

Abstract
Amdinocillin (mecillinam) is a β-lactam antibiotic that is used mainly for the treatment of uncomplicated urinary tract infections. The objectives of this study were to identify mutations that confer amdinocillin resistance on laboratory-isolated mutants and clinical isolates of Escherichia coli and to determine why amdinocillin resistance remains rare clinically even though resistance is easily selected in the laboratory. Under laboratory selection, frequencies of mutation to amdinocillin resistance varied from 8 × 10−8 to 2 × 10−5 per cell, depending on the concentration of amdinocillin used during selection. Several genes have been demonstrated to give amdinocillin resistance, but here eight novel genes previously unknown to be involved in amdinocillin resistance were identified. These genes encode functions involved in the respiratory chain, the ribosome, cysteine biosynthesis, tRNA synthesis, and pyrophosphate metabolism. The clinical isolates exhibited significantly greater fitness than the laboratory-isolated mutants and a different mutation spectrum. The cysB gene was mutated (inactivated) in all of the clinical isolates, in contrast to the laboratory-isolated mutants, where mainly other types of more costly mutations were found. Our results suggest that the frequency of mutation to amdinocillin resistance is high because of the large mutational target (at least 38 genes). However, the majority of these resistant mutants have a low growth rate, reducing the probability that they are stably maintained in the bladder. Inactivation of the cysB gene and a resulting loss of cysteine biosynthesis are the major mechanism of amdinocillin resistance in clinical isolates of E. coli.
INTRODUCTION
Because of the rapid increase in antibiotic resistance, several first-line antibiotics have been rendered less suitable for empirical treatment. For example, in Sweden, the increasing resistance to trimethoprim has led to a shift in the empirical therapy of uncomplicated urinary tract infections (UTIs), with nitrofurantoin and pivmecillinam now being suggested as first-line therapy (1). It is also recommended as a first-line UTI treatment by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases (2). Amdinocillin (Amd; also known as mecillinam) is an extended-spectrum penicillin derivative, a 6β-amidinopenicillanic acid developed in the 1970s (3, 4). It has been widely used in the Scandinavian countries for UTI treatment since the early 1980s (5, 6). A large proportion of the drug is excreted unchanged in the urine (1, 7), leading to high concentrations in urine and a limited effect on the commensal flora (1, 8, 9). Amd binds to and inhibits the transpeptidase activity of penicillin-binding protein 2 (PBP2) (10–13). PBP2 is responsible for the elongation of rod-shaped cells, and cells treated with Amd become enlarged, nondividing spheres that ultimately lyse (11, 14).
Resistance development has remained low in countries where it is used (15–17). This finding is seemingly a paradox, since a large number of genes (Table 1) can, when mutated, confer resistance to Amd, resulting in a high frequency of mutation to resistance during laboratory selection. Resistance mutations found in mutants isolated in laboratory selections affect many different cellular functions, for instance, (i) cell elongation and division (10, 11, 18–22); (ii) cellular amino acid levels and tRNA synthetases (23–25); (iii) composition of lipopolysaccharide in combination with cya/crp (26–28); (iv) the Rcs global regulatory system that controls, for example, capsule production and cell division (29, 30); and (v) cysteine biosynthesis (31, 32). Some of these mutations are associated with an intracellular increase in the concentration of the molecule ppGpp—the signal for the stringent response. It is known that elevated cellular levels of ppGpp render the target of Amd, PBP2, nonessential (33). Extensive work has been done to elucidate how elevated ppGpp levels can cause Amd resistance (10, 21–23, 33–37), but as of now, the mechanism remains unclear. Furthermore, elevated levels of ppGpp have also been shown to upregulate virulence genes in both Escherichia coli and Salmonella enterica serovar Typhimurium (38, 39), suggesting that pathogens could concomitantly become antibiotic resistant and more virulent because of these mutations.
TABLE 1.
Known Amd resistance-encoding genes and the functions of their productsa
| Gene(s) (alias[es]) | [ppGpp] dependence | Reference(s) | Function |
|---|---|---|---|
| mrdA (pbpA) | Unknown | 10, 11, 19 | Cell division and elongation |
| mrdB (rodA) | Unknown | 10, 20 | |
| mreB (envB) | Unknown | 18 | |
| mreC | Unknown | 18 | |
| ftsQ | No | 21, 22 | |
| ftsA | No | 21, 22 | |
| ftsZ | No | 21, 22 | |
| rpoB | Yes | 35 | RNA synthesis |
| cysB | Partly | 31, 32 | Cysteine biosynthesis |
| cysE | Partly | 31, 32 | |
| argS (lov) | Yes | 23 | tRNA synthetases |
| alaS | Yes | 23 | |
| slt | No | 32 | Transglycosylation |
| lon | Unknown | 29 | Rcs regulatory system |
| rcsB | Unknown | 29, 30 | |
| rcsC | Unknown | 29, 30 | |
| yrfF (mucM, igaA) | Unknown | 29 | |
| cyaA | Unknown | 26, 27 | Global regulation |
| crp | Unknown | 26, 27 | |
| spoTb | Yes | 28 | ppGpp degradation and synthesis |
| rfa, rfb, rfcb | 28 | Lipopolysaccharide | |
| galEb | Yes | 28 | |
| aroK | Yes | 34 | Shikimate kinase |
Elevated cellular levels of ppGpp (known as the stringent response) give increased Amd resistance. Some of the known resistance-encoding genes have been shown to confer resistance or not to confer resistance, depending on the cell's capacity to produce ppGpp.
In this study, we isolated and analyzed Amdr E. coli mutants obtained during laboratory selections and Amdr clinical isolates obtained from UTIs where E. coli was the causative agent. Our major objectives were (i) to identify the main genetic determinant(s) of Amd resistance in clinical isolates of E. coli and (ii) to determine why resistance development remains slow in clinical settings, even though Amd resistance emerges very rapidly during selection in the laboratory. Results show that in clinical isolates of Amdr E. coli, inactivation of the cysB gene was the major cause of resistance. In contrast, in laboratory-isolated mutants, many other types of Amd resistance mutations were found. However, many of these mutants showed large reductions in fitness, suggesting that in spite of the high frequency of mutation to resistance in vitro, resistance development remains low in clinical settings because most of these mutants grow too slowly to be stably maintained.
MATERIALS AND METHODS
Bacterial strains and media.
All of the laboratory strains used in this study are derivatives of E. coli MG1655. All of the clinical strains are E. coli UTI isolates kindly provided by Gunnar Kahlmeter, Växjö, Sweden. Clinical strains whose whole genomes have been sequenced were isolated from Swedish patients in 2005 to 2007, and additional strains came from other European countries (United Kingdom, Germany, Finland, and Greece). All of the strains used in this study are listed in Table S1 in the supplemental material. When not specified otherwise, bacteria were grown in Mueller-Hinton (MH) broth (Becton, Dickinson and Company) and plated on MH agar. When more suitable, Luria-Bertani (LB) medium (Sigma-Aldrich) was used instead. Some experiments were performed with urine; morning urine (from E. Thulin) was sterile filtered, samples from several days were pooled, and the urine was sterile filtered again and then frozen in aliquots.
When appropriate, the medium was supplemented with the antibiotics Amd (4 to 32 mg/liter), tetracycline (Tet; 12.5 to 30 mg/liter), chloramphenicol (Cam; 12.5 mg/liter), kanamycin (Kan; 100 mg/liter), and ampicillin (Amp; 100 mg/liter), all from Sigma-Aldrich. Strains were saved by freezing overnight cultures in 10% dimethyl sulfoxide at −80°C.
To screen for cysteine auxotrophy, strains were grown for 48 h on minimal medium, M9 glucose (0.4%) agar plates, and M9 glucose plates supplemented with 0.3 mM cysteine (from Sigma-Aldrich).
MIC assays and cross-resistance testing.
Amd MICs were determined by Etest (AB bioMérieux, Solna Sweden). Bacteria were grown overnight and then diluted 500-fold in phosphate-buffered saline (PBS; 13 mM phosphate, 137 mM NaCl, pH 7.4) before being spread evenly on MH agar plates. An Etest strip was placed on the agar, and the results were analyzed after ∼24 h. When appropriate, the MH agar plates were supplemented with 0.3 or 0.75 mM cysteine. The Amd MIC for E. coli MG1655 was also tested on M9 glucose agar plates.
Potential cross-resistance to 11 antibiotics (Tet, meropenem, cefotaxime, Amp, ciprofloxacin, fosfomycin, trimethoprim, trimethoprim-sulfamethoxazole, erythromycin, Cam, and nalidixic acid) in the reconstructed laboratory-selected mutants was analyzed by Etest as described above.
Isolation of mutants.
Thirty independent overnight cultures in MH medium of E. coli MG1655 were diluted in PBS, and then approximately 107 CFU were spread evenly on MH agar plates with four different concentrations of Amd, 4, 8, 16, and 32 mg/liter, corresponding to 32, 64, 128, and 256 times the MIC for E. coli MG1655, respectively. Colonies were picked after one and two overnight incubations. Single colonies of isolated mutants were restreaked on MH agar plates with the same concentration of Amd as that on which they were isolated. The mutation frequency was calculated as the median number of colonies per plate of each selection concentration divided by the total number of CFU (40).
DNA isolation, sequencing, and bioinformatics.
Isolation of genomic DNA was performed with the Genomic Tip 100G DNA kit (Qiagen) according to the manufacturer's protocol. Nineteen E. coli Amdr mutants and 12 clinical Amdr isolates were sent to BGI (Beijing Genome Institute, China) for whole-genome sequencing (Illumina). The CLC Genomics Workbench software (5.5.1; CLC bio, Aarhus, Denmark) was used to assemble sequence reads and detect regions with sequence variation (quality-based variant detection) and regions with no coverage compared to E. coli MG1655 (NC_000913), respectively. The cutoff frequency was set to 75%.
In order to identify possible Amd resistance mutations in the clinical isolates, the sequences of 17 E. coli genomes (GenBank accession no. AE014075, CP000819, CP000946, CP000970, CP001164, CP001509, CP001637, CP001665, CP001671, CP001855, CP001969, CP002516, CP002167, NC_002695, NC_004431, NC_013353, and NC_013361) were obtained from the National Center for Biotechnology Information. A comparison of amino acid variability in all of the known Amd resistance-encoding genes was done with the sequences from these genomes and the genome of MG1655 by using CLC Main Workbench 6.8.2 (CLC Bio) for translation and alignments of sequences. The alignments were used to find conserved and variable regions of the Amd resistance-encoding genes, and any amino acid change found in a Amd resistance-encoding gene in a clinical strain was considered to be a potential cause of Amd resistance only if it was found in highly conserved regions (that is, regions that were identical in all of the reference strains and E. coli MG1655).
PCR and Sanger sequencing.
PCRs with either Phusion High-Fidelity DNA polymerase (Thermo Fisher) or TaqGold polymerase (Thermo Fisher) were done according to the following protocol: 95°C (TaqGold) or 98°C (Phusion) for 5 min; 29 cycles of 95°C (TaqGold) or 98°C (Phusion) for 30 s, the proper primer annealing temperature for 30 s, and 72°C for the proper length of the sequence of interest (1 min kb−1); and finally 72°C for 7 min before cooling to 4°C. Purification of PCR products was performed with the Gene Jet PCR Purification kit (Thermo Fisher). PCR products were DNA sequenced by Eurofins MWG Operon (Ebersberg, Germany). All of the PCR primers used in this work are listed in Table S2 in the supplemental material.
Strain constructions.
Selected mutations in the laboratory-selected resistant mutants were reconstructed, and gene knockouts were constructed in the E. coli MG1655 genetic background by standard genetic techniques (see Table 3; also see Table S1 in the supplemental material). In brief, a Cam or Kan resistance marker was introduced 10 to 20 kb away from the mutation of interest by temperature-controlled λ Red recombineering (41, 42) with a pSim5-Tet plasmid (43). P1 lysates of the constructed strains were then prepared and used for transduction into MG1655. From each transduction, a congenic strain pair carrying the mutation and the resistance marker or only the resistance marker was saved. The Cam or Kan resistance cassette was subsequently removed by introducing the pCP20 plasmid, which expresses Flp recombinase, which acts on Flp recombination sites flanking the resistance cassette, leaving an 85-nucleotide scar (see Table S1 in the supplemental material) (41, 44). Finally, the linear transformation was confirmed by both PCR (and gel electrophoresis) and sequencing of the scar sequence and the Amd resistance-encoding gene of interest.
TABLE 3.
Characteristics of reconstructed Amdr mutants of E. coli MG1655 strain DA5438 compared to corresponding laboratory-selected mutants
| Strain | Amd resistance-encoding mutation(s) | MIC (mg/liter) | Relative growth rate |
|---|---|---|---|
| DA5438 | None | 0.125 | 1 |
| DA18488 | thrS (E519G) | 48 | 0.31 |
| DA29703 | thrS (E519G)a | 32 | 0.76 |
| DA18480 | aspS (P191S) | 16 | 0.54 |
| DA28434 | aspS (P191S)a | 24 | 0.61 |
| DA18497 | gltX (D180G) | 32 | 0.33 |
| DA30120 | gltX (D180G)a | 24 | 0.36 |
| DA18467 | ubiE (delb bp 346–349 → FSc) | 12 | 0.46 |
| DA28436 | ubiE (del bp 346–349 → FS)a | 12 | 0.54 |
| DA18483 | ubiX (del bp 246 → FS) | 32 | 0.37 |
| DA29707 | ubiX (del bp 246 → FS)a | 32 | 0.40 |
| DA18491 | ispA (IS1 insertion) | 12 | 0.35 |
| DA30122 | ispA (IS1 insertion)a | 16 | 0.33 |
| DA18477 | rplL (promoter −10 and duplication) | 8 | 0.62 |
| DA29710 | rplL (promoter −10)a | 48 | 0.34 |
| DA18481 | ppa (D11A) | 96 | 0.48 |
| DA28432 | ppa (D11A)a | 96 | 0.43 |
| DA18482 | rpoB (H447L), araB (I450L) | 32 | 0.56 |
| DA29712 | rpoB (H447L)a | 48 | 0.61 |
| DA18493 | spoT (S305P) | 24 | 0.75 |
| DA28438 | spoT (S305P)a | 32 | 0.76 |
| DA18489 | mrdA (D389G), trxB (G37D) | 64 | 0.30 |
| DA18490 | mrdA (D389G) | 16 | 0.41 |
| DA29705 | mrdA (D389G)a | 16 | 0.51 |
| DA29715 | trxB (G37D)a | 0.064 | 0.92 |
| DA29717 | mrdA (D389G),a trxB (G37D)a | 64 | 0.29 |
Reconstructed mutant.
del, deletion.
FS, frame shift.
To introduce the cysB mutations identified in the clinical isolates into the E. coli MG1655 background, a cat-sacB marker was introduced to replace the cysB gene in DA5438 by λ Red recombineering as described above. The cat-sacB marker used is part BBa_K864150 from the Registry of Standard Biological Parts, is expressed from the BBa_J23101 promoter (45), and has been assigned GenBank accession number KM018298. PCR-amplified cysB genes from the clinical Amdr strains were then transformed into the cysB::cat-sacB strain and recombinants that replaced the cysB::cat-sacB cassette with the PCR-amplified cysB gene were selected for by growth at high sucrose levels, as sacB is lethal in the presence of sucrose (46). The transformations were confirmed as described above.
All of the PCR primers used for amplification of the resistance cassettes and screening for insertion of the cassettes are listed in Table S3 in the supplemental material.
Transformation of the E. coli MG1655 cysB gene into clinical isolates.
A plasmid carrying the functional cysB gene from MG1655 was introduced into clinical strains. The plasmid used (pEL3c15) was based on BioBrick vector pSB3T5 from the Registry of Standard Biological Parts (47), which additionally carries the red marker mRFP1 in the cloning site to facilitate screening for plasmids with the desired insert and a Camr marker. Briefly, the insert (E. coli MG1655 cysB gene with the native promoter and terminator) was PCR amplified with primers containing XbaI and PstI restriction enzyme cleavage sites, respectively. The insert and the plasmid were first cleaved with Fast Digest XbaI and PstI according to the manual provided by Thermo Fisher and then ligated with Ready-To-Go T4 DNA ligase (GE Health Care Life Sciences). The resulting plasmids were transformed into MG1655 by electroporation and then spread on Cam-supplemented MH agar plates for selection of plasmid uptake. PCR was used to confirm the introduction of cysB into the plasmid in strain DA37697.
For introduction of the plasmid into the clinical strains, it was prepared from DA37697 and then transformed into strains DA14710, DA14713, DA14718, DA14719, and DA24686 and the cysB knockout strain DA28439 as described above, resulting in strains DA37700 to DA37713. The Amd MICs for these clinical strains containing the cysB plasmid were then determined as described above.
Fitness measurements.
Overnight cultures in MH medium were diluted to ∼1 × 106 to 3 × 106 CFU/ml in the same medium, and then 300 μl was added in quadruplicate to a 100-well honeycomb plate. Medium blanks and the reference strain E. coli MG1655 were added (also in quadruplicate) to each plate to enable calculation of the relative growth rate and subtraction of the absorbance value of the medium. Growth of the samples at 37°C with shaking for 16 h was monitored with a Bioscreen C Analyzer (Oy Growth Curves Ab. Ltd.). Measurements of optical density at 600 nm (OD600) were taken every 4 min. Calculations are based on OD600 values between 0.02 and 0.1 wherever growth was observed to be exponential. All of the experiments were run three separate times. The R statistical software (48) was used to calculate relative growth rates by dividing the generation time of the parental strain by the generation time of the mutants from the same experiment. A t test was performed to detect any significant difference in fitness between the E. coli laboratory-isolated mutants and the clinical isolates. The same Bioscreen protocol was used for measurements of fitness in urine.
Fitness in different concentrations of Amd was also examined the same way for a subset of E. coli reconstructed mutants, clinical isolates, and strain MG1655 (DA5438, DA14719, DA24678, DA24686, DA28432, DA28434, DA28436, DA28438, DA28439, DA29705, and DA29712), except that the samples were in triplicate. The Amd concentrations were in a dilution series ranging from 10 to 300 mg/liter for the Amdr strains and from 0.032 to 2 mg/liter for E. coli MG1655.
Nucleotide sequence accession numbers.
The GenBank accession numbers for the sequences of the alaS, argS, aroK, aspS, crp, cyaA, cysB, cysE, ftsA, ftsQ, ftsZ, galE, gltX, ispA, lon, mrdA, mrdB, mreB, mreC, mreD, ppa, rcsB, rcsC, rcsD, rcsF, rplL, rpoB, slt, spoT, thrS, ubiE, and ubiX genes from the clinical Amdr strains are KP670466 to KP670849 (see Table S3 in the supplemental material).
RESULTS
In vitro isolation of Amdr mutants and MIC determinations.
Selection of independent mutants was carried out on MH agar plates supplemented with Amd at 4, 8, 16, or 32 mg/liter. Colonies differing in size and morphology (e.g., mucoidy) and from different selective concentrations were picked to ensure that a wide range of Amdr mutants were isolated. The Amd MICs for isolated E. coli mutants ranged between 4 and 96 mg/liter, compared to the MIC of 0.125 mg/liter for E. coli MG1655, the EUCAST epidemiological cutoff of ≤1 mg/liter, and the EUCAST clinical breakpoints of ≤8 mg/liter for sensitivity and >8 mg/liter for resistance. The mutation frequencies were 8 × 10−8 to 2 × 10−5, depending on the concentration of Amd used in the selection plate (the lowest frequency was at the highest concentration, and the highest frequency was at the lowest concentration). The Amd MICs for the 12 clinical Swedish isolates whose whole genomes have been sequenced, as well as the additional five clinical isolates from other European countries used in this study, were in the same range as those for the laboratory-selected mutants (4 to 64 mg/liter) (see Table 4).
TABLE 4.
Putative Amd resistance-encoding mutations in the clinical isolates used in this study
| Strain | MIC (mg/liter) | Mutation(s) in known Amd resistance-encoding gene(s)a | cysB mutation |
|---|---|---|---|
| DA14704 | 32 | rcsC (T714N), yrfF (R50H), alaS (A515T), mreC (Q324P, delb ntc 975–1035) | V12G |
| DA14709 | 4 | rcsB (K192T), rcsD (A254T), rcsF (P26S), gltX (D301N), aspS (K347N), spoT (T350I), rpoB (G544S) | IS1 insertion |
| DA14710 | 64 | rcsF (P48S), gltX (S65N, T287A), alaS (D712E) | G241E |
| DA14712 | 32 | rcsF (P48S), gltX (S65N, T287A), argS (Y200C), alaS (D712E) | FSd after aae 160 |
| DA14713 | 64 | rcsD (P252Q), yrfF (D307N), aspS (S580G), slt (S2266T, P544L) | Q103stop |
| DA14715 | 16 | thrS (P7H), ftsQ (E262G), ftsZ (V331A) | del nt 278–309 |
| DA14718 | 32 | FS after aa 161 | |
| DA14719 | 16 | crp (T8A, S17F) | FS after aa 87 |
| DA24678 | 48 | rcsD (P490L), rpoB (E625A) | FS after aa 161 |
| DA24686 | 32 | rcsD (P252Q), yrfF (D307N), argS (E326K), mreC (S69L, A327T, P352S), cyaA (Q655L, L693P), slt (T142I) | K76stop |
| DA24690 | 32 | rcsD (P490L), rpoB (E625A) | Q4stop |
| DA24692 | 48 | ubiE (S5L), cyaA (del nt 1428–1434), slt (G58R, G540S), galE (E67K) | FS after aa 242 |
Not found in reference genomes.
del, deletion.
nt, nucleotide.
FS, frame shift.
aa, amino acid.
Whole-genome sequencing and genetic reconstruction of laboratory-isolated mutants.
The whole genomes of 19 independently selected Amdr E. coli mutants were sequenced. To identify the Amd resistance-encoding mutations, genome assembly was done and the results were compared to the E. coli MG1655 genome. Of the 19 mutants, 15 had only one mutation and the other 4 had two mutations. Most of the mutants had mutations in genes that have previously been found to confer Amd resistance, such as mrdA (the gene encoding PBP2), spoT, rpoB, and tRNA synthetase-encoding genes (Table 2 shows the mutated genes and the number of strains with the specific mutations; see Table S1 in the supplemental material). Although some other tRNA synthetases have been connected to Amd resistance before, these specific ones (thrS, aspS, and gltX) were novel. Two strains had the same mutation in mrdA but different MICs (12 and 64 mg/liter, respectively). The strain with the higher MIC had an additional mutation in trxB, which encodes thioredoxin reductase.
TABLE 2.
Characteristics of Amdr laboratory-selected E. coli mutants
| Gene | No. of mutants | No. of different mutationsa | MIC(s) |
|---|---|---|---|
| thrS | 4 | 4 | 16–48 |
| aspS | 7 | 6 (3 in promoter) | 12–16 |
| gltX | 2 | 2 | 16–32 |
| ppa | 3 | 2 | 96 |
| mrdA | 2 | 1 | 12–64 |
| ubiE | 1 | 12 | |
| ubiX | 1 | 32 | |
| ispA | 1 | 12 | |
| rplL | 1 | 8 | |
| rpoB | 1 | 32 | |
| spoT | 1 | 24 | |
| cysB | 1 | 24 |
For the specific mutations in these genes, see Table S1 in the supplemental material.
Some previously unidentified Amd resistance-encoding genes were also found, i.e., the ubiE, ubiX, and ispA genes, whose products are part of the respiratory chain; ppa, the gene encoding inorganic pyrophosphatase; and the rplL gene, which encodes ribosomal protein L7/L12. The rplL mutation was also associated with a large duplication of 38 kbp that included the rplL gene. To confirm that these genes confer Amd resistance, genetic reconstructions were made to introduce each specific mutation singly into the E. coli MG1655 background by means of linear transformation by λ Red recombineering. The MICs for the reconstructed strains and the original mutants and their growth rates are compared in Table 3 and discussed below.
Whole-genome sequencing of clinical isolates.
To identify potential Amd resistance-encoding mutations in clinical isolates, the whole genomes of 12 Amdr E. coli UTI isolates from a collection of UTI isolates obtained in Sweden from 2005 to 2007 were sequenced and assembled to the E. coli MG1655 genome. Potential Amd resistance-encoding genes in these clinical strains, including those previously discovered, as well as the new ones identified here, were compared to those in 17 E. coli reference genomes. Nonsynonymous point mutations in known Amd resistance-encoding genes that were found in the UTI isolates but not in the reference strains were considered potential Amd resistance-conferring mutations.
All of the Amd resistance-encoding genes are listed in Fig. 1, and all of the mutations found in those genes are listed in Table 4. The occurrence of the known Amd resistance-encoding genes (i.e., those identified in laboratory selections) in the resistant clinical isolates were distributed as follows. (i) Mutations in the lon, mrdA, mrdB, ppa, ubiX, ispA, rplL, ftsA, mreB, mreD, and aroK genes were absent from all of the clinical isolates. (ii) Mutations in the rcsB, rcsC, rcsD, rcsF, yrfF, thrS, gltX, aspS, argS, alaS, ubiE, spoT, cysE, ftsQ, ftsZ, mreC, cyaA, crp, galE, rpoB, and slt genes were present in a few strains (one to five). (iii) Mutations in cysB were found in all of the clinical isolates (Table 4). In summary, genes involved in cell division and elongation (mrdAB, ftsQAZ, and mreBCD) or cell respiration (ubiEX, ispA), the Rcs genes (rcsBCDF and yrfF), and those encoding the tRNA synthetases were mutated in a few clinical isolates, whereas the cysB gene was mutated in all of them, suggesting that cysB is of particular importance in generating Amd resistance in clinical E. coli isolates. Furthermore, five additional Amdr clinical isolates obtained from several European countries had a mutated cysB gene (as shown by localized sequencing of the cysB genes); thus, all 17 of the clinical Amdr isolates had cysB mutations. The cysB mutations were of different types; many were point mutations leading to either an amino acid change or a stop codon, several were frame shifts, and there was also one deletion and one IS1 insertion (Table 4).
FIG 1.

Putative Amd resistance-encoding genes in clinical strains. Genes marked by gray shading have a mutation that is not present in any of the reference genomes. Specific mutations are listed in Table 4.
Amd resistance is caused by cysB mutations that prevent production of cysteine.
To confirm that the cysB mutations identified confer Amd resistance, several tests were performed. First, a cysB knockout mutation was constructed and introduced into laboratory strain MG1655. Inactivation of cysB conferred a Amdr phenotype where the cysB knockout mutant for which the MIC was 24 mg/liter (compared to 0.125 for the cysB+ strain), showing that inactivation of the CysB protein causes resistance. This had previously been shown in S. enterica serovar Typhimurium (31). Second, cysteine auxotrophy screening of some 40 Amdr E. coli strains originating from MG1655 in the initial selection of Amdr mutants was done with M9 glucose and M9 glucose-cysteine plates. One strain, DA24053, was a cysteine auxotroph. The cysB gene was PCR amplified and sequenced, showing that the strain had a deletion of bp 148 in cysB. The Amd MIC for this strain was 24 mg/liter, which is identical to that for the cysB knockout strain.
Third, to further demonstrate the role of the cysB mutations in resistance in the clinical isolates, the mutated cysB genes from a subset of the clinical Amdr strains (DA14710, DA14713, DA14718, and DA24686) were genetically transferred into the E. coli MG1655 background by recombineering (generating strains DA34895, DA34896, DA34896, and DA34897, respectively). The effects of these mutations on the MIC and fitness were measured by Etest and growth rate determination. The MICs conferred by the different mutated cysB genes were 12 mg/liter for DA34897, 24 mg/liter for DA34896 and DA34898, and 32 mg/liter for DA34895, demonstrating that these cysB mutations are sufficient to confer high-level Amd resistance. The cysB mutations in strains DA34895 and DA34898 imposed severe fitness costs, with relative growth rates of 0.58 and 0.65, respectively. The fitness costs of the cysB mutations in strains DA34896 and DA34897 were lower, with both strains having a relative growth rate of about 0.9. Finally, to determine if the cysB mutant phenotype is recessive or dominant, we complemented a subset of the clinical cysB mutants (strains DA14710, DA14713, DA14718, DA14719, and DA24686) with a plasmid carrying the functional cysB+ gene from MG1655. Etests of the resulting strains (DA37700 to DA37713) showed that the Amd MIC for all of them was reduced to the MIC for MG1655 (0.125 mg/liter), compared to the between 16 and 64 mg/liter determined for these strains before plasmid introduction. When cysB knockout strain DA28439 was also transformed with the plasmid, a similar result was obtained; the MIC was reduced from 24 to 0.125 mg/liter.
To determine if it was the absence of cysteine or the CysB protein itself that conferred the resistance phenotype, we examined how addition of cysteine influenced the Amd resistance of the clinical isolates and the cysB knockout mutant. The Mecr clinical isolates and the cysB knockout strain (DA28439) were grown on M9 plates with or without 0.3 mM cysteine to screen for cysteine auxotrophy. The cysB knockout and the clinical strains could grow on minimal plates only when they were supplemented with cysteine (Table 5). One clinical strain grew on minimal medium neither without nor with cysteine and was assumed to have another, unknown, auxotrophy. The Amd MICs on MH medium supplemented with two different concentrations of cysteine (0.3 and 0.75 mM) were also measured for the clinical isolates and the cysB knockout. When supplemented with cysteine, these strains lost most of their resistance to Amd (Table 5); a similar result has previously been obtained with S. enterica serovar Typhimurium (31). In fact, at the higher concentration, the MICs for all of the clinical isolates were in the range of the E. coli MG1655 MIC and well below the clinical breakpoint for Amd (8 mg/liter). This result demonstrates that it is the lack of cysteine, and not the absence of the CysB protein, that confers Amd resistance.
TABLE 5.
Effects of cysteine addition on Amd resistance of clinical isolatesa
| Strain | Relevant phenotype | Growth on MM |
Amd MIC (mg/liter) |
|||
|---|---|---|---|---|---|---|
| M9 | M9 + Cys | No Cys | 0.3 mM Cys | 0.75 mM Cys | ||
| DA5438 | Cys+ | Yes | Yes | 0.125 | 0.125 | 0.125 |
| DA28439 | Cys−b | No | Yes | 24 | 2 | 0.25 |
| DA14704 | Cys− | No | Yes | 32 | 0.125 | 0.125 |
| DA14709 | Cys− | No | Yes | 4 | 2 | 0.38 |
| DA14710 | Cys− | No | Yes | 64 | 8 | 0.125 |
| DA14712 | Cys− | No | Yes | 32 | 16 | 0.5 |
| DA14713 | Cys− | No | No | 64 | 4 | 0.125 |
| DA14715 | Cys− | No | Yes | 16 | 2 | 0.19 |
| DA14718 | Cys− | No | Yes | 32 | 8 | 0.38 |
| DA14719 | Cys− | No | Yes | 16 | 0.5 | 0.094 |
| DA24678 | Cys− | No | Yes | 48 | 16 | 0.38 |
| DA24686 | Cys− | No | Yes | 32 | 16 | 0.19 |
| DA24690 | Cys− | No | Yes | 32 | 24 | 0.125 |
| DA24692 | Cys− | No | Yes | 48 | 6 | 0.19 |
Shown are growth on M9 glucose minimal medium (MM) with or without cysteine added and Amd Etest MICs on MH plates with different concentrations of cysteine. DA5438 is E. coli MG1655, DA28439 is a constructed cysB knockout strain, and all of the other strains are Amdr clinical E. coli isolates with cysB mutations.
cysB knockout.
Fitness of Amdr strains.
Fitness was measured as the growth rate relative to that of E. coli MG1655 by determining OD600 over time in the Bioscreen analyzer. The change in OD at exponential growth was recalculated to obtain the growth rate. In MH medium without Amd, all of the resistant mutants isolated in the laboratory were less fit than MG1655; the relative growth rates ranged from 0.25 to 0.7 (Fig. 2). The Amdr E. coli UTI isolates also had lower fitness in MH medium than MG1655 did, with relative growth rates ranging from 0.52 to 0.83 (Fig. 3). The clinical strains had significantly higher growth rates in MH medium than the E. coli laboratory isolates (t test, P = 6 × 10−6). In urine, however, all of the laboratory-isolated Amdr mutants had lower fitness than MG1655 (relative growth rates between 0.26 and 0.81), but all of the clinical isolates had significantly higher fitness than MG1655 (relative growth rates between 1.22 and 1.7) (Fig. 3). The clinical strains also had significantly higher growth rates in urine than the E. coli laboratory isolates (t test, P < 2 × 10−16).
FIG 2.
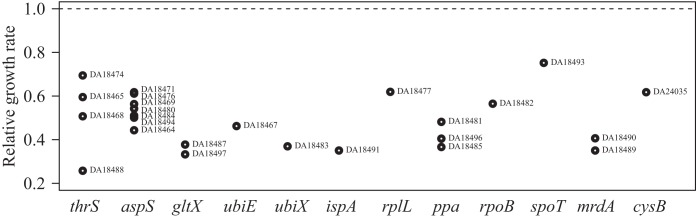
Fitness levels (relative growth rates) of laboratory-selected E. coli mutants, where E. coli MG1655 is set to 1.0. Strain names and mutated genes are shown.
FIG 3.
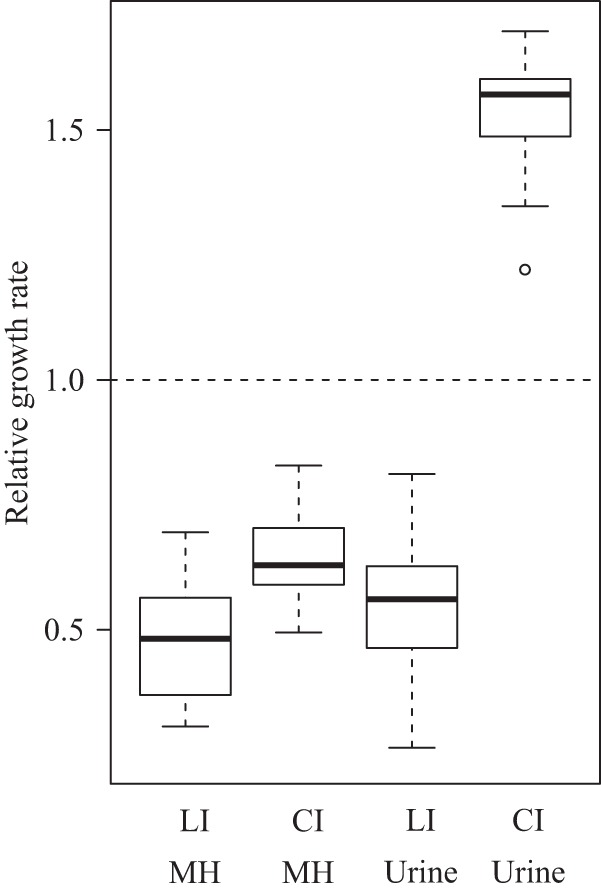
Box plot of fitness of laboratory-selected E. coli isolates (LI) and clinical isolates (CI) in MH medium and urine, showing (from top to bottom) the maximum fitness value, the upper quartile, the median, the lower quartile, and the minimum fitness value. A single outlier is shown as a circle. Fitness was measured as the relative growth rate, where MG1655 strain DA5438 was set to 1.0. t tests showed significant differences in fitness between the E. coli laboratory-isolated mutants and the clinical isolates in both MH medium (P = 6 × 10−6) and urine (P < 10−16).
We determined how the growth of subsets of Amdr clinical isolates and reconstructed mutant strains (DA5438, DA14719, DA24678, DA24686, DA28432, DA28434, DA28436, DA28438, DA28439, DA29705, and DA29712) was affected by increased concentrations of Amd (Fig. 4). All of the resistant mutants had severely reduced growth rates at increased Amd concentrations, and it is notable that none of the resistant isolates could grow at concentrations of >300 mg/liter.
FIG 4.

Relative growth rates of Amdr reconstructed mutants and clinical isolates as a function of the Amd concentration. DA5438 is E. coli MG1655 (with a Amd MIC of 0.125 mg/liter). DA14719 (Amd MIC, 16 mg/liter), DA24678 (Amd MIC, 48 mg/liter), and DA24686 (Amd MIC, 32 mg/liter) are Amdr E. coli clinical isolates. DA28432 (Amd MIC, 96 mg/liter; ppa [D11A]), DA28434 (Amd MIC, 24 mg/liter; aspS [P191S]), DA28436 (Amd MIC, 12 mg/liter; ubiE [deletion of bp 346 to 349 → frame shift]), DA28438 (Amd MIC, 32 mg/liter; spoT [S305P]), DA28439 (Amd MIC, 32 mg/liter; cysB knockout), DA29705 (Amd MIC, 16 mg/liter; mrdA [D389G]), and DA29712 (Amd MIC, 48 mg/liter; rpoB [H447L]) are reconstructed E. coli Amdr mutants.
Cross-resistance in reconstructed E. coli mutants.
The Amdr reconstructed E. coli mutants were examined for cross-resistance to other antibiotics by Etest (Table 6). The antibiotics tested were three other β-lactams (meropenem, cefotaxime, and Amp), five antibiotics that are commonly used to treat UTIs (ciprofloxacin, fosfomycin, nitrofurantoin, trimethoprim, and trimethoprim-sulfamethoxazole), and four other antibiotic groups (erythromycin, Cam, Tet, and nalidixic acid). No large differences from E. coli MG1655 were seen, except for erythromycin, where several Amdr mutants showed higher MICs than MG1655 and some few strain-antibiotic combinations showed a lower MIC than MG1655 (e.g., DA28436 and nitrofurantoin, DA28439 and Tet, and DA29712 and fosfomycin).
TABLE 6.
MICs of antibioticsa for reconstructed Amdr mutants and E. coli MG1655
| Strain | Genotype | MIC (mg/liter) of: |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amd | Mem | Ctx | Amp | Cip | Nit | Fof | Tmp | Sxt | Chl | Nal | Tet | Ery | ||
| DA5438b | Wild type | 0.125 | 0.023 | 0.064 | 2 | 0.016 | 2 | 0.5 | 0.125 | 0.047 | 6 | 3 | 1 | 16 |
| DA28432 | ppa (D11A) | 96 | 0.032 | 0.047 | 0.75 | 0.016 | 3 | 0.19 | 0.94 | 0.032 | 4 | 0.5 | 0.75 | 198 |
| DA28434 | aspS (P191S) | 24 | 0.047 | 0.047 | 2 | 0.016 | 3 | 0.094 | 0.125 | 0.032 | 6 | 3 | 0.5 | 64 |
| DA28436 | ubiE (delc bp 346–349 → FSd) | 12 | 0.023 | 0.032 | 1 | 0.016 | 2 | 0.094 | 0.75 | 0.064 | 1.5 | 3 | 0.5 | 64 |
| DA28438 | spoT (S305P) | 32 | 0.032 | 0.064 | 1.5 | 0.016 | 0.25 | 0.19 | 0.125 | 0.023 | 3 | 2 | 0.094 | 32 |
| DA28439 | cysB KOe | 32 | 0.064 | 0.047 | 2 | 0.023 | 1 | 0.5 | 0.38 | 0.064 | 4 | 3 | 0.75 | >256 |
| DA29703 | thrS (E519G) | 32 | 0.032 | 0.064 | 2 | 0.023 | 4 | 1 | 0.25 | 0.047 | 6 | 3 | 0.75 | 24 |
| DA29705 | mrdA (D389G) | 16 | 0.023 | 0.023 | 0.75 | 0.016 | 1.5 | 0.38 | 0.94 | 0.016 | 6 | 3 | 0.5 | 24 |
| DA29707 | ubiX (del bp 246 → FS) | 32 | 0.047 | 0.032 | 1 | 0.023 | 1 | 0.094 | 0.25 | 0.047 | 3 | 3 | 1 | 96 |
| DA29710 | rplL (promoter −10) | 48 | 0.032 | 0.047 | 0.5 | 0.023 | 2 | 0.125 | 0.125 | 0.032 | 4 | 3 | 0.75 | 24 |
| DA29712 | rpoB (H447L) | 48 | 0.047 | 0.064 | 2 | 0.023 | 4 | 0.064 | 0.094 | 0.047 | 4 | 3 | 0.75 | 16 |
| DA29715 | trxB (G37D) | 0.064 | 0.023 | 0.047 | 2 | 0.016 | 6 | 1 | 0.19 | 0.047 | 4 | 3 | 1 | 24 |
| DA29717 | mrdA (D389G) trxB (G37D) | 64 | 0.016 | 0.023 | 0.75 | 0.023 | 2 | 0.19 | 0.38 | 0.032 | 6 | 6 | 0.5 | 48 |
| DA30120 | gltX (D180G) | 24 | 0.047 | 0.032 | 1 | 0.016 | 0.75 | 0.25 | 0.094 | 0.032 | 4 | 2 | 0.5 | 96 |
| DA30122 | ispA (IS1 insertion) | 16 | 0.032 | 0.023 | 1 | 0.016 | 2 | 0.094 | 0.125 | 0.032 | 1 | 2 | 0.38 | 96 |
Mem, meropenem; Ctx, cefotaxime; Cip, ciprofloxacin; Nit, nitrofurantoin; Fof, fosfomycin; Tmp, trimethoprim; Sxt, trimethoprim-sulfamethoxazole; Nal, nalidixic acid; Ery, erythromycin.
E. coli MG1655.
del, deletion.
FS, frame shift.
KO, knockout.
DISCUSSION
Amd resistance is rapidly acquired by bacteria in laboratory selections but appears rare in clinical isolates (15, 16, 49). Prior to this work, 30 genes that can confer Amd resistance had been found (Table 1), and here we identified eight additional genes. Thus, the fact that at least 38 genes can cause Amd resistance when mutated (often when inactivated) explains the high frequency of mutation to resistance in laboratory selections and the expectation that resistance would also be common in clinical settings. Here, we compared clinical isolates and laboratory-selected resistant mutants to identify resistance mutations and seek explanations for the low incidence of resistance in clinical isolates.
New Amd resistance-encoding genes found in laboratory selections.
Many of the mutations that confer Amd resistance cause a ppGpp concentration increase (Table 1) (10, 21–23, 33–37), but the mechanism by which elevated ppGpp levels confer resistance remains unknown. In this study, several new genes with known or potential involvement in ppGpp metabolism were discovered. First, mutations in three genes coding for tRNA synthetases (thrS, aspS, and gltX) conferred resistance. Although these specific tRNA synthetases have not been associated with Amd resistance previously, others have (23), and the resistance caused by these mutations was due to activation of the stringent response and ppGpp synthesis.
Another example of mutants probably owing their resistance to the stringent response is the ppa mutants. The product of ppa is the enzyme inorganic pyrophosphatase, which catalyzes the cleavage of pyrophosphate (PPi) into two phosphate ions (50, 51). Since PPi is a product of the stringent response, it is plausible that Amd resistance conferred by ppa mutations is connected to the stringent response. In addition, PPi is also an inhibitor of both cell division and tRNA synthetases, which are also possible routes for Amd resistance (52).
Additional new Amd resistance mutations found were in the ribosomal gene rplL and cell respiration genes ubiE, ubiX, and ispA, all of which result in slow growth and might induce the stringent response through the pppGpp synthetase SpoT (38). The rplL mutant might also be connected to the stringent response in another, more direct, way. The main pppGpp synthetase RelA binds to the ribosomal protein L11 (38), which interacts with L12 (the product of rplL) via L10, and it is conceivable that an L12 mutation could affect RelA function via L10 and L11 (53).
We also found that a mutation in trxB could increase the level of resistance of an already resistant mrdA mutant, although not conferring any resistance on its own. The gene trxB encodes thioredoxin, an enzyme that is involved in the cysteine biosynthesis pathway (54). Inactivation of this pathway has previously, as well as in this study, been shown to confer Amd resistance (31, 32).
CysB inactivation is the major mechanism of Amd resistance in clinical isolates of E. coli.
We observed two important differences between the clinical and laboratory-isolated Amdr isolates with regard to mutation spectra. First, the spectrum of resistance-causing mutations appeared much narrower in the clinical isolates than in the laboratory-selected isolates, and second, all of the clinical isolates but only one of the laboratory-selected mutants had a mutated cysB gene. We sequenced the whole genomes or performed cysteine auxotrophy screening of 62 mutants, and since cysB is only 1 of 38 known Amd resistance-encoding genes, many of which confer resistance when having loss-of-function mutations, the observation that 1/62 mutants is defective in cysB is close to the expected result.
CysB protein is the major positive regulator of cysteine biosynthesis in Enterobacteriaceae (54), and mutations in the cysteine biosynthesis pathway genes cysB and cysE have previously been shown to confer Amd resistance on laboratory-selected mutants of S. enterica serovar Typhimurium (31, 32). Although the mechanism by which cysB confers resistance has not been fully elucidated, it is at least partly due to increased intracellular levels of ppGpp (32).
Transfer of different cysB mutations (including amino acid substitutions, stop codons, and frame shifts) from clinical isolates into a laboratory strain conferred high-level resistance, demonstrating that Amd resistance in clinical isolates of E. coli is conferred by inactivation of the cysB gene. Furthermore, introduction of a plasmid containing a functional cysB+ gene from strain MG1655 into the clinical strains resulted in loss of Amd resistance, demonstrating that their resistance results from inactivation of the CysB protein and that the resistant mutant phenotype is recessive. When their medium was supplemented with cysteine, the clinical isolates lost their resistance to Amd, indicating that their resistance is due to the absence of cysteine biosynthesis (rather than loss of the CysB activator per se). We also found clinical isolates that carried cysB mutations with even higher MICs (>256 mg/liter) than that conferred by cysB inactivation alone (16 to 32 mg/liter); these mutants are likely to carry additional, unidentified, resistance mutations.
Why has the frequency of Amd resistance among E. coli UTI isolates remained low?
Even though the frequency of mutation to Amd resistance is very high in in vitro selections, resistance development appears to be relatively uncommon in clinical settings. We propose that this is due to two factors. First, the majority of the resistant mutants that are found in laboratory selections grow slowly, reducing their ability to become fixed in the bladder. Thus, because of frequent repeated micturition, bacteria residing in urine need to grow above a certain rate to be stably maintained, at least during planktonic growth (55). In other words, among all of the mutant types that can be found in vitro, only a small subset (including the cysB mutant) are fit enough to be stably maintained in the bladder. In line with this idea, the type of resistance mutation found in clinical isolates, cysB, imposes a relatively low fitness cost whereas many of the other, highly resistant mutants found in the laboratory selections (e.g., mrdA, ppa, ubiX, ispA, ubiE, and some tRNA synthetase mutants) showed severe growth rate reductions. Other factors that reduce the fixation rate of resistant mutants in clinical settings are the facts that all of the Amdr clinical isolates showed strong growth rate reductions in response to increasing Amd levels and that none of the mutants could grow at a concentration of >300 mg/liter (Fig. 4). Since the concentration of Amd in urine often reaches several hundred mg/liter during treatment (1, 8, 9), this suggests that even highly resistant mutants would have problems remaining in the bladder during treatment since their growth rates are reduced below the threshold level needed for stable maintenance (55).
Finally, an important implication of these findings is that the frequency of mutation to resistance in laboratory selections (as it is often used by the pharmaceutical industry and academic researchers to assess the risk of development of resistance to new antibiotics) might sometimes be a misleading predictor of clinical resistance development. That is, even though the frequency of mutation to Amd resistance in laboratory selections can be very high (10−5), resistance development during treatment remains rare in clinical settings. This implies that a high frequency of mutation to resistance in the laboratory need not necessarily prohibit the development of a promising drug candidate. Instead, assessment of mutant fitness in a relevant physiological context might be a more predictive measure of the risk of resistance evolution in real life.
Supplementary Material
ACKNOWLEDGMENTS
We thank master's degree student Parul Singh for characterizing some of the laboratory-selected E. coli mutants, the staff of Gunnar Kahlmeter's laboratory for generously supplying clinical isolates, Erik Gullberg for generously supplying strains used for reconstruction, Erik Lundin for the pEL plasmid, and Måns Thulin for his assistance with the R software.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04819-14.
REFERENCES
- 1.Anonymous. 2005. UVI_rek1-8. Läkemedelsverket Medical Products Agency, Uppsala, Sweden: http://www.lakemedelsverket.se/malgrupp/Halso---sjukvard/Behandlings--rekommendationer/Behandlingsrekommendation---listan/UVI---Nedre-urinvagsinfektion-hos-kvinnor/. [Google Scholar]
- 2.Gupta K, Hooton TM, Naber KG, Wullt B, Colgan R, Miller LG, Moran GJ, Nicolle LE, Raz R, Schaeffer AJ, Soper DE. 2011. International clinical practice guidelines for the treatment of acute uncomplicated cystitis and pyelonephritis in women: a 2010 update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clin Infect Dis 52:e103–e120. doi: 10.1093/cid/ciq257. [DOI] [PubMed] [Google Scholar]
- 3.Lund F, Tybring L. 1972. 6β-Amidinopenicillanic acids—a new group of antibiotics. Nature 236:135–137. [DOI] [PubMed] [Google Scholar]
- 4.Tybring L, Melchior NH. 1975. Mecillinam (FL 1060), a 6beta-amidinopenicillanic acid derivative: bactericidal action and synergy in vitro. Antimicrob Agents Chemother 8:271–276. doi: 10.1128/AAC.8.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicolle LE. 2000. Pivmecillinam in the treatment of urinary tract infections. J Antimicrob Chemother 46(Suppl 1):35–39. doi: 10.1093/jac/46.suppl_1.35. [DOI] [PubMed] [Google Scholar]
- 6.Clarke PD, Geddes AM, McGhie D, Wall JC. 1976. Mecillinam: a new antibiotic for enteric fever. Br Med J ii:14–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gambertoglio JG, Barriere SL, Lin ET, Conte JE Jr. 1980. Pharmacokinetics of mecillinam in health [sic] subjects. Antimicrob Agents Chemother 18:952–956. doi: 10.1128/AAC.18.6.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kerrn MB, Frimodt-Møller N, Espersen F. 2004. Urinary concentrations and urine ex-vivo effect of mecillinam and sulphamethizole. Clin Microbiol Infect 10:54–61. doi: 10.1111/j.1469-0691.2004.00737.x. [DOI] [PubMed] [Google Scholar]
- 9.Sullivan Å, Edlund C, Nord CE. 2001. Effect of antimicrobial agents on the ecological balance of human microflora. Lancet Infect Dis 1:101–114. doi: 10.1016/S1473-3099(01)00066-4. [DOI] [PubMed] [Google Scholar]
- 10.Tamaki S, Matsuzawa H, Matsuhashi M. 1980. Cluster of mrdA and mrdB genes responsible for the rod shape and mecillinam sensitivity of Escherichia coli. J Bacteriol 141:52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spratt BG. 1977. The mechanism of action of mecillinam. J Antimicrob Chemother 3:13–19. doi: 10.1093/jac/3.suppl_B.13. [DOI] [PubMed] [Google Scholar]
- 12.Typas A, Banzhaf M, Gross CA, Vollmer W. 2012. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 10:123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vollmer W, Bertsche U. 2008. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim Biophys Acta 1778:1714–1734. doi: 10.1016/j.bbamem.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 14.Spratt BG. 1975. Distinct penicillin binding proteins involved in the division, elongation, and shape of Escherichia coli K-12. Proc Natl Acad Sci U S A 72:2999–3003. doi: 10.1073/pnas.72.8.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kahlmeter G. 2003. An international survey of the antimicrobial susceptibility of pathogens from uncomplicated urinary tract infections: the ECO. SENS project. J Antimicrob Chemother 51:69–76. doi: 10.1093/jac/dkg028. [DOI] [PubMed] [Google Scholar]
- 16.Kahlmeter G, Poulsen HO. 2012. Antimicrobial susceptibility of Escherichia coli from community-acquired urinary tract infections in Europe: the ECO.SENS study revisited. Int J Antimicrob Agents 39:45–51. doi: 10.1016/j.ijantimicag.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 17.Naber KG, Schito G, Botto H, Palou J, Mazzei T. 2008. Surveillance study in Europe and Brazil on clinical aspects and antimicrobial resistance epidemiology in females with cystitis (ARESC): implications for empiric therapy. Eur Urol 54:1164–1178. doi: 10.1016/j.eururo.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 18.Wachi M, Doi M, Tamaki S, Park W, Nakajima-Iijima S, Matsuhashi M. 1987. Mutant isolation and molecular cloning of mre genes, which determine cell shape, sensitivity to mecillinam, and amount of penicillin-binding proteins in Escherichia coli. J Bacteriol 169:4935–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwaya M, Jones CW, Khorana J, Strominger JL. 1978. Mapping of the mecillinam-resistant, round morphological mutants of Escherichia coli. J Bacteriol 133:196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuzawa H, Asoh S, Kunai K, Muraiso K, Takasuga A, Ohta T. 1989. Nucleotide sequence of the rodA gene, responsible for the rod shape of Escherichia coli: rodA and the pbpA gene, encoding penicillin-binding protein 2, constitute the rodA operon. J Bacteriol 171:558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vinella D, Joseleau-Petit D, Thévenet D, Bouloc P, D'Ari R. 1993. Penicillin-binding protein 2 inactivation in Escherichia coli results in cell division inhibition, which is relieved by FtsZ overexpression. J Bacteriol 175:6704–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vinella D, Cashel M, D'Ari R. 2000. Selected amplification of the cell division genes ftsQ-ftsA-ftsZ in Escherichia coli. Genetics 156:1483–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vinella D, D'Ari R, Jaffe A, Bouloc P. 1992. Penicillin binding protein 2 is dispensable in Escherichia coli when ppGpp synthesis is induced. EMBO J 11:1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouloc P, Vinella D, D'Ari R. 1992. Leucine and serine induce mecillinam resistance in Escherichia coli. Mol Gen Genet 235:242–246. doi: 10.1007/BF00279366. [DOI] [PubMed] [Google Scholar]
- 25.Jovanovic M, Lilic M, Janjusevic R, Jovanovic G, Savic DJ, Milija J. 1999. tRNA synthetase mutants of Escherichia coli K-12 are resistant to the gyrase inhibitor novobiocin. J Bacteriol 181:2979–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aono R, Yamasaki M, Tamura G. 1979. High and selective resistance to mecillinam in adenylate cyclase-deficient or cyclic adenosine 3′,5′-monophosphate receptor protein-deficient mutants of Escherichia coli. J Bacteriol 137:839–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D'Ari R, Jaffe A, Bouloc P, Robin A. 1988. Cyclic AMP and cell division in Escherichia coli. J Bacteriol 170:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antón DN. 1995. Resistance to mecillinam produced by the cooperative action of mutations affecting iipopolysaccharide, spoT, and cya or crp genes of Salmonella typhimurium. Mol Microbiol 16:587–595. doi: 10.1111/j.1365-2958.1995.tb02421.x. [DOI] [PubMed] [Google Scholar]
- 29.Costa CS, Antón DN. 2001. Role of the ftsA1p promoter in the resistance of mucoid mutants of Salmonella enterica to mecillinam: characterization of a new type of mucoid mutant. FEMS Microbiol Lett 200:201–205. doi: 10.1111/j.1574-6968.2001.tb10716.x. [DOI] [PubMed] [Google Scholar]
- 30.Laubacher ME, Ades SE. 2008. The Rcs phosphorelay is a cell envelope stress response activated by peptidoglycan stress and contributes to intrinsic antibiotic resistance. J Bacteriol 190:2065–2074. doi: 10.1128/JB.01740-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oppezzo OJ, Antón DN. 1995. Involvement of cysB and cysE genes in the sensitivity of Salmonella typhimurium to mecillinam. J Bacteriol 177:4524–4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Costa CS, Antón DN. 2006. High-level resistance to mecillinam produced by inactivation of soluble lytic transglycosylase in Salmonella enterica serovar Typhimurium. FEMS Microbiol Lett 256:311–317. doi: 10.1111/j.1574-6968.2006.00133.x. [DOI] [PubMed] [Google Scholar]
- 33.Joseleau-Petit D, Thévenet D, D'Arl R. 1994. ppGpp concentration, growth without PBP2 activity, and growth-rate control in Escherichia coli. Mol Microbiol 13:911–917. doi: 10.1111/j.1365-2958.1994.tb00482.x. [DOI] [PubMed] [Google Scholar]
- 34.Vinella D, Gagny B, Joseleau-Petit D, D'Ari R, Cashel M. 1996. Mecillinam resistance in Escherichia coli is conferred by loss of a second activity of the AroK protein. J Bacteriol 178:3818–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vinella D, D'Ari R. 1994. Thermoinducible filamentation in Escherichia coli due to an altered RNA polymerase beta subunit is suppressed by high levels of ppGpp. J Bacteriol 176:966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Navarro F, Robin A, D'Ari R, Joseleau-Petit D. 1998. Analysis of the effect of ppGpp on the ftsQAZ operon in Escherichia coli. Mol Microbiol 29:815–823. doi: 10.1046/j.1365-2958.1998.00974.x. [DOI] [PubMed] [Google Scholar]
- 37.D'Ari R. 1997. The Escherichia coli cell cycle, cell division and ppGpp: regulation and mechanisms. Folia Microbiol (Praha) 42:161–164. doi: 10.1007/BF02818972. [DOI] [PubMed] [Google Scholar]
- 38.Magnusson LU, Farewell A, Nyström T. 2005. ppGpp: a global regulator in Escherichia coli. Trends Microbiol 13:236–242. doi: 10.1016/j.tim.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 39.Pizarro-Cerdá J, Tedin K. 2004. The bacterial signal molecule, ppGpp, regulates Salmonella virulence gene expression. Mol Microbiol 52:1827–1844. doi: 10.1111/j.1365-2958.2004.04122.x. [DOI] [PubMed] [Google Scholar]
- 40.Lea DE, Coulson CA. 1949. The distribution of the numbers of mutants in bacterial populations. J Genet 49:264–285. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- 41.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Datta S, Costantino N, Court DL. 2006. A set of recombineering plasmids for Gram-negative bacteria. Gene 379:109–115. doi: 10.1016/j.gene.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 43.Koskiniemi S, Pränting M, Gullberg E, Näsvall J, Andersson DI. 2011. Activation of cryptic aminoglycoside resistance in Salmonella enterica. Mol Microbiol 80:1464–1478. doi: 10.1111/j.1365-2958.2011.07657.x. [DOI] [PubMed] [Google Scholar]
- 44.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. doi: 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- 45.Kelly JR, Rubin AJ, Davis JH, Ajo-Franklin CM, Cumbers J, Czar MJ, de Mora K, Glieberman AL, Monie DD, Endy D. 2009. Measuring the activity of BioBrick promoters using an in vivo reference standard. J Biol Eng 3:4. doi: 10.1186/1754-1611-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ried JL, Collmer A. 1987. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in Gram-negative bacteria by marker exchange-eviction mutagenesis. Gene 57:239–246. doi: 10.1016/0378-1119(87)90127-2. [DOI] [PubMed] [Google Scholar]
- 47.Shetty RP, Endy D, Knight TF. 2008. Engineering BioBrick vectors from BioBrick parts. J Biol Eng 2:5. doi: 10.1186/1754-1611-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.R Development Core Team. 2012. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 49.Hellman J, Norman C, Olsson-Liljequist B. 2010. SWEDRES 2010. A report on Swedish antimicrobial utilisation and resistance in human medicine. Swedish Institute for Communicable Disease Control, Solna, Sweden: http://www.strama.se/uploads/docs/Swedres%202010%20final.pdf. [Google Scholar]
- 50.Chen J, Brevet A, Fromant M, Lévêque F, Schmitter JM, Blanquet S, Plateau P. 1990. Pyrophosphatase is essential for growth of Escherichia coli. J Bacteriol 172:5686–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Madigan MT, Martinko JM, Dunlap PV, Clark DP. 2007. Brock biology of microorganisms, 12th ed Benjamin Cummings, San Francisco, CA. [Google Scholar]
- 52.Lahti R. 1983. Microbial inorganic pyrophosphatases. Microbiol Rev 47:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao H, Sengupta J, Valle M, Korostelev A, Eswar N, Stagg SM, Van Roey P, Agrawal RK, Harvey SC, Sali A, Chapman MS, Frank J. 2003. Study of the structural dynamics of the E. coli 70S ribosome using real-space refinement. Cell 113:789–801. doi: 10.1016/S0092-8674(03)00427-6. [DOI] [PubMed] [Google Scholar]
- 54.Kredich NM. 1996. Biosynthesis of cysteine, p 514–527. In Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed, vol 1 ASM Press, Washington, DC. [Google Scholar]
- 55.Gordon DM, Riley MA. 1992. A theoretical and experimental analysis of bacterial growth in the bladder. Mol Microbiol 6:555–562. doi: 10.1111/j.1365-2958.1992.tb01500.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.