

Abstract
New drugs to treat drug-resistant tuberculosis are urgently needed. Extensively drug-resistant and probably the totally drug-resistant tuberculosis strains are resistant to fluoroquinolones like moxifloxacin, which target gyrase A, and most people infected with these strains die within a year. In this study, we found that a novel aminobenzimidazole, VXc-486, which targets gyrase B, potently inhibits multiple drug-sensitive isolates and drug-resistant isolates of Mycobacterium tuberculosis in vitro (MICs of 0.03 to 0.30 μg/ml and 0.08 to 5.48 μg/ml, respectively) and reduces mycobacterial burdens in lungs of infected mice in vivo. VXc-486 is active against drug-resistant isolates, has bactericidal activity, and kills intracellular and dormant M. tuberculosis bacteria in a low-oxygen environment. Furthermore, we found that VXc-486 inhibits the growth of multiple strains of Mycobacterium abscessus, Mycobacterium avium complex, and Mycobacterium kansasii (MICs of 0.1 to 2.0 μg/ml), as well as that of several strains of Nocardia spp. (MICs of 0.1 to 1.0 μg/ml). We made a direct comparison of the parent compound VXc-486 and a phosphate prodrug of VXc-486 and showed that the prodrug of VXc-486 had more potent killing of M. tuberculosis than did VXc-486 in vivo. In combination with other antimycobacterial drugs, the prodrug of VXc-486 sterilized M. tuberculosis infection when combined with rifapentine-pyrazinamide and bedaquiline-pyrazinamide in a relapse infection study in mice. Furthermore, the prodrug of VXc-486 appeared to perform at least as well as the gyrase A inhibitor moxifloxacin. These findings warrant further development of the prodrug of VXc-486 for the treatment of tuberculosis and nontuberculosis mycobacterial infections.
INTRODUCTION
Tuberculosis (TB) is an important infectious disease threat, with approximately 2 billion infections (mostly latent), 10 million new cases, and 2 million deaths each year (1). HIV coinfection/immunosuppression, ineffective health care management, complex standard-of-care (SOC) regimens, and widespread drug resistance all contribute to the challenge of controlling TB. TB is caused by Mycobacterium tuberculosis, a slow-growing, acid-fast bacillus that withstands a harsh immunological assault by human host macrophages and effector cells, as well as suboptimal chemotherapy, by persisting in a semidormant state of replication. New drugs are urgently needed to shorten the treatment regimen and to more effectively treat drug-sensitive (DS) and, especially, drug-resistant (DR) M. tuberculosis infections.
DNA gyrase and topoisomerase IV are two clinically validated drug targets for bacterial infections. They are highly conserved type II topoisomerases that are essential for DNA replication. Both targets are enzymes with A2B2 heterotetramers, comprising the GyrA and GyrB subunits (DNA gyrase) and the ParC and ParE subunits (topoisomerase IV), respectively; however, a topoisomerase IV homolog has not been identified in M. tuberculosis. In an enzymatic reaction that is coupled with ATP hydrolysis, these enzymes break and rejoin double-stranded DNA. The catalytic subunits (GyrA/ParC) are clinically validated drug targets of the fluoroquinolones, such as moxifloxacin (MXF) (2), while the ATPase subunits (GyrB/ParE) have not been as extensively exploited and may present a new option for treating DR strains of M. tuberculosis (3). Furthermore, several different chemical classes have been described as inhibitors of gyrase B with potent activity against DR bacteria, including M. tuberculosis (4–10).
A novel class of antimicrobials that target the ATPase subunits (GyrB/ParE) are the aminobenzimidazoles, which were optimized using structure-guided design and structure-activity relationship (SAR) studies of potency against both Gram-positive and some Gram-negative bacterial species (11, 12). Further optimization of the metabolic profile led to the identification of VXc-486 (13), and its solubility was later improved by using a phosphate prodrug approach (O'Dowd DS, Chandupatla HK, Bennani Y, Engtrakul J, Ye Z, Yeola S, Liao S, Ewing N, Jones P, Tsao H, Kolaczkowski E, Donahue S, Seliniotakis R, Bao N, Tsai A, Shawgo R, Dixit V, Jones S, McNeil CF, Song B, Macikenas D, Grillot A-L, Charifson P, unpublished data). The purpose of this study was to explore the utility of VXc-486 and its phosphate prodrug (pVXc-486) against M. tuberculosis and nontuberculosis mycobacteria (NTM) in vitro and in M. tuberculosis mouse models in vivo.
MATERIALS AND METHODS
Mycobacterium species isolates.
M. tuberculosis ATCC 35801 (strain Erdman), ATCC 25618 (strain H37Rv), or ATCC 25177 (strain H37Ra) colonies were used for routine MIC analysis. The BK 35 (H37Rv) strain harbors the gyrase A G94A mutation (a G-to-A change at position 94), while the BK49 strain harbors the gyrase A A90V mutation, and both were kindly provided by Nicolas Veziris of the Centre National de Référence des Mycobactéries et de la Résistance des Mycobactéries aux Antituberculeux, CHU Pitié-Salpêtrière, Paris, France. Fluoroquinolone-resistant variants 2D, 2J, and 8D of M. tuberculosis Lvxr were selected by plating M. tuberculosis Erdman on 7H10 agar containing 10% oleic acid-albumin-dextrose-catalase (OADC) and levofloxacin (LVX) at the Central New York Research Corporation. Several colonies were selected, grown in 7H10 broth with 10% OADC, tested for LVX resistance, and frozen at −70°C. The M. tuberculosis isolate Lvxr Gatr 2C was subsequently cultured on 7H10 agar containing gatifloxacin (GAT) to further enhance its quinolone resistance, and the following fluoroquinolone isolates were recovered: 2I1, 8D2, 2C, and 2H2. The extensively drug-resistant (XDR) isolate XDR 5 was provided by Tommie Victor, the TT135, R179, R884, X_3, X_27, X_60, X_61, X_131, and TT149 isolates were provided by Paul Van Helden of Stellenbosch University (14), and the CDC1551, GN9, HH9, H13571, MC19062, H10460, Wg565, LL, W10, 210, W33, C913, AH517, Bw9, and AH13 isolates were provided by Barry Kreiswirth of the Public Health Research Institute, Newark, NJ. Mycobacterium abscessus isolates were provided by Barbara Brown-Elliot of UT Health Northeast, Tyler, TX (6153, 6025, 6005, 5908, 6142, 5931, 5605, 5901, 5812, 5960, 5922, 6111, and 6126), Barbara Body of Lab Corp., Burlington, NC (BB1 to BB9), and the Centers for Disease Control, Atlanta, GA (LT949). Mycobacterium kansasii isolates were obtained from the ATCC (35755 and 12478), Barbara Body (0008, 2242, 4302, 5075, 5983, 1673, 1701, 5076, 2610, 1295, 0009, and 0164), and Betty-Ann Forbes of State University of New York, Syracuse, NY (W5219, PIC, RSL, SHP, 379, 258, and 399). The Mycobacterium avium Far isolate was provided by Betty-Ann Forbes, and the 103 and 3404.4 isolates were provided by Leonid Heifets of National Jewish Hospital, Denver, CO, while Nocardia isolates were provided by Betty-Ann Forbes.
Compounds.
VXc-486 and pVXc-486 were prepared by Vertex Pharmaceuticals Incorporated according to published methods, and other compounds or drugs were either purchased commercially or prepared by published methods. All compounds were dissolved in 100% dimethyl sulfoxide (DMSO) and stored as frozen stocks at a concentration of 10 mM or 1 mg/ml. Before evaluation in vitro, compounds were diluted into DMSO that did not exceed 1% and then resuspended into culture medium consisting of 7H10 broth supplemented with 10% OADC (BBL Microbiology Systems, Cockeysville, MD) and 0.05% Tween 80.
Enzyme assay.
The enzyme assay was performed as previously described (15). The fully assembled heterotetrameric gyrase A2B2 (after the expression of each separate subunit and subsequent reconstitution prior to assay) was used for inhibitor characterization. The M. tuberculosis gyrA (Rv0006) and gyrB genes (Rv0005) were cloned into a pET28b.1 vector, expressed as recombinant proteins, and purified by a method similar to that previously described for the Escherichia coli enzymes. Enzymatic hydrolysis of ATP to ADP was coupled to the conversion of NADH to NAD+. The decrease in NADH absorbance was monitored at 340 nm for 20 min with a microtiter plate reader (Molecular Devices, Sunnyvale, CA).
Growth inhibition and MICs in broth culture.
For microdilution MIC assays, the M. tuberculosis strains were routinely cultured on 7H11 agar, picked, and prepared at 1 × 108 CFU/ml and then diluted 200 times to a final concentration of 5 × 105 CFU/ml in 7H9 broth supplemented with 10% ADC. Compound stock solutions were prepared in DMSO at a 100-fold dilution for each test concentration, and 1 μl of each stock was dispensed into a microtiter well. Compounds were diluted by the addition of 100 μl of the bacterial cell suspension in culture medium. The 96-well plates were then incubated at 37°C in ambient air. For the bactericidal assay, both VXc-486 and MXF were used at 4-fold MICs: VXc-486 was used at a concentration of 0.24 μg/ml, and MXF was used at a concentration of 0.12 μg/ml.
For MIC determination using the M. tuberculosis H37Ra isolate, 30 μl of resazurin detection buffer was added into each well and the plate was returned to the incubator (16). After 24 h of incubation with resazurin, the fluorescence was read using a Biotek Synergy2 at a gain of 35 with an excitation wavelength of 492 nm and an emission wavelength of 595 nm. For MICs of other isolates, wells were visually inspected after 9 days of incubation. The MIC was defined as the lowest concentration of a compound that inhibited the reduction of resazurin to its fluorescent (emission at 595 nm) species by ≥90%. For the low-oxygen-recovery assay (LORA), each compound's growth inhibition against strain H37Rv was evaluated as previously described (17). For M. kansasii testing, Middlebrook 7H9 broth, pH 6.6, supplemented with 10% Middlebrook albumin-dextrose-catalase enrichment was used. M. avium was tested in 7H10 broth supplemented with 10% OADC, while for M. abscessus and Nocardia sp. testing, Mueller-Hinton broth was utilized.
For drug-resistant strains from South Africa, mycobacterial growth was measured by using mycobacterial growth indicator tubes (MGIT). Mycobacterial inocula were prepared from cultures of all strains grown on Lowenstein Jensen (LJ) slants. Cell suspensions were prepared in saline and the turbidity adjusted to 0.5 McFarland units. A 1:5 dilution of the bacterial suspension was prepared, and 0.5 ml of the suspension was inoculated into MGIT tubes containing test and control compounds. For mycobacterial growth evaluation, the MGIT 960 system (Becton Dickinson, Sparks, MD) was used, where M. tuberculosis growth is observed through fluorescent changes due to oxygen consumption during mycobacterial growth (18, 19). One-tenth milliliter of serially diluted compound was added to the MGIT tube containing 7H9 culture medium, with the final DMSO concentration not exceeding 1.2%. Incubation at 37°C was continued in the MGIT system, and the growth units (GU) were monitored daily. For MIC99 evaluations, a 1% bacterial control culture was prepared in a drug-free MGIT tube and the MIC99 of the compound determined relative to the growth units of the control (GU = 400). When the GU of the growth control were 400 and the GU of the drug-containing tube were more than 100, the results were defined as showing resistance, and when the GU of the drug-containing tube were equal to or less than 100, the results were considered to show susceptibility.
Growth inhibition using M. tuberculosis-infected macrophage cultures.
THP-1 stocks were maintained at a culture density of between 2 × 105 and 8 × 105 cells/ml in RPMI 1640 medium (with phenol red, 25 mM HEPES, and 2 mM l-glutamine; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco) and 0.05 mM β-mercaptoethanol (Invitrogen) in 96-well tissue culture plates (Costar 3903; Corning). A cell suspension of sonicated M. tuberculosis H37Ra expressing firefly luciferase (8 × 105 cells/ml) in RPMI 1640 cell culture medium was used to infect phorbol myristate acetate (PMA)-differentiated (100 nM; Sigma Chemicals) THP-1 cells at a multiplicity of infection (MOI) of 2:1 for 2 h at 37°C. The supernatant, containing uningested M. tuberculosis cells, was then carefully removed from each well, and the M. tuberculosis-infected THP-1 cells were replenished with 100 μl of fresh cell culture medium. The cells were then washed a second time and replaced with 50 μl of fresh medium and 50 μl of medium containing test compounds. After 5 days of incubation, 100 μl of Steady-Glo (Promega) reagent was added to each well, the plates were incubated for 15 min at room temperature (RT) and covered with an adhesive top seal, and the luminescence was read, using a Biotek Synergy2 at a gain of 165, at maximum integration time. THP-1 viability was determined by using Celltiter-Glo according to the manufacturer's instructions (Promega). Synergistic (fractional inhibitory concentration [FIC] of ≤0.5), additive (FIC of 0.5 to 4.0), or interfering (FIC of >4.0) effects of compound combinations were determined by calculating the FICs as previously described (20, 21), i.e., FIC = MIC of compound A with B/MIC of compound A alone + MIC of compound B with A/MIC of compound B alone.
Selection of VXc-486-resistant clones.
Four VXc-486-resistant clones were selected at 2× and 4× MICs by using 10e8 to 10e9 CFU of the H37Ra laboratory strain of M. tuberculosis plated on supplemented 7H11 agar plates. The clones were then subcultured in broth medium, MIC shifts (about 20-fold) were verified in repeated MIC assays, and then cultures were selected for gene amplification using nested PCR primers as follows: primers to amplify gyrB open reading frame (ORF) were (i) gyrB_1 For, 5′-CGACACCTACGGATAACACG-3′ (bases 183 to 202), and (ii) gyrB _2 Rev, 5′-CATCTCCTGCTCGATGTCAA-3′ (bases 2574 to 2556), and internal primers were (iii) gyrB_2 For, 5′-CTTCGCCAACACCATCAAC-3′ (bases 1349 to 1367), and (iv) gyrB_1 Rev, 5′-TTCAGTAGCTTGCGGTCCTT-3′ (bases 1456 to 1437). The same primer set was used to amplify the gyrB gene of the XDR TB clinical isolate TT149. The PCR was carried out for 35 cycles with denaturation at 95°C for 15 min, annealing at 52°C for 1 min, and extension with Pyrobest DNA polymerase (TaKaRa, Otsu, Japan) at 72°C for 1.5 min. The PCR products were gel purified using a gel extraction kit (Tianjen, Beijing, China) and sequenced using standard automated methods to determine the genetic variants. The variants and corresponding MICs are detailed in Table S3 in the supplemental material.
Murine M. tuberculosis infection model.
Six-week old female BALB/c mice were purchased from Charles River Laboratories (Wilmington, DE), and C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). All animal procedures were approved by the Institutional Animal Care and Use Committee (Johns Hopkins University) or the Subcommittee for Animal Studies (SAS; Central New York Research Corporation). Aerosol and intranasal infections were performed as previously described (22, 23). Mycobacterial stocks were stored frozen at −70°C until use, and on the day of infection, the culture was thawed and sonicated. The final inoculum size was determined by titration in triplicate on 7H10 agar plates (BD Diagnostic Systems, Sparks, MD) supplemented with 10% OADC enrichment. An early control (EC) group was euthanized at the initiation of therapy to determine the infection load, and a late control (LC) group was used to determine the infection load at the end of the experiment and to confirm virulence.
A previously described paucibacillary model of latent tuberculosis infection in which BALB/c mice are immunized with the Mycobacterium bovis bacillus Calmette-Guérin (BCG) vaccine (Pasteur strain) (24) was used, with the BCG being a recombinant strain that overexpresses the 30-kDa major secretory protein (rBCG30) (25) and with minor modifications to the model (26). Quantitative cultures of lung homogenates were performed in parallel on selective agar plates (7H11 supplemented with 10% OADC), with 2-thiophenecarboxylic acid hydrazide (TCH) (40 μg/ml; Sigma, St. Louis, MO) used to select for M. tuberculosis or hygromycin (40 μg/ml; Roche Diagnostics, Indianapolis, IN) to select for rBCG30.
Chemotherapy.
Mice were block randomized by run to experimental arms of each study. Three weeks postinfection or as indicated otherwise, treatment was typically administered by oral gavage in a 0.2-ml volume 5 times per week for 4 weeks at the following dosages: VXc-486 and pVXc-486, 100 mg/kg of body weight in all experiments unless indicated otherwise; moxifloxacin (MXF), 100 mg/kg; rifampin (RIF) or rifapentine (RPT), 10 mg/kg; isoniazid (INH), 10 or 25 mg/kg; pyrazinamide (PZA), 150 mg/kg; bedaquiline (BDQ), 25 mg/kg; linezolid (LZD), 50 mg/kg; PA-824, 50 mg/kg; clofazimine (CFZ), 20 mg/kg; ethambutol (EMB), 100 mg/kg; and amikacin (AMK), 30 mg/kg. For dose administration, VXc-486 was dissolved in vitamin E TPGS (d-alpha tocopheryl polyethylene glycol 1000 succinate), while the VXc-486 prodrug and all other single-agent compounds administered individually were dissolved in 0.5% carboxymethylcellulose (CMC) in distilled water. In three-compound-combination studies, VXc-486 was dissolved with the other compounds in 50% polyethylene glycol 400, while the INH, RIF, and PZA combination was dissolved in 20% DMSO and BDQ was dissolved in acidified 20% hydroxyl-propyl beta cyclodextrin. At the end of each experiment, mice were euthanized by CO2 inhalation, the lungs were aseptically removed and homogenized, and the numbers of viable organisms were determined by serial dilution and titration on 7H10 agar plates. All animal studies were carried out in accordance with the approval of the Institutional Animal Care and Use Committee and according to the Guide for Care and Use of Laboratory Animals.
Statistical evaluation.
CFU counts were log-transformed before analysis. Group means were compared by one-way analysis of variance with Dunnett's post hoc test (experimental groups versus each control group). Group proportions were compared using Fisher's exact test, adjusted for multiple comparisons. All analyses were performed with GraphPad Prism version 4.01 (GraphPad, San Diego, CA).
Pharmacokinetic studies.
In studies to determine oral and lung exposure, VXc-486 and pVXc-486 were administered by oral gavage to uninfected or M. tuberculosis-infected (Erdman) mice. Whole blood was sampled by retroorbital bleeding (three mice per time point) at 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h after dosing, and plasma was obtained following centrifugation of blood samples at 3,000 × g for 2 min. Lung homogenates were prepared from M. tuberculosis-infected mice at 1 and 6 h at the termination of the 4-week treatment study. Plasma samples were then stored at −20°C in sealed cluster tubes (product number 4413; Costar), and the samples were extracted using acetonitrile (4:1) containing an analytical internal standard. The tubes were mixed thoroughly for 5 min and centrifuged at 3,000 × g for 20 min, and the supernatants were transferred to fresh plates for quantitative liquid chromatography-mass spectrometry (LC-MS). Calibration standards (1 to 5,000 ng/ml) and quality controls (QCs) (20 to 2,000 ng/ml) of VXc-486 were prepared in a plasma matrix and extracted as described above. Samples, standards, and QCs were analyzed using an Agilent 1100 LC system with a Waters Xterra C18 column followed by MS analysis using a SCIEX API 4000 (Applied Biosystems) mass spectrometer in electrospray ionization (ESI) mode and multiple-reaction-monitoring (MRM) scan mode. The standards, QCs, and samples were quantified relative to the results for the analytical internal standard. Pharmacokinetic parameters were determined by noncompartmental analysis of the plasma concentration data using WinNonlin software (Pharsight Corp., Mountain View, CA).
RESULTS
Growth inhibition of M. tuberculosis in vitro.
VXc-486 was observed to have potent antimycobacterial activity against genetically diverse strains of DS and DR M. tuberculosis, with MICs ranging from 0.03 to 0.25 μg/ml (Table 1). The mycobacterial cell potency as measured by MIC was consistent with the level of inhibition of M. tuberculosis gyrase B enzyme using a cell-free assay with the purified enzyme (50% inhibitory concentration [IC50] of <0.16 μg/ml and Ki of <0.39 μg/ml); the potency in this enzymatic assay reached the lower limit of biochemical detection. In a biochemical assay for human topoisomerase II using the alpha isoform, VXc-486 had negligible activity, indicating that inhibition of this host enzyme is highly unlikely. In addition, VXc-486 did not have potency against 66 mammalian kinases in enzymatic assays (data not shown). No cross-resistance to VXc-486 was observed in H37Rv laboratory strains that are resistant to fluoroquinolones (e.g., BK35 and BK49) (Table 1). Furthermore, clinical isolates that were defined as multidrug-resistant (MDR) and extensively drug-resistant (XDR; resistant to fluoroquinolones) (genetic profiles are summarized in Table S1 in the supplemental material) were also susceptible to VXc-486, confirming the activity of the compound against drug-resistant and clinically relevant M. tuberculosis isolates (Table 2).
TABLE 1.
Antimicrobial activity of VXc-486 against M. tuberculosis laboratory strains and clinical isolatesa
| Strain | Phenotype | MIC or MIC range (μg/ml) of: |
|
|---|---|---|---|
| VXc-486 | Moxifloxacin | ||
| Erdman | DS | 0.06–0.125 | 0.015–0.03 |
| H37Rv | DS | 0.12–0.25 | 0.06 |
| CDC1551 | DS | 0.015 | 0.03 |
| HN878 (Beijing type) | DS | 0.015 | 0.015–0.03 |
| GN9 | DS | 0.03 | 0.015–0.03 |
| hh9 | DS | 0.008–0.015 | 0.015 |
| H13571 | DS | 0.03–0.06 | 0.03 |
| MC19062 | DS | 0.06–0.125 | 0.015–0.03 |
| H10460 | MDR | 0.25 | 0.5 |
| Wg565 | MDR | 0.008–0.015 | 1.00 |
| LL | MDR | 0.015 | 0.008–0.15 |
| W10 | MDR | 0.125 | 0.03 |
| 210 | MDR | 0.03–0.06 | 0.015 |
| W33 | MDR | 0.015–0.06 | 0.5 |
| C913 | MDR | 0.06 | 0.03 |
| AH517 | MDR | 0.06 | 0.03 |
| BW9 | MDR | 0.06–0.125 | 0.008 |
| AH13 | MDR | 0.06 | 0.125–0.25 |
| Lvxr variants | |||
| 2J | FQ resistant | 0.03 | 1.0 |
| 2D | FQ resistant | 0.015 | 2.0 |
| 8D | FQ resistant | 0.125 | 4–8 |
| Lvxr Gatr variants | |||
| 2I1 | FQ resistant | 0.125–0.25 | 16 |
| 8D2 | FQ resistant | 0.03 | 8–16 |
| 2C | FQ resistant | 0.06 | 8.0 |
| 2H2 | FQ resistant | 0.125 | 1.0 |
| BK35 | FQ resistant | 0.03 | 0.25–0.5 |
| BK49 | FQ resistant | 0.03 | 2.0 |
| XDR 5 | XDR | 0.125 | 4–8 |
Drug-sensitive (DS), multidrug-resistant (MDR; resistant to INH and RIF), and extensively drug-resistant (XDR) isolates were evaluated. M. tuberculosis Lvxr 2D, 2J, and 8D isolates were selected by plating M. tuberculosis Erdman on 7H10 agar containing levofloxacin (LVX). LVX and gatifloxacin (GAT) originally had MICs of 0.06 μg/ml. After selection, the MIC for LVX was 4 μg/ml and that of GAT was 1 μg/ml for the Lvxr 2D isolate. M. tuberculosis Lvxr 2D was subsequently cultured on 7H10 agar containing GAT to further enhance its quinolone resistance, and the following fluoroquinolone (FQ)-resistant isolates were recovered: 2I1, 2C, 8D2, and 2H2. The M. tuberculosis 2C isolate's MIC for LVX was 2 μg/ml, and its MIC for GAT was 8 μg/ml.
TABLE 2.
Antimicrobial activity of VXc-486 against drug-resistant clinical isolates of M. tuberculosis from South Africaa
| Isolate | Genotype | Susceptibility status | Drug resistance profile | MIC (μg/ml) of VXc-486 |
|---|---|---|---|---|
| TT135 | Atypical Beijing | XDR | H, R, E, Z, ET, S, A, C, O, M, PAS | 0.08 |
| R179 | Atypical Beijing | MDR | H, R, E, ET, S | 0.08 |
| R884 | Low copy number | Pre-XDR | H, R, E, Z, ET, S, O | 0.08 |
| X_3 | Beijing | XDR | H, R, E, K, S, C, O | 0.68 |
| X_27 | Beijing | XDR | H, R, ET, O, A | 0.68 |
| X_60 | Beijing | XDR | H, R, E, A, ET, O, S, C, K | 0.68 |
| X_61 | Beijing | XDR | H, R, ET, A, O, K, S | 0.68 |
| X_131 | F11 | Pre-XDR | H, R, K, S, O | 0.17 |
| TT149 | Atypical Beijing | XDR | H, R, ET, S, A, C, O, M | 5.48 |
Mycobacterial growth was measured by using mycobacterial growth indicator tubes (MGIT), and the MIC (μg/ml) of VXc-486 was determined. The growth units were recorded once the 1:100 controls reached a GU (gas unit) of 400, and the MIC is regarded as a concentration that gives a GU of 0. The isolates grew well in the presence of isoniazid, confirming resistance to this drug. Abbreviations: H, isoniazid; R, rifampin; E, ethionamide; ET, ethambutol; S, streptomycin; A, amikacin; C, capreomycin; O, ofloxacin; M, moxifloxacin; PAS, para-aminosalicylic acid; Z, pyrazinamide.
The bactericidal activity of VXc-486 was also examined in vitro against actively growing cells of M. tuberculosis strain Erdman (MIC of 0.06 μg/ml). VXc-486 achieved the threshold for bactericidal activity (≥3 log reduction of the CFU) after 14 days, and a clear trend toward slow killing of M. tuberculosis was observed (Fig. 1A). As a comparator compound, MXF (MIC of 0.03 μg/ml) was not as bactericidal as VXc-486 but also achieved killing at 3 log CFU after 14 days. Specifically, the mean log CFU value for VXc-486 was 3.991, while for MXF it was 4.885, after 14 days.
FIG 1.

(A) Bactericidal activities of VXc-486 and moxifloxacin (MXF) against Mycobacterium tuberculosis (Erdman strain). Representative results from two independent experiments cultured in triplicate are shown. Both VXc-486 and MXF were used at 4-fold MICs; VXc-486 was used at a concentration of 0.24 μg/ml and MXF was used at a concentration of 0.12 μg/ml. (B) Bactericidal activities of VXc-486, isoniazid (INH), and moxifloxacin (MXF) against Mycobacterium kansasii (strain 0008).
VXc-486 has potent activity against DS M. tuberculosis in an in vitro model system of quiescent bacterial survival, the low-oxygen-recovery assay (LORA; using M. tuberculosis strain H37Rv). In this model, the IC50 of VXc-486 was 0.26 μg/ml, and it was more active than the fluoroquinolones MXF and GAT, which had LORA IC50s of 1.60 μg/ml and 8.81 μg/ml, respectively (Table 3 and as previously reported [17]).
TABLE 3.
VXc-486 inhibited M. tuberculosis replication in macrophages and in a low-oxygen-recovery assaya
| Compound | IC50 (μg/ml) for indicated isolate in macrophages |
IC50 (μg/ml) for H37Rv in LORA | |
|---|---|---|---|
| H37Ra | CDC1551 | ||
| VXc-486 | 0.012 | 0.115 | 0.252 |
| RIF | 0.008 | 0.040 | 0.160 |
| MXF | 0.120 | 0.216 | 1.600 |
| GAT | 0.530 | 2.500 | 8.807 |
Rifampin (RIF), moxifloxacin (MXF), and gatifloxacin (GAT) were used as comparator compounds. The low-oxygen-recovery assay (LORA) was carried out in broth culture without macrophages. The reporter genes used encoded luciferase (for H37Ra), mCherry (for CDC1551), or green fluorescent protein (for H37Rv).
VXc-486 is also active against M. tuberculosis growing within macrophages. The compound inhibited the growth of strain H37Ra cultured in the human macrophage-like cell line THP-1 (IC50 of 0.013 μg/ml and IC90 of 0.3 μg/ml), as well as that of the CDC1551 strain cultured in the mouse macrophage-like cell line J774 (IC50 of 0.15 μg/ml). To assess drug combinations in vitro, a checkerboard MIC assay was used to determine the FIC. We found borderline drug synergy (FIC of 0.5) with linezolid in broth culture and with bedaquiline (BDQ) and clofazimine (CFZ) in M. tuberculosis-infected THP-1 cells (see Table S2 in the supplemental material). The remaining antimycobacterial combinations evaluated with VXc-486 showed an additive effect, and none of the combinations tested showed an antagonistic effect in these assays.
To identify the amino acid residues important in determining target-based resistance to VXc-486, resistant clones were selected in the presence of VXc-486 (2× and 4× MIC) for more than 1 month on agar plates. Four resistant variant clones were confirmed by MIC testing; the colonies were expanded, and genomic DNA for the entire gyrB gene was amplified for gene sequencing. Similar to the results of a previous report that evaluated benzimidazoles against M. tuberculosis (27), we also found that the A92S variant was associated with up to a 20-fold MIC shift (see Fig. S1a in the supplemental material). In addition, we found an S208A variant in one of the resistant clones that had a similar shift in its MIC. In order to understand the impact of this mutation, we performed docking studies using the published crystal structure of M. tuberculosis gyrase B to generate a 3-dimensional model of VXc-486 in complex with the target. The predicted binding mode resulting from these studies was consistent with the experimental binding modes reported for closely related compounds in complex with the highly homologous gyrase B from Staphylococcus aureus (13). The model suggests that the S208A mutation negatively affects the binding of VXc-486 to M. tuberculosis gyrase B by altering the hydrogen bonding network and destabilizing the catalytic water present in the active site (see Fig. S1b).
Growth inhibition of NTM in vitro.
Nontuberculosis mycobacteria (NTM: Mycobacterium abscessus, M. avium, and M. kansasii) and Nocardia spp. are responsible for respiratory infections, localized abscesses, and disseminated diseases in immunocompromised individuals and cystic fibrosis patients. Infections caused by these organisms are difficult to treat because of naturally occurring poor responsiveness to classic antituberculosis drugs and, also, to most other antibiotics. The potencies of VXc-486 and MXF were tested against multiple isolates of M. abscessus and M. kansasii. We found that VXc-486 potently inhibited multiple strains of M. abscessus (MIC50, 1.0 μg/ml, and MIC90, 4.0 μg/ml) (see Table S3 in the supplemental material) and M. kansasii (MIC50, 0.06 μg/ml, and MIC90, 0.5 μg/ml, for VXc-486 and MXF) (see Table S4), while the potency for Mycobacterium smegmatis was substantially less (MIC, 2.4 μg/ml). The activity against M. kansasii was bactericidal and comparable to that of MXF (Fig. 1B). We also tested the in vitro activity of VXc-486 against small subsets of M. avium (MICs of 0.12 to 0.23 μg/ml) and Nocardia (MICs of 0.1 to 1.0 μg/ml) isolates, demonstrating that VXc-486 had significant inhibitory effects against these NTMs as well (see Table S5).
Growth inhibition of M. tuberculosis in vivo.
We next sought to determine if VXc-486 could provide a protective effect in a low-dose, chronic tuberculosis model in mice. pVXc-486, which has improved solubility and is rapidly converted to VXc-486 after oral administration (see Table S6 in the supplemental material), was directly compared to VXc-486 in a dose-response study in vivo. We found that pVXc-486 had dose-dependent bactericidal activity that was more potent than that of VXc-486 and comparable to that of MXF (Fig. 2). Compared with the initial mycobacterial burden prior to treatment (early control [EC]), the median mycobacterial lung burdens after 27 days of treatment with the two higher doses (30 and 100 mg/kg) of VXc-486 and pVXc-486 were significantly reduced (at least −1 log10 difference versus the mycobacterial lung burden in the EC group of mice), indicating that VXc-486 and pVXc-486 had potent bactericidal activities in vivo. We also found substantially higher levels of VXc-486 in the lungs of infected mice after the administration of pVXc-486 than after dosing with the parent compound, suggesting that the improved protection may be due to improved exposure at the site of TB infection in the treated mice (Fig. 3).
FIG 2.
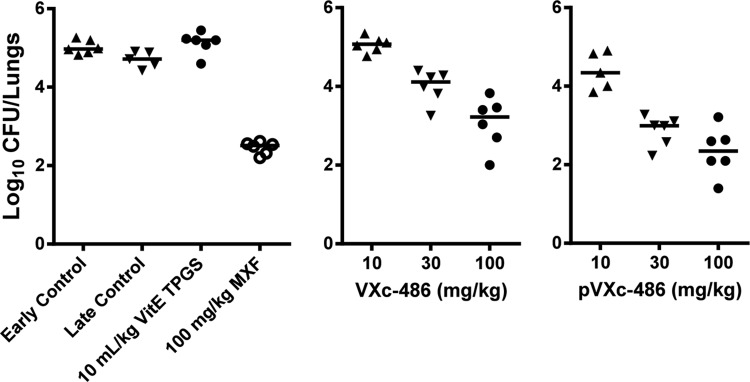
VXc-486 and pVXc-486 reduced the mycobacterial burden in a model of chronic tuberculosis infection in mice (Erdman isolate). The potencies of VXc-486 and pVXc-486, which target gyrase B, were compared with that of moxifloxacin (MXF), which targets gyrase A. VXc-486 and pVXc-486 were administered twice a day, while MXF was administered once a day, by oral gavage for 4 weeks at the doses indicated. Compared to the results for the early and late control groups, VXc-486 at 100 mg/kg, pVXc-486 at 30 and 100 mg/kg, and MXF at 100 mg/kg reduced the mycobacterial burdens by statistically significant amounts (P < 0.05). Mycobacterial burdens were measured by CFU counts from lung homogenates of M. tuberculosis-infected mice at the end of 4 weeks of treatment. Horizontal bars show mean results.
FIG 3.
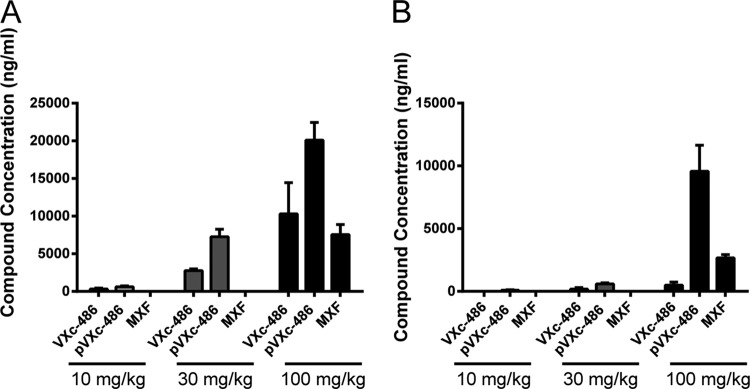
pVXc-486 had increased exposure in the lungs of infected mice compared to the levels of the parent (mesylate salt) compound VXc-486 and moxifloxacin (MXF). The dose administered is noted at the bottom of each group. Samples were collected 1 h (A) and 6 h (B) after administration of the compounds at the termination of the 4-week treatment model (described in the legend to Fig. 2), and lung concentrations were determined. Error bars show standard deviations.
In vivo activity of pVXc-486 in combination with known antimycobacterial agents.
Since tuberculosis is treated with drug combinations and new combinations are needed to treat drug-resistant strains, we sought to determine if pVXc-486 could be used in combination with known TB drugs to sterilize an M. tuberculosis infection in mice. The incorporation of pVXc-486 was evaluated with standard-of-care (SOC) drugs in first- and second-line regimens for DS and DR TB, respectively. When added to the first-line regimen of rifampin (RIF), isoniazid (INH), and pyrazinamide (PZA) or when substituted for INH, pVXc-486 significantly increased the activity of the combination (P < 0.001) over the first 2 months of treatment (Fig. 4). The proportions of mice relapsing after 4 months of treatment were 1 of 15 mice treated with 2 months of RIF-INH-PZA followed by 2 months of RIF-INH, 2 of 15 mice treated with 2 months of RIF–INH–PZA–pVXc-486 followed by 2 months of RIF–INH–pVXc-486, and 1 of 15 mice treated with 2 months of RIF–PZA–pVXc-486 followed by 2 months of RIF–pVXc-486. No relapse occurred in any of these 3 groups after 5 months of treatment (data not shown). In the second-line regimen, the substitution of pVXc-486 for amikacin (AMK) or ethambutol (EMB) also increased the bactericidal activities of MXF, EMB, PZA, and AMK during the initial phase of therapy (P < 0.001), but neither substitution promoted greater sterilizing activity during the relapse phase after PZA was discontinued (see Table S7 in the supplemental material).
FIG 4.

pVXc-486 improved the bactericidal activities of antimycobacterial drugs. Mycobacterial burdens were measured by CFU counts from lung homogenates of M. tuberculosis-infected mice (H37Rv) and were assessed at the end of 8 weeks of treatment. Each compound was administered by the oral route once per day, 5 days per week, for 8 weeks at the following doses: pVXc-486, 100 mg/kg; isoniazid (INH), 10 mg/kg; rifampin (RIF), 10 mg/kg; pyrazinamide (PZA), 150 mg/kg; moxifloxacin (MXF), 100 mg/kg; ethambutol (EMB), 100 mg/kg; and amikacin (AMK), 30 mg/kg. Horizontal bars show mean results.
In another experiment evaluating two-compound combinations with pVXc-486, we found that pVXc-486 was able to replace MXF in combination with RFP or BDQ without loss of efficacy (Fig. 5A). There was only one relapse in the pVXc-486–RFP combination group (1/6), while there were three relapses in the MXF-RFP combination group (3/6). In addition, there were two relapses in the pVXc-486–BDQ group (2/6) compared to one relapse in the MXF-BDQ combination group (1/6). When each of these four groups are compared with each other, the results do not reach statistical significance; larger groups of mice would be needed to achieve statistically significant results that quantify the in vivo performance of pVXc-486 compared to that of MXF in these two-drug combinations. However, based on the results of this study, pVXc-486 and MXF seem to contribute similar activities to these combinations in vivo.
FIG 5.

pVXc-486 was administered to M. tuberculosis-infected mice (100 mg/kg, 5 days per week) in combination with one or two other antimycobacterial drugs at the following doses: isoniazid (INH), 10 mg/kg; rifampin (RIF) or rifapentine (RFP), 10 mg/kg; pyrazinamide (PZA), 150 mg/kg; moxifloxacin (MXF), 100 mg/kg; bedaquiline (BDQ), 25 mg/kg; linezolid (LZD), 50 mg/kg; PA-824, 50 mg/kg; and clofazimine (CFZ), 20 mg/kg. Mycobacterial burdens were measured by CFU counts from lung homogenates of M. tuberculosis-infected mice at the end of the first 8 weeks of treatment (treatment phase) and after an additional 8 weeks without treatment (observation phase). Each compound was administered by oral gavage once per day, 5 days per week, for 8 weeks. (A) Two-drug combinations were used with pVXc-486, and the potencies were compared directly to that of MXF. (B and C) Three-drug combinations in regimens containing PZA were used in two separate experiments. Horizontal bars show mean results.
In a separate experiment, M. tuberculosis bacilli were not recovered from lungs of mice that were treated in a three-compound combination of pVXc-486 with the oxazolidinone linezolid (LZD) and PZA, PA-824 and PZA, or MXF and rifapentine (RFP). However, bacilli were recovered from lungs of mice treated with pVXc-486 combined with PZA and MXF or PZA and CFZ (Fig. 5B). The results from the observation phase (8 weeks posttreatment) showed similar trends, although these findings did not reach statistical significance because of experimental challenges. In a separate study, we found that pVXc-486 sterilized the infection in this mouse model when used in a three-compound combination with PZA and RFP or BDQ. Similarly, MXF also sterilized the infection in mice when used in three-compound combinations with PZA and RFP or BDQ (Fig. 5C).
pVXc-486 in a murine latent TB chemotherapy model.
To compare the potency of pVXc-486 (100 mg/kg) monotherapy to monotherapy with INH and RIF in a model of chemotherapy of latent TB infection, mice were first immunized with M. bovis (BCG vaccine strain) and then challenged with M. tuberculosis (H37Rv strain) to establish a stable paucibacillary infection. After both 1 and 2 months of treatment, all monotherapy regimens were ostensibly superior to no treatment (late control) (P < 0.01 for pVXc-486 and INH and P < 0.001 for RIF), and pVXc-486 demonstrated activity comparable to that of INH in this model (Fig. 6). However, despite a trend toward improved activity with the addition of pVXc-486 to RIF, no statistically significant benefit of combining pVXc-486 with either INH or RIF was demonstrated.
FIG 6.
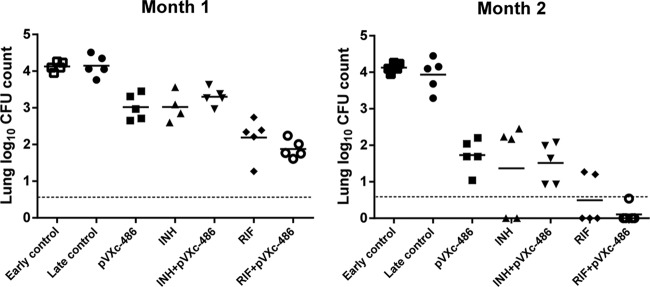
pVXc-486 has activity comparable to that of isoniazid (INH) when used as a single agent and renders mice culture negative in combination with rifampin (RIF) in a mouse model of latent tuberculosis (TB) chemotherapy. BALB/c mice were immunized with Mycobacterium bovis bacillus Calmette-Guérin (BCG) 5 weeks prior to being infected with M. tuberculosis (H37Rv). Mice were then treated 5 weeks later for one or two months with pVXc-486 and control drugs. The M. tuberculosis H37Rv lung log10 CFU counts were 4.13 ± 0.13 (mean ± SD) at the start of drug treatment (early control). The dotted lines represent the lower limit of detection of the assay. Horizontal bars show mean results.
DISCUSSION
Gyrase B is an essential and underexploited enzyme that is an important drug target for eliminating DS and DR bacterial infections. VXc-486 is highly active against M. tuberculosis (0.05 μg/ml to 0.5 μg/ml for most isolates tested) under various mycobacterial culture conditions, including in broth, in macrophages, and in a model of M. tuberculosis dormancy. The potent antimycobacterial activity of VXc-486 against genetically diverse and MDR or XDR M. tuberculosis and NTM strains suggests that this compound has potential utility in the treatment of DR TB (Tables 1 and 2) and NTM (see Tables S3 to S5 in the supplemental material). One isolate, TT149 from South Africa, was intrinsically resistant, likely either by decreased compound permeability or increased compound efflux, since genetic sequencing of this isolate showed no gyrB mutations or variations (Table 2). Importantly, the MXF-resistant isolates of M. tuberculosis and M. kansasii were susceptible to VXc-486, indicating that fluoroquinolone-resistant strains will remain susceptible to antimicrobials targeting gyrase B (see Table S4). The potency observed in the M. tuberculosis gyrase enzyme biochemical assay is consistent with an antibacterial mechanism where DNA replication is inhibited. While the M. tuberculosis killing kinetics of VXc-486 in vitro appears to be relatively slow (Fig. 1), the observed potent activity against quiescent bacteria in the low-oxygen model of dormant M. tuberculosis may also distinguish the aminobenzimidazole series from the fluoroquinolone series (Table 3).
The frequency of resistance to VXc-486 was about 10−8, indicating that a single enzyme is targeted, similar to fluoroquinolones, which target M. tuberculosis gyrase A. Among the VXc-486-resistant clones, we found A92S and S208A variants, which been reported previously by other investigators researching compounds targeting M. tuberculosis gyrase B (see Fig. S1 in the supplemental material). The A92S variant identified in M. tuberculosis was consistent with the results of another study describing aminobenzimidazole GyrB inhibitors (27). The A92S variant introduces a hydroxyl group into the ATP binding site and would predictably cause a conformational change but would not completely obliterate the interaction of VXc-486 with the GyrB target enzyme, which is consistent with a 10- to 20-fold MIC shift. Although M. abscessus possesses a natural A92S mutation in its gyrB gene (27), we found that the potency against this NTM and other NTMs in the MIC assay was greater than that of MXF. The S208A variant is consistent with the results of a study describing the pyrroloamides targeting gyrase B of M. tuberculosis (28). Ser 208 is located in proximity to the urea of VXc-486 in the model of the inhibitor bound to the active site of gyrase B. It does not make a hydrogen bond to the compound, but it makes one to the catalytic water, which in turn is hydrogen bonded to the inhibitor. The switch from serine to alanine alters the hydrogen bonding network and destabilizes the catalytic water.
VXc-486 and its phosphate prodrug, pVXc-486, also protected mice in a low-dose, chronic tuberculosis infection model using the M. tuberculosis Erdman strain, and the administration of pVXc-486 demonstrated activity similar to that of MXF in a 100-mg/kg, once-daily dosing regimen. At a dose of 30 mg/kg, pVXc-486 was superior to VXc-486 (Fig. 2). Previously, several other Vertex aminobenzimidazoles targeting gyrase B were evaluated for their antimycobacterial tuberculosis activities in the high-dose acute infection model using the Erdman strain but did not provide protection to statistically significant levels (unpublished studies). The difference may be due to the slow mycobacterial killing kinetics of VXc-486 and other, similar compounds of the aminobenzimidazole class. These studies highlight the importance of using a chronic infection model rather than an acute infection model to evaluate this compound series. The slow bacterial killing kinetics of this compound series was also noted for M. tuberculosis (Fig. 1) and other bacteria in time-kill experiments in vitro (unpublished data). The acute infection model is most appropriate for compounds with rapid mycobacterial killing kinetics as single agents, such as INH, MXF, or PA-824. Although the acute infection model can provide a relatively rapid experimental result (about 2 months, depending on the length of time for treatment) compared to the time for the low-dose, chronic model (between 3 and 4 months), the acute model is not ideal for compounds with slower mycobacterial killing kinetics, such as PZA (29).
The exposure to VXc-486 at the doses used was adequate to enable pharmacology studies in mouse models of infection. Both the extent and rate of conversion of pVXc-486 were high, and there was no observable exposure to pVXc-486 following oral administration of the prodrug in mice (see Table S6 in the supplemental material). The area under the concentration-time curve (AUC) of VXc-486 increased supraproportional to doses from 3 to 100 mg/kg, and the maximum concentration (Cmax) of VXc-486 increased proportional to doses from 3 to 100 mg/kg. The mechanisms of conversion of pVXc-486 to VXc-486 are not fully understood; however, the results of preliminary studies suggest that bioconversion is likely to occur in the intestinal lumen via epithelial/luminal phosphatases following active uptake (O'Dowd et al., unpublished data).
When pVXc-486 was added to a RIF-INH-PZA combination in a mouse model of DS SOC agents, we found that it increased the bactericidal activity of the regimen (Fig. 4). In addition, the substitution of pVXc-486 for INH increased the bactericidal activity of RIF-PZA, indicating that there was an antagonistic effect of INH on the RIF-PZA combination (with and without pVXc-486). A treatment-shortening effect was not observed in this experiment, since there was a lower than expected relapse rate after RIF-INH-PZA followed by RIF-INH. Similarly, in a mouse model of DR SOC agents, substitution of pVXc-486 for AMK or EMB increased the bactericidal activity of the initial phase of therapy, but neither substitution promoted greater sterilizing activity during the continuation phase, after PZA was discontinued (see Table S7 in the supplemental material).
Since we saw encouraging sterilization results with pVXc-486 combined with LZD and PZA in a three-drug combination study (Fig. 5B) but not when PZA was omitted from the drug combination (Fig. 5A), it may be that another oxazolidinone, such as sutezolid (PNU-100480) or AZD5847, would be a better substitute, since they may be more potent and/or better tolerated than LZD (30–33). One could envision a three-drug combination consisting of pVXc-486 with BDQ or RFP combined with an oxazolidinone as a new combination that may provide a sterilizing effect in the mouse model, and the former combination should be effective against DR strains as well.
In the mouse model of the chemotherapy of latent TB infection (Fig. 6), we found that pVXc-486 was at least as effective as INH and that its activity approached that of RIF. Furthermore, pVXc-486 had an additive activity when it was combined with INH or RIF.
In summary, the in vivo data demonstrate the effectiveness and potency of VXc-486 administered as a phosphate prodrug, pVXc-486. Importantly, pVXc-486 performed well in combination with RFP, BDQ, LZD, and PZA and improved the bactericidal activities of combination regimens in models of DS and DR TB treatment in vivo. pVXc-486 could potentially be used in place of MXF, especially against DR variants and XDR TB. The results from these studies suggest that further development of pVXc-486 is warranted.
Supplementary Material
ACKNOWLEDGMENTS
The team thanks Dave Pais, Doug McConnell, Suzanne Stokes, Hardwin O'Dowd, Tiansheng Wang, and Michael Badia of Vertex Pharmaceuticals Incorporated for project support. We also thank Yuzhou Xu and Jie Wang of Shanghai ChemPartner and Scott Franzblau and Baojie Wang of the University of Illinois Chicago for evaluations of compounds in vitro, David Russell and Brian VanderVen of Cornell University for mycobacteriology assay support, and Michelle DeStefano, Leigha DeStefano, Maria Ackerman, and Mary Sklaney of the Central New York Research Corporation for in vivo and in vitro work.
Funding for this project was provided by Vertex Pharmaceuticals Incorporated.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04347-14.
REFERENCES
- 1.World Health Organization. 2014. Global tuberculosis report 2014. WHO, Geneva, Switzerland: http://www.who.int/tb/publications/global_report/en/. [Google Scholar]
- 2.Woodcock JM, Andrews JM, Boswell FJ, Brenwald NP, Wise R. 1997. In vitro activity of BAY 12-8039, a new fluoroquinolone. Antimicrob Agents Chemother 41:101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mdluli K, Ma Z. 2007. Mycobacterium tuberculosis DNA gyrase as a target for drug discovery. Infect Disord Drug Targets 7:159–168. doi: 10.2174/187152607781001763. [DOI] [PubMed] [Google Scholar]
- 4.Kale MG, Raichurkar A, Hameed S, Waterson D, McKinney D, Manjunatha MR, Kranthi U, Koushik K, Jena L, Shinde V, Rudrapatna S, Barde S, Humnabadkar V, Madhavapeddi P, Basavarajappa H, Ghosh A, Ramya VK, Guptha S, Sharma S, Vachaspati P, Kumar KN, Giridhar J, Reddy J, Panduga V, Ganguly S, Ahuja V, Gaonkar S, Kumar CN, Ogg D, Tucker JA, Boriack-Sjodin PA, de Sousa SM, Sambandamurthy VK, Ghorpade SR. 2013. Thiazolopyridine ureas as novel antitubercular agents acting through inhibition of DNA Gyrase B. J Med Chem 56:8834–8848. doi: 10.1021/jm401268f. [DOI] [PubMed] [Google Scholar]
- 5.Kale RR, Kale MG, Waterson D, Raichurkar A, Hameed SP, Manjunatha MR, Kishore Reddy BK, Malolanarasimhan K, Shinde V, Koushik K, Jena LK, Menasinakai S, Humnabadkar V, Madhavapeddi P, Basavarajappa H, Sharma S, Nandishaiah R, Mahesh Kumar KN, Ganguly S, Ahuja V, Gaonkar S, Naveen Kumar CN, Ogg D, Boriack-Sjodin PA, Sambandamurthy VK, de Sousa SM, Ghorpade SR. 2014. Thiazolopyridone ureas as DNA gyrase B inhibitors: optimization of antitubercular activity and efficacy. Bioorg Med Chem Lett 24:870–879. doi: 10.1016/j.bmcl.2013.12.080. [DOI] [PubMed] [Google Scholar]
- 6.Shirude PS, Madhavapeddi P, Tucker JA, Murugan K, Patil V, Basavarajappa H, Raichurkar AV, Humnabadkar V, Hussein S, Sharma S, Ramya VK, Narayan CB, Balganesh TS, Sambandamurthy VK. 2013. Aminopyrazinamides: novel and specific GyrB inhibitors that kill replicating and nonreplicating Mycobacterium tuberculosis. ACS Chem Biol 8:519–523. doi: 10.1021/cb300510w. [DOI] [PubMed] [Google Scholar]
- 7.Basarab GS, Manchester JI, Bist S, Boriack-Sjodin PA, Dangel B, Illingworth R, Sherer BA, Sriram S, Uria-Nickelsen M, Eakin AE. 2013. Fragment-to-hit-to-lead discovery of a novel pyridylurea scaffold of ATP competitive dual targeting type II topoisomerase inhibiting antibacterial agents. J Med Chem 56:8712–8735. doi: 10.1021/jm401208b. [DOI] [PubMed] [Google Scholar]
- 8.Trzoss M, Bensen DC, Li X, Chen Z, Lam T, Zhang J, Creighton CJ, Cunningham ML, Kwan B, Stidham M, Nelson K, Brown-Driver V, Castellano A, Shaw KJ, Lightstone FC, Wong SE, Nguyen TB, Finn J, Tari LW. 2013. Pyrrolopyrimidine inhibitors of DNA gyrase B (GyrB) and topoisomerase IV (ParE). Part II. Development of inhibitors with broad spectrum, Gram-negative antibacterial activity. Bioorg Med Chem Lett 23:1537–1543. doi: 10.1016/j.bmcl.2012.11.073. [DOI] [PubMed] [Google Scholar]
- 9.Tari LW, Li X, Trzoss M, Bensen DC, Chen Z, Lam T, Zhang J, Lee SJ, Hough G, Phillipson D, Akers-Rodriguez S, Cunningham ML, Kwan BP, Nelson KJ, Castellano A, Locke JB, Brown-Driver V, Murphy TM, Ong VS, Pillar CM, Shinabarger DL, Nix J, Lightstone FC, Wong SE, Nguyen TB, Shaw KJ, Finn J. 2013. Tricyclic GyrB/ParE (TriBE) inhibitors: a new class of broad-spectrum dual-targeting antibacterial agents. PLoS One 8:e84409. doi: 10.1371/journal.pone.0084409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeankumar VU, Renuka J, Kotagiri S, Saxena S, Kakan SS, Sridevi JP, Yellanki S, Kulkarni P, Yogeeswari P, Sriram D. 2014. Gyrase ATPase domain as an antitubercular drug discovery platform: structure-based design and lead optimization of nitrothiazolyl carboxamide analogues. ChemMedChem 9:1850–1859. doi: 10.1002/cmdc.201402035. [DOI] [PubMed] [Google Scholar]
- 11.Charifson PS, Grillot AL, Grossman TH, Parsons JD, Badia M, Bellon S, Deininger DD, Drumm JE, Gross CH, LeTiran A, Liao Y, Mani N, Nicolau DP, Perola E, Ronkin S, Shannon D, Swenson LL, Tang Q, Tessier PR, Tian SK, Trudeau M, Wang T, Wei Y, Zhang H, Stamos D. 2008. Novel dual-targeting benzimidazole urea inhibitors of DNA gyrase and topoisomerase IV possessing potent antibacterial activity: intelligent design and evolution through the judicious use of structure-guided design and structure-activity relationships. J Med Chem 51:5243–5263. doi: 10.1021/jm800318d. [DOI] [PubMed] [Google Scholar]
- 12.Grossman TH, Bartels DJ, Mullin S, Gross CH, Parsons JD, Liao Y, Grillot AL, Stamos D, Olson ER, Charifson PS, Mani N. 2007. Dual targeting of GyrB and ParE by a novel aminobenzimidazole class of antibacterial compounds. Antimicrob Agents Chemother 51:657–666. doi: 10.1128/AAC.00596-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grillot A-L, Le Tiran A, Shannon D, Krueger E, Liao Y, O'Dowd H, Tang Q, Ronkin S, Wang T, Waal N, Li P, Lauffer D, Sizensky E, Tanoury J, Perola E, Grossman TH, Doyle T, Hanzelka B, Jones S, Dixit V, Ewing N, Liao S, Boucher B, Jacobs M, Bennani Y, Charifson PS.. 2014. Second-generation antibacterial benzimidazole ureas: discovery of a preclinical candidate with reduced metabolic liability. J Med Chem 57:8792–8816. doi: 10.1021/jm500563g. [DOI] [PubMed] [Google Scholar]
- 14.Klopper M, Warren RM, Hayes C, Gey van Pittius NC, Streicher EM, Muller B, Sirgel FA, Chabula-Nxiweni M, Hoosain E, Coetzee G, David van Helden P, Victor TC, Trollip AP. 2013. Emergence and spread of extensively and totally drug-resistant tuberculosis, South Africa. Emerg Infect Dis 19:449–455. doi: 10.3201//EID1903.120246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mani N, Gross CH, Parsons JD, Hanzelka B, Muh U, Mullin S, Liao Y, Grillot AL, Stamos D, Charifson PS, Grossman TH. 2006. In vitro characterization of the antibacterial spectrum of novel bacterial type II topoisomerase inhibitors of the aminobenzimidazole class. Antimicrob Agents Chemother 50:1228–1237. doi: 10.1128/AAC.50.4.1228-1237.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franzblau SG, Witzig RS, McLaughlin JC, Torres P, Madico G, Hernandez A, Degnan MT, Cook MB, Quenzer VK, Ferguson RM, Gilman RH. 1998. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J Clin Microbiol 36:362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. 2007. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 51:1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rusch-Gerdes S, Pfyffer GE, Casal M, Chadwick M, Siddiqi S. 2006. Multicenter laboratory validation of the BACTEC MGIT 960 technique for testing susceptibilities of Mycobacterium tuberculosis to classical second-line drugs and newer antimicrobials. J Clin Microbiol 44:688–692. doi: 10.1128/JCM.44.3.688-692.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becton Dickinson and Company. 1999. Bactec MGIT 960 system user's manual, catalog number 445876. Becton Dickinson and Company, Franklin Lakes, NJ. [Google Scholar]
- 20.Chou TC. 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 21.Heifets LB. 1996. Clinical mycobacteriology. Drug susceptibility testing. Clin Lab Med 16:641–656. [PubMed] [Google Scholar]
- 22.Almeida D, Nuermberger E, Tasneen R, Rosenthal I, Tyagi S, Williams K, Peloquin C, Grosset J. 2009. Paradoxical effect of isoniazid on the activity of rifampin-pyrazinamide combination in a mouse model of tuberculosis. Antimicrob Agents Chemother 53:4178–4184. doi: 10.1128/AAC.00830-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shoen CM, DeStefano MS, Sklaney MR, Monica BJ, Slee AM, Cynamon MH. 2004. Short-course treatment regimen to identify potential antituberculous agents in a murine model of tuberculosis. J Antimicrob Chemother 53:641–645. doi: 10.1093/jac/dkh124. [DOI] [PubMed] [Google Scholar]
- 24.Nuermberger EL, Yoshimatsu T, Tyagi S, Bishai WR, Grosset JH. 2004. Paucibacillary tuberculosis in mice after prior aerosol immunization with Mycobacterium bovis BCG. Infect Immun 72:1065–1071. doi: 10.1128/IAI.72.2.1065-1071.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang T, Zhang M, Rosenthal IM, Grosset JH, Nuermberger EL. 2009. Short-course therapy with daily rifapentine in a murine model of latent tuberculosis infection. Am J Resp Crit Care Med 180:1151–1157. doi: 10.1164/rccm.200905-0795OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang T, Li SY, Williams KN, Andries K, Nuermberger EL. 2011. Short-course chemotherapy with TMC207 and rifapentine in a murine model of latent tuberculosis infection. Am J Resp Crit Care Med 184:732–737. doi: 10.1164/rccm.201103-0397OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chopra S, Matsuyama K, Tran T, Malerich JP, Wan B, Franzblau SG, Lun S, Guo H, Maiga MC, Bishai WR, Madrid PB. 2012. Evaluation of gyrase B as a drug target in Mycobacterium tuberculosis. J Antimicrob Chemother 67:415–421. doi: 10.1093/jac/dkr449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hameed S, Solapure S, Mukherjee K, Nandi V, Waterson D, Shandil R, Balganesh M, Sambandamurthy VK, Raichurkar AK, Deshpande A, Ghosh A, Awasthy D, Shanbhag G, Sheikh G, McMiken H, Puttur J, Reddy J, Werngren J, Read J, Kumar M, M R, Chinnapattu M, Madhavapeddi P, Manjrekar P, Basu R, Gaonkar S, Sharma S, Hoffner S, Humnabadkar V, Subbulakshmi V, Panduga V. 2014. Optimization of pyrrolamides as mycobacterial GyrB ATPase inhibitors: structure-activity relationship and in vivo efficacy in a mouse model of tuberculosis. Antimicrob Agents Chemother 58:4993–4994. doi: 10.1128/AAC.03463-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rullas J, Garcia JI, Beltran M, Cardona PJ, Caceres N, Garcia-Bustos JF, Angulo-Barturen I. 2010. Fast standardized therapeutic-efficacy assay for drug discovery against tuberculosis. Antimicrob Agents Chemother 54:2262–2264. doi: 10.1128/AAC.01423-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wallis RS, Dawson R, Friedrich SO, Venter A, Paige D, Zhu T, Silvia A, Gobey J, Ellery C, Zhang Y, Eisenach K, Miller P, Diacon AH. 2014. Mycobactericidal activity of sutezolid (PNU-100480) in sputum (EBA) and blood (WBA) of patients with pulmonary tuberculosis. PLoS One 9:e94462. doi: 10.1371/journal.pone.0094462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw KJ, Barbachyn MR. 2011. The oxazolidinones: past, present, and future. Ann N Y Acad Sci 1241:48–70. doi: 10.1111/j.1749-6632.2011.06330.x. [DOI] [PubMed] [Google Scholar]
- 32.Balasubramanian V, Solapure S, Iyer H, Ghosh A, Sharma S, Kaur P, Deepthi R, Subbulakshmi V, Ramya V, Ramachandran V, Balganesh M, Wright L, Melnick D, Butler SL, Sambandamurthy VK. 2014. Bactericidal activity and mechanism of action of AZD5847, a novel oxazolidinone for treatment of tuberculosis. Antimicrob Agents Chemother 58:495–502. doi: 10.1128/AAC.01903-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Werngren J, Wijkander M, Perskvist N, Balasubramanian V, Sambandamurthy VK, Rodrigues C, Hoffner S. 2014. In vitro activity of AZD5847 against geographically diverse clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:4222–4223. doi: 10.1128/AAC.02718-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.