinct from A/Puerto Rico/8/34 in that it has not been extensively adapted for growth in mice. Prophylactic treatment (2 h prior to infection) with VX-787 at 1, 3, or 10 mg/kg, dosed orally BID, provided complete protection from mortality at all doses and minimal BW loss at 3 or 10 mg/kg (see Fig. S1 and Table S3 in the supplemental material). In contrast, in the group treated with vehicle alone, initial deaths were observed at study day 7, and by study day 12, 18/20 mice succumbed to the disease and the survivors had greater than 35% BW loss on day 9. Oseltamivir at 10 mg/kg also provided protection from mortality, but the mice had BW loss (∼20% on study day 9) that was more severe than that seen with all the VX-787 dose groups, while there was ∼10% BW loss for the group administered the lowest VX-787 dose.
The efficacy of VX-787 was further explored in mice infected with highly pathogenic avian influenza A/Vietnam/1203/2004 (H5N1) virus. All mice treated with vehicle alone succumbed to the disease by study day 11, with 3/10 succumbing on day 7 and 6/10 by day 9. Prophylactic treatment (2 h prior to infection) with VX-787 at 1, 3, or 10 mg/kg dosed orally BID (Fig. 9A; see also Table S4 in the supplemental material) provided complete protection from mortality and minimal BW loss. Oseltamivir at 10 mg/kg also provided protection from mortality.
FIG 9.

Prophylactic or delayed-start-to-treat effectiveness of VX-787 in the mouse influenza A/Viet Nam/1203/2004 (H5N1) virus infection model. Male BALB/c mice (8/group) were infected with a lethal challenge of influenza virus followed by administration of vehicle, oseltamivir, or VX-787 at the indicated doses and start times. The 21-day survival rate and percent BW loss (means ± SEM) are shown. The shaded area represents the treatment period.
The effective delay-to-treat window of VX-787 for a H5N1 strain was investigated in experiments where dosing was started 24 to 96 h after infection with A/Vietnam/1203/2004(H5N1) (Fig. 9B and C; see also Table S4 in the supplemental material). Oseltamivir at 10 mg/kg BID for 10 days was ineffective at protecting mice from mortality. Doses of 3 or 10 mg/kg VX-787 BID for 10 days were effective at providing a survival and BW benefit when administered 24 to 96 h postinfection. VX-787 administered at 3 or 10 mg/kg BID still provided partial protection from death as well as a dose-dependent reduction in BW loss when administered 120 h postinfection (see Table S4). The results of this experiment indicate that VX-787 is highly effective in preventing mortality in mice due to infection with influenza A/Vietnam/1203/2004 (H5N1) virus.
Effect of VX-787 on lung viral titers in the mouse influenza A virus infection model.
The effectiveness of VX-787 in reducing lung viral titers was investigated in three independent experiments. Mice were infected with influenza A/California/04/2009 (H1N1pdm09) or A/Vietnam/1203/2004 (H5N1) virus, and VX-787 at 10 mg/kg or oseltamivir at 10 mg/kg was administered BID, beginning 2 h before infection. Additionally, mice were infected with A/Puerto Rico/8/34 and VX-787 or oseltamivir was administered BID beginning 24 h after infection. In all cases, the lungs were harvested on day 6 postinfection and the level of infectious virus was determined (Table 4).
TABLE 4.
Summary of lung viral titer experiments in influenza virus-infected mice treated with VX-787 or oseltamivir
| Strain | Treatment start time relative to infection time (h) | Dose (mg/kg; BID) |
Day 6 log viral titer/lung (mean ± SD)a | Log10 reduction vs vehicle | |
|---|---|---|---|---|---|
| VX-787 | Oseltamivir | ||||
| A/California/04/2009 (H1N1pdm09)b | −2 | 10 | <2.60 | >5.3 | |
| 10 | 7.90 ± 0.20 | 0.00 | |||
| 0 | 7.90 ± 0.40 | NAd | |||
| A/Viet Nam/1203/2004 (H5N1)b | −2 | 10 | 1.60 ± 0.50*** | 2.95 | |
| 10 | 3.80 ± 0.62 | 0.85 | |||
| 0 | 4.65 ± 1.22 | NA | |||
| A/Puerto Rico/8/34 (H1N1)c | +24 | 10 | 5.20 ± 0.53*** | 1.00 | |
| 10 | 6.05 ± 0.44 | 0.15 | |||
| 0 | 6.20 ± 0.31 | NA | |||
Data represent the results of two-way ANOVA with a Bonferroni posttest. ***, P < 0.001.
The experiments were performed at Utah State University.
The experiments were performed at Vertex Pharmaceuticals Inc.
NA, not applicable.
All the VX-787 treatment groups showed significant reductions in lung viral titers, with log reductions compared to vehicle control of >5.3, 2.95, and 1.0 for the A/California/04/2009 (H1N1pdm09), A/Vietnam/1203/2004 (H5N1), and A/Puerto Rico/8/34 virus strains, respectively. These reductions in lung viral titer were significantly greater than that observed with oseltamivir in every case.
DISCUSSION
New anti-influenza drugs are needed due to the limitations of approved agents, primarily, the variable emergence of neuraminidase inhibitor resistance and the limited time window after infection (48 h) within which these agents are active (44, 45).
PB2 is an attractive target for antiviral development because it shows less annual variation (46) than hemagglutinin and neuraminidase, which are the principal components of the host immune response targeted by vaccines. Other investigators have examined PB2 as a target (47, 48), but no cellular or animal antiviral activity was reported to our knowledge. We initiated a search for new anti-influenza agents using a phenotypic-based approach (28), aware that this represented a high bar for screening because influenza produces a rapid and clear CPE followed by cell death in vitro (49–53). As the screen employed measured protection from cell death, compounds that were directly cytotoxic were eliminated. The approach could have identified either a host target protein or a viral target (54–56). In this report, we describe VX-787, a small-molecule inhibitor that binds the purified central domain of PB2, occupies the cap-binding site as defined by X-ray crystallography, and blocks cap-dependent (+)-strand viral RNA production in time course experiments.
VX-787 shows consistent activity against all influenza A virus strains tested, and we show its activity against the H1N1pdm09 strain and a highly pathogenic avian influenza virus A/Viet Nam/1203/2004 strain in this report. PB2 has been reported to play a significant role in host restriction (57, 58), with a K627E substitution observed in avian strains relative to human-derived strains. VX-787 is equally effective against strains containing this K627E variant, which is outside the PB2 cap-binding site (data not shown). Targeting PB2 instead of neuraminidase in the influenza virus replication cycle provides several unique opportunities. At any point during the course of disease in vivo, there are both infected cells that need to be controlled and as-yet-uninfected cells that need to be protected; VX-787 has the potential to perform both functions. The means by which VX-787 blocks CPE in infected cells could simply be inhibition of (+)-strand viral RNA synthesis, but it is possible that other mechanisms, such as interference with influenza virus-induced host cell shutoff, are involved (59, 60). Many details of the function of PB2 remain to be elucidated. Hopefully, the availability of a highly efficacious inhibitor such as VX-787 will help further our understanding.
In vitro selection experiments identified influenza virus variants with reduced susceptibility to VX-787 and provided further support for data regarding the mechanism and target of this compound. These variants had amino acid substitutions in the PB2 gene, but not in other influenza virus polymerase complex genes, and these substitutions were rare in the database of naturally occurring human isolates. The issue of resistance to a direct-acting antiviral agent such as VX-787 is expected to some degree and can be managed in a treatment setting by proper dose selection and/or possibly by the combination of VX-787 with drugs in another class of antiviral agent such as the neuraminidase inhibitors (61). We observed in vitro synergy between VX-787 and oseltamivir, zanamivir, or favipiravir in this study. Further, we did not observe any patterns suggestive of emergence of resistance in our mouse efficacy experiments.
Though no structural information is available for influenza B virus PB2, the amino acid sequence shows several differences from influenza A virus among m7GTP contact residues F323Q, R355G, H357W, S337G, N429S, and H432Y. These differences could explain the lack of activity of VX-787 against influenza B virus. There is significant amino acid divergence between influenza A virus PB2 and the other characterized eukaryotic host cell cap-binding proteins eIF4E, nuclear cap-binding complex protein CBC, or vaccinia virus VP39 (47), reducing the likelihood of undesired effects in uninfected host cells.
Mouse pharmacology studies employing oseltamivir typically report doses of 10 to 20 mg/kg/day (33, 62–72). The lower end of this range is equivalent to the human dose based upon AUC matching (data not shown). This dose of oseltamivir has demonstrated a survival benefit in numerous studies, though it is accompanied with severe weight loss, usually peaking at approximately 20% to 30% by day 8. Interestingly, studies of the effect of oseltamivir on lung viral loads have been inconsistent, with TCID50 values of as low as 0.5 logs being reported (33, 63, 67, 68, 73). In contrast to these studies, VX-787 demonstrated a significant survival benefit in prophylactic studies, with 100% survival observed at 1 mg/kg BID and as little as 2.8% BW loss at the 10 mg/kg BID dose. Importantly, VX-787 showed effectiveness in delayed-start-to-treat experiments, with 100% survival at up to day 4 postinfection and partial protection at up to day 5 postinfection. This result exceeds any obtained with the neuraminidase inhibitors and exceeds results reported to date for favipiravir, the RNA polymerase inhibitor (73).
The effect of VX-787 on lung viral loads in mice is superior to that observed with oseltamivir. At several different doses, VX-787 showed a 1 to >5 log viral load reduction relative to vehicle controls, in contrast to the 0.15 to 0.85 log viral load reduction observed with oseltamivir. Consistent with previously published studies and our own unpublished observations, oseltamivir (10 mg/kg BID) did not provide a significant reduction in lung viral titers at day 6 or at any other time points examined (results not shown). Administration of VX-787 provides a rapid reduction in the amount of influenza A virus in the lungs of infected mice and suggests a direct effect on viral replication. This result is consistent with the observed MOI independence of VX-787; initially infected lung cells may create a local high-MOI environment that would remain under antiviral suppression by VX-787 but not be suppressed by oseltamivir. The results from this series of studies represent the first evidence that a pharmacologic inhibitor of influenza A virus PB2 effectively inhibits viral replication in vivo and successfully abrogates or attenuates influenza virus infection in mouse models of the disease.
In summary, VX-787 is a highly efficacious influenza A virus PB2 inhibitor that represents a distinct and novel class of antiviral agent. The properties of this inhibitor, as represented by both the in vitro and in vivo data, indicate that VX-787 is an exciting candidate for further evaluation in the clinic with several potential advantages over current antiviral agents used to treat influenza virus infection.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Paul Caron for discussions regarding potential targets for early screening hits and Kwame Nti-Addae for preparation of dosing solutions for in vivo testing. We thank Chao Lin, Min Jiang, and Thomas Pfeiffer for additional antiviral studies. We also thank Donald Smee and Dale Barnard of the Institute for Antiviral Research, Utah State University, Logan, UT, for performing in vivo studies with highly pathogenic influenza virus strains. We finally thank Mark Namchuk, Youssef Bennani, Scott Raybuck, and Ray Winquist for useful discussions throughout the course of this work.
This work was funded by Vertex Pharmaceuticals, Incorporated.
P.S.C., R.A.B., S.M.J., M.P.C., and C.M.B. directed aspects of the preclinical research. P.S.C. and R.A.B. interpreted data and wrote the majority of the manuscript. We all contributed to the writing of the manuscript. M.W.L., E.P., M.P.C., and P.S.C. conceived of or synthesized the compounds described in the manuscript. A.D.K. and M.A.M. initiated this work and provided critical early guidance. M.D.J. generated the X-ray structures; A.N. and K.S. generated the biophysical PB2 ITC and SPR data. R.A.B. and R.R. designed and directed the antiviral studies. J.R.L., H.B.B., and Y.Z. carried out many of the virology-related experiments. S.M.J. and C.F.M. generated in vivo pharmacology data. A.W.T. and S.M.J. performed the pharmacokinetics and pharmacodynamics (PK/PD) experiments.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04623-14.
REFERENCES
- 1.Morens DM, Taubenberger JK, Folkers GK, Fauci AS. 2010. Pandemic influenza's 500th anniversary. Clin Infect Dis 51:1442–1444. doi: 10.1086/657429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention. 2010. Estimates of deaths associated with seasonal influenza—United States, 1976–2007. MMWR Morb Mortal Wkly Rep 59:1057–1062. [PubMed] [Google Scholar]
- 3.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. 2003. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289:179–186. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 4.Fiore AE, Bridges CB, Cox NJ. 2009. Seasonal influenza vaccines. Curr Top Microbiol Immunol 333:43–82. doi: 10.1007/978-3-540-92165-3_3. [DOI] [PubMed] [Google Scholar]
- 5.Lambert LC, Fauci AS. 2010. Influenza vaccines for the future. N Engl J Med 363:2036–2044. doi: 10.1056/NEJMra1002842. [DOI] [PubMed] [Google Scholar]
- 6.Flannery B, Thaker SN, Clippard J, Monto AS, Ohmit SE, Zimmerman RK, Nowalk MP, Gaglani M, Jackson ML, Jackson LA, Belongia EA, McLean HQ, Berman L, Foust A, Sessions W, Spencer S, Fry AM, Centers for Disease Control and Prevention. 2014. Interim estimates of 2013–14 seasonal influenza vaccine effectiveness—United States, February 2014. MMWR Morb Mortal Wkly Rep 63:137–142. [PMC free article] [PubMed] [Google Scholar]
- 7.Janjua NZ, Skowronski DM, De Serres G, Dickinson J, Crowcroft NS, Taylor M, Winter AL, Hottes TS, Fonseca K, Charest H, Drews SJ, Sabaiduc S, Bastien N, Li Y, Gardy JL, Petric M. 2012. Estimates of influenza vaccine effectiveness for 2007–2008 from Canada's sentinel surveillance system: cross-protection against major and minor variants. J Infect Dis 205:1858–1868. doi: 10.1093/infdis/jis283. [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization. 2009, posting date Pandemic influenza vaccine manufacturing and process timeline: pandemic (H1N1) briefing note 7. WHO, Geneva, Switzerland. http://www.who.int/csr/disease/swineflu/notes/h1n1_vaccine_20090806/en/.
- 9.Carrat F, Flahault A. 2007. Influenza vaccine: the challenge of antigenic drift. Vaccine 25:6852–6862. doi: 10.1016/j.vaccine.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 10.Partridge J, Kieny MP. 2013. Global production capacity of seasonal influenza vaccine in 2011. Vaccine 31:728–731. doi: 10.1016/j.vaccine.2012.10.111. [DOI] [PubMed] [Google Scholar]
- 11.Li Q, Zhou L, Zhou M, Chen Z, Li F, Wu H, Xiang N, Chen E, Tang F, Wang D, Meng L, Hong Z, Tu W, Cao Y, Li L, Ding F, Liu B, Wang M, Xie R, Gao R, Li X, Bai T, Zou S, He J, Hu J, Xu Y, Chai C, Wang S, Gao Y, Jin L, Zhang Y, Luo H, Yu H, Gao L, Pang X, Liu G, Shu Y, Yang W, Uyeki TM, Wang Y, Wu F, Feng Z. 2014. Epidemiology of the avian influenza A (H7N9) outbreak in China. N Engl J Med 370:520–532. doi: 10.1056/NEJMoa1304617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fouchier RA, Schneeberger PM, Rozendaal FW, Broekman JM, Kemink SA, Munster V, Kuiken T, Rimmelzwaan GF, Schutten M, Van Doornum GJ, Koch G, Bosman A, Koopmans M, Osterhaus AD. 2004. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proc Natl Acad Sci U S A 101:1356–1361. doi: 10.1073/pnas.0308352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koopmans M, Wilbrink B, Conyn M, Natrop G, van der Nat H, Vennema H, Meijer A, van Steenbergen J, Fouchier R, Osterhaus A, Bosman A. 2004. Transmission of H7N7 avian influenza A virus to human beings during a large outbreak in commercial poultry farms in the Netherlands. Lancet 363:587–593. doi: 10.1016/S0140-6736(04)15589-X. [DOI] [PubMed] [Google Scholar]
- 14.Chen H, Yuan H, Gao R, Zhang J, Wang D, Xiong Y, Fan G, Yang F, Li X, Zhou J, Zou S, Yang L, Chen T, Dong L, Bo H, Zhao X, Zhang Y, Lan Y, Bai T, Dong J, Li Q, Wang S, Zhang Y, Li H, Gong T, Shi Y, Ni X, Li J, Zhou J, Fan J, Wu J, Zhou X, Hu M, Wan J, Yang W, Li D, Wu G, Feng Z, Gao GF, Wang Y, Jin Q, Liu M, Shu Y. 2014. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: a descriptive study. Lancet 383:714–721. doi: 10.1016/S0140-6736(14)60111-2. [DOI] [PubMed] [Google Scholar]
- 15.Abdelwhab EM, Veits J, Mettenleiter TC. 2014. Prevalence and control of H7 avian influenza viruses in birds and humans. Epidemiol Infect 142:896–920. doi: 10.1017/S0950268813003324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Longini IM Jr, Nizam A, Xu S, Ungchusak K, Hanshaoworakul W, Cummings DA, Halloran ME. 2005. Containing pandemic influenza at the source. Science 309:1083–1087. doi: 10.1126/science.1115717. [DOI] [PubMed] [Google Scholar]
- 17.Siston AM, Rasmussen SA, Honein MA, Fry AM, Seib K, Callaghan WM, Louie J, Doyle TJ, Crockett M, Lynfield R, Moore Z, Wiedeman C, Anand M, Tabony L, Nielsen CF, Waller K, Page S, Thompson JM, Avery C, Springs CB, Jones T, Williams JL, Newsome K, Finelli L, Jamieson DJ; Pandemic H1N1 Influenza in Pregnancy Working Group. 2010. Pandemic 2009 influenza A(H1N1) virus illness among pregnant women in the United States. JAMA 303:1517–1525. doi: 10.1001/jama.2010.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Louie JK, Yang S, Acosta M, Yen C, Samuel MC, Schechter R, Guevara H, Uyeki TM. 2012. Treatment with neuraminidase inhibitors for critically ill patients with influenza A (H1N1)pdm09. Clin Infect Dis 55:1198–1204. doi: 10.1093/cid/cis636. [DOI] [PubMed] [Google Scholar]
- 19.Muthuri SG, Venkatesan S, Myles PR, Leonardi-Bee J, Al Khuwaitir TS, Al Mamun A, Anovadiya AP, Azziz-Baumgartner E, Baez C, Bassetti M, Beovic B, Bertisch B, Bonmarin I, Booy R, Borja-Aburto VH, Burgmann H, Cao B, Carratala J, Denholm JT, Dominguez SR, Duarte PA, Dubnov-Raz G, Echavarria M, Fanella S, Gao Z, Gerardin P, Giannella M, Gubbels S, Herberg J, Iglesias AL, Hoger PH, Hu X, Islam QT, Jimenez MF, Kandeel A, Keijzers G, Khalili H, Knight M, Kudo K, Kusznierz G, Kuzman I, Kwan AM, Amine IL, Langenegger E, Lankarani KB, Leo YS, Linko R, Liu P, Madanat F, et al. 2014. Effectiveness of neuraminidase inhibitors in reducing mortality in patients admitted to hospital with influenza A H1N1pdm09 virus infection: a meta-analysis of individual participant data. Lancet ii:395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jefferson T, Jones MA, Doshi P, Del Mar CB, Hama R, Thompson MJ, Spencer EA, Onakpoya I, Mahtani KR, Nunan D, Howick J, Heneghan CJ. 2014. Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children. Cochrane Database Syst Rev 4:CD008965. doi: 10.1002/14651858.CD008965.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michiels B, Van Puyenbroeck K, Verhoeven V, Vermeire E, Coenen S. 2013. The value of neuraminidase inhibitors for the prevention and treatment of seasonal influenza: a systematic review of systematic reviews. PLoS One 8:e60348. doi: 10.1371/journal.pone.0060348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinstock DM, Zuccotti G. 2009. The evolution of influenza resistance and treatment. JAMA 301:1066–1069. doi: 10.1001/jama.2009.324. [DOI] [PubMed] [Google Scholar]
- 23.Hurt AC. 2014. The epidemiology and spread of drug resistant human influenza viruses. Curr Opin Virol 8:22–29. doi: 10.1016/j.coviro.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 24.Thorlund K, Awad T, Boivin G, Thabane L. 2011. Systematic review of influenza resistance to the neuraminidase inhibitors. BMC Infect Dis 11:134. doi: 10.1186/1471-2334-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chao DL, Bloom JD, Kochin BF, Antia R, Longini IM Jr. 2012. The global spread of drug-resistant influenza. J R Soc Interface 9:648–656. doi: 10.1098/rsif.2011.0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.WHO. 2002. WHO manual on animal influenza diagnosis and surveillance. WHO, Geneva, Switzerland. [Google Scholar]
- 27.Wathen MW, Barro M, Bright RA. 2013. Antivirals in seasonal and pandemic influenza–future perspectives. Influenza Other Resp Vir 7(Suppl 1):S76–S80. doi: 10.1111/irv.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark MP, Ledeboer MW, Davies I, Byrn RA, Jones SM, Perola E, Tsai A, Jacobs M, Nti-Addae K, Bandarage UK, Boyd MJ, Bethiel RS, Court JJ, Deng H, Duffy JP, Dorsch WA, Farmer LJ, Gao H, Gu W, Jackson K, Jacobs DH, Kennedy JM, Ledford B, Liang J, Maltais F, Murcko M, Wang T, Wannamaker MW, Bennett HB, Leeman JR, McNeil C, Taylor WT, Memmott C, Jiang M, Rijnbrand R, Bral C, Germann U, Nezami A, Zhang Y, Salituro FG, Bennani YL, Charifson PS. 2014. The discovery of a novel, first-in-Class, orally bioavailable azaindole inhibitor (VX-787) of influenza PB2. J Med Chem 57:6668–6678. doi: 10.1021/jm5007275. [DOI] [PubMed] [Google Scholar]
- 29.Guilligay D, Tarendeau F, Resa-Infante P, Coloma R, Crepin T, Sehr P, Lewis J, Ruigrok RW, Ortin J, Hart DJ, Cusack S. 2008. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat Struct Mol Biol 15:500–506. doi: 10.1038/nsmb.1421. [DOI] [PubMed] [Google Scholar]
- 30.Fechter P, Mingay L, Sharps J, Chambers A, Fodor E, Brownlee GG. 2003. Two aromatic residues in the PB2 subunit of influenza A RNA polymerase are crucial for cap binding. J Biol Chem 278:20381–20388. doi: 10.1074/jbc.M300130200. [DOI] [PubMed] [Google Scholar]
- 31.Palese PS, Shaw ML. 2007. Orthomyxoviridae: the viruses and their replication, p 1647–1689. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 32.Egawa H, Furuta Y, Sugita J, Uehara S, Hamamoto S, Yonezawa T. October 2004. Pyrazine derivatives or salts thereof, pharmaceutical composition containing the same, and production intermediates thereof. US patent 6,800,629.
- 33.Sidwell RW, Smee DF. 2000. In vitro and in vivo assay systems for study of influenza virus inhibitors. Antiviral Res 48:1–16. doi: 10.1016/S0166-3542(00)00125-X. [DOI] [PubMed] [Google Scholar]
- 34.Noah JW, Severson W, Noah DL, Rasmussen L, White EL, Jonsson CB. 2007. A cell-based luminescence assay is effective for high-throughput screening of potential influenza antivirals. Antiviral Res 73:50–59. doi: 10.1016/j.antiviral.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 35.Division of AIDS, National Institute of Allergy and Infectious Diseases. 1997. DAIDS virology manual for HIV laboratories, p 87, publication NIH-97-3828 U.S. Department of Health and Human Services, Washington, DC. [Google Scholar]
- 36.Wagaman PC, Leong MA, Simmen KA. 2002. Development of a novel influenza A antiviral assay. J Virol Methods 105:105–114. doi: 10.1016/S0166-0934(02)00088-5. [DOI] [PubMed] [Google Scholar]
- 37.Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hughes M, Perez DR, Donis R, Hoffmann E, Hobom G, Kawaoka Y. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci U S A 96:9345–9350. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lutz A, Dyall J, Olivo PD, Pekosz A. 2005. Virus-inducible reporter genes as a tool for detecting and quantifying influenza A virus replication. J Virol Methods 126:13–20. doi: 10.1016/j.jviromet.2005.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okomo-Adhiambo M, Sleeman K, Ballenger K, Nguen HT, VP M. 2010. Neuraminidase inhibitor susceptibility testing in human influenza viruses: a laboratory surveillance perspective. Viruses 2:2269–2289. doi: 10.3390/v2102269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldoni M, Johansson C. 2007. A mathematical approach to study combined effects of toxicants in vitro: evaluation of the Bliss independence criterion and the Loewe additivity model. Toxicol In Vitro 21:759–769. doi: 10.1016/j.tiv.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 41.Prichard MN, Shipman C Jr. 1990. A three-dimensional model to analyze drug-drug interactions. Antiviral Res 14:181–205. [DOI] [PubMed] [Google Scholar]
- 42.Chou TC, Talalay P. 1984. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 43.Ilyushina NA, Khalenkov AM, Seiler JP, Forrest HL, Bovin NV, Marjuki H, Barman S, Webster RG, Webby RJ. 2010. Adaptation of pandemic H1N1 influenza viruses in mice. J Virol 84:8607–8616. doi: 10.1128/JVI.00159-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Clercq E. 2006. Antiviral agents active against influenza A viruses. Nat Rev Drug Discov 5:1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gong J, Fang H, Li M, Liu Y, Yang K, Liu Y, Xu W. 2009. Potential targets and their relevant inhibitors in anti-influenza fields. Curr Med Chem 16:3716–3739. doi: 10.2174/092986709789104984. [DOI] [PubMed] [Google Scholar]
- 46.Gorman OT, Donis RO, Kawaoka Y, Webster RG. 1990. Evolution of influenza A virus PB2 genes: implications for evolution of the ribonucleoprotein complex and origin of human influenza A virus. J Virol 64:4893–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hooker L, Sully R, Handa B, Ono N, Koyano H, Klumpp K. 2003. Quantitative analysis of influenza virus RNP interaction with RNA cap structures and comparison to human cap binding protein eIF4E. Biochemistry 42:6234–6240. doi: 10.1021/bi027081r. [DOI] [PubMed] [Google Scholar]
- 48.Pautus S, Sehr P, Lewis J, Fortune A, Wolkerstorfer A, Szolar O, Guilligay D, Lunardi T, Decout JL, Cusack S. 2013. New 7-methylguanine derivatives targeting the influenza polymerase PB2 cap-binding domain. J Med Chem 56:8915–8930. doi: 10.1021/jm401369y. [DOI] [PubMed] [Google Scholar]
- 49.Severson WE, McDowell M, Ananthan S, Chung DH, Rasmussen L, Sosa MI, White EL, Noah J, Jonsson CB. 2008. High-throughput screening of a 100,000-compound library for inhibitors of influenza A virus (H3N2). J Biomol Screen 13:879–887. doi: 10.1177/1087057108323123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maddry JA, Chen X, Jonsson CB, Ananthan S, Hobrath J, Smee DF, Noah JW, Noah D, Xu X, Jia F, Maddox C, Sosa MI, White EL, Severson WE. 2011. Discovery of novel benzoquinazolinones and thiazoloimidazoles, inhibitors of influenza H5N1 and H1N1 viruses, from a cell-based high-throughput screen. J Biomol Screen 16:73–81. doi: 10.1177/1087057110384613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerritz SW, Cianci C, Kim S, Pearce BC, Deminie C, Discotto L, McAuliffe B, Minassian BF, Shi S, Zhu S, Zhai W, Pendri A, Li G, Poss MA, Edavettal S, McDonnell PA, Lewis HA, Maskos K, Mortl M, Kiefersauer R, Steinbacher S, Baldwin ET, Metzler W, Bryson J, Healy MD, Philip T, Zoeckler M, Schartman R, Sinz M, Leyva-Grado VH, Hoffmann HH, Langley DR, Meanwell NA, Krystal M. 2011. Inhibition of influenza virus replication via small molecules that induce the formation of higher-order nucleoprotein oligomers. Proc Natl Acad Sci U S A 108:15366–15371. doi: 10.1073/pnas.1107906108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Atkins C, Evans CW, White EL, Noah JW. 2012. Screening methods for influenza antiviral drug discovery. Expert Opin Drug Discov 7:429–438. doi: 10.1517/17460441.2012.674510. [DOI] [PubMed] [Google Scholar]
- 53.Shih SR, Chu TY, Reddy GR, Tseng SN, Chen HL, Tang WF, Wu MS, Yeh JY, Chao YS, Hsu JT, Hsieh HP, Horng JT. 2010. Pyrazole compound BPR1P0034 with potent and selective anti-influenza virus activity. J Biomed Sci 17:13. doi: 10.1186/1423-0127-17-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Min JY, Subbarao K. 2010. Cellular targets for influenza drugs. Nat Biotechnol 28:239–240. doi: 10.1038/nbt0310-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee SM, Yen HL. 2012. Targeting the host or the virus: current and novel concepts for antiviral approaches against influenza virus infection. Antiviral Res 96:391–404. doi: 10.1016/j.antiviral.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaw ML. 2011. The host interactome of influenza virus presents new potential targets for antiviral drugs. Rev Med Virol 21:358–369. doi: 10.1002/rmv.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heiny AT, Miotto O, Srinivasan KN, Khan AM, Zhang GL, Brusic V, Tan TW, August JT. 2007. Evolutionarily conserved protein sequences of influenza A viruses, avian and human, as vaccine targets. PLoS One 2:e1190. doi: 10.1371/journal.pone.0001190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamada S, Hatta M, Staker BL, Watanabe S, Imai M, Shinya K, Sakai-Tagawa Y, Ito M, Ozawa M, Watanabe T, Sakabe S, Li C, Kim JH, Myler PJ, Phan I, Raymond A, Smith E, Stacy R, Nidom CA, Lank SM, Wiseman RW, Bimber BN, O'Connor DH, Neumann G, Stewart LJ, Kawaoka Y. 2010. Biological and structural characterization of a host-adapting amino acid in influenza virus. PLoS Pathog 6:e1001034. doi: 10.1371/journal.ppat.1001034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vreede FT, Chan AY, Sharps J, Fodor E. 2010. Mechanisms and functional implications of the degradation of host RNA polymerase II in influenza virus infected cells. Virology 396:125–134. doi: 10.1016/j.virol.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vreede FT, Fodor E. 2010. The role of the influenza virus RNA polymerase in host shut-off. Virulence 1:436–439. doi: 10.4161/viru.1.5.12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hayden FG. 2013. Newer influenza antivirals, biotherapeutics and combinations. Influenza Other Respir Viruses 7(Suppl 1):63–75. doi: 10.1111/irv.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takahashi K, Furuta Y, Fukuda Y, Kuno M, Kamiyama T, Kozaki K, Nomura N, Egawa H, Minami S, Shiraki K. 2003. In vitro and in vivo activities of T-705 and oseltamivir against influenza virus. Antivir Chem Chemother 14:235–241. [DOI] [PubMed] [Google Scholar]
- 63.Govorkova EA, Leneva IA, Goloubeva OG, Bush K, Webster RG. 2001. Comparison of efficacies of RWJ-270201, zanamivir, and oseltamivir against H5N1, H9N2, and other avian influenza viruses. Antimicrob Agents Chemother 45:2723–2732. doi: 10.1128/AAC.45.10.2723-2732.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mendel DB, Tai CY, Escarpe PA, Li W, Sidwell RW, Huffman JH, Sweet C, Jakeman KJ, Merson J, Lacy SA, Lew W, Williams MA, Zhang L, Chen MS, Bischofberger N, Kim CU. 1998. Oral administration of a prodrug of the influenza virus neuraminidase inhibitor GS 4071 protects mice and ferrets against influenza infection. Antimicrob Agents Chemother 42:640–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sidwell RW, Huffman JH, Barnard DL, Bailey KW, Wong MH, Morrison A, Syndergaard T, Kim CU. 1998. Inhibition of influenza virus infections in mice by GS4104, an orally effective influenza virus neuraminidase inhibitor. Antiviral Res 37:107–120. doi: 10.1016/S0166-3542(97)00065-X. [DOI] [PubMed] [Google Scholar]
- 66.Sidwell RW, Bailey KW, Bemis PA, Wong MH, Eisenberg EJ, Huffman JH. 1999. Influence of treatment schedule and viral challenge dose on the in vivo influenza virus-inhibitory effects of the orally administered neuraminidase inhibitor GS 4104. Antivir Chem Chemother 10:187–193. [DOI] [PubMed] [Google Scholar]
- 67.Leneva IA, Roberts N, Govorkova EA, Goloubeva OG, Webster RG. 2000. The neuraminidase inhibitor GS4104 (oseltamivir phosphate) is efficacious against A/Hong Kong/156/97 (H5N1) and A/Hong Kong/1074/99 (H9N2) influenza viruses. Antiviral Res 48:101–115. doi: 10.1016/S0166-3542(00)00123-6. [DOI] [PubMed] [Google Scholar]
- 68.Sidwell RW, Smee DF, Huffman JH, Barnard DL, Bailey KW, Morrey JD, Babu YS. 2001. In vivo influenza virus-inhibitory effects of the cyclopentane neuraminidase inhibitor RJW-270201. Antimicrob Agents Chemother 45:749–757. doi: 10.1128/AAC.45.3.749-757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sidwell RW, Smee DF, Huffman JH, Barnard DL, Morrey JD, Bailey KW, Feng WC, Babu YS, Bush K. 2001. Influence of virus strain, challenge dose, and time of therapy initiation on the in vivo influenza inhibitory effects of RWJ-270201. Antiviral Res 51:179–187. doi: 10.1016/S0166-3542(01)00149-8. [DOI] [PubMed] [Google Scholar]
- 70.Yen HL, Monto AS, Webster RG, Govorkova EA. 2005. Virulence may determine the necessary duration and dosage of oseltamivir treatment for highly pathogenic A/Vietnam/1203/04 influenza virus in mice. J Infect Dis 192:665–672. doi: 10.1086/432008. [DOI] [PubMed] [Google Scholar]
- 71.Smee DF, Wong MH, Bailey KW, Sidwell RW. 2006. Activities of oseltamivir and ribavirin used alone and in combination against infections in mice with recent isolates of influenza A (H1N1) and B viruses. Antivir Chem Chemother 17:185–192. [DOI] [PubMed] [Google Scholar]
- 72.Yun NE, Linde NS, Zacks MA, Barr IG, Hurt AC, Smith JN, Dziuba N, Holbrook MR, Zhang L, Kilpatrick JM, Arnold CS, Paessler S. 2008. Injectable peramivir mitigates disease and promotes survival in ferrets and mice infected with the highly virulent influenza virus, A/Vietnam/1203/04 (H5N1). Virology 374:198–209. doi: 10.1016/j.virol.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sidwell RW, Barnard DL, Day CW, Smee DF, Bailey KW, Wong MH, Morrey JD, Furuta Y. 2007. Efficacy of orally administered T-705 on lethal avian influenza A (H5N1) virus infections in mice. Antimicrob Agents Chemother 51:845–851. doi: 10.1128/AAC.01051-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.